Back to Journals » Journal of Inflammation Research » Volume 14
Methylglyoxal Exacerbates Lipopolysaccharide-Induced Acute Lung Injury via RAGE-Induced ROS Generation: Protective Effects of Metformin
Authors Medeiros ML ![]() , Oliveira AL, de Oliveira MG, Monica FZ, Antunes E
, Oliveira AL, de Oliveira MG, Monica FZ, Antunes E
Received 1 September 2021
Accepted for publication 15 November 2021
Published 2 December 2021 Volume 2021:14 Pages 6477—6489
DOI https://doi.org/10.2147/JIR.S337115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Matheus L Medeiros, Akila L Oliveira, Mariana G de Oliveira, Fabíola Z Mónica, Edson Antunes
Department of Pharmacology, University of Campinas (UNICAMP), Campinas, Sao Paulo, Brazil
Correspondence: Edson Antunes
Department of Pharmacology, Faculty of Medical Sciences, University of Campinas (UNICAMP), Campinas, Sao Paulo, 13084-971, Brazil
Tel +55 19-9-9601-1516
Email [email protected]; [email protected]
Purpose: Methylglyoxal (MGO) is a highly reactive dicarbonyl species implicated in diabetic-associated diseases. Acute lung injury (ALI) symptoms and prognosis are worsened by diabetes and obesity. Here, we hypothesized that elevated MGO levels aggravate ALI, which can be prevented by metformin. Therefore, this study evaluated the lung inflammation in lipopolysaccharide (LPS)-exposed mice pretreated with MGO.
Methods: C57Bl/6 male mice treated or not with MGO for 12 weeks were intranasally instilled with LPS (30 μg) to induce ALI, and metformin (300 mg/kg) was given as gavage in the last two weeks of treatment. After 6 h, bronchoalveolar lavage fluid (BALF) and lung tissues were collected to quantify the cell infiltration, cytokine levels, reactive-oxygen species (ROS) production, and RAGE expression.
Results: LPS exposure markedly increased the neutrophil infiltration in BALF and lung tissue, which was accompanied by higher levels of IFN-γ, TNF-α and IL-1β compared with untreated group. MGO treatment significantly increased the airways neutrophil infiltration and mRNA expressions of TNF-α and IL-1β, whereas COX-2 expression remained unchanged. In lung tissues of LPS-exposed mice, MGO treatment significantly increased the immunostaining and mRNA expression of RAGE, and the ROS levels. Serum MGO concentration achieved after 12-week intake was 9.2-fold higher than control mice, which was normalized by metformin treatment. Metformin also reduced the inflammatory markers in response to MGO.
Conclusion: MGO intake potentiates the LPS-induced ALI, increases RAGE expression and ROS generation, which is normalized by metformin. MGO scavengers may be a good adjuvant therapy to reduce ALI in patients with cardiometabolic diseases.
Keywords: advanced glycated end products, neutrophil, airways, immunohistochemistry
Introduction
Acute lung injury (ALI) and its clinical correlate, the acute respiratory distress syndrome (ARDS), are highly prevalent diseases associated with elevated morbidity and mortality rates in the critically ill patient population.1 The acute changes at the onset of ALI/ARDS consist of severe hypoxemia and reduced lung compliance that is accompanied by increased endothelial and epithelial permeabilities, leading to pulmonary edema and leukocyte infiltration.2 There are many cells and factors that affect the immune response to ALI/ARDS, but airways accumulation of neutrophils represents a hallmark of this disease.3 Neutrophil infiltration amplifies the signals to increase the production of pro-inflammatory cytokines like interferon gamma (IFN-γ), interleukin 1β (IL-1β) and tumor necrosis factor-α (TNF-α).4 Th17 cells also participate in clearing pathogens during host defense reactions and have been implicated in the ALI physiopathology.5 Furthermore, IL-10 and cyclooxygenase-2 (COX-2) metabolites are reported to exert protective effects in ALI.6,7
Methylglyoxal (MGO) is a highly reactive dicarbonyl compound formed endogenously from 3-carbon glycolytic intermediates of glycolysis,8 which has emerged as a key player in the pathophysiology of diabetes-associated complications.9 High levels of MGO are found in plasma and urine of pre-diabetic, diabetic and obese individuals, achieving concentrations ranging from 233 nM to 400 µM.10–16 Obese mice also exhibit elevated levels of MGO in plasma and urine.17 Evidence shows that MGO modifies free amino groups such as lysine and arginine to yield the advanced glycation end products (AGEs),18 which interacts with RAGE, a transmembrane receptor of the immunoglobulin superfamily, initiating the immune response to tissue injury and inflammation.19 RAGE in turn activates multiple intracellular signaling pathways, including increased production of pro-inflammatory mediators and generation of reactive oxygen species (ROS), thus providing a pro-oxidant environment that is usually implicated in obesity-associated pathological conditions.20 Administration of MGO to healthy rodents has been shown to mimic some the complications of the diabetes.21–24
Metformin (N, N-dimethylbiguanide) is the first-line oral anti-hyperglycemic drug used for the treatment of type 2 diabetes. Metformin increases tissue sensitivity to insulin and decreases the levels of glycated hemoglobin (HbA1c), an effect partially mediated by activation of the adenosine monophosphate-activated protein kinase (AMPK) signaling pathway.25 Besides these metabolic actions, metformin has been shown to prevent MGO formation8,26 and to scavenger MGO,27,28 thereby reducing its plasma levels in type 2 diabetic patients.29–31 Considering that obese/diabetic patients may be at greater risk of developing ALI/ARDS than normal weight individuals,32–34 in the present study, we hypothesized that elevated MGO levels contribute to their greater susceptibility to ALI, which may be reversed by metformin. Therefore, male mice were treated with 0.5% MGO for 12 weeks in the absence and the presence of metformin, after which they were intranasally instilled with lipopolysaccharide (LPS) to induce ALI. The cell infiltration in bronchoalveolar lavage (BAL) fluid and lung tissue, and levels of pro- and anti-inflammatory cytokines were evaluated in this study. Immunohistochemistry and mRNA expressions for RAGE and dihydroethidine (DHE) assays as a marker of ROS production were also performed in the lung tissues.
Materials and Methods
Animals
Male C57BL/6 mice, 4-week-old, were housed in cages (three per cage) located in a ventilated cage shelters with constant humidity of 55 ± 5% and temperature of 24 ± 1 °C under a 12 h light-dark cycle. Animals received standard food ad libitum. The experimental protocols were approved by the Animal Welfare Ethics of State University of Campinas (CEUA-UNICAMP; Protocol No. 5200–1/2019) and followed the “Brazilian Guidelines for The Production, Maintenance and Use of Animals for Teaching or Research” from the National Council of Control in Animal Experimentation (CONCEA).
Treatments with MGO and Metformin
Mice received 0.5% MGO (Sigma Aldrich, MI, USA) in the drinking water for 12 weeks, according to previous studies in rodents.22,35 Metformin was given as a daily gavage at 300 mg/kg in the last two weeks of MGO treatment.36 Thus, our experimental protocols resulted in four experimental groups of 7 to 9 mice each, as follows: Control, MGO, Metformin, and MGO + metformin. In each group, mice were instilled with saline or LPS, making a total of eight groups, as detailed below.
Airways LPS Instillation
Mice were intranasally instilled with either sterile saline (50 µL) or LPS (30 µg/50 µL), which was obtained from E. coli (0111:B4, Sigma Aldrich, Missouri, USA). At 6 h thereafter, the mice were euthanized with overdose of isoflurane (>5%) and cervical dislocation was then performed to confirm the euthanasia. After exsanguination, lungs were harvested with phosphate-buffered saline (PBS) to obtain bronchoalveolar lavage (BAL) fluid, and lung were collected for the morphometric analysis. Briefly, the trachea was exposed and cannulated with a polyethylene tube connected to a syringe. The lungs were washed by flushing five times with PBS (300 µL) through the tracheal cannula, and the fluid recovered after each wash was combined, making a total BAL volume of approximately 1500 µL. BAL fluid was centrifuged (500 g for 10 min at 4°C) and the supernatant fluid was stored at −80°C until use whereas the cell pellet was resuspended in 200 µL of PBS to perform the cell counting. The counts of total inflammatory cells were done using a Neubauer chamber, whereas Diff-Quick stain was used for determination of cell differential counts. A minimum of 300 cells were counted and classified as neutrophils, eosinophils and mononuclear cells based on normal morphological criteria.
Morphometrical Lung Analysis
Lungs were immersed in 10% phosphate buffered formalin for 24 h and kept in 70% ethanol until embedding in paraffin. Tissues were sliced (5-μm sections) and stained with hematoxylin/eosin for light microscopy examination. Morphometrical analysis was performed using a Leica DM 5000B digital camera and Leica Q Win Image Processing and Analysis Software. For each different staining, the area of positivity was measured in mm2 for 3 bronchioles per slide.
Measurements of Cytokines in BAL Fluid and MGO in Serum
Levels of IFN-γ, TNF-α, IL-1β, IL-17 and IL-10 in BAL fluid were measured using commercially available DuoSet ELISA kits (R&D, Minneapolis, USA). For the measurement of MGO levels in serum, samples were deproteinized using the Deproteinizing Sample Preparation Kit – TCA (Cat. No. Ab204708, Abcam, Cambridge, UK) following the manufacturer’s instructions, and then quantified using an ELISA competitive kit for OxiSelect™ Methylglyoxal (Catalog No. STA-811, Cell Biolabs, San Diego, CA, USA).
Immunohistochemistry for RAGE
Lung tissue from mice were obtained and processed for a posterior immunohistochemical procedures. Briefly, lung tissues were excised, immersed in 10% formalin fixative solution for 48 h and embedded in paraffin. Serial 5-μm tick sections were mounted onto aminopropyltriethoxysilane-coated glass slides. Sections were deparaffinized with xylol, rehydrated and washed with 0.05 M Tris buffer solution (TBS) at pH 7.4. Subsequently, sections were treated with 0.01 M Tris-EDTA buffer containing 0.05% Tween-20 (pH 9.0) for 24 min at 98°C for antigen retrieval. Before blocking the non-specific sites with 5% bovine serum albumin (BSA) solution containing 0.1% Tween-20 for 60 min, endogenous peroxidase activity was inhibited with 0.3% hydrogen peroxide (H2O2) solution diluted in TBS for 30 min at room temperature. Sections were incubated with mouse monoclonal anti-RAGE primary antibody (1:500; Cat. no. ab54741, Abcam, Cambridge, UK) diluted in TBS containing 3% BSA overnight at 4°C. Next, sections were washed with TBS and incubated with biotinylated goat anti-mouse IgG, avidin and biotinylated HRP (1:20; cat. no. EXTRA2, Sigma Aldrich, St Louis, MO, USA) following all the manufacturer’s instructions. The area stained for RAGE was detected using 3.3′ diaminobenzidine solution (DAB; cat. no. D4293, Sigma Aldrich). To ensure the specificity of the IHC, a section as a negative control was tested, in which the primary antibody was omitted. All slides were counterstained with hematoxylin and mounted for observation by microscopy. Images were acquired using a light microscope Leica DM 5000B coupled to a digital camera, with a 40× objective. One filed of each section was captured, and three bronchi were analyzed. The brown-stained area in the sections incubated with mouse anti-RAGE was automatically detected by the free software ImageJ (http://imagej.nih.gov/ij). The data are presented as percentage of positive area for staining for RAGE in the section area.
Quantitative Real-Time RT-PCR (qPCR)
Total RNA was extracted from freshly homogenized lungs using TRIzol® reagent (Invitrogen, Mississippi, USA) according to the manufacturer’s protocol. DNase treated RNA samples were then transcribed with High-Capacity Reverse Transcription Kit® (Applied Biosystems, California, USA). cDNA samples concentrations were quantified using a spectrophotometer (Nanodrop Lite®, Thermo Scientific, Massachusetts, USA). Synthetic oligonucleotide primers (Table 1) were obtained from Integrated DNA Technologies (Iowa, USA) and Qiagen (Hilden, Germany). The reactions were performed with 10 ng cDNA, 6 µL SYBR Green Master Mix® (Life Technologies, California, USA) and the optimal primer concentration, in a total volume of 10 μL. Real-time PCR was performed in the equipment StepOne-Plus® Real Time PCR System (Applied Biosystems). The reaction program was 95°C for 10 min, followed by 40 cycles of 95°C for 15s then 60°C for 1 min. At the end of a normal amplification, a degradation time was added, during which the temperature increased gradually from 60°C to 95°C. Threshold cycle (Ct) was defined as the point at which the fluorescence rises appreciably above the background fluorescence. To determine the specificity of the amplification, the melting curve analysis of the PCR products were performed to ensure that only one fragment was amplified. The 2−ΔΔCt method was utilized to analyze the results, which were expressed by the difference between Ct values of chosen genes and the housekeeping gene, the 18S ribosomal RNA (18S rRNA), which signal strength did not differ between groups (Ct: 9.8 ± 0.2 and 9.6 ± 0.2 for control and MGO, respectively).
 |
Table 1 Primer Sequences Used for Real-Time PCR Amplifications |
Measurement of ROS Levels
The lungs were emerged in Tissue-Tek OCT freezing medium (Sakura, California, USA) and frozen in liquid nitrogen. Using a cryostat, we obtained 12-μm lung sections and two different lung sections were obtained from each animal. Sections were then placed on glass slides, maintained at 37°C for 20 min, followed by the incubation with dihydroethidium (DHE, 2 μM, Sigma Aldrich, Missouri, USA) diluted in phosphate buffer for 30 min at 37°C in a humid chamber. The sections were observed with a fluorescence microscope (Eclipse 80i, Nikon, Japan) equipped with a camera (DS-U3, Nikon, Japan) and using a rhodamine filter. At 200× magnification, two different images were randomly acquired from each section. The intensity of fluorescence was determined using the ImageJ Software (National Institutes of Health, Bethesda, MD, USA).
Statistical Analysis
Data were expressed as means ± SEM. The program GraphPad version 6.0 software was used for statistical analysis. Statistically significant differences were determined using one-way analysis of variance (ANOVA) for multiple comparisons followed by Tukey’s test. A value of P < 0.05 was accepted as significant.
Results
MGO Intake Increases LPS-Induced Airway Neutrophil Migration
Total and differential cell counts were evaluated in BAL fluid of four groups of mice, namely: (i) untreated animals instilled with sterile saline, (ii) untreated animals instilled with LPS, (iii) MGO-treated animals instilled with saline and (iv) MGO-treated animals instilled with LPS.
In untreated mice intranasally instilled with saline (control group), the BAL fluid consisted mainly of mononuclear cells with inflammatory cells nearly absent (Figure 1A-C; n = 7–9). However, LPS instillation in these animals markedly increased (P < 0.05) the number of total leukocytes, neutrophils and mononuclear cells in BAL fluid compared with saline group (Figure 1A–C). The analysis of the histological sections of lungs in both saline groups confirmed the presence of normal tissue with no inflammatory cells surrounding the peribronchiolar regions and lung parenchyma (Figure 2A–C). As opposed, the lung histology in LPS-exposed mice showed a large number of inflammatory cells in peribronchiolar regions of the lung, consisting mainly of neutrophils (P < 0.05; Figure 2A–C).
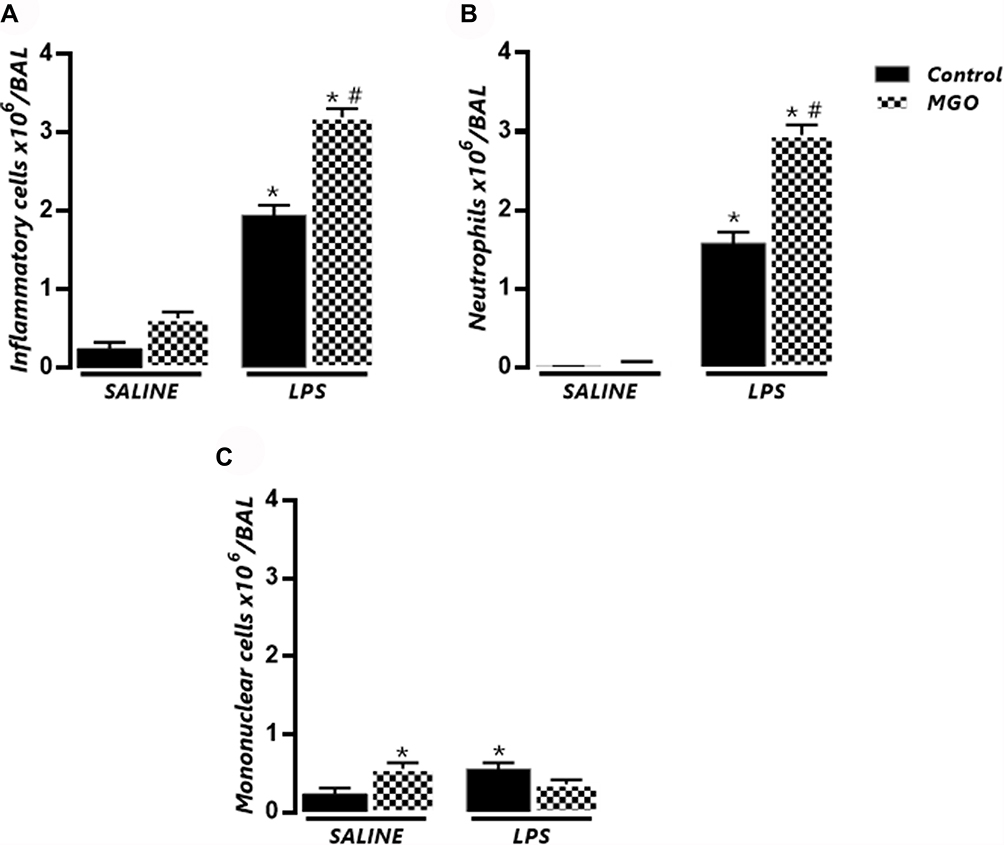 |
Figure 1 Methylglyoxal (MGO) increases the number of total cells and neutrophils in bronchoalveolar lavage fluid (BAL) of mice intranasally instilled with lipopolysaccharide (LPS). Mice were treated or not with 0.5% MGO in the drinking water for 12 weeks, and then intranasally instilled with LPS (30 µg). (A–C) show total cells, neutrophils, and mononuclear cells in BAL fluid at 6 h following LPS exposure (or instillation with saline). Data are expressed as mean ± SEM. *P < 0.05 compared with respective saline group; #P < 0.05 compared with LPS in control mice. |
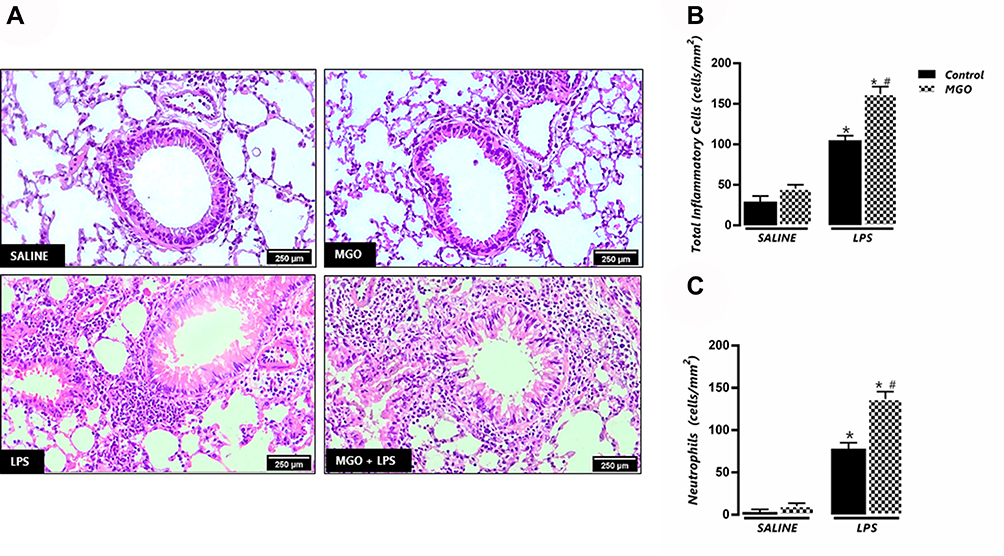 |
Figure 2 Methylglyoxal (MGO) increases the neutrophil infiltration in lung connective tissue surrounding the bronchial and bronchiolar segments in lipopolysaccharide (LPS)-exposed mice. (A) shows representative photomicrographs of bronchiolar segments at 6 h following LPS exposure (or instillation with saline) for each experimental group. (B) and (C) show the number of total inflammatory cells and neutrophils, respectively. Each column represents mean ± SEM of the number of cells mm2 for each group. Haematoxylin–eosin, high magnification (bar represents 200 μm). *P < 0.05 compared with the respective control group; #P < 0.05 compared with LPS in control group. |
In MGO-treated mice, saline instillation did not significantly affect the number of total leukocytes and neutrophils but caused a small (despite significant) increase of the mononuclear cell counts in BAL fluid compared with the untreated animals (Figure 1A–C). Intranasal instillation of LPS in MGO-treated animals markedly elevated the content of total inflammatory cells and neutrophils at significantly higher numbers than the untreated animals (68% and 95% higher, respectively; P < 0.05; Figure 1A and B). The number of mononuclear cells in BAL fluid in LPS-instilled mice remained unchanged between control and MGO groups (Figure 1C). The histological sections of lungs confirmed the higher infiltration of inflammatory cells and neutrophils compared with the untreated animals (Figure 2A–C).
Levels of Cytokines in BAL Fluid
Levels of IFN-γ, TNF-α, IL-1β, IL-17 and IL-10 were measured in BAL fluid after saline or LPS instillation in both untreated and MGO-treated mice. Higher levels of IFN-γ, TNF-α and IL-1β were detected in untreated mice exposed with LPS compared with saline group, which were further increased in MGO-treated mice (P < 0.05; Figure 3A–C). The levels of IL-17 were higher in LPS-exposed mice (P < 0.05), but MGO treatment did not further elevate this cytokine level compared to its respective saline group (Figure 3D). In addition, reduced levels of IL-10 were detected in MGO-treated mice exposed with LPS in comparison with the other groups (P < 0.05; Figure 3E).
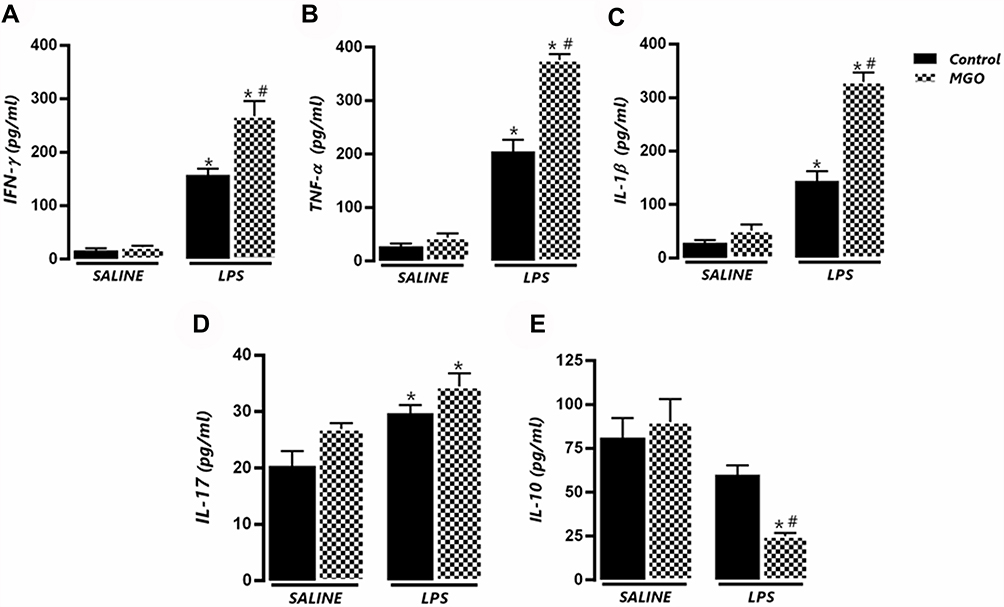 |
Figure 3 Methylglyoxal (MGO) increases the levels of pro-inflammatory cytokines in bronchoalveolar lavage (BAL) fluid of lipopolysaccharide (LPS)-exposed mice. (A–E) show IFN-γ, TNF-α, IL-1β, IL-17 and IL-10, respectively, in BAL fluid at 6 h following LPS exposure (or instillation with saline). Each column represents the mean ± SEM. Data are expressed as mean ± SEM. *P < 0.05 compared with respective saline group; #P < 0.05 compared with LPS in control group. |
qPCR Analysis of TNF-α, IL-1β and COX-2 in Lung Tissues
LPS exposure led to significant increases of the mRNA expressions of TNF-α and IL-1β, which were further increased by MGO treatment (P < 0.05; Figure 4A and B). The COX-2 expression was also increased by LPS exposure, but MGO treatment did not lead to additional increases in this enzyme expression (Figure 4C). In lung tissues of all saline groups, MGO treatment did not affect the mRNA expressions of TNF-α, IL-1β, and COX-2 (Figure 4A–C).
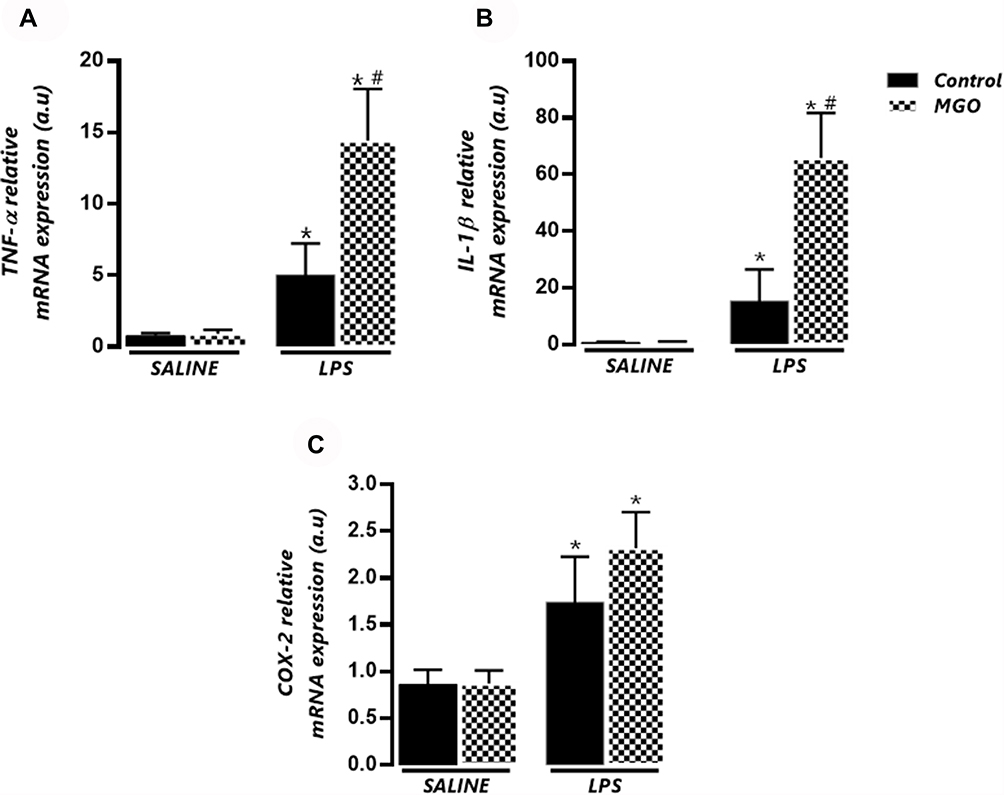 |
Figure 4 Effects of methylglyoxal (MGO) on the mRNA expressions of TNF-α (A), IL-1β (B) and cyclooxygenase-2 (COX-2; C) in lung tissues of lipopolysaccharide (LPS)-exposed mice. The mRNA expression levels of TNF-α, IL-1β and COX-2 were evaluated at 6 h following LPS exposure (or instillation with saline). Each gene was normalized to 18 S rRNA expression levels, and the values are expressed in arbitrary units (a.u). Data are means ± SEM. *P < 0.05 compared with control group; #P < 0.05 compared with LPS in control group. |
Immunohistochemistry and qPCR Analysis of RAGE in Lung Tissues
Immunohistochemistry analysis for RAGE in saline-instilled mice from control and MGO groups revealed a weak immunostaining for RAGE in the peribronchiolar areas of (Figure 5A and B). In contrast, exposure to LPS mice significantly increased the RAGE expression in the airway epithelium (P < 0.05), which was further enhanced by treatment with MGO (P < 0.05; Figure 5A and B).
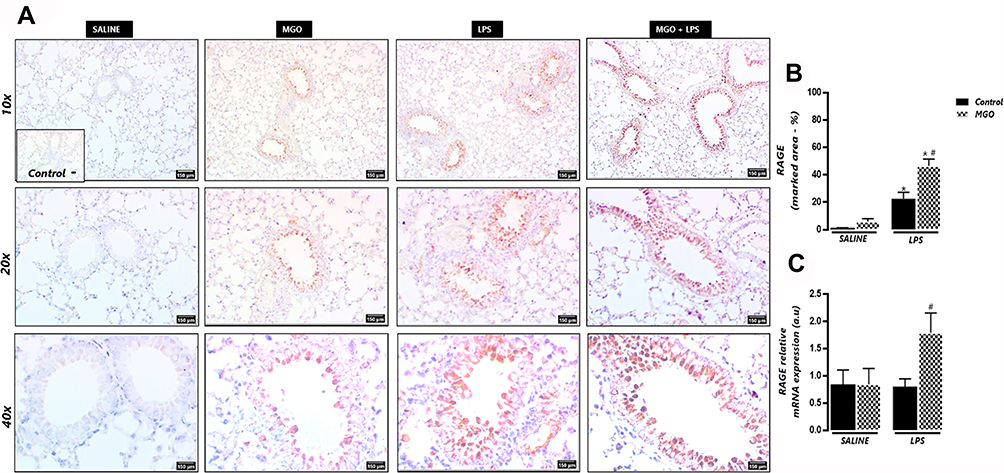 |
Figure 5 Methylglyoxal (MGO) increases immunostaining and mRNA expression of RAGE in airways of lipopolysaccharide (LPS)-exposed mice. (A) shows representative photomicrographs of immunohistochemistry of bronchiolar segments at 6 h following LPS exposure (or instillation with saline), as evidenced by the brown color in the bronchial epithelium. (B) shows the RAGE immunostaining. (C) shows the RAGE mRNA expression (gene normalized to 18 SrRNA) and data expressed in arbitrary units (a.u). Micrographs recorded at 150× magnification. Data are means ± SEM. *P < 0.05 compared with control group; #P < 0.05 compared with LPS in control group. |
In addition, LPS exposure in MGO-treated mice significantly increased the mRNA expression of RAGE when compared with the untreated group (P < 0.05; Figure 5C). The RAGE mRNA expression in saline groups treated or not with MGO did not differ between each other (Figure 5C).
Serum Levels of MGO in Control and Metformin-Treated Mice
Figure 6 shows that MGO given in the drinking water at 0.5% solution for 12 weeks yielded a serum concentration of about of 20 µg/mL (equivalent to approximately 290 nM), which was 9.2-fold higher (P < 0.05) than basal levels achieved in control animals (31 nM). Metformin, given at 300 mg/kg as a daily gavage for two weeks, returned the MGO concentration to the levels of control mice. Metformin alone had no effects on the basal levels of MGO (Figure 6).
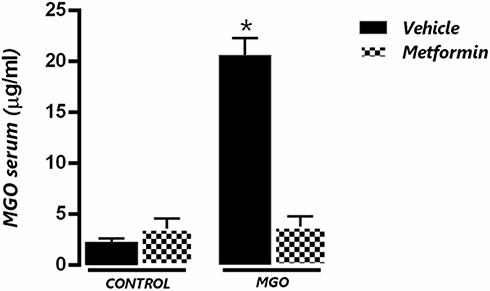 |
Figure 6 Serum levels of methylglyoxal (MGO) in control (CT) and MGO-treated mice in association or not with metformin (MET). Mice were treated or not with 0.5% MGO in the drinking water for 12 weeks, and metformin was given (300 mg/kg) in the last two weeks of MGO treatment. *P < 0.05 compared with MGO+MET group. |
Effects of Metformin on Airway Cell Infiltration, Cytokine Levels, and RAGE Expression
We next evaluated the airway cell infiltration, cytokine levels (TNF-α, IFN-δ and IL-1β) and RAGE expression in BAL and/or lung tissue of MGO-treated mice associated or not with metformin. The increases of total cell infiltration and neutrophils in response to LPS instillation in control group were unaffected by metformin treatment (Figure 7A and B). However, metformin fully normalized the exacerbation of LPS-induced cell infiltration by MGO treatment (P < 0.05; Figure 7A and B). Likewise, in control group of LPS-instilled mice, metformin interfered neither with the increased production of TNF-α, IL-β and IFN-δ in BAL fluid (Figure 7C, E and G) nor with the mRNA expressions of TNF-α and IL-β in lung tissues (Figure 7D and F); however, metformin returned these cytokine levels in the MGO group to the levels of the control group (P < 0.05; Figure 7C–G). The increased mRNA RAGE expression in MGO group was also restored by metformin (Figure 7H). In saline-instilled mice of control and MGO groups, metformin did not significantly affect any basal parameter.
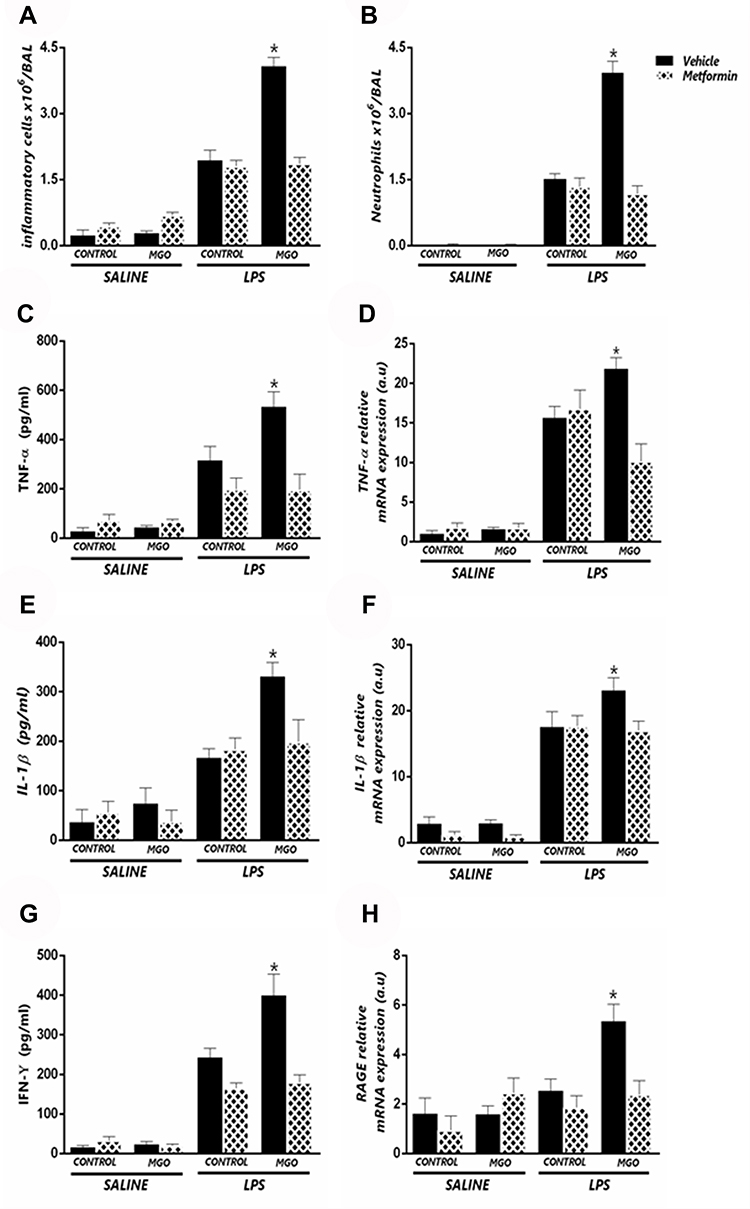 |
Figure 7 Metformin suppresses the potentiation by methylglyoxal (MGO) of LPS-induced lung inflammation. Mice were intranasally instilled saline or LPS (30 µg). Panels show respectively number of total inflammatory cells (A) neutrophil number (B) TNF-α levels in BALF (C) TNF-α mRNA expression in lung tissue (D) IL-1β levels in BALF (E) IL-1β mRNA expression in lung tissue (F) IFN-γ levels in BALF (G) and RAGE mRNA expression (H). Data are expressed as mean ± SEM. *P < 0.05 compared with the LPS + metformin group. |
Effects of Metformin on ROS Levels in Lung Tissues
The ROS levels in lung tissues were about of 2.0-fold higher in LPS-instilled mice than its respective saline group (P < 0.05), which were further increased by MGO treatment (P < 0.05; Figure 8A and B). Treatment with MGO also caused a small, despite significant (P < 0.05), increase of ROS levels in the saline-instilled mice compared with respective untreated group. We then evaluated the effects of metformin treatment on ROS levels in all groups. In metformin-treated mice, LPS exposure also significantly increased the ROS levels in the same magnitude as the vehicle group. However, in MGO-treated mice, metformin but fully prevented the increased ROS levels by LPS exposure (Figure 8A and B).
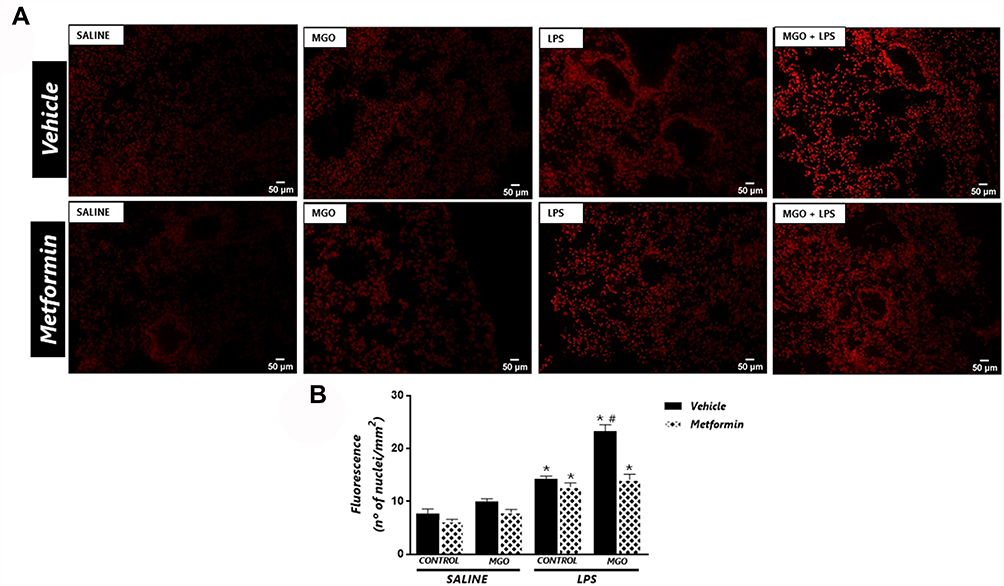 |
Figure 8 Metformin reduces the increased the reactive-oxygen species (ROS) levels in lungs of lipopolysaccharide (LPS)-exposed mice treated with methylglyoxal (MGO). (A) shows representative photomicrographs of the lung sections, and (B) shows quantitative ROS levels through dihydroethidium (DHE)-induced fluorescence in all groups. Data are means ± SEM. *P < 0.05 compared with respective saline groups; #P < 0.05 compared with LPS-MGO group. |
Discussion
We hypothesized here that excess of MGO take part of the machinery that exacerbates ALI in obese/diabetic individuals. We then treated healthy mice with 0.5% MGO for 12 weeks to elevate the MGO levels in plasma, after which animals were intranasally instilled LPS to induce ALI. Airways instillation with LPS has been shown to induce airway inflammation mainly due to neutrophil influx, which cause epithelium and endothelium disruption mostly via release of cytotoxic molecules.37 Interferon-γ, IL-1β and TNF- α exert important roles in human and experimental ALI.38,39 We then sought that the higher levels of these inflammatory cytokines by MGO treatment might worsen LPS-induced ALI. As expected, LPS-exposed mice exhibit elevated protein levels of these cytokines in BAL fluid and/or mRNA expressions in lung tissues, the levels of which were all potentiated by the MGO treatment. Interleukin-17 is another pro-inflammatory cytokine produced by the CD4+ T cell implicated in LPS-induced mouse cell recruitment.40 We found higher IL-17 levels in BAL fluid of LPS-exposed mice, but MGO treatment failed to significantly elevate its levels, suggesting that this cytokine may not be critical to the MGO actions. Interleukin-10 classically exerts anti-inflammatory effects by negatively balancing the release of pro-inflammatory cytokines.41 The IL-10 levels in BAL fluid did not change between saline and LPS groups, but interestingly MGO significantly reduced the IL-10 levels in LPS-exposed mice, indicating that ALI aggravation by MGO treatment may be at least in part due to removal of the lung protection by reducing the IL-10 release. Of interest, loss-of-function mutations in IL-10 or IL-10 receptor has been shown to increase immunoinflammation during infections.42 Up-regulation of intrapulmonary COX-2 expression and its increased enzyme activity are reported to contribute to resolution of ALI/ARDS, possibly via prostaglandin E2 generation.6,7 Here, we found that the increases of COX-2 mRNA expressions in lungs of LPS-exposed mice were of the same magnitude in control and MGO groups, suggesting that the deleterious actions of MGO are not accompanied by further protective generation of COX-2-derived lipid metabolites.
Methylglyoxal has been identified as the major precursor of AGE formation, which usually results from normal metabolism in healthy individuals; however, in pathological states of pre-diabetes, diabetes, and obesity, the elevated MGO levels act to further increase the AGE generation.19,43 AGEs bind to its receptor RAGE, activating different intracellular signaling pathways, including increased ROS production.44 In different pathological conditions, lung tissues express supraphysiological levels of RAGE and AGEs, leading to excessive pulmonary oxidative stress.20,45 Recently, LPS-induced ALI in mice was accompanied by increased expressions of RAGE and its ligands high-mobility group box 1 protein (HMGB1), HSP70 and S100b, which were attenuated by the RAGE inhibitors FPS-ZM1 or Azeliragon.46 However, despite high levels of RAGE and ROS are described in lung diseases, no previous studies explored the role of MGO in ALI conditions. We found here that MGO treatment markedly potentiated the immunostaining and mRNA expression of RAGE and increased the ROS levels (DHE technique) in the lung tissues of LPS-exposed mice, indicating that activation of the AGE-RAGE-ROS axis by MGO plays a crucial role in exacerbating ALI.
Inactivating MGO has been recognized as an interesting approach to reduce deleterious effects of AGEs.47,48 Among the molecules reported to inhibit MGO formation or sequestration, guanidino compounds such as the antihyperglycemic agent metformin are reported to react with MGO forming a guanidine-dicarbonyl adduct, reducing AGE formation.27,28 Therefore, in the second part of this study, we evaluated the protective effects of metformin on the deleterious actions of MGO in LPS-induced lung inflammation. We then evaluated the serum levels of MGO in the MGO-treated mice receiving or not metformin. Serum concentration of MGO achieved by the 12-week oral dosage regimen (290 nM) was 9.2-fold higher than the basal levels found in control mice (31 µM), which is consistent with the plasma concentration found in diabetic individuals10 and high-fat diet-fed mice.17 In the MGO-treated mice, association of metformin (given in the last week of MGO treatment) fully restored the MGO concentration to the levels of control mice. Next, we evaluated the main markers of LPS-induced ALI. Our data showed that metformin itself did not affect the LPS-induced ALI, but completely abrogated the exacerbation of the lung inflammation in MGO-treated animals. Our present findings strongly suggest that metformin acts by scavenging MGO in the circulation, reducing its plasma levels, thereby preventing the activation of AGE-RAGE-ROS axis. Metformin can also prevent MGO formation,8,26 but the findings MGO intake does not affect the fasting glucose levels or the insulin resistance35 exclude the possibility of metformin preventing endogenous MGO formation.
Conclusion
Prolonged oral intake of MGO potentiates LPS-induced mouse airway inflammation and release of inflammatory cytokines that is accompanied by increased RAGE expression and excessive ROS generation. In MGO-treated mice, association of metformin normalizes the serum MGO levels and suppresses the pro-inflammatory effects of MGO. It is likely that metformin scavenges MGO in vivo, preventing the formation of AGEs. Therefore, pharmacological treatments aiming to scavenger MGO from the circulation may be of therapeutic value in reducing ALI in patients with cardiometabolic diseases.
Abbreviations
AGEs, advanced glycation end products; ALI, acute lung injury; ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage; COX-2, cyclooxygenase-2; DAB, 3.3′ diaminobenzidine solution; DHE, dihydroethidine; H2O2, hydrogen peroxide; IFN-γ, interferon gamma; IL-1β, interleukin; IL-10, interleukin 10; IL-17, interleukin-17; LPS, lipopolysaccharide; MGO, methylglyoxal; RAGE, receptor for advanced glycation end products; ROS, reactive-oxygen species; TNF-α, tumor necrosis factor-α.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare they have no competing financial interests and no conflicts of interest for this work.
References
1. Bellani G, Laffey JG, Pham T, et al.; LUNG SAFE Investigators; ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi:10.1001/jama.2016.0291
2. Liu H, Yu X, Yu S, Kou J. Molecular mechanisms in lipopolysaccharide-induced pulmonary endothelial barrier dysfunction. Int Immunopharmacol. 2015;29(2):937–946. doi:10.1016/j.intimp.2015.10.010
3. Lee L, Downey GP. Neutrophil activation and acute lung injury. Curr Opinion Crit Care. 2001;7(1):1–7. doi:10.1097/00075198-200102000-00001
4. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol. 2015;194(3):855–860. doi:10.4049/jimmunol.1402513
5. Righetti RF, Dos Santos TM, Camargo LDN, et al. Protective effects of anti-IL17 on acute lung injury induced by LPS in mice. Front Pharmacol. 2018;9:1021. doi:10.3389/fphar.2018.01021
6. Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174(8):5033–5039. doi:10.4049/jimmunol.174.8.5033
7. Jinzhou Z, Tao H, Wensheng C, et al. Cyclooxygenase-2 suppresses polymorphonuclear neutrophil apoptosis after acute lung injury. J Trauma. 2008;64(4):1055–1060. doi:10.1097/TA.0b013e318047c07c
8. Schalkwijk CG, Stehouwer CDA. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev. 2020;100(1):407–461. doi:10.1152/physrev.00001.2019
9. Hanssen NMJ, Westerink J, Scheijen JLJM, van der Graaf Y, Stehouwer CDA, Schalkwijk CG. Higher plasma methylglyoxal levels are associated with incident cardiovascular disease and mortality in individuals with type 2 diabetes. Diabetes Care. 2018;41(8):1689–1695. doi:10.2337/dc18-0159
10. Odani H, Shinzato T, Matsumoto Y, Usami J, Maeda K. Increase in three alpha, beta-dicarbonyl compound levels in human uremic plasma: specific in vivo determination of intermediates in advanced Maillard reaction. Biochem Biophys Res Commun. 1999;256(1):89–93. doi:10.1006/bbrc.1999.0221
11. Lapolla A, Flamini R, Dalla Vedova A, et al. Glyoxal and methylglyoxal levels in diabetic patients: quantitative determination by a new GC/MS method. Clin Chem Lab Med. 2003;41(9):1166–1173. doi:10.1515/CCLM.2003.180
12. Ogawa S, Nakayama K, Nakayama M, et al. Methylglyoxal is a predictor in type 2 diabetic patients of intima-media thickening and elevation of blood pressure. Hypertension. 2010;56(3):471–476. doi:10.1161/HYPERTENSIONAHA.110.156786
13. Senda M, Ogawa S, Nako K, Okamura M, Sakamoto T, Ito S. The strong relation between post-hemodialysis blood methylglyoxal levels and post-hemodialysis blood glucose concentration rise. Clin Exp Nephrol. 2015;19(3):527–533. doi:10.1007/s10157-014-1018-6
14. Sukkar MB, Ullah MA, Gan WJ, et al. RAGE: a new frontier in chronic airways disease. Br J Pharmacol. 2012;167(6):1161–1176. doi:10.1111/j.1476-5381.2012.01984.x
15. de la Cruz-ares S, Cardelo MP, Gutiérrez-Mariscal FM, et al. Endothelial dysfunction and advanced glycation end products in patients with newly diagnosed versus established diabetes: from the CORDIOPREV Study. Nutrients. 2020;12(1):238. doi:10.3390/nu12010238
16. Gutierrez-Mariscal FM, Cardelo MP, de la Cruz S, et al. Reduction in circulating advanced glycation end products by Mediterranean diet is associated with increased likelihood of type 2 diabetes remission in patients with coronary heart disease: from the Cordioprev Study. Mol Nutr Food Res. 2021;65(1):e1901290. doi:10.1002/mnfr.201901290
17. Tang Y, Zhao Y, Wang P, Sang S. Simultaneous determination of multiple reactive carbonyl species in high fat diet-induced metabolic disordered mice and the inhibitory effects of rosemary on carbonyl stress. J Agric Food Chem. 2021;69(3):1123–1131. doi:10.1021/acs.jafc.0c07748
18. Kimzey MJ, Kinsky OR, Yassine HN, et al. Site specific modification of the human plasma proteome by methylglyoxal. Toxicol Appl Pharmacol. 2015;289(2):155–162. doi:10.1016/j.taap.2015.09.029
19. Ahmed N, Thornalley PJ. Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes Metab. 2007;9(3):233–245. doi:10.1111/j.1463-1326.2006.00595.x
20. Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflamm Res. 2020;13:1057–1073. doi:10.2147/JIR.S275595
21. Berlanga J, Cibrian D, Guillén I, et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin Sci. 2005;109(1):83–95. doi:10.1042/CS20050026
22. Sena CM, Matafome P, Crisóstomo J, et al. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol Res. 2012;65(5):497–506. doi:10.1016/j.phrs.2012.03.004
23. Crisóstomo J, Matafome P, Santos-Silva D, et al. Methylglyoxal chronic administration promotes diabetes-like cardiac ischaemia disease in Wistar normal rats. Nutr Metab Cardiovasc Dis. 2013;23(12):1223–1230. doi:10.1016/j.numecd.2013.01.005
24. de Oliveira MG, Medeiros ML, Tavares EBG, Mónica FZ, Antunes E. Methylglyoxal, a reactive glucose metabolite, induces bladder overactivity in addition to inflammation in mice. Front Physiol. 2020;11:290. doi:10.3389/fphys.2020.00290
25. Kothari V, Galdo JA, Mathews ST. Hypoglycemic agents and potential anti-inflammatory activity. J Inflamm Res. 2016;9:27–38. doi:10.2147/JIR.S86917
26. Battah S, Ahmed N, Thornalley PJ. Kinetics and mechanism of the reaction of metformin with methylglyoxal. Int Congr Ser. 2002;1245:355–356. doi:10.1016/S0531-5131(02)00889-0
27. Kinsky OR, Hargraves TL, Anumol T, et al. Metformin scavenges methylglyoxal to form a novel imidazolinone metabolite in humans. Chem Res Toxicol. 2016;29(2):227–234. doi:10.1021/acs.chemrestox.5b00497
28. Ruggiero-Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol. 1999;58(11):1765–1773. doi:10.1016/S0006-2952(99)00263-4
29. Kender Z, Fleming T, Kopf S, et al. Effect of metformin on methylglyoxal metabolism in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2014;122(5):316–319. doi:10.1055/s-0034-1371818
30. Brings S, Fleming T, De Buh S, et al. A scavenger peptide prevents methylglyoxal induced pain in mice. Biochim Biophys Acta Mol Basis. 2017;1863(3):654–662. doi:10.1016/j.bbadis.2016.12.001
31. Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999;48(1):198–202. doi:10.2337/diabetes.48.1.198
32. McCallister JW, Adkins EJ, O’Brien JM
33. Gong MN, Bajwa EK, Thompson BT, Christiani DC. Body mass index is associated with the development of acute respiratory distress syndrome. Thorax. 2010;65(1):44–50. doi:10.1136/thx.2009.117572
34. Stapleton RD, Suratt BT. Obesity and nutrition in acute respiratory distress syndrome. Clin Chest Med. 2014;35(4):655–671. doi:10.1016/j.ccm.2014.08.005
35. Medeiros ML, de Oliveira MG, Tavares EG, et al. Long-term methylglyoxal intake aggravates murine Th2-mediated airway eosinophil infiltration. Int Immunopharmacol. 2020;81:106254.
36. Calixto MC, Lintomen L, André DM, et al. Metformin attenuates the exacerbation of the allergic eosinophilic inflammation in high fat-diet-induced obesity in mice. PLoS One. 2013;8(10):e76786. doi:10.1371/journal.pone.0076786
37. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17(3–4):293–307. doi:10.2119/molmed.2010.00138
38. Pittet JF, Mackersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med. 1997;155(4):1187–1205. doi:10.1164/ajrccm.155.4.9105054
39. Kabir K, Gelinas JP, Chen M, et al. Characterization of a murine model of endotoxin-induced acute lung injury. Shock. 2002;17(4):300–303. doi:10.1097/00024382-200204000-00010
40. Ding Q, Liu GQ, Zeng YY, et al. Role of IL-17 in LPS-induced acute lung injury: an in vivo study. Oncotarget. 2017;8(55):93704–93711. doi:10.18632/oncotarget.21474
41. Neumann C, Scheffold A, Rutz S. Functions and regulation of T cell-derived interleukin-10. Semin Immunol. 2019;44:101344. doi:10.1016/j.smim.2019.101344
42. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180(9):5771–5777. doi:10.4049/jimmunol.180.9.5771
43. Delbin MA, Davel AP, Couto GK, et al. Interaction between advanced glycation end products formation and vascular responses in femoral and coronary arteries from exercised diabetic rats. PLoS One. 2012;7:e53318.
44. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69(1):349–364. doi:10.1146/annurev-med-041316-085215
45. Sanders KA, Delker DA, Huecksteadt T, et al. RAGE is a critical mediator of pulmonary oxidative stress, alveolar macrophage activation and emphysema in response to cigarette smoke. Sci Rep. 2019;9(1):1–16. doi:10.1038/s41598-018-36163-z
46. Li J, Wang K, Huang B, et al. The receptor for advanced glycation end products mediates dysfunction of airway epithelial barrier in a lipopolysaccharides-induced murine acute lung injury model. Int Immunopharmacol. 2021;93:107419. doi:10.1016/j.intimp.2021.107419
47. Matafome P, Rodrigues T, Sena C, Seiça R. Methylglyoxal in metabolic disorders: facts, myths, and promises. Med Res Rev. 2017;37(2):368–403. doi:10.1002/med.21410
48. Tate M, Higgins GC, De Blasio MJ, et al. The mitochondria-targeted methylglyoxal sequestering compound, mitoGamide, is cardioprotective in the diabetic heart. Cardiovasc Drugs Ther. 2020;34(2):223. doi:10.1007/s10557-019-06929-2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.