Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
MicroRNA Let-7 Induces M2 Macrophage Polarization in COPD Emphysema Through the IL-6/STAT3 Pathway
Authors Liu T, Zhang Z ![]() , Shen W, Wu Y, Bian T
, Shen W, Wu Y, Bian T
Received 14 January 2023
Accepted for publication 6 April 2023
Published 13 April 2023 Volume 2023:18 Pages 575—591
DOI https://doi.org/10.2147/COPD.S404850
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Tingting Liu, Zheming Zhang, Weiyu Shen, Yan Wu, Tao Bian
Department of Respiratory Medicine, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, 214023, People’s Republic of China
Correspondence: Tao Bian; Yan Wu, Department of Respiratory Medicine, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, 214023, People’s Republic of China, Email [email protected]; [email protected]
Background: M2 polarized macrophages are involved in the occurrence and development of emphysema in COPD patients. However, the molecular mechanism of M2 macrophage polarization is still unclear. This study investigated the molecular mechanism of let-7 differentially expressed in bronchial epithelial cells of COPD patients participating in COPD emphysema by regulating the expression of IL-6 and inducing M2 polarization of alveolar macrophages (AM).
Materials and Methods: We measured let-7c expression in human lung tissue, serum and the lung tissue of cigarette smoke (CS)-exposed mice by qRT‒PCR. We observed the M1/M2 AM polarization in the lungs of COPD patients and COPD model mice by immunofluorescence analysis. Western blotting was used to determine the expression of MMP9/12 in the lung tissue of COPD patients and CS-exposed mice. An in vitro experiment was performed to determine the molecular mechanism of let-7c-induced macrophage polarization.
Results: Let-7c expression was downregulated in COPD patients, CS-exposed mice, and CS extract (CSE)-treated human bronchial epithelial (HBE) cells. AMs in COPD patients and CS-exposed mice were dominated by the M2 type, and the release of MMP9/12 was increased. In vitro, the transfection of mimics overexpressing let-7 or the use of tocilizumab to block signal transduction between HBE cells and macrophages inhibited the IL-6/STAT3 pathway. M2 macrophage polarization was inhibited, and MMP9/12 release was reduced.
Conclusion: Our results indicate that CS decreased let-7c expression in HBE cells, and M2 AM polarization was dominant in COPD. In HBE cells, let-7c could inhibit M2 polarization of AMs through the IL-6/STAT3 pathway, providing potential diagnostic and therapeutic value for slowing COPD emphysema.
Keywords: let-7c, macrophage polarization, interleukin-6, IL-6/signal transducer and activator of transcription 3, STAT3, signaling pathway, emphysema, COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is a common chronic airway disease. At present, COPD has become the third leading cause of death worldwide. It has been reported that the prevalence of COPD is approximately 11.7% in people over 30 years old. Patients may have symptoms such as wheezing, shortness of breath, chest tightness, and dyspnea. The pathogenesis of COPD includes inflammatory reactions, oxidative stress, protease antiprotease imbalance, muscle dysfunction, and pulmonary microorganisms.1–4 Emphysema is a common feature of COPD and refers to a decrease in the elasticity of the distal airway of the terminal bronchioles of the lung, excessive inflation of the airway, an increase in lung volume and damage to the airway. It is generally recognized that the development of COPD emphysema is associated with an imbalance in proteases and antiproteases. Proteases can destroy the lung parenchyma, affecting tissue remodeling and repair, while antiproteases play a protective role by inhibiting protease activity.5 When proteolytic activity exceeds the inhibitory capacity of the lung, it will lead to the destruction of the lung parenchyma and the development of emphysema, which will then lead to the occurrence of COPD. Although it is clear that the imbalance between proteases and antiproteases causes COPD emphysema, the factors and mechanisms affecting this imbalance are still unclear.
Macrophages are important immune cells that release cytokines and proteases in response to antigen transmission and phagocytosis and play a role in regulating the immune system. Macrophages are highly plastic and can be polarized into M1 and M2 phenotypes.6–9 M1 macrophages produce inducible nitric oxide synthase (iNOS) and tumor necrosis factor (TNF-α), which play proinflammatory and immune defense roles. M2 macrophages, which express CD206, Arg1 and secrete transforming growth factor beta (TGF-β), inhibit inflammation and are involved in tissue remodeling and angiogenesis. Macrophage polarization plays an important role in the pathogenesis of COPD.10–12 Previously, some scholars believed that macrophages in COPD tended to M1 type, while others believed that they tended to M2 type. Recently, it has been considered that alveolar macrophages (AMs) tend to exhibit M1 polarization in the early stage but more M2 polarization in the later stage.13 It is also believed that macrophages can also be bipolarized and express M1 and M2 at the same time.14 However, the specific dynamic process is still unclear. A large number of studies have shown that smoke stimulation can cause emphysema in mice and that macrophages are polarized into the M2 type and release a large number of matrix metalloproteinases (MMPs).15 MMPs are proteolytic enzymes. When the level of proteases increases and the levels of antiproteases are relatively insufficient, various proteins in the extracellular matrix (ECM) are degraded, leading to emphysema. It has been found that activation of the matrix metalloproteinase-9 (MMP-9) gene leads to continuous deterioration of airway remodeling and airway obstruction, which has been confirmed by many researchers.16–18 Although we know that macrophage polarization is involved in COPD emphysema, the effect of macrophage polarization on the pathogenesis of COPD is still unclear and needs further study.
Let-7 was the second miRNA family to be discovered after Lin-4, and there are a total of 13 members. Let-7 can target the mRNA expression of many genes and regulate cell proliferation, differentiation, metabolism and apoptosis.19 It was found that the expression of let-7 in whole blood, serum, bronchoalveolar lavage fluid and lung tissue of COPD patients was significantly lower than that of normal subjects, especially let-7a, let-7b, let-7c and let-7d. The expression of let-7 correlates with the number of neutrophils in the lung and may be involved in the recruitment of immune cells to the lung or airway.20 Let-7 participates in the pathogenesis of COPD through various mechanisms. For example, let-7 can directly target arginase 2 (ARG2) in BEAS-2B cells, reduce the level of intracellular reactive oxygen species (ROS) and the percentage of apoptotic cells, and participate in the oxidative stress process of COPD. Let-7 can also target signal transducer and activator of transcription 3 (STAT3) in macrophages to inhibit inflammation induced by LPS and play an anti-inflammatory role.21,22 Furthermore, let-7 was expressed in human bronchial epithelial (HBE) cells and was differentially expressed when the cells were stimulated by CSE. In Di’s study, let-7 mimics were transfected into HBE cells, which were cocultured with fibroblasts. It was confirmed that let-7 participates in COPD airway remodeling by targeting the expression of interleukin (IL)-6 and inhibiting fibroblast differentiation and ECM deposition.23 Although the mechanism by which let-7 affects COPD pulmonary fibrosis has been determined, the effect of let-7 on COPD emphysema and the polarization of AMs mediated by CS exposure is still unclear.
Here, we hypothesized that let-7 expression was downregulated in CSE-treated HBE cells. IL-6 was released by HBE cells and used to connect HBE cells with AMs. When IL-6 binds to IL-6R on the surface of AMs, it transmits signals to the cells. It can induce M2 polarization in AMs by affecting STAT3 phosphorylation and release large amounts of MMP-9/12. It is important to understand the mechanism by which COPD emphysema develops by examining the regulatory effect of let-7 on AM polarization.
Materials and Methods
Reagents and Cell Culture
HBE cells (Chi Scientific, Jiangsu, China), a normal HBE cell line, were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) (Gibco Life Technologies, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific, USA).
THP-1 cells, which are human myeloid leukemia mononuclear cells, were obtained from the National Collection of Authenticated Cell Cultures. THP-1 cells were cultured in RPMI 1640 (Gibco Life Technologies, USA) with 10% FBS. Both cell lines were cultured in a humidified incubator containing 95% air and 5% CO2 at 37°C.
In the coculture system, THP-1 cells were seeded on six-well plates at a density of 1×106 cells per well and treated with 10 µM PMA for 48 h to form THP-1 macrophages (THP-M cells). Then, THP-M cells were cocultured with normal, CSE-treated or let-7 mimic-transfected HBE cells seeded on 24 mm diameter inserts with 0.4 μm pores (#3412, Corning, USA). HBE cells were transfected with mimics for 48 h and treated with 5% CSE, while THP-M cells were treated with tocilizumab24,25 (HY-P9917, MedChemExpress, China), an anti-IL-6 receptor antibody before they were cocultured.
Human Sample Collection
Peripheral lung tissues were collected from COPD patients undergoing lung transplantation and patients with lung nodules undergoing pulmonary lobectomy at Wuxi People’s Hospital. All tissue donors provided informed consent in accordance with the Declaration of Istanbul. Peripheral lung tissues were stored in several tubes at −80°C.
Serum samples were collected from COPD patients and healthy volunteers for medical examination between 2020 and 2022 at Wuxi People’s Hospital. All serum donors provided informed consent in accordance with the Declaration of Istanbul. Serum samples were stored at −80°C.
This study was approved by the Ethics Committee of Wuxi People’s Hospital Affiliated to Nanjing Medical University. Informed consent was obtained from all donors in accordance with the Declaration of Istanbul. All COPD patients met the diagnosis criteria of GOLD 2021. Lung function and other characteristics of subjects participating in the study are reported in Table 1. As shown in Table 1, the median age and sex ratio were similar in all groups of subjects.
 |
Table 1 Clinical Characteristics of the Subjects |
Establishment of Murine COPD Model
Male 6-to-8 week old C57BL6J mice (Changzhou Kawensi Experimental Animal Co., Ltd. China) were divided into two groups; the experimental group was exposed to smoke, and the normal control group was exposed to the air. The mice were exposed to smoke from 20 Da Qian Men (10 mg tar and 0.8 mg nicotine/cigarette, Shanghai, China) cigarettes in a tempered glass box for 2 h twice per day, 6 h apart, 7 days a week for a total of 6 months. The control group that was not exposed to smoke was kept in a similar environment. The animals in this study were treated humanely according to a protocol approved by the Institutional Animal Care and Use Committee of Wuxi People’s Hospital, Jiangsu Province, in compliance with the law on the administration of experimental animals.
Preparation of Cigarette Smoke Extract (CSE)
CSE was prepared according to the experimental model previously reported.23,26 The smoke from two cigarettes (Daqianmen, China) was dissolved in 10 mL of serum-free DMEM (Gibco, USA) to form a CSE solution with a pH of 7.4. Then, the CSE solution was filtered through a 0.22-μm pore filter (Merck Millipore, USA) to remove insoluble particles and was standardized by measuring the absorbance at 320 and 540 nm, which was defined as 100% CSE. The CSE was diluted to a concentration of 5% with DMEM and used within 30 min.
Cell Transfection
The let-7c mimic and negative control (NC) mimic plasmids were designed by Jikai Gene (Shanghai, China). HBE cells were seeded into six-well plates. The plasmids were transfected into cells when the cell density reached 70%-80%. According to the instructions of Lipofectamine 3000 reagent (Thermo Fisher Scientific, USA), 2500 ng of plasmid, 7.5 µL of Lipo3000 and 5 µL of p3000 were dissolved in 250 µL of opti-MEM and mixed for 30 min in each well. Then, the mixture was added to six-well plates and supplemented with opti-MEM medium to a volume of 2 mL per well. After 4–6 h of transfection, the medium was replaced with DMEM supplemented with 10% FBS. After 48 h of transfection, CSE was added according to the experimental group.
RNA Extraction and Real-Time PCR
RNA was isolated from tissues and cells by using RNAiso Plus (TaKaRa, Japan). According to the manufacturer’s recommendations, 1000 ng of RNA was transcribed into cDNA by using a reverse transcription kit (TaKaRa, Japan), and then the cDNA was stored at −20°C. The primers were synthesized by Sangon Biotech (Shanghai, China), and the primer sequences are listed in Table 2. The RT‒PCR assay was performed with TB Green™ Premix Ex Taq™ II (Takara, Japan) and an ABI 9600 real-time PCR detection system (Applied Biosystems). The microRNA and mRNA internal controls were U6 and GAPDH, respectively. The ratio of the expression level of each gene was compared using the formula 2—(ΔΔCt) by the comparative threshold cycle (Ct) method. Statistical data were obtained from at least three independent experiments.
 |
Table 2 Primers Used in the Study |
Western Blot Analysis
Total protein was extracted from cells using RIPA buffer (KeyGEN, China) supplemented with a protease inhibitor cocktail (CWBIO CW2200, China) and phosphatase inhibitor (CWBIO CW2383, China). Equal amounts (20 μg-40 µg) of protein were separated via SDS‒PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA). After being blocked for 1 h in 5% milk, the membranes were incubated with antibodies targeting GAPDH (Proteintech, China), MMP9 (Abcam), MMP12 (Proteintech), STAT3 (Cell Signaling Technology), and phospho-STAT3 (Cell Signaling Technology) at 4°C overnight. After being incubated with secondary antibodies (Jackson ImmunoResearch, USA) for 1 h at room temperature, the protein bands were measured with the Immobilon ECL system (Millipore, S.p.A., Italy). ImageJ software was used for densitometric analyses.
Elisa Assay
The cell supernatant was centrifuged at 3000 rpm for 5 min and analyzed with a human IL-6 ELISA kit (R & D Systems, USA) according to the manufacturer’s instructions. The absorbance was measured at wavelengths of 450 nm and 540 nm. The release of IL-6 was calculated according to the standard curve, and at least three independent experiments were performed.
Dual-Luciferase Reporter Assay
To investigate whether let-7c (GAGGUA) has a binding sequence with IL-6 mRNA, IL-6-WT (TACCTC) and IL-6-MT (ATGGAG) were designed by Jikai Gene (Shanghai, China). The let-7c, NC, IL-6-WT and IL-6-MT mimics were transfected into HBE cells for 48 h by using Lipofectamine 3000 (Thermo Fisher Scientific, USA). The cells were harvested, and firefly and Renilla activities were measured using a dual-luciferase reporter assay system (Promega, USA) with an Infinite 200 PRO multimode microplate reader (Tecan Group, Ltd., Switzerland).
Immunofluorescence Analysis
Lung tissue sections were incubated with antibodies against CD68 (Proteintech), F4/80 (Santa Cruz Biotechnology), iNOS (Abcam) and CD206 (Abcam) overnight at 4°C. Then, the sections were incubated with Alexa 594-labeled goat anti-mouse and Alexa 488-labeled goat anti-rabbit secondary antibodies for 1 h at 37°C. DAPI (Beyotime, Shanghai, China) was added for nuclear staining. The fluorescent staining of CD68+ cells (total macrophages), iNOS+CD68+ cells (M1 macrophages)27,28 and CD206+CD68+ cells (M2 macrophages)29 was observed by a fluorescence microscope. Use ImageJ software to extract each single channel from the merge image, and analyze the average fluorescence intensity of each channel for quantitative analysis. Average fluorescence intensity=total fluorescence intensity/area.
Hematoxylin and Eosin (H&E) Staining
Mouse lung tissue sections were stained with hematoxylin for 5 min, hydrochloric acid-ethanol for 2–3 s, and eosin for 2 min and fixed with neutral resin. Pathological changes in the different groups were observed under an Olympus microscope. The alveolar space distance was measured by the mean linear intercept (MLI) and mean alveolar number (MAN).
Flow Cytometry
Cocultured cells were collected with trypsin EDTA and centrifuged at 2500 r for 5 min. The cell pellets were resuspended in 200 µL of PBS containing 10% FBS. Nonspecific staining was blocked with Fc Block (564219, BD). The cells were incubated with fluorescence-conjugated antibodies against APC CD86 (564544, BD) and BB515 CD206 (550889, BD) at 4°C for 30 min. After the addition of 300 µL of ice-cold PBS and centrifugation at 2500 r for 5 min, the cells were resuspended in 500 µL of PBS containing 10% FBS. M1 (CD86+) and M2 (CD206+) cells were distinguished by flow cytometry, and the data were analyzed by FlowJo software.
Lung Function Measurement
Mouse lung function was measured with whole-body plethysmography (Buxco Electronics, Ltd., USA) at Jiangsu Provincial Center for Disease Control and Prevention. The enhanced pause (Penh) and apnea index (PAU) were recorded using FinePoint software (Buxco Electronics Ltd., USA) to determine pulmonary resistance.
Statistical Analysis
All data are shown as the mean ± SD of at least three independent experiments and were analyzed with SPSS 2.0 software. Comparisons of mean values were analyzed using one-way ANOVA (Dunnett’s t-test) and two-tailed Student’s t-test. Values of P < 0.05 were considered statistically significant (*P<0.05; **P < 0.01).
Results
M2-Polarized Macrophages are Dominant in the Lung Tissues of COPD Patients
Lung function and the clinical characteristics of the subjects are shown in Table 1. The study was performed on 23 subjects who were categorized into three groups (healthy nonsmoking controls, non-COPD smokers and COPD patients). In addition to clinically distinct respiratory symptoms, the total number of AMs in COPD patients and smokers was increased compared with that in healthy nonsmoking controls, which was a pathological manifestation. The number in the COPD group was approximately 7-fold more than that in the healthy group (Figure 1A and B). Macrophages are classified into M1 and M2 phenotypes. To investigate which macrophage phenotype was predominant in the alveoli of COPD patients, the total macrophage surface marker CD68 was labeled with green fluorescence, and iNOS and CD206 were labeled with red fluorescence to indicate M1 and M2 macrophages, respectively. The immunofluorescence results indicated no significant differences between iNOS+/CD68+ and CD206+/CD68+ cells in healthy controls. However, the CD206+/CD68+ ratio in the smoking group was approximately 3-fold higher than the iNOS+/CD68+ ratio, while in the COPD group, it was approximately 8-fold higher, indicating an M2-polarized phenotype (Figure 1C). Furthermore, M2 macrophages release MMPs, which degrade the ECM to destroy the alveolar septum. The expression of MMP9/12 in COPD lung tissues was 2-3-fold higher than that in the control group, as shown by Western blot analysis (Figure 1D and E), indicating the presence of substantial numbers of M2 macrophages in COPD patients.
 |
Figure 1 Increased M2 macrophage polarization in COPD patients. Con-NS, nonsmokers without COPD; Con-S, smokers without COPD; COPD, COPD patients. (A) Lung macrophages were observed by immunofluorescence analysis in the Con-NS (n=5), Con-S (n=4), and COPD (n=3) groups. Scale bars, 50 µm. CD68: green; iNOS: red; CD206: red. (B) Immunofluorescence analysis of the total number of AMs. (C) Quantification of the percentages of M2 and M1 macrophages among total macrophages. Total macrophages: CD68+. M1%: iNOS+/CD68+. M2%: CD206+/CD68+. (D) Western blots. (E) Relative protein levels of MMP9 and MMP12 in the lung. The data are the mean ± SD (n=5). **P < 0.01. |
Emphysema and M2 Polarization Occur in CS-Exposed Mice
The COPD animal model was constructed according to the experimental method, and noninvasive assessment of pulmonary function was performed. The Penh value was obtained as an index of airway responsiveness in mice, and higher Penh values indicated enhanced airway resistance. The Penh value of the COPD group was 2- to 3-fold higher than that of the control group, suggesting that the airway resistance of mice was increased after stimulation with CS (Figure 2A). Histological analysis by H&E staining also confirmed the development of emphysema in CS-exposed mice; the MLI level was significantly increased and the MAN level was significantly decreased in lung tissues (Figure 2B and C). To analyze the polarization of AMs in mice, immunofluorescence experiments were performed, and the results showed that the total number of AMs in CS-exposed mice was increased approximately 5-fold compared with that in the control group (Figure 2D and E), while the proportion of M2 macrophages among total macrophages in CS-exposed mice was twice as high as that in air-exposed mice, and the proportion of M1 macrophages was reduced (Figure 2F). The transcription levels of M1 and M2 polarization markers in the lung tissues of mice were measured by qRT‒PCR. We found that the transcription levels of the M2-related markers Arg1 and TGF-β in CS-exposed mice were significantly upregulated compared with those in the control group, while no significant changes in the transcription of the M1-related markers iNOS and TNF-α were detected (Figure 2G). In addition, the protein expression of MMP9/12 in CS-exposed mice was 1.5–1.7 times higher than that in the control group (Figure 2H and I). Collectively, these data indicate that the polarization of AMs in CS-exposed mice was shifted toward the M2 phenotype, which is consistent with the phenomenon in COPD patients.
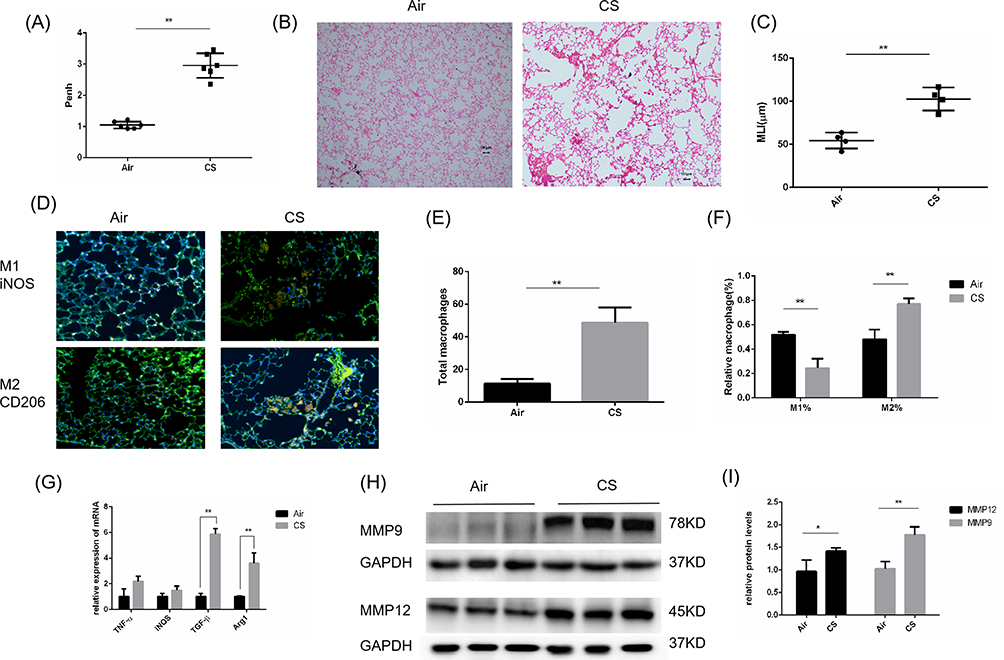 |
Figure 2 Emphysema and increased M2 macrophages in CS-exposed mice. Male C57BL/6J mice (6–8 weeks of age) were exposed to CS for 24 weeks. (A) Pulmonary function is shown as Penh in air-exposed mice and CS-exposed mice (n=6). (B) H&E staining of lung sections (scale bars, 100 µm) and (C) quantification of the MLI. (D) Lung macrophages in air-exposed mice (n=4) and CS-exposed mice (n=5) were observed by immunofluorescence analysis (scale bars, 100 µm). (E) Immunofluorescence analysis of the total number of macrophages and (F) quantification of the percentages of M2 and M1 macrophages among total macrophages in mouse sections. Total macrophages: F4/80+. M1%: iNOS+/F4/80+. M2%: CD206+/F4/80+. (G) The mRNA levels of M2 macrophage markers (Arg1 and Tgf-β1) and M1 macrophage markers (iNOS and TNF-α) were measured by quantitative RT–PCR. (H) Western blotting was performed, and (I) the relative protein levels of MMP9 and MMP12 were determined. The data are the mean ± SD (n=4). *P<0.05; **P < 0.01. |
The Induction of M2 Macrophage Polarization by CSE-Treated HBE Cells in vitro and the Downregulation of Let-7 Expression in vitro and in vivo
Considering that patients in the COPD group were generally in a severe condition, we tried to extend the duration of CSE exposure in cellular experiments to mimic the physiological environment in patients. According to the preliminary experiments, the longest time that HBE cells could withstand 5% CSE treatment was 48 h. We chose 5% CSE treatment for 48 h as the experimental conditions in vitro. To simulate the interaction between cells after smoke enters the human respiratory tract, HBE cells that were treated with 5% CSE (CHBE) were cocultured with macrophages induced by THP-1 and supplemented with PMA for 48 h (Figure 3A). TNF-α and iNOS were used as markers of M1 polarization, while Arg1 and TGF-β were used as markers of M2 polarization. The transcription level of macrophage polarization markers was measured by qRT‒PCR. The transcription levels of Arg1 and TGF-β in macrophages that were cocultured with CHBE cells were elevated by 1.5–2 times compared with those in the control group, while the M1 polarization marker levels showed no significant differences between the two groups (Figure 3B). The M2/M1 macrophage ratio in the coculture model was examined by flow cytometry. The results showed that the M2/M1 ratio of macrophages cocultured with CHBE was approximately 1.4 times higher than that of the control group (Figure 3C and D). These results indicated that 5% CSE treatment of HBE cells for 48 h induced M2 macrophage polarization, which was consistent with the phenomenon in severe COPD patients. Thus, the crosstalk between HBE cells and macrophages could facilitate M2 macrophage polarization during the occurrence and progression of COPD.
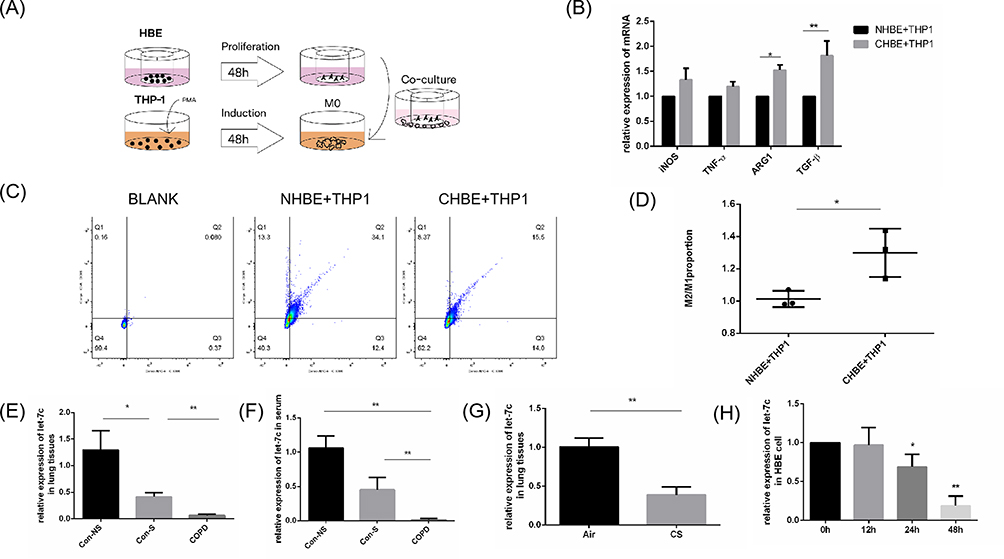 |
Figure 3 CHBE induced polarization of M2 type macrophages and expression of let-7c. (A) Scheme illustrating co culture experimental set-up. (B) The mRNA levels of macrophage markers in macrophages that were cocultured with CHBE cells were measured by quantitative RT–PCR.(C) Flow cytometry was performed, and (D) the M2/M1 ratio in the coculture cell model was determined. (E and F) Let-7 expression in human lung tissues (n=4) and serum (n=6). (G) Let-7 expression in the lung tissues of mice (n=4). (H) The mRNA levels of macrophage markers in HBE cells stimulated with 5% CSE for 0, 12, 24 and 48 h were measured. The data are the mean ± SD. *P<0.05; **P < 0.01. |
To investigate whether let-7c, which is a microRNA, was involved in the crosstalk between HBE cells and macrophages to enhance COPD progression, we initially performed qRT‒PCR to validate the expression level of let-7c in the lung tissues (Figure 3E) and serum (Figure 3F) of nonsmokers, smokers and COPD patients. We found that the expression level of let-7c in the smoking group was lower than that in the nonsmoking group. In particular, let-7c expression was barely detectable in the COPD group. Similarly, the expression level of let-7c in the lung tissues of COPD mice decreased by approximately half compared with that in the control group (Figure 3G). Furthermore, HBE cells were stimulated with 5% CSE for 0, 12, 24 and 48 h. We found that the let-7c expression level was downregulated at 24 h, and it declined by approximately 5-fold at 48 h after CSE administration (Figure 3H). It can be concluded that CS stimulation reduced let-7c expression, indicating that let-7c may participate in the crosstalk between HBE cells and macrophages to promote the development of COPD.
IL-6 is Released from CHBE Cells and Regulated by Let-7c
Many studies have shown that IL-6 expression is increased in the sputum and plasma of COPD patients. IL-6 is a well-known inflammatory factor that can be produced and released by HBE cells. We stimulated HBE cells with 2% and 5% CSE for 0.12.24.48 h. The results showed that the release of IL-6 increased at 24 h (Figure 4A). Furthermore, 5% CSE stimulation for 48 h facilitated substantial IL-6 generation and release from HBE cells (Figure 4A and B). In addition, under the same duration of smoking, the release of IL-6 stimulated by 5% CSE was higher than that stimulated by 2% CSE. This result indicated that the release of IL-6 was regulated by the duration and concentration of CSE.
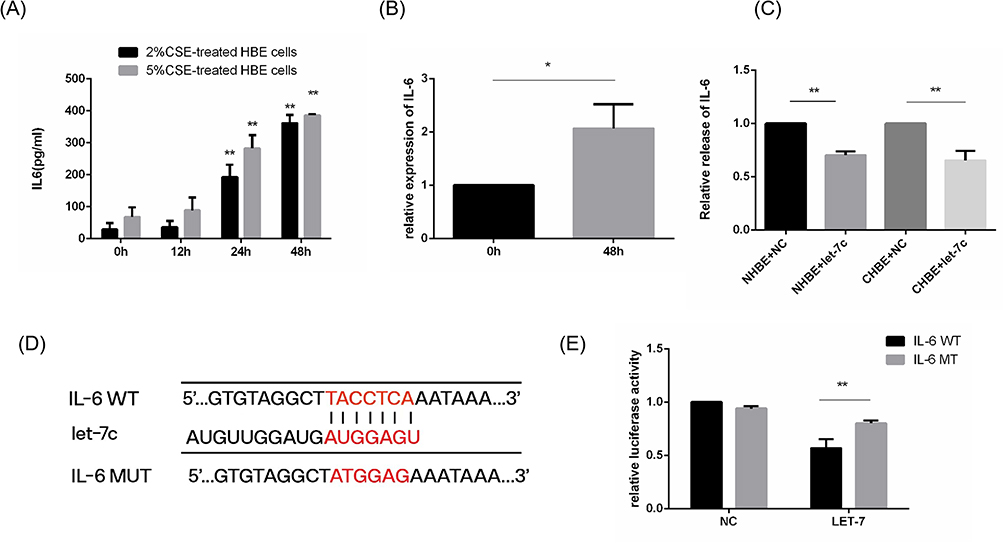 |
Figure 4 The release of IL-6 was stimulated by CSE and regulated by let-7c. NC, HBE cells treated with the NC mimic for 48 h. Let-7c, HBE cells treated with the let-7c mimic for 48 h. (A) IL-6 derived from 2% and 5% CSE-treated HBE cells induced for different times and measured by ELISA. (B) Relative mRNA expression of IL-6 in 5% CSE-treated HBE cells. (C) ELISA was performed to determine the relative release of IL-6 by HBE cells. (D) Schematic showing the putative let-7c target sites in the 3’UTR of IL-6 and the sequences of the mutant UTRs. (E) A luciferase reporter assay was performed after cotransfection with the let-7c mimic or NC mimic and IL-6-wt mimic or IL-6-mut mimic. The data are the mean ± SD (n=3). *P<0.05; **P < 0.01. |
These results suggest that IL-6 and let-7c in HBE cells are differentially expressed in response to smoking. We hypothesized that the release of IL-6 was regulated by let-7. Next, we transfected let-7c mimics into HBE cells and determined the levels of IL-6 in the cell culture supernatant by ELISA. The results showed that the release of IL-6 from HBE cells in the let-7c mimic group was less than that in the control group (Figure 4C), demonstrating that let-7c could directly regulate the expression of IL-6. Bioinformatics analysis showed that let-7c (GAGGUA) has a potential binding site in the 3’UTR (TACCTC) of IL-6 (Figure 4D). We cotransfected IL-6MT mimics or IL-6WT mimics with let-7c mimics or NC mimics into HBE cells. The dual-luciferase reporter assay results revealed that the luciferase activity in the IL-6WT+let-7c group was one-half less than that in the control group, while there was no significant difference between the other groups (Figure 4E). These results indicated that Let-7c could bind to IL-6 and directly regulate the expression of IL-6.
IL-6 Mediates HBE Cells and Macrophages and Induces STAT3 Phosphorylation in Macrophages
IL-6 was proven to be involved in cell-to-cell signaling.30,31 Next, we examined the downstream pathway in macrophages that was regulated by IL-6 signaling. The IL-6 receptor has been reported to exist on the surface of macrophages and can be activated by binding with IL-6, subsequently inducing STAT3 phosphorylation in macrophages. When tocilizumab,24,25 an IL-6 receptor inhibitor, was added to the macrophage culture medium, the p-STAT3/STAT3 ratio was significantly reduced to 1/3 of that in the control group (Figure 5A and B). It was reported that p-STAT3 was transferred to the nucleus in the form of a dimer, where it functioned as a transcription factor and regulated the transcription and expression of related genes by binding to DNA sequences.32 Our previous data showed that M2 macrophage polarization was promoted by CSE. To examine the effect of IL-6/STAT3 pathway activation on the polarization of macrophages, qRT‒PCR and flow cytometry experiments were performed. The results showed that the transcription levels of TNF-α and iNOS in the tocilizumab-treated group were upregulated, while the mRNA levels of Arg1 and TGF-β were downregulated by half (Figure 5C). These results suggested that a compromised IL-6 receptor might inhibit M2 macrophage polarization and promote M1 polarization, which was further confirmed by flow cytometry (Figure 5D). The differential levels of MMP9/12 released by M2 macrophages were confirmed by Western blot analysis. Because of the inhibition of M2 polarization by tocilizumab, the expression of MMP9/12 was reduced by half (Figure 5E and F). These results indicated that IL-6 transferred signals to macrophages to regulate STAT3 phosphorylation. In summary, the IL-6/STAT3 pathway was involved in inducing M2 macrophage polarization.
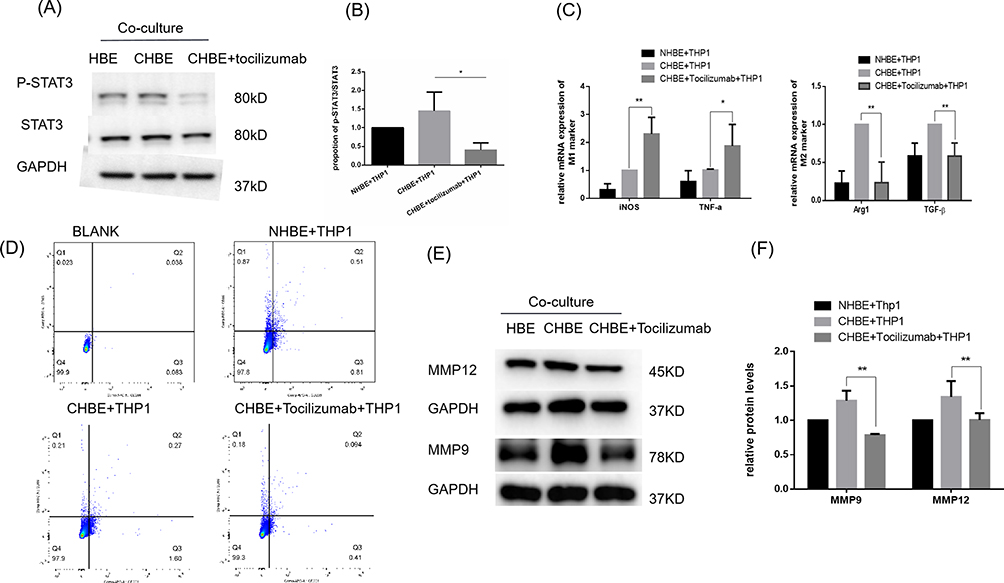 |
Figure 5 IL-6 induced STAT3 phosphorylation in M2 macrophages in vitro. CHBE, HBE cells that were treated with 5% CSE for 48 h; CHBE+tocilizumab, HBE cells that were pretreated with tocilizumab (4 µg/mL) for one hour and then treated with 5% CSE for 48 h. THP-1 cells were cocultured with HBE, CHBE and CHBE+tocilizumab cells for 48 h. Band densities were quantified by ImageJ software. GAPDH levels were measured in parallel and served as controls. (A) Western blotting was performed, and (B) the relative protein levels of STAT3 and p-STAT3 were determined. (C) The mRNA levels of M1 and M2 macrophage markers were measured by quantitative RT–PCR. (D) Quantification of the proportions of M2/M1 macrophages was assessed by flow cytometry. (E) Western blotting was performed, and (F) the relative protein levels of MMP9 and MMP12 were determined. The data are the mean ± SD. *P<0.05; **P < 0.01. |
MicroRNA Let-7 Inhibits M2 Macrophage Polarization via the IL-6/STAT3 Pathway
To further verify the effect of let-7 on macrophage polarization and the important role of the IL-6/STAT3 pathway in this process, we conducted an in vitro intervention experiment. The let-7c and NC mimics were transfected into HBE cells, and the transfection efficiency was evaluated by PCR. The results showed that the expression of let-7c in the let-7c-transfected group was more than twice that in the control group (Figure 6A). It is thought that the transfection rate is consistent with the standard, which provides a reliable prerequisite for subsequent experiments. In the in vitro rescue experiment, we used let-7c or NC plasmid-transfected HBE cells, added 5% CSE according to the groupings and cocultured the cells with macrophages for 48 h. After the cocultured macrophages were collected, the phosphorylation level of STAT3 protein and the expression of macrophage polarization markers were measured. The results showed that let-7 overexpression in HBE cells had little effect on macrophages, while let-7 overexpression in CHBE cells significantly reduced the phosphorylation level of STAT3 in macrophages (Figure 6B and C). Next, we observed the effect of let-7c on macrophage polarization and found that the proportion of M2/M1 macrophages in the let-7c overexpression group was significantly lower than that in the NC transfection control group, as determined by flow cytometry (Figure 6D and E). Furthermore, we confirmed that let-7 overexpression in CHBE cells increased the mRNA expression of iNOS and TNF-α in macrophages, but the difference was not significant. However, the mRNA expression of TGF-β and Arg1 decreased 0.6 times relative to that in the NC plasmid group (Figure 6F). The level of MMP9/12 released by macrophages was measured by Western blotting, which indirectly showed that let-7 inhibited the M2 macrophage polarization (Figure 6G and H). Collectively, these data indicate that smoke reduces let-7c in HBE cells and promotes M2 macrophage polarization by activating the IL-6/STAT3 pathway. However, when let-7c is overexpressed in CHBE cells, the IL-6/STAT3 pathway is inhibited to reduce the formation of M2 macrophages, thereby reducing MMP-mediated damage to alveoli.
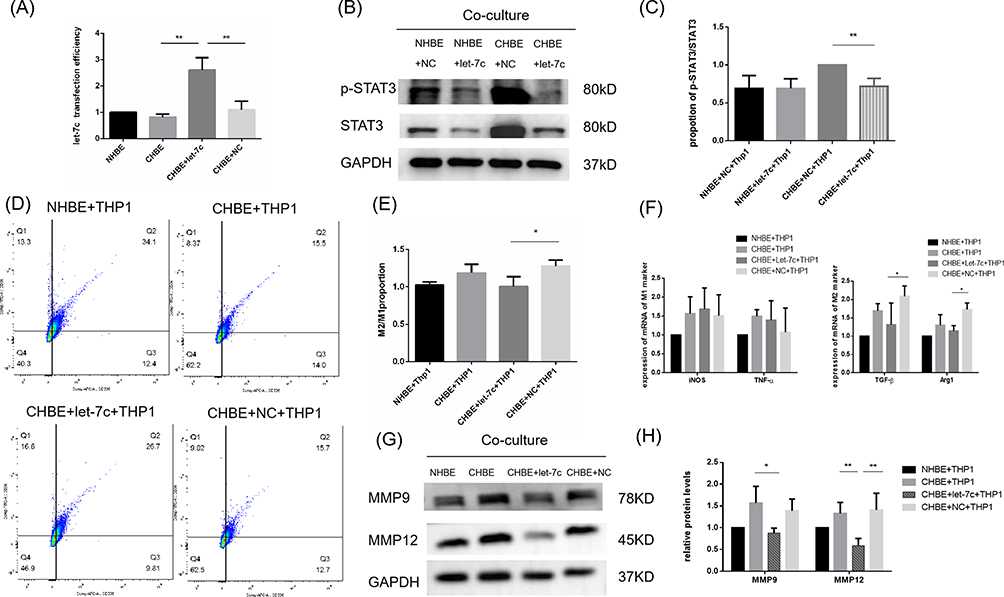 |
Figure 6 MicroRNA let-7 inhibits M2 macrophage polarization through the IL-6/STAT3 pathway. (A) PCR was performed to measure the transfection efficiency of the let-7c mimics. (B) Western blotting was performed, and (C) the relative protein levels of STAT3 and p-STAT3 were determined. (D) Flow cytometry was performed, and (E) the ratio of M2/M1 in the coculture cell model was determined. (F) The mRNA levels of M1 and M2 macrophage markers were measured by quantitative RT–PCR. (G) Western blotting was performed, and (H) the relative protein levels of MMP9 and MMP12 were determined. The data are the mean ± SD. *P<0.05; **P < 0.01. |
Discussion
COPD is the main cause of the global incidence and mortality of chronic diseases, and the overall global prevalence of COPD is on the rise.33 Among the risk factors related to COPD, smoking has always been the main factor causing the disease burden of COPD in the past decade. COPD is a chronic airway disease characterized by incomplete reversible airflow restriction. This airflow restriction is comprehensively affected by small airway diseases (such as chronic obstructive bronchitis) and lung parenchyma destruction (emphysema).34 Emphysema is anatomically defined as abnormal permanent enlargement of terminal bronchioles accompanied by the destruction of alveolar walls. The decrease in alveolar elastic retraction force is the main pathological mechanism of COPD caused by emphysema. Smoking can affect different types of emphysema.35 Our research shows that let-7 is induced by CS and can affect the polarization of AMs by reducing the release of IL-6 from bronchial epithelial cells and reduce the release of MMPs to slow the progression of COPD emphysema.
With the progression of COPD, emphysema becomes more obvious. Macrophages play an important role in promoting emphysema.36 The relative number of M2 macrophages is not only closely related to the severity of emphysema but also affects macrophage phagocytosis. To study the dynamic process of macrophage polarization, we used in vitro experiments to simulate the cell‒cell interactions after CS enters the human airway. We cocultured HBE cells that were treated with 5% CSE and macrophages induced by Thp-1 with PMA for 0.12, 24, and 48 h. The mRNA expression of M1 polarization genes (iNOS and TNF-α) increased from 0–24 h and decreased at 48 h. In contrast, the mRNA expression of M2 polarization genes (Arg1 and TGF-β) did not change much within 24 h but increased significantly at 48 h (Figure 7). We concluded that with increasing smoking time, the polarization of macrophages induced by bronchial epithelial cells gradually shifted from M1 to M2. It is generally believed that in the early stage of the disease, macrophages polarize into the M1 phenotype, which has proinflammatory functions and is beneficial for eliminating invading pathogens and stimulating adaptive immunity. In the late stage of the disease, when the inflammatory reaction is too strong and damages the tissues, macrophages shift to an anti-inflammatory M2 phenotype to neutralize the inflammatory reaction, protect the host from inflammatory damage and trigger wound healing. This finding is also consistent with our in vitro experimental results. A large number of studies have focused on the polarization of macrophages in COPD. Clinical studies have shown that CS can cause M2 polarization of macrophages in COPD patients. The ratio of M2 cells to monocytes in the sputum and bronchoalveolar lavage fluid of COPD patients is much higher than that of M1 cells.36,37 CS-induced M2 macrophages play a major role in COPD and secrete large amounts of MMP9/12 to exacerbate emphysema.10,38 Our experimental results are consistent with these views. First, the number of macrophages in the lungs of COPD patients and COPD model mice was significantly increased, and the proportion of M2 macrophages was greater than that of M1 macrophages. Second, the expression of MMP9/12 in the COPD group was significantly higher than that in the smoking control group and nonsmoking control group. MMP9/12 is a matrix metalloproteinase that is mainly released by M2 macrophages and can destroy the alveolar septum and promote the formation and progression of emphysema.39–41 Studies have shown that the level of MMP-12 in the alveolar lavage fluid of COPD patients is approximately 4–10 times higher than that of healthy people. Moreover, the addition of an MMP-12 inhibitor or the elimination of MMP12 expression can slow the expansion of the air cavity and ultimately prevent the occurrence of emphysema.42,43 COPD emphysema is closely related to M2 macrophage polarization.
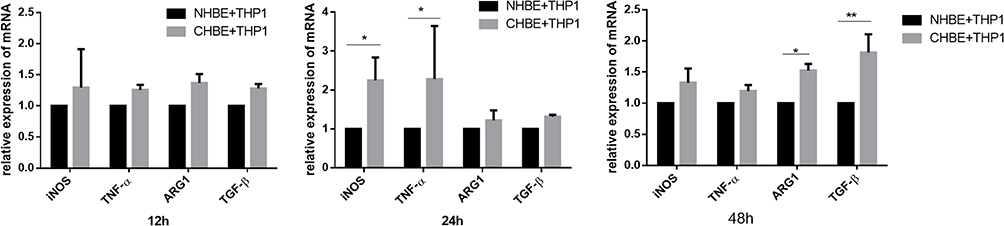 |
Figure 7 The mRNA levels of macrophage markers were measured in macrophages co-cultured with CHBE for 0.12.24.48 h by quantitative RT–PCR. The data are the mean ± SD. *P<0.05; **P < 0.01. |
The IL-6/STAT3 signaling pathway affects neutrophil activity, contributes to the severity of inflammation and immune regulation, and causes lung tissue damage.44–46 In recent years, it has been found that IL-6/STAT3 is involved in regulating the proliferation and apoptosis of human lung cancer cells. In addition, the IL-6/STAT-3 pathway can promote the polarization of M2 macrophages.47,48 In physiological conditions, the STAT-3 protein exists in the cytoplasm in an inactive form. In response to IL-6, stimulation the binding of IL-6 and its receptor IL6R induces the dimerization of the receptor subunit gp130, which causes JAK tyrosine kinase to cross-phosphorylate, become activated and directly phosphorylate the STAT3 protein. Then, the IL-6/STAT-3 pathway is activated.49,50 Subsequently, p-STAT3 forms a dimer, translocates from the cytoplasm to the nucleus and binds to the promoter regions of target genes such as PPAR to induce gene expression and promote M2 polarization.51,52 To verify the correlation between IL-6 and STAT3, we used tocilizumab to block the IL-6 receptor on the surface of macrophages in vitro. As a result, the phosphorylation of the STAT3 protein in cells was inhibited, and the mRNA expression of M2 polarization marker genes was reduced. Furthermore, the release of MMP-9/12 was reduced. At the protein level, flow cytometry showed that the M2/M1 ratio of macrophages that were treated with tocilizumab was decreased. Some studies have shown that IL-6 is significantly increased in the plasma of COPD patients, which is involved in the expression of intercellular adhesion factor in the alveolar epithelium and the initiation of the early pulmonary inflammatory response.53 IL-6 is also a predictor of the severity and exacerbation frequency of COPD.54,55 Most studies suggests that the level of IL-6 in the sputum of COPD patients is increased, and Kiszalkiewicz et al found that the levels of IL-6 in the sputum of patients with acute exacerbation of COPD were higher than those in patients with stable COPD.56,57 However, different researchers have different results, which may be affected by other basic diseases or by ignoring smoking factors in the control group. The pathophysiological mechanism is complex, and pathways interact with each other. The expression level of IL-6 is affected by many factors. Clarifying the role of the IL-6/STAT3 pathway in the pathogenesis of COPD still needs further analysis.
The let-7 family is a microRNA family that was discovered early. The differential expression of let-7 is involved in the pathogenesis of many diseases. Among the 627 miRNAs in the sputum of COPD patients, let-7c was significantly lower than that in the sputum of nonsmokers. The expression of let-7c is negatively correlated with the expression of tumor necrosis factor receptor type II (TNFR2), which is involved in the pathogenesis of COPD.58,59 The expression of let-7 family was measured in the lung tissue, serum and primary bronchial epithelial cells of COPD patients. It was found that the expression of let-7a, let-7c, and let-7d were significantly down regulated, among which let-7c was the most down regulated.23 This finding is consistent with the significant decrease in let-7c in the lung tissue and serum of COPD patients and the lung tissue of COPD model mice compared with the control group in our study. Furthermore, we verified that a longer the smoking time resulted in lower let-7c expression in vitro. IL-6 can be generated and released by HBE cells and can mediate communication between HBE cells and AMs. Our study also confirmed that the expression of IL-6 was regulated by let-7, and the release of IL-6 increased with increasing smoke concentration and smoking time. IL-6 participates in COPD emphysema by connecting HBE cells and macrophages. According to the research results of our research group, let-7 is not only involved in the process of airway remodeling, but also in the pathogenesis of COPD emphysema.
In conclusion, our results demonstrate that smoke stimulation can downregulate the expression of let-7 in the HBE cells and induce the formation of M2 macrophages. In addition, let-7c can reduce M2 polarization in AMs by inhibiting the IL-6/STAT3 pathway, thereby reducing the release of MMPs and slowing the progression of severe COPD emphysema (Figure 8). Therefore, let-7 is expected to provide a new therapeutic target for the prevention and treatment of COPD emphysema. However, our research has limitations. First, we lack in vivo experiments to directly prove that let-7 can inhibit the occurrence of COPD emphysema. Second, we only studied patients with severe COPD, but the polarization of macrophages is a dynamic process. To understand the dynamic changes in macrophage polarization from the onset of COPD and the turning point of macrophage M1 to M2 polarization, more research is needed. Moreover, it is unclear whether let-7c also mediates other important molecular mechanisms to participate in the pathogenesis of COPD.
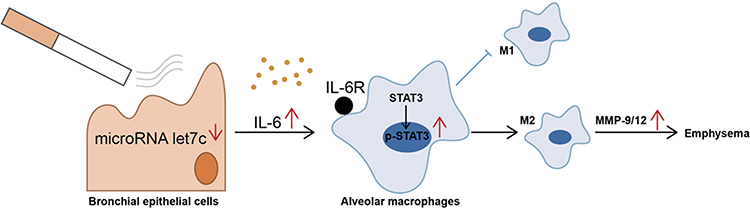 |
Figure 8 Schematic representation of the potential role of let-7c in COPD emphysema. Let-7c is negatively regulated in CHBE, promoting more releasable IL-6 to bind to IL-6R on the surface of alveolar macrophages, which induces phosphorylation of STAT3 in alveolar macrophages and polarization of M2 macrophages. M2 macrophages release more MMPs, leading to COPD emphysema. |
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Ethics Approval and Consent to Participate
All experimental work was approved by the ethical review board of Wuxi People’s Hospital Affiliated to Nanjing Medical University. This study was conducted in accordance with the tenets of the Declaration of Helsinki. The procedures for the care and use of animals were approved by the Ethics Committee of Nanjing Medical University (KY21033). The animals used in this study were maintained in accordance with the ethical guidelines of the Guiding.
Principles in the Care and Use of Animals (China) and the Policy of Animal Care and Use Committee of Nanjing Medical University. Humane care was provided according to the 3R principles of animal experiments.
Author Contributions
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all of these areas. All authors took part in drafting, revising or critically reviewing the article, gave final approval of the version to be published, have agreed on the journal to which the article has been submitted and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundations of China (82173472), the project of the Translational Medicine Program of Wuxi City, Jiangsu Province (2020ZHYB13).
Disclosure
The authors declare that there are no conflicts of interest.
References
1. Maltais F, Decramer M, Casaburi R, et al. An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(9):e15–e62. doi:10.1164/rccm.201402-0373ST
2. Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52(8):2813–2823. doi:10.1128/JCM.00035-14
3. Wang L, Hao K, Yang T, Wang C. Role of the lung microbiome in the pathogenesis of chronic obstructive pulmonary disease. Chin Med J. 2017;130(17):2107–2111. doi:10.4103/0366-6999.211452
4. Gea J, Martinez-Llorens J. Muscle dysfunction in chronic obstructive pulmonary disease: latest developments. Arch Bronconeumol. 2019;55(5):237–238. doi:10.1016/j.arbres.2018.07.016
5. Mohan A, Sharma M, Uniyal A, et al. Variability in proteinase-antiproteinase balance, nutritional status, and quality of life in stable chronic obstructive pulmonary disease due to tobacco and nontobacco etiology. Lung India. 2016;33(6):605–610. doi:10.4103/0970-2113.192859
6. Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi:10.1002/jcp.26429
7. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15(12):731–744. doi:10.1038/nri3920
8. Kadomoto S, Izumi K, Mizokami A. Macrophage polarity and disease control. Int J Mol Sci. 2021;23(1):144. doi:10.3390/ijms23010144
9. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154(2):186–195. doi:10.1111/imm.12910
10. Liu J, Zhang Z, Yang Y, Di T, Wu Y, Bian T. NCOA4-mediated ferroptosis in bronchial epithelial cells promotes macrophage M2 polarization in COPD emphysema. Int J Chron Obstruct Pulmon Dis. 2022;17:667–681. doi:10.2147/COPD.S354896
11. Liu L, Qin Y, Cai Z, et al. Effective-components combination improves airway remodeling in COPD rats by suppressing M2 macrophage polarization via the inhibition of mTORC2 activity. Phytomedicine. 2021;92:153759. doi:10.1016/j.phymed.2021.153759
12. Li N, Liu Y, Cai J. LncRNA MIR155HG regulates M1/M2 macrophage polarization in chronic obstructive pulmonary disease. Biomed Pharmacother. 2019;117:109015. doi:10.1016/j.biopha.2019.109015
13. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
14. Bazzan E, Turato G, Tine M, et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir Res. 2017;18(1):40. doi:10.1186/s12931-017-0522-0
15. Eapen MS, Hansbro PM, McAlinden K, et al. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci Rep. 2017;7(1):13392. doi:10.1038/s41598-017-13888-x
16. Li Y, Lu X, Li W, et al. The circRERE/miR-144-3p/TLR2/MMP9 signaling axis in COPD pulmonary monocytes promotes the EMT of pulmonary epithelial cells. Biochem Biophys Res Commun. 2022;625:1–8. doi:10.1016/j.bbrc.2022.07.119
17. Le Y, Cao W, Zhou L, et al. Infection of Mycobacterium tuberculosis promotes both M1/M2 polarization and MMP production in cigarette smoke-exposed macrophages. Front Immunol. 2020;11:1902. doi:10.3389/fimmu.2020.01902
18. Wells JM, Gaggar A, Blalock JE. MMP generated matrikines. Matrix Biol. 2015;44–46:122–129. doi:10.1016/j.matbio.2015.01.016
19. Xie D, Chen F, Zhang Y, et al. Let-7 underlies metformin-induced inhibition of hepatic glucose production. Proc Natl Acad Sci U S A. 2022;119(14):e2122217119. doi:10.1073/pnas.2122217119
20. Qian Y, Mao ZD, Shi YJ, Liu ZG, Cao Q, Zhang Q. Comprehensive analysis of miRNA-mRNA-lncRNA networks in non-smoking and smoking patients with chronic obstructive pulmonary disease. Cell Physiol Biochem. 2018;50(3):1140–1153. doi:10.1159/000494541
21. Song L, Li D, Gu Y, Li X, Peng L. Let-7a modulates particulate matter (</= 2.5 mum)-induced oxidative stress and injury in human airway epithelial cells by targeting arginase 2. J Appl Toxicol. 2016;36(10):1302–1310. doi:10.1002/jat.3309
22. Yu JH, Long L, Luo ZX, Li LM, You JR. Anti-inflammatory role of microRNA let-7c in LPS treated alveolar macrophages by targeting STAT3. Asian Pac J Trop Med. 2016;9(1):72–75. doi:10.1016/j.apjtm.2015.12.015
23. Di T, Yang Y, Fu C, et al. Let-7 mediated airway remodelling in chronic obstructive pulmonary disease via the regulation of IL-6. Eur J Clin Invest. 2021;51(4):e13425. doi:10.1111/eci.13425
24. Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol. 2020;16(6):335–345. doi:10.1038/s41584-020-0419-z
25. Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125–139. doi:10.1016/j.pharmthera.2013.09.004
26. Li E, Xu Z, Liu F, et al. Continual exposure to cigarette smoke extracts induces tumor-like transformation of human nontumor bronchial epithelial cells in a microfluidic chip. J Thorac Oncol. 2014;9(8):1091–1100. doi:10.1097/JTO.0000000000000219
27. Paul S, Chhatar S, Mishra A, Lal G. Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor. J Immunother Cancer. 2019;7(1):208. doi:10.1186/s40425-019-0697-7
28. Klug F, Prakash H, Huber PE, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. doi:10.1016/j.ccr.2013.09.014
29. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Corrigendum: macrophage polarization: different gene signatures in M1(LPS+) vs classically and M2(LPS-) vs alternatively activated macrophages. Front Immunol. 2020;11:234. doi:10.3389/fimmu.2020.00234
30. Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64(6):1403–1415. doi:10.1016/j.jhep.2016.02.004
31. Epstein SG, Brook E, Bardenstein-Wald B, Shitrit D. TGF-beta pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir Res. 2020;21(1):56. doi:10.1186/s12931-020-1319-0
32. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248. doi:10.1038/nrclinonc.2018.8
33. Song J, Zeng M, Wang H, et al. Distinct effects of asthma and COPD comorbidity on disease expression and outcome in patients with COVID-19. Allergy. 2021;76(2):483–496. doi:10.1111/all.14517
34. Singh D, Agusti A, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019;53(5):1900164. doi:10.1183/13993003.00164-2019
35. Chauhan NS, Sood D, Takkar P, Dhadwal DS, Kapila R. Quantitative assessment of airway and parenchymal components of chronic obstructive pulmonary disease using thin-section helical computed tomography. Pol J Radiol. 2019;84:e54–e60. doi:10.5114/pjr.2019.82737
36. Lu J, Xie L, Liu C, Zhang Q, Sun S. PTEN/PI3k/AKT regulates macrophage polarization in emphysematous mice. Scand J Immunol. 2017;85(6):395–405. doi:10.1111/sji.12545
37. Kunz LI, Lapperre TS, Snoeck-Stroband JB, et al. Smoking status and anti-inflammatory macrophages in bronchoalveolar lavage and induced sputum in COPD. Respir Res. 2011;12(1):34. doi:10.1186/1465-9921-12-34
38. John AE, Wilson MR, Habgood A, et al. Loss of epithelial Gq and G11 signaling inhibits TGFbeta production but promotes IL-33-mediated macrophage polarization and emphysema. Sci Signal. 2016;9(451):a104. doi:10.1126/scisignal.aad5568
39. Staitieh BS, Malik S, Auld SC, et al. HIV increases the risk of cigarette smoke-induced emphysema via MMP-9. J Acquir Immune Defic Syndr. 2022;92(3):263–270. doi:10.1097/QAI.0000000000003125
40. Gharib SA, Manicone AM, Parks WC. Matrix metalloproteinases in emphysema. Matrix Biol. 2018;73:34–51. doi:10.1016/j.matbio.2018.01.018
41. Hou HH, Wang HC, Cheng SL, Chen YF, Lu KZ, Yu CJ. MMP-12 activates protease-activated receptor-1, upregulates placenta growth factor, and leads to pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol. 2018;315(3):L432–L442. doi:10.1152/ajplung.00216.2017
42. Shibata S, Miyake K, Tateishi T, et al. Basophils trigger emphysema development in a murine model of COPD through IL-4-mediated generation of MMP-12-producing macrophages. Proc Natl Acad Sci U S A. 2018;115(51):13057–13062. doi:10.1073/pnas.1813927115
43. Demedts IK, Morel-Montero A, Lebecque S, et al. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax. 2006;61(3):196–201. doi:10.1136/thx.2005.042432
44. Chen IC, Wang SC, Chen YT, et al. Corylin ameliorates LPS-induced acute lung injury via suppressing the MAPKs and IL-6/STAT3 signaling pathways. Pharmaceuticals. 2021;14(10):1046. doi:10.3390/ph14101046
45. Ruwanpura SM, McLeod L, Miller A, et al. Deregulated Stat3 signaling dissociates pulmonary inflammation from emphysema in gp130 mutant mice. Am J Physiol Lung Cell Mol Physiol. 2012;302(7):L627–L639. doi:10.1152/ajplung.00285.2011
46. Fielding CA, McLoughlin RM, McLeod L, et al. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008;181(3):2189–2195. doi:10.4049/jimmunol.181.3.2189
47. Yang Y, Ding L, Hu Q, et al. MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol Cancer. 2017;16(1):141. doi:10.1186/s12943-017-0710-z
48. Kuo IY, Yang YE, Yang PS, et al. Converged Rab37/IL-6 trafficking and STAT3/PD-1 transcription axes elicit an immunosuppressive lung tumor microenvironment. Theranostics. 2021;11(14):7029–7044. doi:10.7150/thno.60040
49. Jafarzadeh A, Nemati M, Jafarzadeh S. Contribution of STAT3 to the pathogenesis of COVID-19. Microb Pathog. 2021;154:104836. doi:10.1016/j.micpath.2021.104836
50. Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A. 2013;110(42):16975–16980. doi:10.1073/pnas.1315862110
51. Liu Z, Meng Y, Miao Y, et al. Propofol ameliorates renal ischemia/reperfusion injury by enhancing macrophage M2 polarization through PPARgamma/STAT3 signaling. Aging. 2021;13(11):15511–15522. doi:10.18632/aging.203107
52. Stark JM, Coquet JM, Tibbitt CA. The role of PPAR-gamma in allergic disease. Curr Allergy Asthma Rep. 2021;21(11):45.
53. Liu CW, Lee TL, Chen YC, et al. PM(2.5)-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-kappaB-dependent pathway. Part Fibre Toxicol. 2018;15(1):4. doi:10.1186/s12989-018-0240-x
54. Singh R, Narang M, Dawson L, Kamra N, Singh G, Bahamania KK. Could disease severity and inflammatory markers (IL-6, Hs-CRP, TNF-alpha) be related to frailty in COPD? A prospective study. J Assoc Physicians India. 2022;70(4):11–12.
55. Huang H, Huang X, Zeng K, Deng F, Lin C, Huang W. Interleukin-6 is a strong predictor of the frequency of COPD exacerbation within 1 year. Int J Chron Obstruct Pulmon Dis. 2021;16:2945–2951. doi:10.2147/COPD.S332505
56. Kiszalkiewicz JM, Majewski S, Piotrowski WJ, et al. Evaluation of selected IL6/STAT3 pathway molecules and miRNA expression in chronic obstructive pulmonary disease. Sci Rep. 2021;11(1):22756. doi:10.1038/s41598-021-01950-8
57. Grubek-Jaworska H, Paplinska M, Hermanowicz-Salamon J, et al. IL-6 and IL-13 in induced sputum of COPD and asthma patients: correlation with respiratory tests. Respiration. 2012;84(2):101–107. doi:10.1159/000334900
58. Shin JI, Brusselle GG. Mechanistic links between COPD and lung cancer: a role of microRNA let‑7? Nat Rev Cancer. 2014;14(1):70. doi:10.1038/nrc3477-c1
59. Van Pottelberge GR, Mestdagh P, Bracke KR, et al. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(7):898–906. doi:10.1164/rccm.201002-0304OC
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Phenotyping COPD Patients with Emphysema Distribution Using Quantitative CT Measurement; More Severe Airway Involvement in Lower Dominant Emphysema
Park J, Kim EK, Lee SH, Kim MA, Kim JH, Lee SM, Lee JS, Oh YM, Lee SD, Lee JH
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2013-2025
Published Date: 31 August 2022
Association Between Empirical Anti-Pseudomonal Antibiotics for Recurrent Lower Respiratory Tract Infections and Mortality: A Retrospective Cohort Study
Shiroshita A, Yamamoto S, Anan K, Suzuki H, Takeshita M, Kataoka Y
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2919-2929
Published Date: 17 November 2022
Impact of Interstitial Lung Abnormalities on Disease Expression and Outcomes in COPD or Emphysema: A Systematic Review
Liu Y, Tang J, Sun Y
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:189-206
Published Date: 2 March 2023
Effect of the Lipoxin Receptor Agonist BML-111 on Cigarette Smoke Extract-Induced Macrophage Polarization and Inflammation in RAW264.7 Cells
Cao E, Xu J, Gong Y, Yuan J, Chen A, Liu J, Fan Y, Fan X, Kuang X
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:919-932
Published Date: 19 May 2023
Prevalence of Cardiovascular Disease and Rate of Major Adverse Cardiovascular Events in Severe Alpha-1 Antitrypsin Deficiency COPD
Ellis P, Bailey E, Choate R, Holm KE, Sandhaus RA, Turner AM, Newnham M
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:149-159
Published Date: 17 January 2024