Back to Journals » Journal of Inflammation Research » Volume 19
Microglial Epigenetic Memory is Associated with Accelerated Resolution of Inflammatory Pain Induced by Prophylactic Macrophage-Derived Small Extracellular Vesicles
Authors Luo X, Wickman JR, DaCunza JT, Tian Y, Sacan A, Ajit SK ![]()
Received 16 February 2026
Accepted for publication 15 June 2026
Published 9 July 2026 Volume 2026:19 598095
DOI https://doi.org/10.2147/JIR.S598095
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dharmappa Krishnappa
Xuan Luo,1 Jason R Wickman,1 Jason T DaCunza,1 Yuzhen Tian,1 Ahmet Sacan,2 Seena K Ajit1
1Department of Pharmacology & Physiology, Drexel University College of Medicine, Philadelphia, PA, USA; 2School of Biomedical Engineering, Science & Health Systems, Drexel University, Philadelphia, PA, USA
Correspondence: Seena K Ajit, Pharmacology & Physiology, Drexel University College of Medicine, 245 North 15th Street, Mail Stop 488, Room 8223, Philadelphia, PA, 19102, Tel +1 267 359 2614, Email [email protected]
Introduction: Small extracellular vesicles (sEVs) are cell-released lipid vesicles that facilitate intercellular communication by transferring bioactive cargo to recipient cells. We previously showed that a single intrathecal administration of RAW 264.7 macrophage–derived sEVs, given two weeks prior to complete Freund’s adjuvant (CFA)-induced inflammation, resulted in earlier recovery from mechanical and thermal hypersensitivity. How this long-term memory develops, and how sEVs regulate immune responses, are unknown. Recent studies have shown that priming microglia with inflammatory stimuli can enhance or suppress responses to a delayed secondary insult via epigenetic modifications. We hypothesized that prophylactic intrathecal administration of macrophage-derived sEVs confers accelerated resolution of inflammatory pain by reprogramming epigenetic memory in spinal microglia.
Methods: Microglia were ablated using the colony-stimulating factor 1 receptor (CSF1R) inhibitor PLX5622 prior to sEV administration. Pain behaviors were assessed following CFA-induced inflammation. Chromatin immunoprecipitation sequencing (ChIP-seq) was performed on spinal microglia isolated 14 days after sEV administration. The role of epigenetic modification was evaluated by pharmacological inhibition of the H3K4 mono-methyltransferase SETD7.
Results: Prophylactic sEV administration accelerated the resolution of inflammatory pain hypersensitivity. This effect was abolished in mice treated with PLX5622, indicating that microglia are required during sEV exposure. ChIP-seq analysis revealed enrichment of H3K4me1-marked loci in spinal microglia 14 days after sEV administration, consistent with induction of innate immune memory. Inhibition of SETD7 eliminated the protective effect of sEVs, demonstrating a requirement for H3K4 mono-methylation.
Discussion: Macrophage-derived sEVs induce a microglia-dependent, epigenetically mediated form of pain prophylaxis. These findings support a model in which sEVs establish a primed, memory-like state in spinal microglia, characterized by enhancer-associated chromatin changes that confer latent regulatory potential and enhance resolution of subsequent inflammatory pain. This work links extracellular vesicles to microglial epigenetic remodeling and suggests a potential strategy for non-addictive, preventive pain therapeutics.
Keywords: extracellular vesicles, exosomes, inflammatory pain, microglia, epigenetics, H3K4me1, SETD7
Introduction
Neuroimmune interactions in pain engage multiple signaling pathways and cell types, including neurons, glia, and infiltrating immune cells, that together regulate both peripheral and central mechanisms of nociception.1 These interactions rely on coordinated, cell type and stimulus-specific transcriptional programs involving the dynamic regulation of large gene networks. Transcriptional responses are governed by the accessibility of RNA polymerase and transcription factors to defined genomic regions, a process tightly regulated by chromatin structure through histone modifications and DNA methylation.2,3 Inflammatory stimulation can induce epigenetic changes in cells, some of which are rapidly reversible, while others appear to be long-lasting and poised for gene transcription in response to secondary triggers. These persistent epigenetic alterations have been increasingly recognized as a mechanism by which immune cells retain a functional memory of prior inflammatory experiences and linked to innate immune memory.4,5
Traditionally, immunological memory was thought to exist only in the adaptive immune system. T and B lymphocytes, major components of the adaptative immunity, express a diverse repertoire of highly specific antigen receptors generated through the rearrangement of the antigen receptor genes, allowing these cells to mount tailored immune responses against various antigens.6 When naïve, antigen-specific lymphocytes recognize an antigen, they undergo clonal expansion and produce memory cells that can persist for years, ready to respond swiftly and effectively to future exposures of the same antigen. However, recent findings show that innate myeloid and lymphoid cells also retain memory of prior pathogen exposure and become primed to elicit an enhanced response to a secondary stimulus.7,8 Unlike the specificity of the adaptive immunity driven by antigen receptors like immunoglobulins and T cell receptors, innate immune cells can provide non-specific responses to homologous (same as the primary encounter) or heterologous (different from the prior stimulus) pathogens or non-microbial ligands. This phenomenon termed innate immune memory or trained immunity is a vital survival mechanism for organisms like plants and invertebrates that lack an adaptive immune system, and for mammals that are deficient in functional T and B cells.9 Besides trained immunity, innate immune memory can manifest as immune tolerance, when trained cells produce a diminished response to subsequent stimulation. The enhanced or suppressed effects of this response largely depend on the magnitude and duration of the initial stimulus. For instance, microglia treated with a low dose of lipopolysaccharide (LPS), exhibit immune training via increased pro-inflammatory cytokine release upon re-stimulation with a fixed dose of LPS. Conversely, a high initial dose of LPS triggers immune tolerance in microglia, resulting in reduced pro-inflammatory but increased anti-inflammatory cytokine release upon rechallenge.10
Immune stimulation induces dynamic changes in histone modifications, including histone H3 lysine 4 mono- and tri-methylation (H3K4me1, H3K4me3) and histone H3 lysine 27 acetylation (H3K27ac), leading to lasting reprogramming of the enhancer landscape.11–13 A hallmark of innate immune memory is the acquisition and persistence of H3K4me1 at enhancers after stimulus withdrawal, even as H3K27ac and transcription factor occupancy are lost, leaving enhancers in a primed state poised for rapid gene induction upon restimulation.12 In macrophages, these so-called latent enhancers are unmarked at baseline but acquire enhancer features following inflammatory stimulation, retaining H3K4me1 after resolution and enabling accelerated transcriptional responses to secondary challenges.12 Importantly, similar stimulus-dependent chromatin remodeling has been described in other innate immune cells, including microglia, which can exhibit altered responsiveness to delayed secondary insults through persistent epigenetic modifications.14,15 Collectively, these persistent enhancer and promoter changes constitute a form of innate immune memory, commonly referred to as trained immunity.8,12,15
Small extracellular vesicles (sEVs), including exosomes, are key mediators of intercellular communication and can exert potent immunomodulatory effects. However, the mechanisms by which sEVs influence pain hypersensitivity remain poorly understood. Innate immune cell–derived sEVs are known to regulate both innate and adaptive immune responses16 and accumulating evidence suggests that sEVs can be protective in a range of painful conditions.17–21 We previously demonstrated that a single prophylactic intrathecal administration of RAW 264.7 macrophage-derived sEVs two weeks prior to injury accelerates the resolution of mechanical allodynia and thermal hyperalgesia in a complete Freund’s adjuvant (CFA) mouse model of inflammatory pain.20
Notably, the mechanisms by which sEVs confer long-lasting prophylactic effects in naïve mice remain unknown. Microglia, the tissue-resident macrophages of the central nervous system (CNS), play a central role in the development and maintenance of both neuropathic22 and inflammatory pain23,24 through sustained activation and production of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α.25,26 Emerging evidence indicates sex-dependent differences in microglial structure and function.27 Microglia have been reported to play a prominent role in the maintenance of pain in male mice,28,29 although their contribution may vary depending on the experimental context.30 The present study was performed in male mice.
Despite growing evidence that extracellular vesicles modulate immune and neural function, it remains unclear whether macrophage-derived sEVs induce durable epigenetic changes in microglia that influence pain resolution. Here, we hypothesized that prophylactic intrathecal administration of RAW 264.7 macrophage-derived sEVs promotes faster resolution of CFA-induced pain hypersensitivity by establishing a memory-like, epigenetically mediated state in spinal microglia.
Materials and Methods
Cell Culture
Murine mouse macrophage cell line RAW 264.7 (#TIB-71, American Type Culture Collection) was cultured in complete Dulbecco’s Modified Eagle’s Medium (DMEM; Corning, New York, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS; #35-011-CV, Corning) and 1% penicillin-streptomycin (P/S; ThermoFisher Scientific, Waltham, MA) in a humidified incubator at 37°C with 5% CO2.
Preparation of Exosome-Depleted FBS
Heat-inactivated FBS was subjected to ultracentrifugation to remove larger vesicles and aggregates, including exosomes. Briefly, FBS was centrifuged in an Optima LE-80K ultracentrifuge (Beckman Coulter) with a Type 45 Ti fixed-angle rotor (Beckman Coulter) in 70-mL ultracentrifuge tubes (#355622, Beckman Coulter, Brea, CA) at 120,000×g for 18 h at 4°C. The supernatant was syringe filtered (0.22µm) and stored at −20°C for further use.
Isolation of Small Extracellular Vesicles (sEVs) from Conditioned Medium
sEV-conditioned medium was obtained from cultures of RAW 264.7 cells plated in T-175 flasks (Corning) at a density of 29,000 cells/cm2. At 70–80% confluency, cells were washed thrice with 10 mL of phosphate-buffered saline (PBS; Corning), followed by addition of 35 mL of DMEM with 10% exosome-depleted FBS and 1% P/S. Conditioned medium was collected 24 hours later in 50-mL conical tubes and centrifuged at 500×g for 10 min at 4°C. Supernatants were transferred to new conical tubes and stored at −80°C until further processing. We followed the Minimal Information for Studies of Extracellular Vesicles (MISEV) guidelines to increase purity31 and employed a multi-step approach utilizing filter concentration, size-exclusion chromatography (SEC), and differential ultracentrifugation. All centrifugations were performed at 4°C. Stored media was thawed and centrifuged at 12,000×g for 35 min (Sorvall RC-5C Plus centrifuge with Sorvall SA-600 fixed-angle rotor, ThermoFisher Scientific) and the supernatant was syringe filtered (0.22μm). The supernatant was concentrated using 100 kDa Amicon centrifugal filters (#UFC9100, Millipore, Burlington, MA) to less than 500 µL, centrifuging at 5000×g for 35 min. Retentate was diluted with PBS to 500 µL and further purified by SEC using a qEV original 35 nm Legacy column (#SP5, Izon Science, New Zealand) according to the manufacturer’s instructions. Fractions were separated by 0.5 mL, and fractions 7–10 were collected, containing the EV enriched material, and subjected to ultracentrifugation at 110,000×g for 70 min (Optima TLX ultracentrifuge with TLA 100.4 rotor, Beckman Coulter). The final pellet, representing sEVs, was resuspended in 35 µL of PBS and stored at −80 °C. Protein concentrations of sEVs were quantified using the microassay protocol for the DC protein assay (Bio-Rad, Hercules, CA).
Nanoparticle Tracking Analysis (NTA) of sEVs
sEV size distributions and concentrations were measured using a NanoSight LM10 instrument equipped with a 405 nm laser (Malvern Panalytical), which utilizes laser illumination to track Brownian motion. We used 0.1µm filtered PBS to dilute samples before use to achieve an optimal concentration of 20–90 particles/frame. Samples were injected into the sample chamber, and five 60-second videos were captured for each sample, and the temperature was also recorded. For the ZetaView S/N 18–390 (ParticleMetrix), samples were diluted to approximately 1×108 particles/mL and analyzed using the ZetaView software (version 8.05.12 SP2).
Western Blot for sEV Markers
Protein lysates of RAW 264.7 cells and sEVs were prepared in radioimmunoprecipitation assay (RIPA) buffer (ThermoFisher Scientific) with 1X protease inhibitor cocktail (ThermoFisher Scientific) and protein concentrations were determined using the DC protein assay. Five μg of cell lysates and sEVs were resolved on 4–12% gradient SDS-PAGE gel (ThermoFisher Scientific), transferred to 0.45 μm PVDF membranes and blocked with 5% non-fat dry milk (NFDM) in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 hour at RT. Membranes were washed thrice with TBST and incubated with primary antibodies (TBST + 5% NFDM) overnight at 4°C. Membranes were again washed thrice in TBST and incubated with secondary HRP-conjugated antibodies (TBST + 5% NFDM) at room temperature for 1 h. SuperSignal West Dura Substrate (ThermoFisher Scientific) was added to membranes and incubated for 5 min at room temperature. Membranes were visualized using the Odyssey Fc imaging system (LI-COR Biosciences). The following primary antibodies and concentrations were used: mouse anti-CD81 Clone B-11 (#sc-166029, Santa Cruz Biotechnology, 1:500), mouse anti-Alix Clone 1A12 (#sc-53540, Santa Cruz Biotechnology, 1:300), and rabbit anti-Calnexin (#PA5-34754, Santa Cruz Biotechnology, 1:5000). Secondary HRP-conjugated antibodies used were Goat anti-rabbit IgG-HRP and Goat anti-mouse IgG-HRP (#1706515 and #1706516, Bio-Rad).
Detection of Immune Surface Markers of sEVs via Flow Cytometry
A bead-based immunodetection assay identified 37 immune markers on sEVs using 10 µg of input material, performed according to the manufacturer’s protocol as previously described32 (mouse MACSPlex EV Kit, #130-122-211, Miltenyi Biotec, Germany). Acquisition was performed on aBD LSRFortessa flow cytometer (BD Biosciences, San Diego, CA). sEVs are identified by CD9+, CD63+, and CD81+ APC-labelled capture beads, which are then labelled with marker specific antibodies. For markers with %Pos ≥ 60%, the weighted expression is calculated as: 0.5 * %Pos * (1 + MFI / Highest MFI). Samples are represented in the form of a heat map. Three individual sEV preparations were used.
Mice
Wild-type (WT) C57BL/6J male mice aged 6–9 weeks were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed socially (2–5 mice per cage) in individually ventilated cages in a temperature- and humidity-controlled room with a 12-h light/dark cycle. Mice were provided with irradiated rodent diet (#5053, LabDiet, St. Louis, MO) and water ad libitum unless otherwise specified. Upon arrival, mice were acclimated in the animal facility for at least one week prior to experiments.
Intrathecal Administration of sEVs and PFI-2
Mice received intrathecal injections at 8–11 weeks of age. For single sEV injections, a Hamilton microliter syringe equipped with a 30-gauge needle was inserted between the L5 and L6 vertebrae at a 30° angle. Successful entry into the intradural space was confirmed by observing a tail flick response. One µg of sEVs resuspended in 10 µL of PBS (0.1 µg/µL) or an equal volume of PBS, was injected at a slow and steady rate to avoid rapid changes in pressure.
To enable repeated intrathecal drug delivery, a polyurethane intrathecal catheter (32 G tip, 6 cm long, 22 G connection; #MIT-02, SAI Infusion, Lake Villa, IL) was inserted between the L5 and L6 vertebrae into the subarachnoid space and extended to lumbar L4 to L5 regions as previously reported.33 Sutures underneath the skin (4–0) were used to secure the catheter, and the catheter was flushed with 7 µL of PBS, followed by heat sealing of the tube opening. Mice recovered for 5–7 days before experimentation. PFI-2 (#S7294, Selleck Chemicals, Houston, TX), a specific inhibitor of SET domain-containing 7 histone lysine methyltransferase (SETD7), was used to inhibit the methyltransferase activity of SETD7. PFI-2 was reconstituted in dimethyl sulfoxide (DMSO; G-Biosciences, St. Louis, MO) at a concentration of 10 mM and stored at −20°C. PBS was administered between drugs and after delivery to avoid direct PFI-2-sEV interactions and to ensure circulation in cerebrospinal fluid. The injection regimen was as follows. Day 1: 5 µL of 20 µM PFI-2 in 1% DMSO, 3 µL of PBS, 5 µL of sEVs containing 1 µg of sEVs (0.2 µg/µL), and 7 µL of PBS. Day 2–4: 10 µL of 10 µM PFI-2 in 0.1% DMSO, followed by 7 µL of PBS. DMSO and 5 µL of PBS were used as controls for PFI-2 and sEVs, respectively. The catheter was heat-sealed after injections each day. Mice were anesthetized with 2% isoflurane gas during catheterization and injections. Additionally, mice were housed individually until the completion of intrathecal drug delivery to prevent catheter displacement.
Complete Freund’s Adjuvant (CFA) Model of Inflammatory Pain
To induce inflammatory pain in the hind paw of mice, an emulsion comprised of 20 µL of 50% CFA (10 µg, #F5881, Sigma-Aldrich, St. Louis, MO) diluted in 0.9% saline was administered subcutaneously into the plantar surface of the right hind paw using a 30-gauge needle and syringe. For studies in which the prophylactic effects of sEVs were evaluated, mice received a CFA injection two weeks after a single dose of 1 µg sEVs.20
Pain Behavioral Assessment of Mechanical Hypersensitivity
Mechanical sensitivity of the hind paws was assessed by von Frey filaments as previously described.20 Habituation of mice for von Frey testing was performed with mice individually placed into plexiglass chambers on an elevated mesh grid platform for 1 h, 1–2 days prior to baseline testing. Mice were additionally habituated for 30 min each day prior to behavioral testing. A series of von Frey filaments (#BIO-VF-M, Bioseb, France) ranging from 1.4 to 2 grams of force (ascending order) were sequentially applied to the center of the hind paw plantar surface. Positive responses were recorded when mice withdrew, licked, or flinched the testing paw. Filament was applied five times for 2–3 sec each. The paw withdrawal threshold was determined as the force at which three out of five stimuli elicited positive responses. Measurement of sensitivity in unaffected paws (naïve mice or the contralateral (left) paw of CFA model mice) began with a 0.16 g filament, while the ipsilateral (right) paw of CFA model began with a 0.02 g filament. The experimenter was blinded to the treatment conditions.
PLX5622-Mediated Microglia Depletion
Upon arrival from the Jackson Laboratory, mice were fed control AIN-76A rodent diet (#D10001i, Research Diets, New Brunswick, NJ) ad libitum. Microglial ablation was induced by pharmacological means using CSF1R inhibitor, PLX5622 (#HY-114153, MedChemExpress, Monmouth Junction, NJ), formulated (1200 parts per million) in AIN-76A chow (#D19101002i, Research Diets). PLX5622 diet was fed to 8–9 week-old mice starting 10 days prior to the sEV administration, for a duration of 14 days. These mice were reverted to the control diet for the remainder of the study period.
Perfusion, Tissue Fixation, and Immunohistochemistry (IHC)
Intraperitoneal (i.p.) injection of 100 mg/kg body weight of ketamine and 10 mg/kg body weight of xylazine was used to anesthetize mice, with subsequent transcardial perfusion with room temperature PBS, followed by ice-cold 4% (w/v) paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA) in 0.1 M phosphate buffer (PB) for 5 min. Lumbar spinal cord segments (L4-L5) were dissected and stored in in 4% PFA in PB overnight at 4°C and then cryoprotected in 30% sucrose in PB for at least 24 h. O.C.T. compound was used to embed tissues, which were then sectioned at 25 µm in PB (free-floating) at −20°C using a cryostat. For immunostaining, sections were washed thrice for 5 min with wash buffer (0.3% Triton X-100 in PB), then blocked 5% normal goat serum (NGS; #S-1000, Vector Laboratories) in wash buffer for 2 h at room temperature. Sections were incubated with primary antibodies diluted in blocking buffer on a shaker overnight at 4°C. The next day, sections were washed thrice in wash buffer, and incubated with secondary antibodies in PB supplemented with 5% NGS for 2 h at room temperature on a shaker. Sections were washed three times in wash buffer and counterstained with 1 µg/mL DAPI (4’,6-diamidino-2-phenylindole; ThermoFisher Scientific) for 10 min at room temperature. Following a final wash step, tissue sections were mounted on Superfrost Plus Gold slides and cured with mountant (Invitrogen) overnight in the dark. Confocal images were acquired using an Olympus FV3000 microscope. Antibodies were used as follows: Iba1 (1:2000, #019-19741, Wako Chemicals) and goat anti-rabbit Alexa Fluor 594 (1:1000, #A-11012, Invitrogen).
Preparation of Single-Cell Suspension from Adult Mouse Spinal Cord
Spinal cords from PBS-perfused mice were hydraulically extruded into ice-cold Dulbecco’s Phosphate-Buffered Saline (DPBS) supplemented with calcium and magnesium (DPBS++; #14287072, Gibco) using a trimmed 200-µL pipette tip and a 10-mL syringe filled with cold DPBS++. Tissue dissociation was carried out using the Adult Brain Dissociation Kit for mouse (#130-107-677, Miltenyi Biotec). In brief, spinal cord was minced into 2 mm2 pieces and placed in a gentleMACS C tube (Miltenyi Biotec) containing Enzymes P and A. The C tube was then placed on a gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec) and subjected to appropriate programs based on tissue weight (20–100 mg: 37C_ABDK_02; >100 mg: 37C_ABDK_01). Dissociated cells were then passed through a 70-µm cell strainer into a 50-mL conical tube and centrifuged at 300xg for 10 min at 4 °C. Cells were resuspended in an appropriate buffer for downstream experiments.
Isolation of Spinal Cord Microglia Using Magnetic-Activated Cell Sorting (MACS)
Single cells from adult mouse spinal cord were resuspended in 3.1 mL of DPBS++ and the suspension was then transferred to a 15-mL conical tube. To remove debris such as myelin, 0.9 mL of debris removal solution provided by the dissociation kit was thoroughly mixed with the suspension. Subsequently, 4 mL of DPBS++ was carefully overlaid on the cell suspension, followed by centrifugation at 3000×g for 10 min at 4°C. The top two phases were aspirated, and the remaining suspension was diluted to 15 mL with DPBS++ followed by centrifugation at 1000×g for 10 min at 4°C. Debris-free cells were incubated with CD11b MicroBeads (10 µL beads per 107 cell; #130-049-601, Miltenyi Biotec) in PBS + 0.5% bovine serum albumin (BSA) for 15 min in a refrigerator (2−8 °C). Unbound antibody was removed by adding 1–2 mL of 0.5% BSA in PBS per 107 cells and centrifuging at 300xg for 10 min at 4°C. The magnetic-labeled positive fraction (CD11b+ cells) was isolated using an LS column (Miltenyi Biotec). CD11b+ cells were resuspended in a suitable buffer for subsequent experiments. Each MACS isolation utilized a pool of 6–8 mice. For RNA isolation, final cell pellets were dissolved in 300 µL of RNA Lysis/Binding buffer and snap-frozen in liquid nitrogen.
Percoll Density Gradients
Isolation of mononuclear cells from spinal cord using Percoll gradients was performed as described.34 First, a stock isotonic Percoll (SIP) was prepared by mixing 9 parts of Percoll (Cytiva, Marlborough, MA) with one part of 10X HBSS without calcium, magnesium, or phenol red (Gibco). SIP was diluted to 30% with complete RPMI 1640 medium (Gibco) containing phenol red, 10% FBS and 1% P/S, and diluted to 70% with 1x HBSS (Avantor, Radnor, PA). After dissociation, cells were resuspended in 10 mL of 30% SIP, transferred to a 15-mL conical tube, and underlaid with 2.5 mL of 70% SIP. Gradients were centrifuged at 500×g for 30 min at 18°C with no brake. The supernatant containing the myelin was discarded. Subsequently, 4 mL of cell suspension was collected at the 30%/70% interphase into a new 15-mL conical tube containing 8 mL of 1X HBSS and centrifuged at 500×g for 7 min at 18°C. Cell pellets were then resuspended in an appropriate buffer.
Isolation of Spinal Microglia Using Fluorescence-Activated Cell Sorting (FACS)
For isolating microglial cells using FACS, each sample was comprised of a pool of 6–8 mice (n=2). Spinal mononuclear cells isolated using Percoll gradients were washed in FACS buffer (PBS containing 2% FBS, 1% P/S, 25 mM HEPES (Gibco), and 2.5 mM EDTA (Invitrogen)). After buffer exchange, cells were resuspended in 50 µL FACS buffer containing FcR block (1:100; #156603, BioLegend) for 5 min at room temperature, followed by 50 µL of live/dead staining in PBS (1:500; #L10119, Invitrogen) for 15 min at 4°C. Cells were washed and resuspended in FACS buffer. Cells were then co-stained with 55 µL of antibody cocktail in stain buffer (#566349, BD Biosciences) for 45 min at 4°C. Cells were washed and incubated with 100 µL of Brilliant Violet (BV) 785 Streptavidin in FACS buffer (1:100; #405249, BioLegend) for 5 min at room temperature to label biotinylated antibodies (CD3, CD19, and NK1.1) in a dump channel to exclude T cells, B cells, and natural killer (NK) cells. Following a final wash, cells were resuspended in 300 µL of FACS buffer. For each washing step, samples were filled up to 1.2 mL with FACS buffer and centrifuged at 1500 rpm for 5 min at 4°C. Finally, cells were filtered through a 35-µm cell strainer (#352235, Falcon) immediately before sorting on a Cytek Aurora CS cell sorter (63.6 psi sheath pressure; Cytek Biosciences, Fremont, CA) with SpectroFlo software. All procedures were conducted with minimal light exposure. Primary antibodies were used as follows: BUV395 CD45 (1:50; #564279, BD Biosciences), APC CD11b (1:50; #561690, BD Biosciences), PE/Dazzle 594 Ly6C (1:50; #128043, BioLegend), PerCP/Cyanine5.5 Ly6G (3:50; #127615, BioLegend), biotin CD3 (3:50; #100243, BioLegend), biotin CD19 (1:25; #115504, BioLegend), and biotin NK1.1 (1:50; #108703, BioLegend). Microglia were sorted as CD45+CD11b+Ly6C−Ly6G−Dump. For each collection of microglia from CFA model mice, ipsilateral lumbar (L1–L6) spinal cords were pooled from 6–8 mice.
Tissue Homogenization and RNA Isolation
Dissected L4-L5 spinal cord was snap-frozen in 1.5-mL nuclease-free tubes in dry ice or liquid nitrogen and stored at −80°C. Immediately before disruption, 3–4 ceramic homogenizer beads and ice-cold RNA Lysis/Binding buffer were added to each sample. Tissues were homogenized using an Omni Bead Ruptor (Omni International) for two cycles of 30 sec disruption until no large tissue chunks were observed. Samples were then centrifuged at 10,000×g for 20 min and the supernatant was collected for RNA extraction. Total RNA was isolated using the mirVana RNA isolation kit (#AM1561, Invitrogen) according to the manufacturer’s instructions, with on-column DNAse treatment (#12185010, Invitrogen) performed during Wash I. After RNA concentrations were determined using a NanoDrop ND1000 spectrophotometer, RNA samples were stored at −80°C.
RNA Sequencing (RNA-Seq) and Bioinformatics Analysis
RNA-seq was done as previously described.20 Briefly, total RNA was isolated from the dorsal horn of the lumbar spinal cord using the miRVana kit (Applied Biosystems). RNA concentration and integrity were assessed with the Agilent RNA 6000 Nano kit on a Bioanalyzer 2100 (Agilent Technologies). Libraries were generated by reverse transcription to double-stranded cDNA, followed by end repair, 3′ adenylation, adaptor ligation, purification, and PCR amplification. Sequencing was carried out on the BGISEQ-500 platform to obtain ~30 million 50 bp single-end reads per sample. Raw reads were filtered using SOAPnuke to remove low-quality reads, adaptor sequences, and reads with ambiguous bases. Clean reads were assembled into unigenes, annotated, and analyzed for expression levels and SNPs. Reads were mapped to the reference transcriptome with HISAT.35
For the microglia RNAseq experiment, the samples were collected in two batches and sequencing was conducted by Azenta Life Sciences. To minimize technical variation, batch correction was applied to raw read counts using ComBat-seq.36 Low-abundance transcripts (TPM < 1 across all samples) were excluded from further analysis. Data is visualized using principal component analysis and hierarchical clustering with Pearson correlation distance metric. Differentially expressed genes (DEGs) were identified from normalized log2-TPM values using a two-tailed t-test (p ≤ 0.01) with a fold-change cutoff of ≥ 2. Adjusted p values are reported using Benjamini and Hochberg false discovery rate multiple testing correction. DEGs were submitted to enrichr,37 to determine the biological annotations and pathways they are involved in. Specifically, we tested for enrichment of terms available in the ChEA, Reactome, GO biological process, transcription factor perturbations, Wiki pathway, and KEGG pathway libraries. Significant enrichments were determined with p ≤ 0.01 and at least two DEGs annotated.
Chromatin Immunoprecipitation (ChIP) and Bioinformatics Analysis
We conducted ChIP-seq using protocols adapted from Covaris truChIP chromatin shearing kit tissue SDS and Millipore magna ChIP G tissue kits as described before33 with an H3K4me1 antibody (Abcam ab8895) on spinal microglia 14 days after sEV injection from the two biological replicates per condition. Each replicate was generated by pooling 6–8 mice and analyzed as an independent biological replicate. ChIP-seq library preparation and sequencing were performed by Azenta Life Sciences. Libraries were generated from ChIP DNA using Azenta’s Illumina ChIP DNA Library Preparation workflow. Sequencing was carried out on an Illumina platform using a 2×150 bp paired-end configuration with single indexing, yielding approximately 350 million raw paired-end reads per lane (~105 GB). A minimum quality threshold of ≥80% bases ≥Q30 was achieved for all libraries. Azenta provided raw FASTQ files along with initial quality reports, adapter trimming, read mapping, and peak-calling outputs. For downstream analysis, raw sequencing reads were processed using a standardized computational pipeline. Sequencing adapters and low-quality bases were trimmed with Trimmomatic,38 and the resulting reads were aligned to the mm10 reference genome using Bowtie2.39 Alignments were filtered with samtools40 to retain primary concordant reads with a mapping quality ≥30. Peak calling was performed using MACS2,41 and peaks overlapping genome-specific blacklist regions were removed. Valid peaks across samples were merged; to ensure high stringency, only peaks consistently identified in both biological replicates were retained for further analysis.
Statistical Analysis
Data analysis was performed with GraphPad Prism 10.2.0. Sample sizes (n) represent biological replicates unless otherwise indicated. Behavioral data were analyzed using repeated measures two-way analysis of variance (ANOVA), followed by Bonferroni post-hoc test for multiple comparisons and presented as mean ± standard error of the mean (SEM). For RNA-seq analyses, adjusted p values were calculated using the Benjamini-Hochberg false discovery rate correction. Assumptions for statistical analyses were assessed where applicable.
Results
sEV Characterization
Typical diameter of sEVs range from 50–150 nm42 and our NTA analysis with standard beads of 70, 125, 200, and 400 nm in diameter, showed that the sEVs had an average diameter of 149.2 ± 4.9 nm (Figure 1A). We previously reported that sEVs from RAW 264.7 cells had a mean diameter of 113.6 ± 7.9 nm.20 Western blotting of sEVs confirmed the expression of the exosome marker protein CD81 but not the negative control protein calnexin (Figure 1B). We also characterized sEV markers using a MACSPlex flow cytometry bead-based capture assay and detected the common sEV markers including tetraspanins CD81, CD9, and CD63. sEV composition reflects characteristics of their parent cell43 and we observed expression of monocytic/macrophage markers in RAW 264.7 derived sEVs including CD115 (CSF1R) and CD86 (Figure 1C). These results indicate that sEVs isolated meet the MISEV guidelines31 for purity.
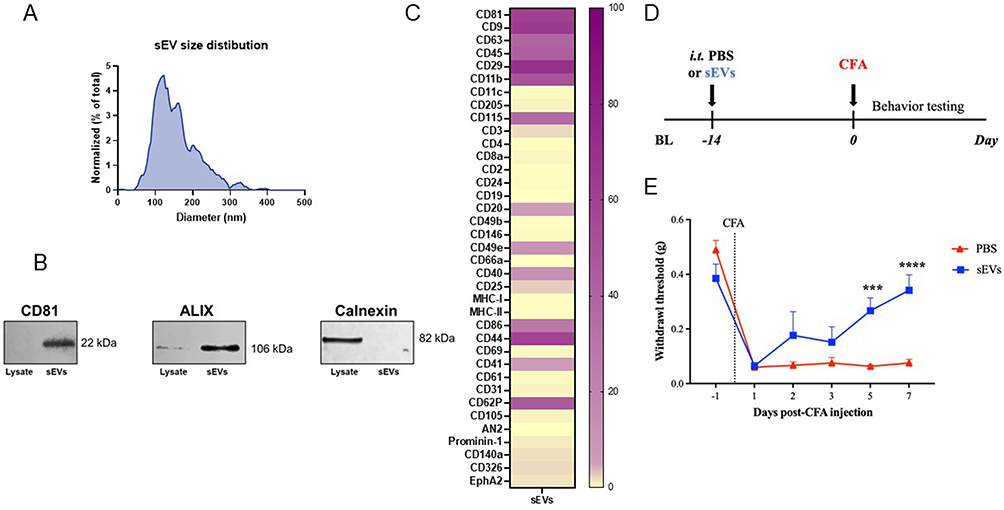 |
Figure 1 Confirmation of purity and in vivo efficacy of small extracellular vesicles (sEVs) from RAW 264.7 cells. (A) Nanoparticle tracking analysis (NTA) showing an average diameter of 149.2 ± 4.9 nm in sEVs isolated from the conditioned medium of RAW 264.7 cells. (B) Western blotting showing the presence of EV markers CD81 and ALIX in sEV preparations, with the negative marker calnexin only expressed in cell lysate. (C) Bead-based flow cytometry confirmed the presence of sEV markers such as tetraspanins CD81, CD9, and CD63 and monocytic/macrophage markers in RAW 264.7-derived sEVs including CD115 (CSF1R) and CD86. Percent weighted expression calculated from fraction of positive beads and normalized mean fluorescent intensity. Purple indicates high expression and yellow indicates low expression. (D) Schematic representation of prophylactic injection of sEVs in a mouse model of CFA-induced inflammatory pain. (E) Prophylactic intrathecal administration of 1µg sEVs 14 days before CFA injection in the hind paw induces faster resolution of mechanical sensitivity. Macrophage sEVs significantly alleviate inflammatory pain starting from day 5. Data shown as mean ± SEM, n=9 for PBS and n=6 for sEVs. ***, ****p < 0.001, 0.0001 repeated measures two-way ANOVA with Bonferroni test. Abbreviations: BL, baseline; CFA, complete Freund’s adjuvant; sEVs, small extracellular vesicles. |
Prophylactic Intrathecal sEV Administration Attenuates Inflammatory Pain
In our previous studies, prophylactic intrathecal administration of 1 µg of macrophage-derived sEVs two weeks prior effectively attenuated CFA-induced inflammatory pain in male C57BL/6J mice, starting from two days post-CFA injection.20 To independently confirm and replicate these effects in CFA model mice we followed the same paradigm (Figure 1D). Our data showed that macrophage-derived sEVs significantly reduced inflammatory pain hypersensitivity on days 5 and 7 after CFA injection (Figure 1E). Overall, our behavioral results demonstrate that macrophage-derived sEVs accelerate recovery from CFA-induced pain.
Microglial Depletion and Repopulation Using PLX5622 Does Not Influence Normal Sensitivity in Mice
To study the role of microglia in sEV-mediated pain resolution, we used a selective colony stimulating factor 1 receptor (CSF1R) inhibitor PLX5622. CSF1R serves as a key regulator of microglia and macrophages and is essential for myeloid cell survival.44 Numerous studies have validated the use of CSF1R inhibitors in microglial ablation.45 Interestingly, microglia can be fully repopulated in the CNS within 7 days after the cessation of CSF1R inhibition.46 This is particularly advantageous for our study, as the absence of microglia may impact the development and maintenance of inflammatory pain,24,28 making it difficult to interpret behavioral data. To induce microglial ablation, we fed the mice with PLX5622 (1200 ppm) in AIN-76A diet (PLX). AIN-76A diet (control diet or CD) was used as the control. We examined microglial depletion in lumbar spinal cord in mice fed with PLX for 7 and 11 days. IHC was performed on lumbar segments (L4-L5) collected from these mice using the microglial-specific marker Iba1 (shown in red) and counterstained with DAPI (blue) to identify microglia (Figure 2). Confocal images confirmed that microglia were ablated in spinal cord 7 days after PLX diet, with further depletion over an additional 4 days of PLX diet (Figure 2A). To ensure complete ablation of microglia at the time of sEV injection, we extended the feeding schedule of PLX diet to 14 days. After depletion, the PLX diet was replaced with CD for a minimum of 10 days, allowing sufficient time for microglial repopulation to occur prior to CFA injection. Microglia were repopulated in the lumbar spinal cord 10 days after cessation of the PLX5622 diet (Figure 2B).
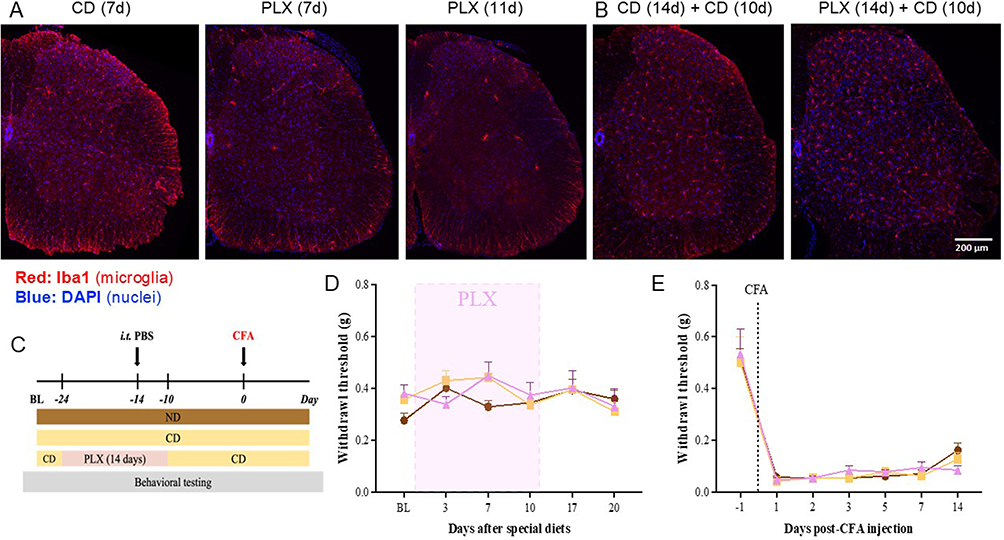 |
Figure 2 Depletion and repopulation of microglia in lumbar spinal cord and its effect on CFA-induced inflammatory pain. (A) IHC images of lumbar spinal cord (L4-L5) confirm depletion of microglia 7 days and 11 days after PLX5622 diet. (B) IHC also confirms microglial repopulation in L4–L5 spinal cord when mice were fed for 14 days with PLX5622 diet, followed by 10 days of control diet. Iba1 shown in red was used as microglial marker and counterstained with DAPI (blue). Scale bar = 200 µm. (C) Schematic of experiment to test if repopulated microglia alter CFA-induced hypersensitivity. In PLX group, mice were fed for 14 days with PLX5622 diet (before and after intrathecal injection of PBS), followed by 10 days of control diet. Comparison of the effects of three types of diets on normal sensitivity (D) and CFA-induced pain hypersensitivity (E) No differences in mechanical allodynia were observed among these three diets before and after CFA injections (repeated measures two-way ANOVA, Bonferroni post-test). Data presented as mean ± SEM (n=6 for ND; n=7 for CD and PLX). Abbreviations: CD, control AIN-76A diet; PLX, AIN-76A diet containing PLX5622. |
To investigate whether microglial depletion and repopulation affect basal sensitivity and CFA-induced pain hypersensitivity, we conducted behavioral testing using von Frey filaments on mice fed with different diets before and after CFA injection. We also included a group of mice that were fed with a normal diet (ND) provided by our animal facility for comparison with CD and PLX diets. The schematic of experimental design is shown in Figure 2C. Mice on CD and PLX diets showed no significant alteration in basal pain thresholds (Figure 2D). Moreover, mice on all diets demonstrated similar pain sensitivity in response to CFA injection (Figure 2E). We conclude that microglial ablation and repopulation does not influence normal mechanical sensitivity in mice.
Microglia are Indispensable for Macrophage sEV-Induced Prophylaxis
We next investigated whether prophylactic sEVs could still confer early pain resolution when microglia were absent during sEV administration. After 10 days on PLX diet, mice were injected i.t. with 1 µg of sEVs or PBS. Microglial depletion continued for an additional 4 days, after which PLX diet was replaced with CD for the rest of the study (Figure 3A). Control mice were fed with CD throughout the experiment. Prophylactic sEVs provided protection against CFA-induced pain in CD-fed mice, starting from day 5 and continuing until day 14 (Figure 3B). However, the early resolution of mechanical hypersensitivity induced by sEVs was completely abolished in PLX-fed mice (Figure 3B), indicating that microglia at the time of sEV administration were necessary for sEVs to promote faster resolution of inflammatory pain in CFA model mice.
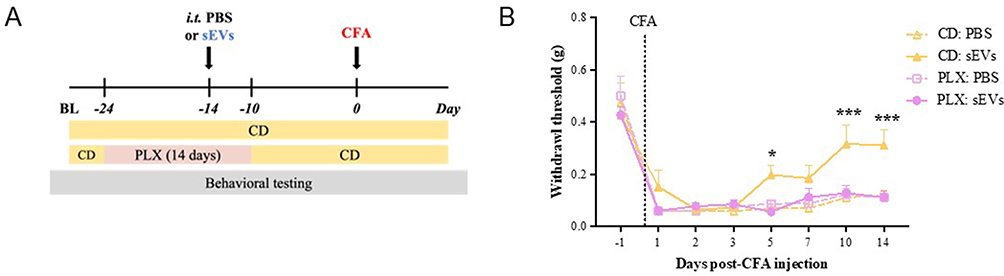 |
Figure 3 Spinal cord microglia are essential for macrophage sEV-induced prophylaxis. (A) Experimental design. Mice were fed with PLX5622 diet (PLX) or AIN-76A control diet for 14 days (10 days before and 4 days after) sEV injection. (B) sEV-induced prophylaxis was diminished in the mice fed with PLX diet during sEV injection. von Frey test for mechanical sensitivity showed that pain attenuation conferred by sEVs is abolished in PLX-fed mice. All mice received CFA. Data presented as the mean ± SEM (n=7-8; *p < 0.05, ***p < 0.001 vs. PLX: sEVs, repeated measures two-way ANOVA with Bonferroni post-hoc test). Abbreviations: BL, baseline; CFA, complete Freund’s adjuvant; sEVs, small extracellular vesicles; CD, control AIN-76A diet; PLX, AIN-76A diet containing PLX5622. |
sEVs Induce Gene Expression Changes in Lumbar Spinal Cord 7 Days After I.t. Injection
To evaluate the transcriptional effects of sEV treatment on spinal cord tissue, we performed bulk RNA-seq on lumbar spinal cord segments (L4-L5) collected 7 days after intrathecal injection of sEVs or PBS. We identified 3 upregulated and 34 downregulated genes in sEV-treated spinal cords compared to PBS controls. The top 10 DEGs are listed in Table 1 and the whole list is shown in Supplementary Table 1. Notably, several small nucleolar RNAs (eg, Snord116, Snord35a, and Snord89) and microRNAs (eg, mir-9-2, mir-453) were among the most significantly altered, suggesting that sEVs may influence non-coding RNA regulation in the spinal cord. Three genes, somatostatin receptor 4, carbonic anhydrase 1, and regulatory subunit of type II PKA R-subunit (RIIa) domain containing 1 have been implicated in regulating pain.47–49 To further investigate the biological relevance of the transcriptional changes, we performed gene set enrichment analysis (GSEA), which revealed significant associations with oxidative stress and neuroendocrine signaling. Genes altered by sEV treatment were enriched in NFE2L2 knockout signatures, suggesting modulation of antioxidant pathways. Additional enrichment was observed in pathways related to peptide hormone secretion and neuropeptide signaling, indicating potential effects on hormonal and synaptic regulation within the spinal cord (Supplementary Table 1).
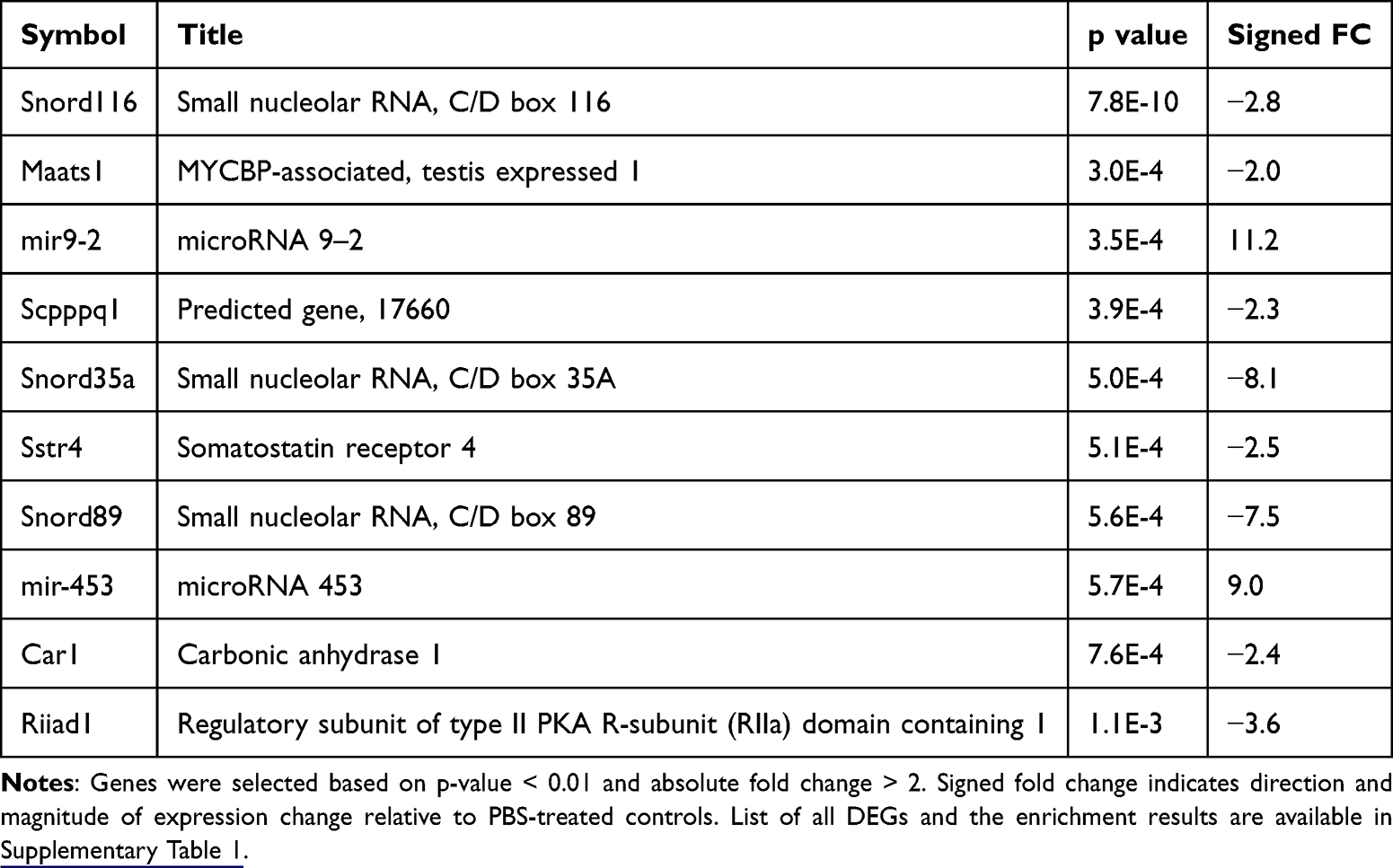 |
Table 1 The Top 10 Differentially Expressed Genes in Lumbar Spinal Cord Tissue 7 Days After sEV Treatment |
sEV Treatment Alters Microglial Gene Expression in Lumbar Spinal Cord at 14 Days
Next we used isolated microglia from the whole spinal cord of mice. Mice that received PBS were used as control. To obtain sufficient RNA yield for RNA-seq, spinal cord samples were pooled. Each sequencing replicate consisted of a pool of 6–8 mice. Each pooled sample was treated as one biological replicate for downstream analyses. We used two MACS and two FACS-sorted CD11b+ microglia. The gating strategy is shown in Supplementary Figure 1. One sEV sample (sEV2) was determined to be an outlier via PCA and clustering and was excluded from the analysis (Supplementary Figure 2A and B). Differential expression analysis of the remaining samples identified 24 upregulated and 26 downregulated genes in sEV-treated microglia compared to PBS treated control microglia. Most of these DEGs were predicted or unannotated genes; top 10 annotated DEGs are listed in Table 2 and the full list is included in Supplementary Table 2. Gene set enrichment analysis revealed that the DEGs were significantly associated with immune and inflammatory signaling pathways. Notably, enriched terms included cytokine-cytokine receptor interaction, chemokine signaling, and malaria-related pathways, driven by genes such as CD40LG, CCL8, XCL1, and CCR1L1. These findings suggest that sEVs modulate microglial immune activity and chemokine-mediated communication in the spinal cord.
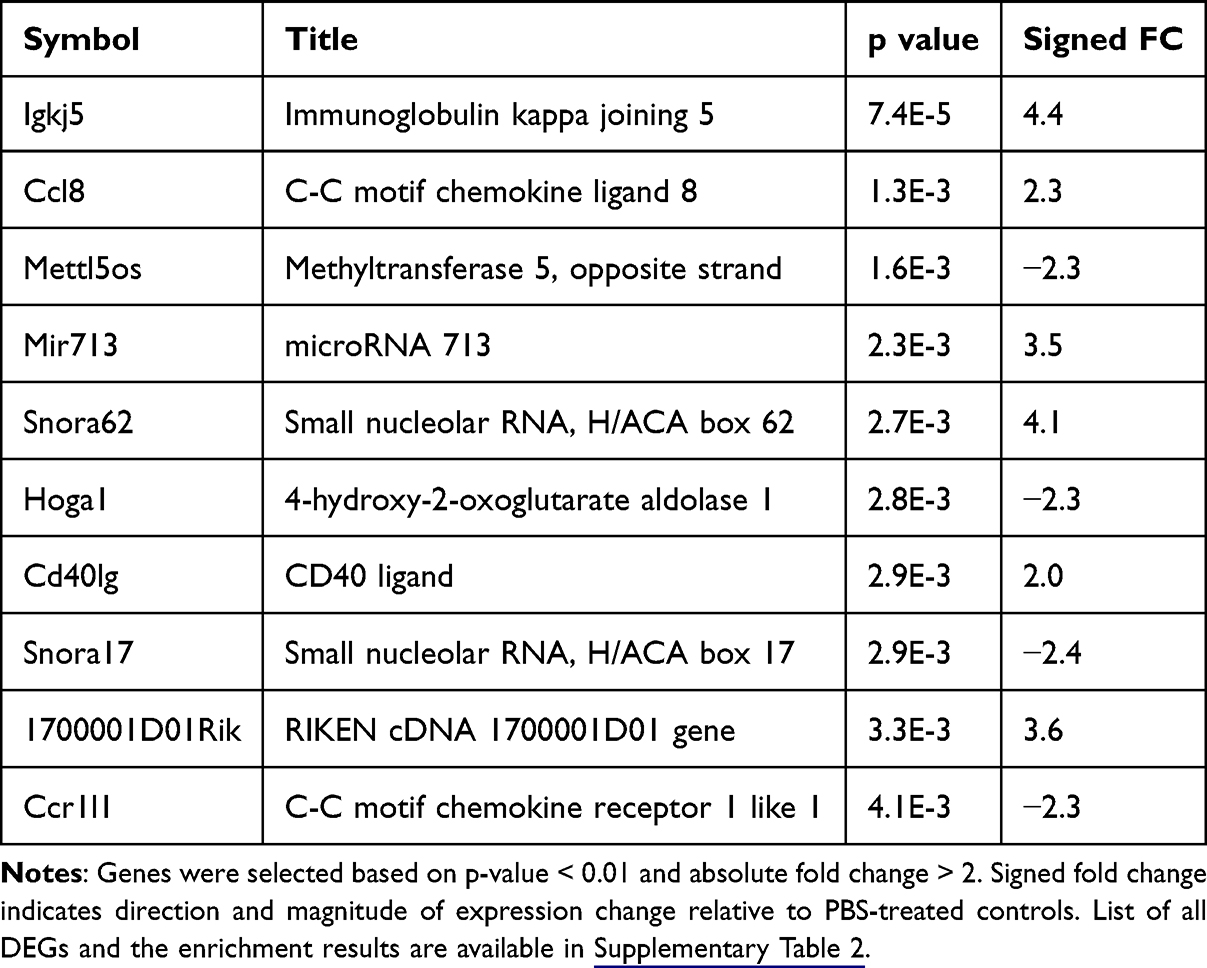 |
Table 2 The Top 10 Differentially Expressed Genes (Excluding Predicted or Unannotated Genes) in Spinal Cord Microglia Isolated 14 Days After sEV Treatment |
sEVs Alter H3K4me1 Enrichment in Spinal Microglia
To determine if the alterations in H3K4me1 enrichment following sEV treatment contribute to the resolution of CFA-induced inflammatory pain, we conducted chromatin immunoprecipitation followed by sequencing (ChIP-seq) with an H3K4me1 antibody on MACS-sorted CD11b+ microglia from ipsilateral lumbar L1-L6 spinal cord of CFA model mice that prophylactically received PBS or sEVs 14 days prior to CFA injection. Each replicate consisted of a pool of 6–8 mice and from the two biological samples per condition, we identified 12 merged annotated peaks in spinal microglia from PBS-treated mice and 85 merged annotated peaks in sEV-treated samples (Supplementary Table 3). There were no overlapping peaks between the two conditions. In the PBS-treated sample, most peaks (83.33%) were annotated to the promoter regions within 1 kb of the transcription start site (TSS), while the remainder were located in the distal intergenic regions (> 1.5 kb from TSS). Similarly, in the sEV-treated group, the majority of the peaks were also annotated to the promoter regions, and the average signal profiles indicated that the binding of H3K4me1 was enriched at TSSs. To explore the functional relevance of these sEV-specific H3K4me1-enriched regions, we performed GSEA using the genes associated with peaks present only in the sEV-treated group (Supplementary Table 3). The analysis revealed strong enrichment for targets of Polycomb group proteins and chromatin regulators, including SUZ12, MTF2, JARID2, and EZH2, suggesting that sEVs may influence epigenetic remodeling in spinal microglia. Additionally, enriched pathways included neuronal signaling, chemokine signaling, and synaptic regulation, with genes such as Gata2, Cacna1e, Doc2b, Pde10a, and Bcl11b contributing to these signatures. These findings support the hypothesis that sEVs modulate microglial function through epigenetic mechanisms that prime transcriptional programs relevant to neuronal and immune signaling.
Cross-Experiment Comparisons Reveal Distinct Gene and Pathway Responses to sEV Treatment, with Transcription Factor-Mediated Links
To assess the relationship between transcriptional and epigenetic responses to sEV treatment, we compared results across bulk RNA-seq (7 days post sEV treatment lumbar spinal cord), purified microglial RNA-seq (14 days post sEV treatment), and microglia ChIP-seq (14 days post sEV treatment) experiments. Because these datasets represent distinct biological layers, including tissue-level transcription, cell-specific transcriptional responses, and chromatin-associated regulatory states, direct overlap between datasets was not necessarily expected. As shown in Figure 4A, there were no overlapping genes among the datasets, indicating that sEVs modulate distinct gene sets depending on cell type and time point. Similarly, gene set enrichment analysis revealed no shared pathways across experiments (Figure 4B), suggesting functional divergence in sEV-induced responses. To identify potential regulatory relationships despite limited direct overlap between datasets, we focused on transcription factors identified as either differentially expressed or enriched in one dataset and mapped their known targets in other datasets. Notably, only transcription factors differentially expressed or enriched in the ChIP-seq dataset were found to target the differentially expressed genes in other datasets. These relationships, visualized in Figure 5, highlight transcription factor-mediated links across experiments and suggest that sEVs orchestrate coordinated but compartmentalized regulatory programs in spinal cord microglia and tissue.
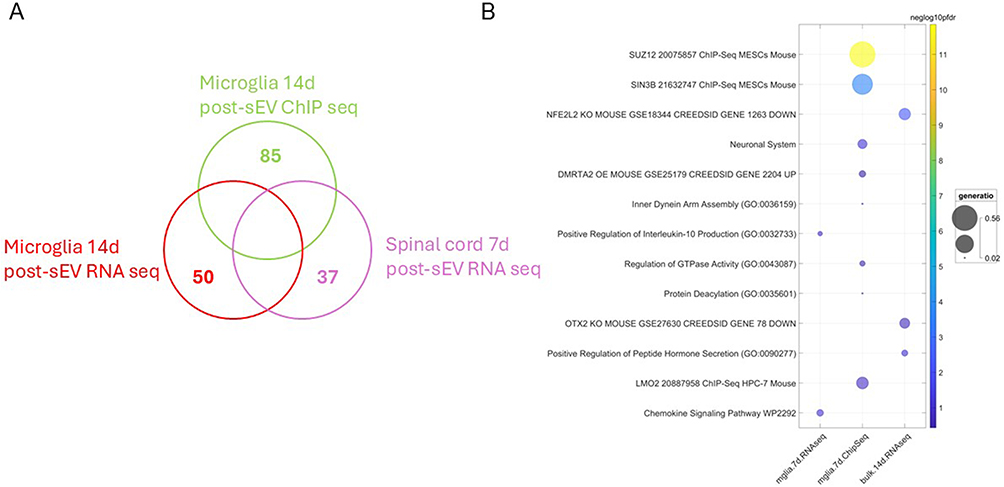 |
Figure 4 Cross-experiment comparisons reveal distinct gene and pathway responses to sEV treatment. (A) Venn diagram showing the uniqueness and lack of overlap of genes identified across bulk RNA-seq, microglia RNA-seq, and ChIP-seq experiments. Each set represents genes significantly altered in spinal cord tissue or microglia after sEV treatment. (B) Bubble chart of enriched gene sets from different experiments. Bubble size is proportional to the fraction of enriched DEGs annotated to that term (gene ratio) and the color reflects statistical significance of enrichment, scaled by the negative log10 of the false discovery rate (FDR). To reduce redundancy, gene sets sharing more than 50% of their genes were filtered, retaining only the most statistically significant term. |
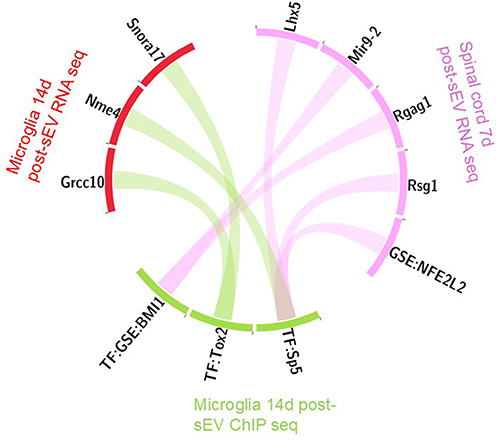 |
Figure 5 Circos diagram illustrating transcription factor-mediated regulatory connections across datasets. Transcription factors (TF) that were either differentially expressed or were enriched in the ChIP-seq dataset were mapped to their known target genes identified in RNA-seq experiments, linking chromatin-associated regulatory programs with tissue-level and microglia-specific transcriptional responses. There were no such connections originating from the RNA-seq experiments. |
Methyltransferase Activity of SETD7 is Essential for Prophylaxis Conferred by sEVs
To determine the necessity of H3K4me1 deposition for sEVs to confer faster resolution of pain hypersensitivity, we treated the mice with PFI-2, a selective methyltransferase inhibitor which blocks the methyltransferase activity of SETD750 (Figure 6A). PFI-2 was given via intrathecal catheters immediately after sEV delivery and repeated every day for an additional four days. Detailed experimental design and grouping are listed in Supplementary Table 4. Mice treated with PFI-2 showed no difference in mechanical thresholds post-CFA as compared to vehicle, indicating PFI-2 treatment alone did not affect the induction of the CFA model (Figure 6B). Mice treated with sEVs followed by PFI-2 however showed significantly lower mechanical thresholds at days 7, 10, and 14 post-CFA than those treated with sEVs and vehicle. However, it is worth noting that multiple prophylactic injections of DMSO in the control groups (VEH: PBS, VEH: sEVs, and PFI-2: PBS), also facilitated faster recovery from CFA-induced pain as DMSO has anti-inflammatory effects.51 Despite the confounding effects of DMSO, VEH: sEVs were still able to provide significant pain protection at day 14 post-CFA compared to VEH: PBS.
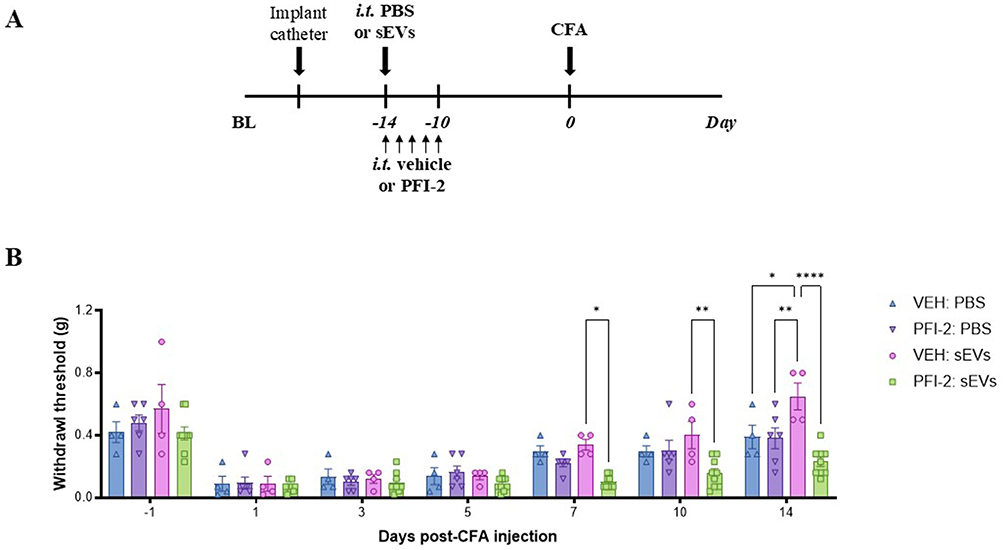 |
Figure 6 Inhibition of SETD7 mono-methylation activity blocks sEV-induced pain attenuation. (A) Schematic representations of the experimental design. Mice were implanted with intrathecal catheters for repeated intrathecal injections. On the first injection day, mice received either sEVs or PBS. Additionally, all mice were administered PFI-2 or DMSO (VEH) for five consecutive days. Two weeks after sEV or PBS injection, CFA was injected into the hind paw. (B) Mechanical sensitivity tested in mice with distinct intrathecal injections. Data shown as mean ± SEM (n=4-9) and analyzed by repeated measures two-way ANOVA with Bonferroni multiple comparison test. *, **, ****p < 0.05, 0.01, 0.0001, respectively. Abbreviations: BL, baseline; sEVs, small extracellular vesicles; CFA, complete Freund’s adjuvant; VEH, vehicle. |
Discussion
The concept of innate immune memory is largely unexplored in the context of sEV biology52 and pain. Based on our observation that 14-day prophylactic administration of sEVs from RAW 264.7 macrophage leads to faster resolution of mechanical and thermal hypersensitivity induced by CFA, we investigated whether these macrophage-derived sEVs confer long-lasting memory to induce protection against a subsequent painful insult. Recent findings suggest that microglia, the tissue-resident long-lived macrophages and primary innate immune cells of the CNS, can develop this form of immune memory.14,53 Our previous work illustrated that spinal microglia take up macrophage-derived sEVs when injected intrathecally.20,54 Thus, we hypothesized that microglia in the spinal cord are involved in sEV-induced prophylaxis. To test this, we used PLX5622, a highly selective CSF1R inhibitor, to ablate microglia.44
PLX5622 can be formulated into a chow diet, which minimizes the stress on mice from multiple injections. We first confirmed microglial ablation in the spinal cord with the PLX5622 diet. IHC results showed that microglia were ablated in the lumbar (L4–L5) spinal cord 7 days after initiation of PLX5622 diet compared to control with an additional qualitative decrease in microglia numbers by day 11. This is in line with previous studies that a 14-day PLX5622 treatment is required to induce a stable microglial depletion in CNS.55,56 Therefore, we adhered to a 14-day PLX5622 treatment for subsequent studies. Interestingly, cessation of PLX5622 leads to the repopulation of microglia.46 Since microglia play a significant role in regulating inflammatory pain,24 we switched mice back to a control diet for at least 10 days following the PLX5622 diet to allow for maximal microglia repopulation in the spinal cord which we also confirmed by IHC. To rule out any confounding factors from the PLX5622 diet, we also confirmed that neither the PLX5622 nor control diet affected basal sensation. To ensure microglia depletion at the time of intrathecal PBS/sEV injection but sufficient time for microglial repopulation prior to CFA we injected PBS/sEVs intrathecally 10 days after initiation of PLX5622 treatment.
Behavioral data demonstrated that sEV-mediated pain prophylaxis was completely lost in mice fed with PLX5622 diet during sEV delivery. This finding suggests the crucial role of microglia in the prophylactic effects of sEVs in the CFA model. However, although PLX5622 is widely used to eliminate microglia in the CNS, and depleted cells are generally identified as microglia, it should be noted that CSF1R is also expressed in other myeloid cells, including macrophages and myeloid progenitors.57 Even short-term PLX5622 treatment can induce persistent changes in myeloid and lymphoid cells.58 In addition, it is possible that sEVs indirectly modulate microglia by acting on other cell types within the spinal cord, which then impart epigenetic changes in microglia during the early phase (4 days after sEV injection when PLX5622 was discontinued). The timing of depletion and repopulation poses difficulties for conducting short-term depletion/repopulation in the late phase after administering sEVs. Moreover, a recent study has shown that depleting microglia during stimulation can lead to morphologically distinct microglia with altered transcriptomes.59 We cannot rule out that administrating sEVs during microglial depletion may influence the repopulated microglia, although we saw no effect of microglia depletion alone on mechanical thresholds post-CFA induction.
One characteristic of innate immune memory is that innate cells, once triggered by a primary stimulus for a transient response, typically return to their basal transcription level, leaving the changes at the epigenetic level and/or in cellular metabolism. These modifications are responsible for subsequent transcriptional alterations upon later or secondary triggers to form a heightened and rapid response.60 Active enhancers are identified by enrichments of both H3K27ac and H3K4me1, while H3K4me1 alone indicates an enhancer in an inactive, poised state. Immune stimulation results in de novo deposition of H3K27ac at distal regulatory regions and leads to increased H3K4me1 marks, which persist even after the primary stimulus and H3K27ac marks are removed.12 Enhancers carrying only H3K4me1 facilitate a more rapid histone acetylation and gene induction upon subsequent stimulation. Thus, the retention of H3K4me1 contributes to an epigenomic memory of the initial stimulation.15 We first investigated transcriptomic changes in PBS- and sEV-treated mice by performing bulk RNA-seq on lumbar spinal cord collected 7 days after sEV injection. We observed 3 upregulated and 34 downregulated genes. Several small nucleolar RNAs and miRNAs (miR-9-2, miR-453) were among the most altered, suggesting sEV-mediated modulation of noncoding RNA expressions. We then performed RNA-seq on CD11b+ microglia isolated from the whole spinal cord and identified 24 upregulated and 26 downregulated genes in sEV-treated microglia compared to control. GSEA showed that the DEGs were associated with immune and inflammatory signaling pathways suggesting that sEVs modulate microglial immune activity and chemokine-mediated communication in the spinal cord.
As gene expression changes in sEV treated mice at day 7 and 14 post-CFA were limited compared to controls, the factors that contribute to subsequent protection may be due to regulatory events at the epigenetic level. Enhancers are enriched for DNA motifs recognized by TFs61 and serve as platforms that integrate transcription factor binding and, through chromatin looping, communicate with gene promoters to modulate transcriptional output.62 RNA polymerase II gene transcription burst frequency is primarily encoded in enhancers and burst size in core promoters.63 Enhancers can thus fine-tune gene expression by bridging transcription factors and the RNA polymerase II complex, thereby linking regulatory activity to gene transcription. Our data suggest that sEVs modulate microglial function through epigenetic priming of neuronal and immune signaling programs. It is important to note that the current analysis is unable to identify H3K4me1 peaks in enhancer regions. Enhancers can be located up to 1 million bp (Mbp) away from the TSS. Therefore a combination of other histone marks, such as H3K4me3 and H3K27ac,64 or more sophisticated algorithms65 are necessary to determine if putative enhancer regions with long-lasting H3K4me1 depositions are present.
To examine the interplay between transcriptional and epigenetic responses to sEV treatment, we integrated RNA-seq data (lumbar spinal cord 7 days), microglia (14 days), and microglia ChIP-seq (14 days). There were no overlapping genes detected across these datasets, and gene set enrichment analysis revealed no shared pathways, indicating that sEVs influence distinct gene networks depending on cell type and temporal context. To explore potential regulatory connections, we focused on transcription factors differentially expressed or enriched within each dataset and mapped their known targets across experiments. Notably, only transcription factors with peaks identified in the ChIP-seq dataset were predicted to regulate differentially expressed genes in the RNA-seq datasets. These relationships suggest that sEVs may exert coordinated yet compartmentalized control of transcriptional programs in spinal microglia and surrounding tissue. The absence of overlap between bulk spinal cord (RNA-seq) and microglia-specific (RNA-seq/ChIP-seq) datasets or shared pathway enrichment may suggest temporal differences but is also in alignment with emerging evidence that microglial epigenomic remodeling is frequently uncoupled from steady-state transcriptional output. This is consistent with prior work showing that microglia-specific transcriptional signatures are often masked in heterogeneous cells66,67 Additionally, microglia undergo extensive enhancer reorganization in response to environmental cues or injury without corresponding immediate gene expression changes. Microglial enhancer landscapes are highly plastic and respond to cytokines, neuronal signals, and environmental perturbations, yet many new or primed enhancers remain transcriptionally silent until secondary stimulation.68,69 Similarly, microglia exposed to chronic stimuli or peripheral inflammation can acquire latent or trained epigenetic states that do not manifest in baseline transcriptomes but alter future responsiveness.12,14 Temporal dynamics are also critical as microglial transcription typically shows rapid, transient waves after injury or immune stimulation,66,67 whereas enhancer remodeling marked by H3K4me1 can persist for weeks after initial activation. Together, these observations suggest that the sEV-induced microglial chromatin changes we observe at 14 days may represent a long-lasting regulatory state that does not necessarily coincide with stable transcriptional changes at the same time point or in the surrounding tissue environment.
We next tested if inhibiting the H3K4 methyltransferase SETD7 to prevent H3K4me1 deposition abolished sEV-induced pain attenuation. SETD7 has been identified for its role in mono-methylation of H3K470,71 in the context of innate immune memory. Increased expression of SETD7 in spinal microglia from mice with chronic constriction injury (CCI) correlated with elevated H3K4me1 levels.72 We utilized the selective SETD7 inhibitor (R)-PFI-2 to inhibit SETD7 and reduce H3K4me1 deposition, following the same pharmacological approach that mitigated CCI-induced pain.72 SETD7 inhibition for 5 consecutive days at the time of sEV administration significantly reduced the mechanical thresholds post-CFA as compared to vehicle treated mice, suggesting the importance of H3K4me1 deposition for mediating prophylactic sEV mediated pain resolution.
While these findings provide important insights, this study has limitations. Although we performed ChIP-seq for H3K4me1 to identify putative enhancer elements in spinal microglia following sEV treatment, we did not perform complementary ChIP-seq for H3K27ac. It is known that H3K4me1 alone marks primed enhancers, whereas H3K27ac is required to identify enhancers that are transcriptionally active or dynamically regulated in response to environmental cues.73 Because active enhancers are typically defined by the combinatorial presence of H3K4me1 and H3K27ac, the absence of H3K27ac profiling limits our ability to distinguish poised from active enhancer states and to comprehensively map stimulus-responsive regulatory regions. Thus, incorporating H3K27ac ChIP-seq in future work will be essential for a more complete understanding of enhancer activation in microglia in response to sEVs. In addition, performing RNA-seq studies after CFA induction, and correlating them with ChIP-seq results would aid in confirming functional alterations in a secondary response for genes with altered H3K4me1 deposition. Another limitation is that only male mice were used in this study. Although a male-specific microglial contribution to neuropathic pain is reported,28 subsequent work has shown that this dichotomy is not universal. Several studies have reported microglial activation and functional involvement in females as well, depending on the pain model, injury type, and inflammatory context.29 Microglia exhibit sex differences not only in pain mechanisms but also in their transcriptional and epigenomic landscapes. Prior studies have shown that male and female microglia differ in baseline chromatin accessibility, enhancer usage, and responsiveness to inflammatory stimuli, which can shape divergent pain pathways.27,74 Though sEVs are efficacious in both sexes, future work will determine whether sEV-mediated microglial epigenomic responses differ between males and females.
Overall, our findings showed that intrathecal injection of macrophage-derived sEVs required microglia to exert their prophylactic effects on accelerating inflammatory pain resolution. Inhibition of H3K4 mono-methyltransferase SETD7 during prophylactic sEV administration also prevented accelerated pain resolution. Given sEV induced alterations in H3K4me1 enrichment on specific gene loci in microglia, these changes may contribute to the prophylactic effects against CFA-induced pain.
Our data support a chromatin-based mechanism in which sEV exposure induces persistent changes in the microglial epigenetic landscape, including enrichment of H3K4me1-marked loci. Inhibition of SETD7 abrogated this effect, implicating H3K4 monomethylation. Integrative analysis of bulk spinal cord RNA-seq, microglia-specific RNA-seq, and H3K4me1 ChIP-seq datasets revealed modest overlap between transcriptional and chromatin signatures. Rather than indicating discordance, this pattern is consistent with enhancer–transcription uncoupling, where H3K4me1-marked primed enhancers confer transcriptional competence without immediate gene expression.11 In this context, sEV exposure appears to establish a latent, memory-like regulatory state in microglia. This framework may explain the divergence across datasets, reflecting temporal separation between enhancer priming and transcriptional activation, cell-type specificity not captured in bulk tissue, and latent regulatory potential of H3K4me1-marked loci. These findings align with concepts of tolerance, suggesting that sEV-induced chromatin remodeling may facilitate resolution-phase responses rather than amplify inflammation. While direct links between specific enhancers and transcriptional outputs were not established, the data support a model in which SETD7-dependent chromatin remodeling encodes regulatory potential that may be mobilized upon subsequent challenge.
Our findings suggest that macrophage-derived sEVs promote an enhancer-primed microglial state associated with accelerated resolution of inflammatory pain. Limitations of the current study include the absence of secondary stimulus-response analyses and the potential contribution of other cell types. In addition, while several differentially expressed genes, non-coding RNAs, and regions exhibiting altered H3K4me1 enrichment in spinal microglia may contribute to the observed phenotype, including a few previously implicated in pain regulation,47–49 their functional relevance was not examined in the current study and will require future mechanistic investigation. Future studies incorporating temporally resolved and stimulus-response paradigms, as well as defining the molecular mediators downstream of sEV-induced chromatin remodeling, will be important to directly link enhancer dynamics with transcriptional outputs and functional outcomes. Although chronic pain is prevalent, a prophylactic strategy has not yet been tested in chronic pain models. Other prophylactic strategies that induce innate immune memory such as administration of live vaccines, induce protection against non-target diseases,75 which suggests that prophylactic macrophage sEVs could offer non-specific protection against subsequent pain insults. This opens the possibility for such prophylactic strategies to be extended to other pain and non-pain conditions and decrease opioid prescriptions. Our findings here provide a mechanistic basis that lays the foundation for these possibilities.
Data Sharing Statement
The data supporting the findings of this study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was conducted and reported in accordance with the ARRIVE 2.0 guidelines for animal research. All procedures involving animals were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and were approved by the Institutional Animal Care and Use Committee (IACUC) of Drexel University College of Medicine (Record Number LA-25-036). All anesthesia and euthanasia procedures were performed in accordance with institutional IACUC approval and the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. For non-perfusion studies, mice were euthanized by isoflurane inhalation anesthesia followed by decapitation as a secondary physical method to ensure death. For immunohistochemistry studies requiring transcardial perfusion, mice were deeply anesthetized with intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg). Adequate depth of anesthesia was confirmed prior to perfusion. Mice were then transcardially perfused with phosphate-buffered saline followed by fixative solution. Death occurred as a result of exsanguination during perfusion.
Acknowledements
A version of this paper was made available as a pre-print: https://www.biorxiv.org/content/10.64898/2026.01.15.699551v1.
Author Contributions
XL: Data curation, Funding acquisition, Investigation, Project administration, Validation, Visualisation, Writing - Original draft.
JRW: Funding acquisition, Investigation, Validation, Visualisation, Writing - Review and editing.
JTD: Funding acquisition, Investigation, Visualisation, Writing - Review and editing.
YT: Investigation, Validation, Writing - Review and editing.
AS: Data curation, Funding acquisition, Visualisation, Supervision, Writing - Original draft.
SKA: Conceptualisation, Funding acquisition, Project administration, Supervision, Visualisation, Writing - Original draft.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is supported by grant from NIH NINDS R01NS129191 to Seena Ajit. Xuan Luo and Jason Wickman are recipients of Dean’s Fellowship for Excellence in Collaborative or Themed Research from Drexel University College of Medicine.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Ji -R-R, Chamessian A, Zhang Y-Q. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354(6312):572–20. doi:10.1126/science.aaf8924
2. Dogan N, Wu W, Morrissey CS, et al. Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenet Chromatin. 2015;8:16. doi:10.1186/s13072-015-0009-5
3. Ramaker RC, Hardigan AA, Goh ST, et al. Dissecting the regulatory activity and sequence content of loci with exceptional numbers of transcription factor associations. Genome Res. 2020;30(7):939–950. doi:10.1101/gr.260463.119
4. Cirovic B, de Bree LCJ, Groh L, et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe. 2020;28(2):322–334e5. doi:10.1016/j.chom.2020.05.014
5. de Laval B, Maurizio J, Kandalla PK, et al. C/EBPbeta-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell. 2020;26(5):793. doi:10.1016/j.stem.2020.03.014
6. Litman GW, Anderson MK, Rast JP. Evolution of antigen binding receptors. Annu Rev Immunol. 1999;17:109–147. doi:10.1146/annurev.immunol.17.1.109
7. Netea MG, Latz E, Mills KH, O’Neill LA. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol. 2015;16(7):675–679. doi:10.1038/ni.3178
8. Netea MG, Joosten LA, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098. doi:10.1126/science.aaf1098
9. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. 2011;9(5):355–361. doi:10.1016/j.chom.2011.04.006
10. Lajqi T, Lang G-P, Haas F, et al. Memory-like inflammatory responses of microglia to rising doses of LPS: key role of PI3Kγ. Original Research. Front Immunol. 2019. doi:10.3389/fimmu.2019.02492
11. Ostuni R, Piccolo V, Barozzi I, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152(1–2):157–171. doi:10.1016/j.cell.2012.12.018
12. Saeed S, Quintin J, Kerstens HHD, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. doi:10.1126/science.1251086
13. Quintin J, Saeed S, Martens JHA, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12(2):223–232. doi:10.1016/j.chom.2012.06.006
14. Wendeln AC, Degenhardt K, Kaurani L, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556(7701):332–338. doi:10.1038/s41586-018-0023-4
15. Bekkering S, Domínguez-Andrés J, Joosten LAB, Riksen NP, Netea MG. Trained immunity: reprogramming innate immunity in health and disease. Annu Rev Immunol. 2021. doi:10.1146/annurev-immunol-102119-073855
16. Groot Kormelink T, Mol S, de Jong EC, Wauben MHM. The role of extracellular vesicles when innate meets adaptive. Semin Immunopathol. 2018;40(5):439–452. doi:10.1007/s00281-018-0681-1
17. Kim SH, Kim S, Oligino TJ, Robbins PD. Effective treatment of established mouse collagen-induced arthritis by systemic administration of dendritic cells genetically modified to express FasL. Mol Ther. 2002;6(5):584–590.
18. McDonald MK, Tian Y, Qureshi RA, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain®. 2014;155(8):1527–1539. doi:10.1016/j.pain.2014.04.029
19. Shiue SJ, Rau RH, Shiue HS, et al. Mesenchymal stem cell exosomes as a cell-free therapy for nerve injury-induced pain in rats. Pain. 2019;160(1):210–223. doi:10.1097/j.pain.0000000000001395
20. Jean-Toussaint R, Lin Z, Tian Y, et al. Therapeutic and prophylactic effects of macrophage-derived small extracellular vesicles in the attenuation of inflammatory pain. Brain Behav Immun. 2021;94:210–224. doi:10.1016/j.bbi.2021.02.005
21. Mianehsaz E, Mirzaei HR, Mahjoubin-Tehran M, et al. Mesenchymal stem cell-derived exosomes: a new therapeutic approach to osteoarthritis? Stem Cell Res Ther. 2019;10(1):340. doi:10.1186/s13287-019-1445-0
22. Inoue K, Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci. 2018;19(3):138–152. doi:10.1038/nrn.2018.2
23. Zhou LJ, Peng J, Xu YN, et al. Microglia are indispensable for synaptic plasticity in the spinal dorsal horn and chronic pain. Cell Rep. 2019;27(13):3844–3859e6. doi:10.1016/j.celrep.2019.05.087
24. Gu N, Yi MH, Murugan M, et al. Spinal microglia contribute to sustained inflammatory pain via amplifying neuronal activity. Mol Brain. 2022;15(1):86. doi:10.1186/s13041-022-00970-3
25. Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20(2):467–473. doi:10.1111/j.1460-9568.2004.03514.x
26. Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. 2018;100(6):1292–1311. doi:10.1016/j.neuron.2018.11.009
27. Guneykaya D, Ivanov A, Hernandez DP, et al. Transcriptional and translational differences of microglia from male and female brains. Cell Rep. 2018;24(10):2773–2783e6. doi:10.1016/j.celrep.2018.08.001
28. Sorge RE, Mapplebeck JC, Rosen S, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18(8):1081–1083. doi:10.1038/nn.4053
29. Mogil JS, Parisien M, Esfahani SJ, Diatchenko L. Sex differences in mechanisms of pain hypersensitivity. Neurosci Biobehav Rev. 2024;163:105749. doi:10.1016/j.neubiorev.2024.105749
30. Huck NA, Siliezar-Doyle J, Haight ES, et al. Temporal contribution of myeloid-lineage TLR4 to the transition to chronic pain: a focus on sex differences. J Neurosci. 2021;41(19):4349–4365. DOI:10.1523/JNEUROSCI.1940-20.2021
31. Welsh JA, Goberdhan DCI, O’Driscoll L, et al. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J Extracell Vesicles. 2024;13(2):e12404. doi:10.1002/jev2.12404
32. Lin Z, Luo X, Wickman JR, et al. Inflammatory pain resolution by mouse serum-derived small extracellular vesicles. Brain Behav Immun. 2025;123:422–441. doi:10.1016/j.bbi.2024.09.032
33. Manners MT, Ertel A, Tian Y, Ajit SK. Genome-wide redistribution of MeCP2 in dorsal root ganglia after peripheral nerve injury. Epigenet Chromatin. 2016;9(1):23. doi:10.1186/s13072-016-0073-5
34. Pino PA, Cardona AE. Isolation of brain and spinal cord mononuclear cells using percoll gradients. J Visual Exp. 2011;(48). doi:10.3791/2348
35. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Meth. 2015;12(4):357–360. doi:10.1038/nmeth.3317
36. Zhang Y, Parmigiani G, Johnson WE. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform. 2020;2(3). doi:10.1093/nargab/lqaa078
37. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–7. doi:10.1093/nar/gkw377
38. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
39. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9(4):357–359. doi:10.1038/nmeth.1923
40. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2). doi:10.1093/gigascience/giab008
41. Gaspar JM. Improved peak-calling with MACS2. bioRxiv. 2018;496521. doi:10.1101/496521
42. Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116–125. doi:10.1016/j.ceb.2014.05.004
43. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Rev Cell Dev Biol. 2014;30:255–289. doi:10.1146/annurev-cellbio-101512-122326
44. Elmore MR, Najafi AR, Koike MA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi:10.1016/j.neuron.2014.02.040
45. Green KN, Crapser JD, Hohsfield LA. To Kill a Microglia: a Case for CSF1R Inhibitors. Trends Immunol. 2020;41(9):771–784. doi:10.1016/j.it.2020.07.001
46. Huang Y, Xu Z, Xiong S, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21(4):530–540. doi:10.1038/s41593-018-0090-8
47. Szolcsányi J, Pintér E, Helyes Z, Petho G. Inhibition of the function of TRPV1-expressing nociceptive sensory neurons by somatostatin 4 receptor agonism: mechanism and therapeutical implications. Curr Top Med Chem. 2011;11(17):2253–2263. doi:10.2174/156802611796904852
48. Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother. 2016;16(8):961–968. doi:10.1080/14737175.2016.1193009
49. Isensee J, Diskar M, Waldherr S, et al. Pain modulators regulate the dynamics of PKA-RII phosphorylation in subgroups of sensory neurons. J Cell Sci. 2014;127(1):216–229. doi:10.1242/jcs.136580
50. Barsyte-Lovejoy D, Li F, Oudhoff MJ, et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proceed Nat Acad Sci. 2014;111(35):12853–12858. doi:10.1073/pnas.1407358111
51. Elisia I, Nakamura H, Lam V, et al. DMSO represses inflammatory cytokine production from human blood cells and reduces autoimmune arthritis. PLoS One. 2016;11(3):e0152538. doi:10.1371/journal.pone.0152538
52. Lajqi T, Köstlin-Gille N, Hillmer S, et al. Gut microbiota-derived small extracellular vesicles endorse memory-like inflammatory responses in murine neutrophils. Biomedicines. 2022;10(2). doi:10.3390/biomedicines10020442
53. Neher JJ, Cunningham C. Priming microglia for innate immune memory in the brain. Trends Immunol. 2019;40(4):358–374. doi:10.1016/j.it.2019.02.001
54. Gupta R, Luo X, Lin Z, Tian Y, Ajit SK. Uptake of fluorescent labeled small extracellular vesicles in vitro and in spinal cord. J Visual Exp. 2021;171(171). doi:10.3791/62537
55. Brennan FH, Li Y, Wang C, et al. Microglia coordinate cellular interactions during spinal cord repair in mice. Nat Commun. 2022;13(1):4096. doi:10.1038/s41467-022-31797-0
56. Sawicki CM, Kim JK, Weber MD, et al. Microglia promote increased pain behavior through enhanced inflammation in the spinal cord during repeated social defeat stress. J Neurosci. 2019;39(7):1139–1149. doi:10.1523/jneurosci.2785-18.2018
57. Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119(8):1810–1820. doi:10.1182/blood-2011-09-379214
58. Lei F, Cui N, Zhou C, Chodosh J, Vavvas DG, Paschalis EI. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proceed Nat Acad Sci United States Am. 2020;117(38):23336–23338. doi:10.1073/pnas.1922788117
59. Donovan LJ, Bridges CM, Nippert AR, et al. Repopulated spinal cord microglia exhibit a unique transcriptome and contribute to pain resolution. Cell Rep. 2024;43(2):113683. doi:10.1016/j.celrep.2024.113683
60. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe. 2019;25(1):13–26. doi:10.1016/j.chom.2018.12.006
61. Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16(3):144–154. doi:10.1038/nrm3949
62. Meng H, Bartholomew B. Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J Biol Chem. 2018;293(36):13786–13794. doi:10.1074/jbc.R117.813485
63. Larsson AJM, Johnsson P, Hagemann-Jensen M, et al. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565(7738):251–254. doi:10.1038/s41586-018-0836-1
64. Blinka S, Reimer MH Jr, Pulakanti K, Pinello L, Yuan GC, Rao S. Identification of transcribed enhancers by genome-wide chromatin immunoprecipitation sequencing. Meth Mol Biol. 2017;1468:91–109. doi:10.1007/978-1-4939-4035-6_8
65. Butt AH, Alkhalifah T, Alturise F, Khan YD. A machine learning technique for identifying DNA enhancer regions utilizing CIS-regulatory element patterns. Sci Rep. 2022;12(1):15183. doi:10.1038/s41598-022-19099-3
66. Masuda T, Sankowski R, Staszewski O, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566(7744):388–392. doi:10.1038/s41586-019-0924-x
67. Hammond TR, Dufort C, Dissing-Olesen L, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50(1):253–271.e6. doi:10.1016/j.immuni.2018.11.004
68. Gosselin D, Link VM, Romanoski CE, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159(6):1327–1340. doi:10.1016/j.cell.2014.11.023
69. Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. doi:10.1016/j.cell.2014.11.018
70. Keating ST, Groh L, van der Heijden C, et al. The Set7 lysine methyltransferase regulates plasticity in oxidative phosphorylation necessary for trained immunity induced by beta-glucan. Cell Rep. 2020;31(3):107548. doi:10.1016/j.celrep.2020.107548
71. Benjaskulluecha S, Boonmee A, Pattarakankul T, Wongprom B, Klomsing J, Palaga T. Screening of compounds to identify novel epigenetic regulatory factors that affect innate immune memory in macrophages. Sci Rep. 2022;12(1):1912. doi:10.1038/s41598-022-05929-x
72. Shen Y, Ding Z, Ma S, et al. SETD7 mediates spinal microgliosis and neuropathic pain in a rat model of peripheral nerve injury. Brain Behav Immun. 2019;82:382–395. doi:10.1016/j.bbi.2019.09.007
73. Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceed Nat Acad Sci United States Am. 2010;107(50):21931–21936. doi:10.1073/pnas.1016071107
74. Hanamsagar R, Alter MD, Block CS, Sullivan H, Bolton JL, Bilbo SD. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia. 2017;65(9):1504–1520. doi:10.1002/glia.23176
75. Netea MG, Dominguez-Andres J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020;20(6):375–388. doi:10.1038/s41577-020-0285-6
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Tailored Extracellular Vesicles from Dental Stem Cells: Advances in Specific Modifications for Enhanced Therapeutic Applications
Liu X, Li Z, Fu J, Wang R, He J, Yao J, Ye Q, He Y
International Journal of Nanomedicine 2025, 20:8327-8341
Published Date: 26 June 2025
Regenerative Potential of Exosomes from 3D Spheroid-Cultured Stem Cells Compared with 2D Culture in Dentin–Pulp Regeneration: A Systematic Review
Jos Erry HW, Margono A, Bagio DA, Suniarti DF, Kusuma I, Budi HS, Mauludin R, Dalimunthe RS, Tambun DV, Amir LR
Clinical, Cosmetic and Investigational Dentistry 2026, 18:614885
Published Date: 2 June 2026
Extracellular Vesicle-Associated Non-Coding RNAs in Preeclampsia: Mechanistic Insights, Biomarker Discovery, and Emerging Nanomedicine Concepts
Zhang Z, Du J, Chen T, Teng Y, Chen J, Li Y, Zhang Y, Long Y, Li C, Wu Z, Li N, Yang Z
International Journal of Nanomedicine 2026, 21:612044
Published Date: 29 June 2026