Back to Journals » Journal of Inflammation Research » Volume 14
Metal-Stimulated Interleukin-6 Production Through a Proton-Sensing Receptor, Ovarian Cancer G Protein-Coupled Receptor 1, in Human Bronchial Smooth Muscle Cells: A Response Inhibited by Dexamethasone
Authors Kadowaki M, Sato K ![]() , Kamio H, Kumagai M, Sato R, Nyui T, Umeda Y, Waseda Y, Anzai M, Aoki-Saito H, Koga Y
, Kamio H, Kumagai M, Sato R, Nyui T, Umeda Y, Waseda Y, Anzai M, Aoki-Saito H, Koga Y ![]() , Hisada T, Tomura H, Okajima F, Ishizuka T
, Hisada T, Tomura H, Okajima F, Ishizuka T ![]()
Received 28 June 2021
Accepted for publication 1 December 2021
Published 18 December 2021 Volume 2021:14 Pages 7021—7034
DOI https://doi.org/10.2147/JIR.S326964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Maiko Kadowaki,1,* Koichi Sato,2,* Hisashi Kamio,3 Makoto Kumagai,3 Rikishi Sato,3 Takafumi Nyui,3 Yukihiro Umeda,1 Yuko Waseda,1 Masaki Anzai,1 Haruka Aoki-Saito,4 Yasuhiko Koga,4 Takeshi Hisada,5 Hideaki Tomura,6 Fumikazu Okajima,3 Tamotsu Ishizuka1
1Third Department of Internal Medicine, Faculty of Medical Sciences, University of Fukui, Fukui, 910-1193, Japan; 2Laboratory of Signal Transduction, Institute for Molecular and Cellular Regulation, Gunma University, Maebeshi, 371-8512, Japan; 3Laboratory of Signal Transduction, Faculty of Pharmaceutical Sciences, Aomori University, Aomori, 030-0943, Japan; 4Department of Respiratory Medicine, Gunma University Graduate School of Medicine, Maebeshi, 371-8511, Japan; 5Gunma University Graduate School of Health Sciences, Maebeshi, 371-8514, Japan; 6Laboratory of Cell Signaling Regulation, Division of Life Science, School of Agriculture, Meiji University, Kawasaki, 214-8571, Japan
*These authors contributed equally to this work
Correspondence: Tamotsu Ishizuka
Third Department of Internal Medicine, Faculty of Medical Sciences, University of Fukui, 23-3 Matsuoka-Shimoaizuki, Eiheiji, Fukui, 910-1193, Japan
Tel +81-776-61-8355
Fax +81-776-61-8111
Email [email protected]
Purpose: Human bronchial smooth muscle cells (BSMCs) contribute to airway obstruction and hyperresponsiveness in patients with bronchial asthma. BSMCs also generate cytokines and matricellular proteins in response to extracellular acidification through the ovarian cancer G protein-coupled receptor 1 (OGR1). Cobalt (Co) and nickel (Ni) are occupational agents, which cause occupational asthma. We examined the effects of Co and Ni on interleukin-6 (IL-6) secretion by human BSMCs because these metals may act as ligands of OGR1.
Methods: Human BSMCs were incubated in Dulbecco’s Modified Eagle Medium (DMEM) containing 0.1% bovine serum albumin (BSA) (0.1% BSA-DMEM) for 16 hours and stimulated for the indicated time by exchanging the medium with 0.1% BSA-DMEM containing any of the metals or pH-adjusted 0.1% BSA-DMEM. IL-6 mRNA expression was quantified via reverse transcription polymerase chain reaction (RT-PCR) using the real-time TaqMan technology. IL-6 was measured using an enzyme-linked immunosorbent assay. Dexamethasone (DEX) was added 30 minutes before each stimulation. To knock down the expression of OGR1 in BSMCs, small interfering RNA (siRNA) targeting OGR1 (OGR1-siRNA) was transfected to the cells and non-targeting siRNA (NT-siRNA) was used as a control.
Results: Co and Ni both significantly increased IL-6 secretion in human BSMCs at 300 μM. This significant increase in IL-6 mRNA expression was observed 5 hours after stimulation. BSMCs transfected with OGR1-siRNA produced less IL-6 than BSMCs transfected with NT-siRNA in response to either Co or Ni stimulation. DEX inhibited Co- and Ni-stimulated IL-6 secretion by human BSMCs as well as pH 6.3-stimulated IL-6 secretion in a dose-dependent manner. DEX did not decrease phosphorylation of ERK1/2, p38 MAP kinase, and NF-κB p65 induced by either Co or Ni stimulation.
Conclusion: Co and Ni induce secretion of IL-6 in human BSMCs through activation of OGR1. Co- and Ni-stimulated IL-6 secretion is inhibited by DEX.
Keywords: cobalt, nickel, proton, cytokine, glucocorticoid, asthma
Introduction
Human bronchial smooth muscle cells (BSMCs) directly contribute to bronchial contraction in patients with bronchial asthma. An increase in airway smooth muscle mass also plays a pivotal role in airway obstruction and hyperresponsiveness.1 Ovarian cancer G protein-coupled receptor 1 (OGR1), also known as G protein-coupled receptor 68 (GPR68), has previously been identified as a proton-sensing G protein-coupled receptor (GPCR).2 OGR1 seems to regulate the contraction and proliferation of airway smooth muscle (ASM).3,4 Modest reductions in extracellular pH induce Ca2+ mobilization and contraction through OGR1 in human ASM cells.3 The benzodiazepine drug lorazepam was identified as a non-selective OGR1 positive allosteric modulator by yeast-based screens against OGR1.5 Among benzodiazepines, Gs-biased agent sulazepam, which selectively activates the Gs of the G protein signaling pathway through OGR1, promotes ASM relaxation but the balanced lorazepam does not.6,7 In future, the synthesis of OGR1 modulators, which avoids pro-contractile Gq/11 but activates pro-relaxant Gs may lead to the treatment of obstructive airway diseases such as asthma.4
Human BSMCs generate cytokines under the stimulation of different environmental factors, suggesting that BSMCs may be directly involved in airway inflammation.8–17 IL-6 is a pleiotropic cytokine that can be produced by human BSMCs in response to various inflammatory stimuli8,13,18,19 and cigarette smoke20 as well as by many other kinds of cells. IL-6 induces mast cell proliferation,21 expansion of Th2 cells,22,23 and mucus hypersecretion.24 Increased levels of IL-6 have been found in the serum,25 induced sputum,26 and bronchoalveolar lavage fluid (BALF)27,28 of asthmatic patients. Serum IL-6 concentration is negatively associated with pulmonary function in patients with obese asthma.29 Moreover, the level of IL-6 in BALF is higher in patients with non-allergic asthma than in patients with allergic asthma.30 These findings suggest that IL-6 may play an important role in the pathogenesis of asthma, particularly in obese or non-allergic asthma. Interestingly, extracellular acidification induces secretion of interleukin-6 (IL-6),8 interleukin-8 (IL-8)/ C-X-C motif chemokine ligand 8 (CXCL8),9 and connective tissue growth factor (CTGF)10 in addition to cell contraction3 through OGR1 activation in human BSMCs.
Hard metal is an alloy of tungsten carbide in a matrix of cobalt (Co). Nickel (Ni) is sometimes added to hard metal as a matrix in addition to Co. Hard metal dust is known to be associated with occupational asthma (OA), which has been termed hard metal asthma, since the first report of the development of asthma in a patient working in a tungsten carbide plant.31 Occupational exposure to low-molecular-weight (LMW) agents such as acid anhydrides and platinum salts induces specific IgE production and these agents seem to cause OA via IgE-mediated mechanisms.32 Asthma caused by Co sensitization is observed among Co production workers, diamond polishers, glassware manufacturers, and manufacturer of automotive engine valves.33 Evidence suggests that Co and Ni may be etiologic agents of hard metal asthma. Bronchial provocation challenges with Co induce either early or late asthmatic responses or both.34,35 These facts suggest that Co and Ni induce the pathogenesis of asthma by immunological mechanisms. Although IgE specific for these metals can be demonstrated in some patients with hard metal asthma,36,37 their involvements in the pathogenesis are unclear. This is because IgE-independent immunological mechanisms are expected to contribute to the pathogenesis of OA caused by LMW sensitizers such as diisocyanate although specific IgE is detected in diisocyanate asthmatics.38 Cell-mediated immunity39,40 has also been suggested to play a role in pathogenesis of hard metal asthma. However, the clinical features and pathogenesis of hard metal asthma are still largely unknown. Although either eosinophilic or neutrophilic inflammation can be predominant in induced sputum samples from patients with OA, neutrophilic inflammation is more common in OA induced by LMW agents.41
Recently, metals including iron (Fe), zinc (Zn), Co, Ni, and manganese (Mn) have been shown to induce intracellular Gq-coupled inositol phosphate signals in OGR1-expressing cells and osteoclasts through OGR1.42–44 This suggests that metals such as Fe, Zn, Co, Ni, and Mn are novel OGR1 agonists, which can individually activate OGR1 in neutral pH conditions. These metals may directly act on BSMCs and induce airway inflammation.
This study was initiated to elucidate the non-immunological mechanisms of OA caused by metals. Specifically, we examined OGR1-mediated IL-6 production from human BSMCs stimulated by Co and Ni and the effect of dexamethasone (DEX) on IL-6 production to determine the effect of glucocorticoids (GCs), i.e., the most effective therapeutic agents for asthma.
Materials and Methods
Cells and Reagents
Human BSMCs (catalog No. CC-2576) originated from non-diseased individuals were purchased from Lonza (Walkersville, MD). Cells were grown in the complete culture medium (Lonza), which was composed of smooth muscle cell basal medium supplemented with human epidermal growth factor, insulin, human fibroblast growth factor β, 5% fetal bovine serum, gentamicin, and amphotericin B, under a humidified atmosphere of 95% air plus 5% CO2 at 37°C, as previously reported.9,10 Three to six passages of BSMCs were used in the experiments. Human BSMCs were cultured in collagen I-coated 12-well plates (BioCoatTM, Corning International K.K., Tokyo, Japan) for enzyme-linked immunosorbent assay (ELISA) and in collagen I-coated 6-well plates (BioCoatTM) for real-time RT-PCR using TaqMan probes. Fatty acid-free bovine serum albumin (BSA) was purchased from EMD chemicals (San Diego, CA). Trypan blue solution, YM-254890, cobalt (II) chloride hexahydrate, nickel (II) chloride hexahydrate, chromium (Cr) (III) chloride hexahydrate, manganese (II) chloride tetrahydrate, and iron (III) chloride hexahydrate were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan) and dissolved in ultra-pure water (H2O). Goat anti-rabbit IgG-HRP was from GE Healthcare Japan Corporation (Tokyo, Japan). Antibodies for phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E), phospho-p38 MAPK (Thr180/Tyr182) (D3F9), phospho-NF-κB p65 (Ser536) (93H1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (14C10) were from Cell Signaling Technology (Danvers, MA). Dexamethasone (DEX), Dulbecco’s Modified Eagle Medium (DMEM, low glucose) and all other chemicals were purchased from Sigma-Aldrich Japan (Tokyo, Japan).
Cell Stimulation and ELISA for IL-6 and IL-8
Cultured human BSMCs were serum deprived for 16 hours in 0.1% BSA-DMEM and then stimulated by replacing the medium with 0.1% BSA-DMEM containing any of the metals: Co, Ni, Mn, Cr, and Fe or the control vehicle (0.1% H2O). To investigate the effect of DEX on cytokine secretion, DEX or a control vehicle (0.1% ethanol) was added to the medium 30 minutes before replacing the medium containing Co, Ni, and DEX or control vehicles. To evaluate the time course of IL-6 secretion, BSMCs were incubated in 0.1% BSA-DMEM containing Ni (300 μM), Co (300 μM), or the control vehicle for 6, 12, and 24 hours. We selected 300 μM as a concentration of Ni and Co that would induce sufficient cytokine production without toxicity. In some experiments, human BSMCs were stimulated by replacing the medium with pH-adjusted 0.1% BSA-DMEM (pH 6.3 or 7.4) instead of the medium containing metals. In these experiments, the pH of the DMEM solution containing 25 mM HEPES, 27 mM NaHCO3, and 0.1% BSA was adjusted to pH 6.3 or 7.4 by titration with HCl or NaOH. To investigate the effects of DEX on IL-6 secretion, DEX was also added 30 minutes before stimulation. The cell culture supernatants were collected and stored at −30°C. Concentrations of IL-6 and IL-8 were measured by ELISA (Duo Set ® ELISA development system, R&D Systems, Minneapolis, MN).
Quantitative RT-PCR Using Real-Time TaqMan Technology
To examine IL-6 mRNA expression, human BSMCs were incubated for 5 hours in 0.1% BSA-DMEM containing 300 μM Ni, 300 μM Co, or the control vehicle. To investigate the effect of DEX on IL-6 mRNA expression, DEX (100 nM) or the control vehicle was added to the medium 30 minutes before replacing the medium containing Ni, Co, and DEX or control vehicles. Total RNA was isolated from BSMCs in 6-well plates using the RNeasyⓇ Plus Mini Kit (Qiagen, Hilden, Germany) or Direct-zolTM RNA MiniPrep (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. Up to 2.5 g of total RNA was reverse-transcribed using random priming and reverse transcriptase (SuperScriptTM VILOTM Master Mix, Thermo Fisher Scientific, Waltham, MA). To evaluate expression levels of OGR1, IL-6, IL-8, and GAPDH mRNA, quantitative RT-PCR was performed using real-time TaqMan technology with a StepOneTM Real-Time PCR system (Thermo Fisher Scientific). TaqMan probes specific for human OGR1 (Hs00268858_s1), IL-6 (Hs00174131_m1), IL-8 (Hs00174103_m1), and GAPDH (Hs02758991_g1) were purchased from Thermo Fisher Scientific. The expression levels of IL-6, IL-8, and OGR1 mRNA were normalized to GAPDH mRNA expression levels, as previously reported.8
Transfection of Small Interfering RNA
Small interfering RNA (siRNA) targeting OGR1 (OGR1-siRNA, L-005591-00-0005) and non-targeting siRNA (NT-siRNA, D-001810-10-05) as a control were purchased from Thermo Fisher Scientific. OGR1-siRNA and NT-siRNA were transfected into cells at a final concentration of 3 nM using RNAiMAX reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. BSMCs suspended in the complete culture medium without gentamicin and amphotericin B were mixed with siRNA and the RNAiMAX reagent and then cultured in 6- or 12-well plates for 46 hours before further experiments.
Evaluation of Cell Viability and Cell Proliferation
Cell viability was assessed by trypan blue staining. Cell proliferation was measured by detecting bromodeoxyuridine (BrdU) incorporated into DNA during cell proliferation. Cell-based ELISA kit for measuring BrdU incorporation in situ (CycLex Cellular BrdU ELISA Kit Ver.2, Medical & Biological Laboratories, Tokyo, Japan) was used according to the manufacturer’s protocol. Namely, BSMCs cultured in 96 well plates were incubated in 0.1% BSA-DMEM for 16 hours, then Ni, Co, or control vehicle was added, and the cells were incubated for additional 24 hours. BrdU was added simultaneously when BSMCs were stimulated with metals. The incorporation of BrdU into DNA was measured at an absorbance of 450 nm.
Measurement of Intracellular Ca2+ Concentration
Intracellular Ca2+ concentration was measured as previously described.45–47 After a 20 minutes incubation of the cells with 1 μM Fura-2/AM at 37°C in Ham’s F-10 medium containing 0.1% BSA, the cells were washed twice with ice-cold HEPES-buffered medium composed of 20 mM HEPES, 134 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 2.5 mM NaHCO3, 5 mM glucose, and 0.1% BSA and suspended in the same medium. Intracellular Ca2+ concentration ([Ca2+]i) was measured in cell suspension under gentle stirring condition in HEPES-buffered medium. The Fura 2-loaded cells were warmed for 3 minutes at 37°C and the [Ca2+]i change was monitored in the intensities of 540 nm fluorescence obtained by the two excitation wavelengths (340 nm and 380 nm), which were monitored by CAF-100 fluorometer (JASCO, Tokyo, Japan).47 The [Ca2+]i change by the addition of Ni or Co was shown as Δfluorescence ratio (difference between peak and basal fluorescence ratio).
Western Blotting
The incubation was terminated by adding 0.1 mL of the cold lysis buffer composed of 50 mM Tris (pH 8.0), 150 mM NaCl2, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% NP-40 substitute, 1.04 mM AEBSF, 0.8 μM aprotinin, 0.04 mM bestatin, 14 μM E-64, 20 μM leupeptin, and 15 μM pepstatin A and immediately harvested from the 6-well plates with cell scrapers. The recovered lysate was incubated for 30 minutes on ice and centrifuged at 17,800 xg for 15 minutes. The supernatant was then analyzed by Western blotting with specific antibodies for phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), phospho-NF-κB p65 (Ser536), and GAPDH as previously described.8,9
Statistical Analysis
Almost all experiments were performed independently at least three times. The results of multiple observations are expressed as means ± standard error of the mean (SEM).
When raw data were standardized in each independent experiment, the standardized data were also shown as means ± SEM. The data were analyzed using Excel statistics software (SSRI, Tokyo, Japan). Differences between the mean values of two independent groups were determined using Student’s t-test. The Mann-Whitney test was also used to compare the two groups of nonparametric data. In analyses of more than two groups, analysis of variance was used to examine the significance of differences, and post-hoc analysis (Bonferroni test) was performed when significance was found. The Kruskal-Wallis test was also used to compare the nonparametric data in multiple groups. Steel test was performed when significance was found. P values less than 0.05 were considered significant.
Results
Effects of Co, Ni, Fe, Mn, and Cr on IL-6 and IL-8 Production by Human BSMCs
After 24 hours, as measured by ELISA, Co and Ni induced significant secretion of IL-6 compared with the control vehicle. Mn induced a low and non-significant amount of IL-6. Neither Cr nor Fe induced substantial IL-6 secretion (Figure 1A). A dose dependency of the metal effects was observed for Ni and Co (Figure 1B and C). A significant high level of IL-6 secretion was observed at 6 hours after the addition of either Co or Ni and gradually increased until 24 hours (Figure 1D). The absolute values of IL-6 secreted by Co- or Ni-stimulated BSMCs varied across experiments according to the cell condition or the cell lot number. The mean value of IL-6 secreted by BSMCs was 1.42 ng/mL under Co stimulation and 1.36 ng/mL under Ni stimulation. The mean value of IL-6 secreted by BSMCs under incubation with the control vehicle was 0.09 ng/mL. As measured by RT-PCR, significant increases in IL-6 mRNA expression were observed in Co- and Ni-stimulated human BSMCs compared to that in control vehicle-treated cells (Figure 1E). Co- and Ni- stimulation significantly enhanced IL-8 production in human BSMCs compared to unstimulated controls (Figure 2A). The mean value of IL-8 in the supernatant 24 hours after Co (300 μM)- or Ni (300 μM)-stimulation were 2.56 ng/mL (Co) and 1.56 ng/mL (Ni) respectively, while that in the control vehicle was 0.34 ng/mL. The expression levels of IL-8 mRNA were also significantly increased 5 hours after Co and Ni stimulation compared to the unstimulated control (Figure 2B).
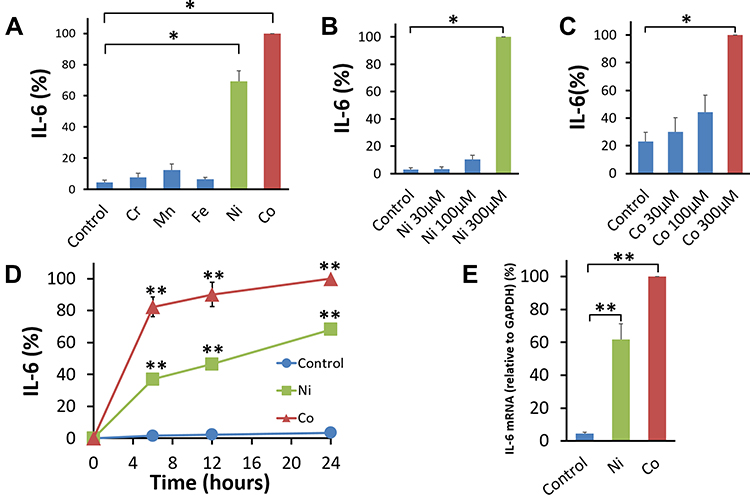 |
Figure 1 Effect of metals on IL-6 secretion by human bronchial smooth muscle cells. (A) Human bronchial smooth muscle cells (BSMCs) were stimulated with 300 μM of the metal Cr, Mn, Fe, Ni, or Co for 24 hours. Co and Ni induced substantial secretion of IL-6, but not Mn, Cr, and Fe compared with the control vehicle. The amount of IL-6 secreted by each individual metal-stimulated BSMCs was standardized to that secreted by Co-stimulated cells (mean ± SEM, n=4; *P<0.05). Ni (B) and Co (C) induced substantial secretion of IL-6 in a dose-dependent manner after 24-hour-incubation. The amount of IL-6 in each culture condition was standardized to that secreted by Ni (300 μM) or Co (300 μM)-stimulated cells (mean ± SEM, n=4; *P<0.05). (D) Co (300 μM) and Ni (300 μM) induced a substantial amount of IL-6 secretion at 6 hours. The level of IL-6 in the cell culture supernatant gradually increased until 24 hours after either stimulation. The IL-6 amount under Co and Ni stimulation was significantly higher than that secreted by non-stimulated BSMCs (Control: control vehicle alone) at 6, 12, and 24 hours after either stimulation. Data were standardized to the amount of IL-6 secreted by Co-stimulated BSMCs at 24 hours (mean ± SEM, n=6; **P<0.01). (E) The expression of IL-6 mRNA in Co (300 μM) and Ni (300 μM)-stimulated human BSMCs significantly increased at 5 hours compared with that in the cells incubated with the control vehicle. IL-6 mRNA was normalized to the expression of GAPDH mRNA. Data were standardized to IL-6 mRNA expression in Co (300 μM)-stimulated BSMCs (mean ± SEM, n=5; **P<0.01). |
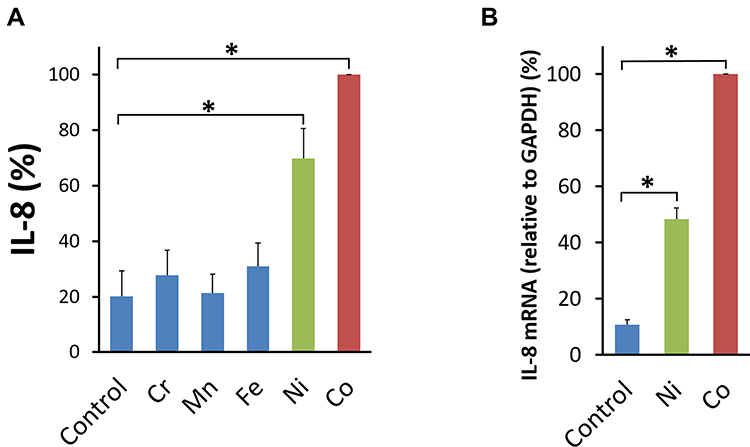 |
Figure 2 Effects of metals on IL-8 production by human BSMCs. (A) Human bronchial smooth muscle cells (BSMCs) were stimulated with 300 μM of the Metals: Cr, Mn, Fe, Ni, or Co for 24 hours. Co and Ni induced substantial secretion of IL-8, but not Mn, Cr, and Fe compared with the control vehicle (Control). The amount of IL-8 secreted by each individual metal-stimulated BSMCs was standardized to that secreted by Co-stimulated cells (mean ± SEM, n=4; *P<0.05). (B) The expression of IL-8 mRNA in Co (300 μM) and Ni (300 μM)-stimulated human BSMCs significantly increased at 5 hours compared with that in the cells incubated with the control vehicle (Control). IL-8 mRNA was normalized to the expression of GAPDH mRNA. Data were standardized to IL-8 mRNA expression in Co (300 μM)-stimulated BSMCs (mean ± SEM, n=4; *P<0.05). |
OGR1 is Involved in Co- and Ni-Stimulated IL-6 Production in Human BSMCs
In our previous study, only OGR1 mRNA was practically detected in human BSMCs among four kinds of GPCRs, namely OGR1, G protein-coupled receptor 4 (GPR4), T-cell death-associated gene 8 (TDAG8), and G2A8 and the expression level of OGR1 mRNA after treatment with OGR1-siRNA was decreased to about 5% of the level found after treatment with NT-siRNA in BSMCs.9 Human BSMCs under Co and Ni stimulation were transfected with OGR1-siRNA to decrease OGR1 expression in this study. We confirmed the knockdown efficacy of OGR1-siRNA as well. OGR1 mRNA expression in BSMCs transfected with OGR1-siRNA was considerably decreased compared with NT-siRNA transfected BSMCs (Figure 3A). IL-6 secretion under Co and Ni stimulation by human BSMCs transfected with OGR1-siRNA was significantly decreased compared with that of human BSMCs transfected with NT-siRNA (Figure 3B and C). The effect of OGR1 knockdown on IL-8 production from BSMCs was also examined similarly. In this case, IL-8 secretion seemed to be lower in BSMCs with OGR1 knockdown than in cells without knockdown in both Ni- and Co-stimulation. However, these differences were not necessarily significant, at least with respect to IL-8 production by Ni-stimulation (Figure 3D and E).
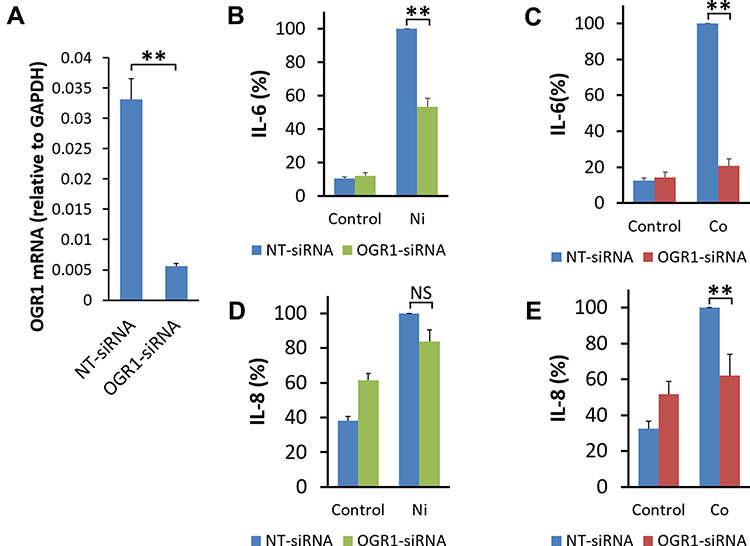 |
Figure 3 Possible involvement of OGR1 in Co- or Ni-stimulated IL-6 secretion by human bronchial smooth muscle cells. Human bronchial smooth muscle cells (BSMCs) were transfected with non-targeting small interfering RNA (NT-siRNA) or siRNA specific for ovarian cancer G protein-coupled receptor 1 (OGR1-siRNA). (A) The expression level of OGR1 mRNA 46 hours after the siRNA transfection in BSMCs was analyzed by quantitative RT-PCR performed using real-time TaqMan technology. OGR1 mRNA expression in BSMCs transfected with OGR1-siRNA was significantly decreased to less than 20% compared with that in NT-siRNA-transfected BSMCs. OGR1 mRNA was normalized to the expression of GAPDH mRNA (mean ± SEM, n=3; **P<0.01). At 46 hours after transfection, human BSMCs were incubated in 0.1% BSA-DMEM containing Ni (300 μM), Co (300 μM), or the control vehicle (0.1% H2O: Control) for an additional 24 hours. (B) Ni-stimulated IL-6 secretion was decreased in human BSMCs transfected with OGR1-siRNA compared with that in human BSMCs transfected with NT-siRNA. (C) Co-stimulated IL-6 secretion was also suppressed in human BSMCs transfected with OGR1-siRNA compared with that transfected with NT-siRNA. Data are expressed as percentages of the IL-6 values in the culture supernatant of BSMCs transfected with NT-siRNA after 24-hour stimulation by Ni or Co (mean ± SEM, n=8; **P<0.01). (D) Ni-stimulated IL-8 secretion tended to decrease, but the suppression was not significant in human BSMCs transfected with OGR1-siRNA compared with that in human BSMCs transfected with NT-siRNA. (E) Co-stimulated IL-8 secretion was significantly suppressed in human BSMCs transfected with OGR1-siRNA compared with those transfected with NT-siRNA. Data are expressed as percentages of the IL-8 values in the culture supernatant of BSMCs transfected with NT-siRNA after 24-hour stimulation by Ni or Co (mean ± SEM, n=8; NS, not significant; **P<0.01). |
Toxicity of Co and Ni on Human BSMCs in Serum-Deprived Media
To investigate toxicity or lethal effects of Co or Ni on BSMCs in serum-deprived media, BSMCs were stained by 0.2% trypan blue after 16 hour-preincubation in 0.1% BSA-DMEM and further 24 hour-incubation with 300 μM Ni, 300 μM Co, or the control vehicle (H2O). Cell viability (mean, n=6) was 98% after incubation with 300 μM Ni, 96% with 300 μM Co, and 95% with the control vehicle.
Effects of Co and Ni on the Proliferation of Human BSMCs
Proliferation of human BSMCs was assessed by BrdU assay. The incorporation of BrdU into DNA after 24 hours of Ni- or Co-stimulation did not increase. On the contrary, Ni and Co significantly decreased the incorporation of BrdU into DNA compared to the control vehicle (Figure 4).
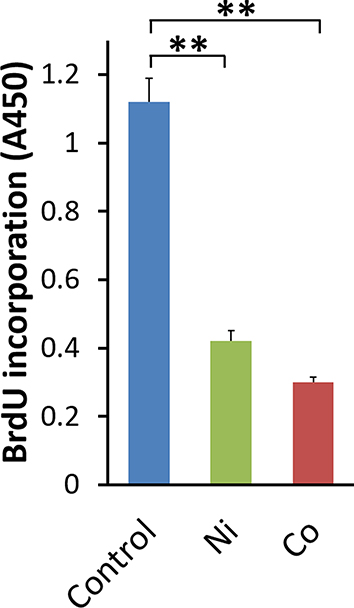 |
Figure 4 Effects of Ni and Co on the incorporation of BrdU into DNA in BSMCs. BrdU incorporation for 24 hours in Ni- or Co-stimulated BSMCs was measured as absorbance at 450 nm (A450). As a control (Control), BrdU incorporation in the control vehicle (0.1% H2O)-stimulated BSMCs was used (mean ± SEM, n=9; **P<0.01). |
Effects of DEX on IL-6 Production by Human BSMCs Stimulated by Co and Ni
In our previous study, DEX inhibited acidic pH-stimulated IL-8 production by human BSMCs in a dose-dependent manner. The inhibition was significant at a DEX concentration ≥1 nM. DEX also significantly inhibited IL-8 mRNA expression in pH 6.3-stimulated human BSMCs.9 DEX inhibited Ni- and Co-stimulated IL-6 production in a dose-dependent manner. This inhibition was significant at a DEX concentration ranging between 1 and 1000 nM (Figure 5A and B). The expression of IL-6 mRNA in human BSMCs increased after a 5 hour-incubation with the medium containing Ni or Co. DEX significantly inhibited Ni- and Co-induced IL-6 mRNA expression in human BSMCs (Figure 5C and D).
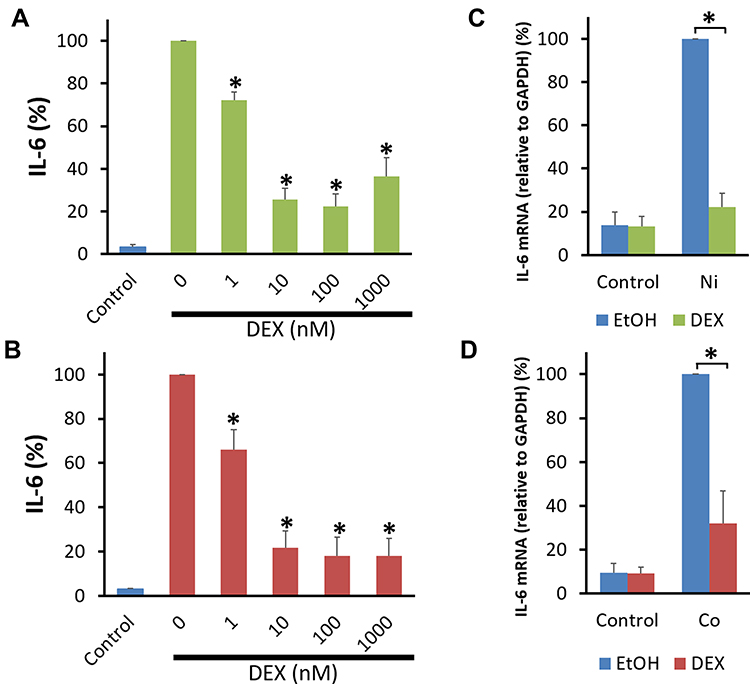 |
Figure 5 Effect of DEX on Co- and Ni-stimulated IL-6 production by human bronchial smooth muscle cells. Human bronchial smooth muscle cells (BSMCs) were pretreated with dexamethasone (DEX, 1–1000 nM) or 0.1% ethanol for 30 min. The cells were further incubated in 0.1% BSA-DMEM containing Ni (300 μM), Co (300 μM), or the control vehicle (0.1% H2O: Control) with DEX or 0.1% ethanol for 24 hours. DEX inhibited IL-6 secretion by Ni (A)- and Co (B)-stimulated human BSMCs in a dose-dependent manner. The inhibition was significant at a DEX concentration of 1–1000 nM. Data are expressed as percentages of IL-6 values in the culture supernatant of Ni- and Co-stimulated BSMCs without DEX (mean ± SEM, n=4; *P<0.05). Total RNA was extracted from BSMCs at 5 hours after incubation with Ni (300 μM), Co (300 μM), or the control vehicle (0.1% H2O). DEX (100 nM) significantly inhibited IL-6 mRNA expression in human BSMCs at 5 hours after Ni (C)- and Co (D)-stimulation. The expression levels of IL-6 mRNA were standardized to expression levels of GAPDH mRNA. Data are expressed as percentages of IL-6 mRNA expression at 5 hours after Ni- or Co-stimulation (mean ± SEM, n=4; *P<0.05). Abbreviations: EtOH, 0.1% ethanol as a solvent for DEX. |
Comparison of IL-6 Secretion by Human BSMCs Under Co, Ni, and Acidic pH Stimulation and the Effect of DEX on IL-6 Production by Acidic pH-Stimulated Cells
The amount of IL-6 secreted by both Co- and Ni-stimulated cells seemed slightly lower than that secreted by acidic pH-stimulated cells, but the difference was not significant (Figure 6A). DEX inhibited IL-6 secretion by acidic pH-stimulated human BSMCs as well as that by Ni- and Co-stimulated cells in a dose-dependent manner. The inhibition was significant at a 1–1000 nM DEX concentration (Figure 6B).
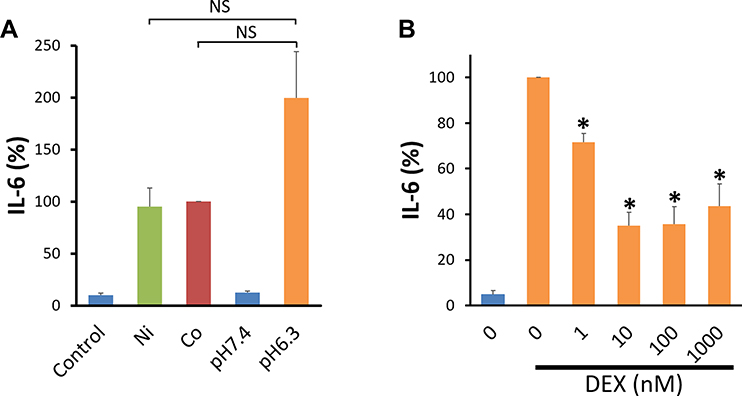 |
Figure 6 Comparison of IL-6 secretion by human bronchial smooth muscle cells stimulated by acidic pH, Ni, and Co. (A) Human bronchial smooth muscle cells (BSMCs) cultured in collagen-coated 12-well plates were serum deprived for 16 hours in 0.1% BSA-DMEM. The cells were stimulated by replacing the culture medium with 0.1% BSA-DMEM containing Ni (300 μM), Co (300 μM), the control vehicle (0.1% H2O: Control), or pH-adjusted 0.1% BSA-DMEM (pH 6.3 or 7.4) and incubated for additional 24 hours. Data are expressed as percentages of the IL-6 values in the culture supernatant of Co-stimulated human BSMCs. About half the amount of IL-6 appeared to be secreted by Ni- and Co-stimulated cells compared with acidic pH-stimulated cells, although the differences were not significant (mean ± SEM, n=9; NS, not significant). (B) DEX inhibited acidic pH-stimulated IL-6 secretion of human BSMCs in a dose-dependent manner. The inhibition was significant at a DEX concentration of 1–1000 nM (mean ± SEM, n=4; *P<0.05). |
Effects of Co and Ni on [Ca2+]i Changes and the Involvement of Gq/11-Proteins in [Ca2+]i Changes
The [Ca2+]i change was monitored in the intensities of 540 nm fluorescence obtained by the two excitations (340 nm and 380 nm) and the increase of [Ca2+]i was shown as Δfluorescence ratio (340/380). Addition of Ni (300 μM) or Co (300 μM) increased [Ca2+]i of BSMCs and Gq/11 inhibitor, YM-254890 (100 nM) significantly inhibited the increase of [Ca2+]i (Figure 7A–C).
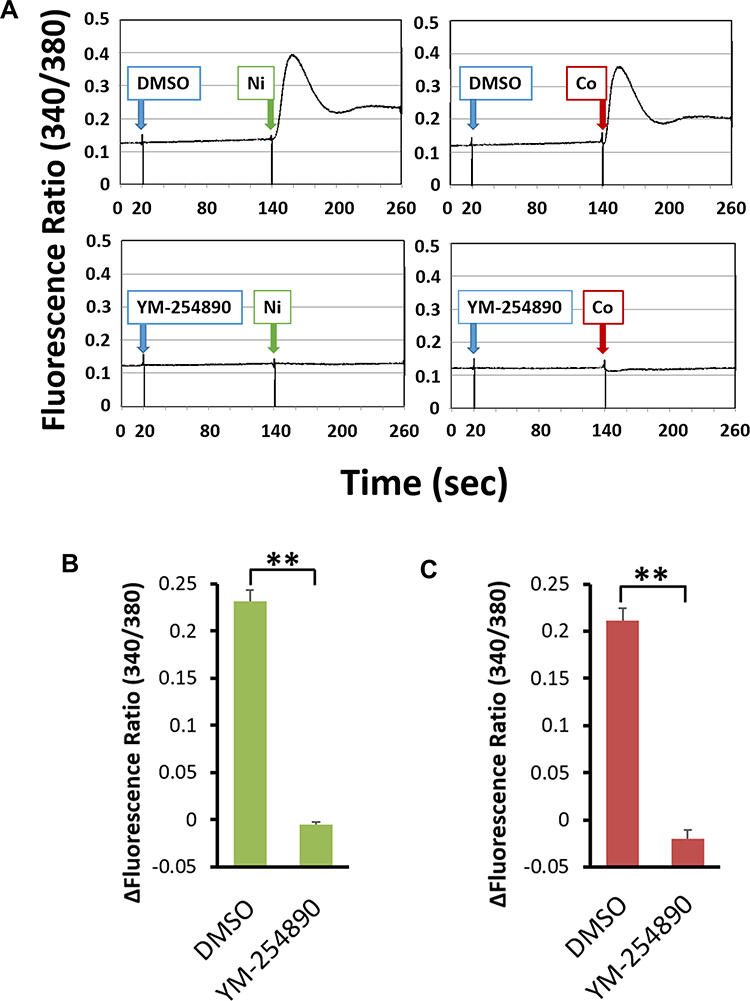 |
Figure 7 The [Ca2+]i increase induced by the addition of Ni or Co in human bronchial smooth muscle cells and the involvement of Gq/11 proteins. After the Fura2-loaded BSMCs were pre-incubated with in HEPES-buffered medium (pH 7.4) for 2 minutes with either 100 nM YM-254890 or DMSO as a vehicle, the cells were stimulated by addition of Ni (300 μM) or Co (300 μM). Ni and Co increased [Ca2+]i (shown as fluorescence ratio). YM-254890 (100 nM) almost completely inhibited this increase. Representative data are shown (A). The increase of [Ca2+]i shown as Δfluorescence ratio was significantly inhibited by YM-254890 in Ni (B)- and Co (C)-stimulated BSMCs (mean±SEM, n=3; **P<0.01). |
Activation of Extracellular Signal-Regulated Kinase 1/2 (ERK1/2), p38 Mitogen-Activated Protein Kinase (p38 MAPK), and Nuclear Factor-Kappa B (NF-κB) by Ni- and Co-Stimulation
Phosphorylation of ERK1/2 and p38 MAPK was assessed by Western blotting using specific antibodies for phosphorylated ERK1/2, phosphorylated p38 MAPK, phosphorylated NF-κB (p65), and GAPDH. ERK1/2 and p38 MAPK are activated by their phosphorylation. We examined endogenous levels of ERK1 and ERK2 when they were dually phosphorylated at Thr202 and Tyr204 of ERK1 (Thr185 and Tyr187 of ERK2) and singly phosphorylated at Thr202. Endogenous levels of p38 MAPK only when phosphorylated at Thr180 and Tyr182 were also analyzed. Addition of Ni (300 μM) and Co (300 μM) increased phosphorylated ERK1/2 and phosphorylated p38 MAPK. The level of these phosphorylated kinases reached a maximum at 10 minutes after each stimulation and decreased at 30 and 60 minutes. NF-κB p65 was detected only when it was phosphorylated at Ser536. Addition of Ni (300 μM) and Co (300 μM) also increased the amount of NF-κB p65 phosphorylated at Ser536. IκB kinase-α and IκB kinase-β have been implicated in the direct phosphorylation of p65 at Ser536 and this phosphorylation seems to be associated with transcriptional activity.48 The phosphorylated NF-κB p65 reached the maximum level at 30 minutes after Ni- or Co-stimulation (Figure 8A).
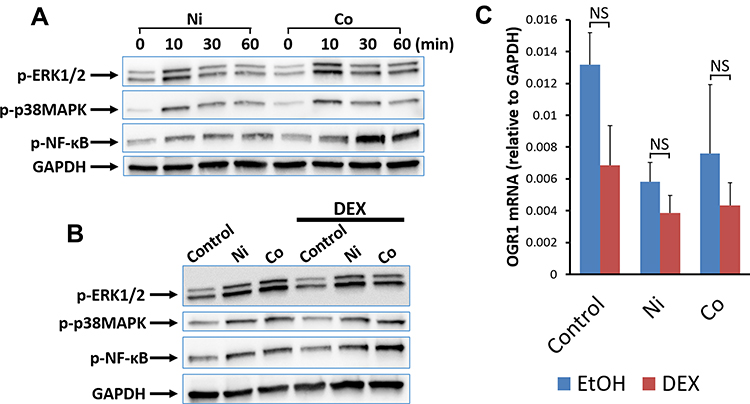 |
Figure 8 Effects of DEX on Ni- and Co-stimulated phosphorylation of ERK1/2, p38 MAPK, and NF-κB p65 and the expression of OGR1 mRNA in human bronchial smooth muscle cells. Phosphorylated ERK1/2, p38 MAPK, and NF-κB p65 were analyzed by Western blotting using specific antibodies for phospho-ERK1/2, phospho-p38 MAPK, and phospho-NF-κB p65 at 0, 10, 30, and 60 minutes after Ni- or Co-stimulation. GAPDH was used as a control protein in Western blotting. Ni (300 μM) and Co (300 μM) increased the phospho-ERK1/2, phospho-p38 MAPK, and phospho-NF-κB p65 after each metal stimulation. Phosphorylated ERK1/2 and phosphorylated p38 MAPK were maximal at 10 minutes after Ni and Co stimulation, and phosphorylated NF-κB p65 was maximal at 30 minutes after the stimulation. Representative results from two independent experiments are shown (A). DEX (100 nM) added 30 minutes before stimulation; it did not inhibit ERK1/2 phosphorylation at 10 min, p38 MAPK phosphorylation at 10 minutes, or NF-κB p65 phosphorylation at 30 minutes after Ni- and Co-stimulation. Representative results from three independent experiments are shown (B). The addition of DEX (100 nM) 30 minutes before Ni- and Co- stimulation did not decrease OGR1 mRNA expression after 5 h of Ni and Co stimulation. In BSMCs without metal stimulation (Control), there was a downward trend in OGR1 mRNA expression after 5.5 hours of DEX treatment compared to that without DEX treatment, but the difference was not significant (mean±SEM, n=4; NS, not significant) (C). Abbreviations: EtOH, 0.1% ethanol as a solvent for DEX. |
Effects of DEX on Ni- and Co-Stimulated Activation of ERK1/2, p38 MAPK, and NF-κB p65
After 30 minutes incubation with DEX (100 nM) or control vehicle (0.1% ethanol), BSMCs were stimulated by Ni (300 μM) or Co (300 μM). Ni and Co increased phosphorylated ERK1/2, p38 MAPK at 10 minutes and phosphorylated NF-κB p65 at 30 minutes after the metal stimulation. DEX did not decrease the phosphorylation (Figure 8B).
Effects of DEX on the Expression of OGR1 mRNA in Human BSMCs
OGR1 mRNA expression was analyzed by quantitative RT-PCR 5 hours after Ni, Co, or control vehicle (H2O) stimulation. DEX (100 nM) or the control vehicle (0.1% ethanol) was added 30 minutes before metal stimulation. OGR1 mRNA expression was slightly decreased after 5.5 hours in the presence of DEX compared to the absence of DEX, but the differences were not significant (Figure 8C).
Discussion
It has been demonstrated that GPCRs, including OGR1, GPR4, and TDAG8/GPR65, sense extracellular protons and mediate the cellular actions induced by an alkaline and an acidic pH ranging from 8 to 6 through histidine residues in a variety of cell types.2,49–51 We have previously reported that OGR1 expression is required to develop allergic asthma in mice.52 Bronchial asthma is a disease characterized by chronic airway inflammation, airway hyperresponsiveness, and reversible airway obstruction. The pathogenesis in most asthmatics is characterized by eosinophilic airway inflammation, so-called type 2 inflammation, and inhaled GCs (inhaled corticosteroids: ICS) are the most effective anti-inflammatory therapy for asthma.53 However, in some asthmatics, especially obese asthmatics and some occupational asthmatics, there is a phenotype wherein the eosinophilic airway inflammation is not prominent but neutrophilic airway inflammation is predominant.54
Human BSMCs are increasingly recognized as an important source of inflammatory cytokines, as well as the effector cells of bronchoconstriction.55–62 Among these cytokines, IL-6, IL-8, and IL-17 may be particularly associated with non-type 2 airway inflammation.54,63 Our previous studies showed that extracellular acidification induced human BSMCs to generate IL-6, IL-8, and CTGF via OGR1 activation.8–10 In the present study, BSMCs, in which the expression of OGR1 was decreased by OGR1-targeting siRNA, released less IL-6 in response to Co or Ni than the control BSMCs, suggesting that metals such as Co and Ni may stimulate human BSMCs and induce IL-6 secretion through OGR1-mediated intracellular signal transduction. However, the results for IL-8 were not as clear as those for IL-6. Additionally, the degree of cytokine suppression by OGR1-siRNA seemed to be weaker in Ni stimulation than in Co stimulation. These results suggest that Ni-stimulated cytokine production in BSMCs, especially IL-8 production, may well be mediated by mechanisms other than OGR1 activation. The amount of IL-6 released by Co- and Ni-stimulated cells was substantial and was about half of that produced by acidic pH (pH 6.3)-stimulated BSMCs, suggesting that both Co and Ni can activate OGR1 in human BSMCs to a level comparable to pH 6.3. Altogether, our results suggest that Co and Ni act as OGR1 ligands in the airway and may elicit non-eosinophilic or non-type 2 inflammation independent of sensitization-mediated immune response and environmental acidification. The findings shed light on new mechanisms mediating the development of OA caused by Co or Ni exposure.
Interestingly, certain alleles in the IL-6 receptor gene are associated with increased asthma susceptibility.64 Activation of the IL-6 pathway has been suggested to be involved in neutrophilic and mixed granulocytic airway inflammation in asthmatic patients.65,66 A longitudinal analysis confirmed an exacerbation-prone asthma phenotype characterized by features of metabolic dysfunction. Blood measures of IL-6 concentrations but not eosinophil counts, were significantly associated with exacerbation-prone asthma, and baseline levels of IL-6 and eosinophils predicted exacerbations.67 In a recent study, the classification of asthmatic patients with bronchial epithelial IL-6 trans-signaling pathway activation as a novel subset was advocated. This phenotype is characterized by an increased exacerbation rate, type 2 airway inflammation-independent eosinophilia, and epithelial dysfunction.68
GCs are the most effective anti-inflammatory drugs for chronic airway inflammation in asthmatics, and ICS are widely used for the long-term management of bronchial asthma. GC resistance in asthmatic patients not responding well to GC therapy is important for understanding the pathogenesis of severe asthma. We have shown that DEX inhibited IL-8 secretion by acidic pH-stimulated human BSMCs.9 Similarly, DEX inhibited IL-6 secretion by Co- and Ni-stimulated BSMCs as well as that by acidic pH-stimulated BSMCs in a dose-dependent manner. Moreover, like IL-6 protein, IL-6 mRNA expression in Co- and Ni-stimulated BSMCs was inhibited by DEX, showing that DEX inhibited IL-6 gene transcription following OGR1 activation. Since the inhibition of cytokine production by DEX is not specific to OGR1-mediated stimulation, and since the pretreatment time with DEX was 30 minutes prior to the application of Ni or Co stimulation in the present study, it is unlikely that DEX reduced IL-6 production by decreasing the expression of OGR1 in BSMCs. In fact, at the mRNA level, the expression of OGR1 mRNA might be slightly decreased in unstimulated BSMCs 5.5 hours after the start of DEX treatment, but the difference was not significant. There was no DEX-induced decrease in OGR1 mRNA expression in Co- or Ni-stimulated BSMCs. Co and Ni stimulation elicited activation of OGR1-linked Gq, increased intracellular Ca concentration and phosphorylation of ERK1/2, p38MAPK, and NF-κB p65. Since these phosphorylation pathways are likely to be involved in OGR1-mediated IL-6 production, we examined the effects of DEX on phosphorylation. Phosphorylation of ERK1/2, p38 MAPK, and NF-κB p65 was not inhibited by DEX. Considering the mechanism of action of DEX in suppressing IL-6 production, it is expected that it mainly acts in the step after the transcription factor binds to the target DNA by extracellular stimulation,53 but this will be a subject of future research.
Hypertrophy and hyperplasia of BSMCs are characteristic pathological findings of severe asthma.1,69 In the present study, ERK1/2 in BSMCs were phosphorylated and activated by Co and Ni stimulation, suggesting that these metal stimuli might promote cell proliferation of BSMCs. However, Co (300 μM) and Ni (300 μM) did not promote the incorporation of BrdU into DNA, and we could not prove that these metals promote the proliferation of BSMCs. Although the concentrations of Co and Ni used in the serum-free culture medium in this experiment did not show toxicity that induced cell death, we cannot deny the possibility that they had an adverse effect on the intracellular mechanisms involved in cell proliferation, and we believe that this issue requires further investigation.
One limitation in our study is that the experiments were conducted using human BSMCs originated from a non-diseased individual. No data were obtained to show that Co or Ni is associated with proliferation or hypertrophy of BSMCs, which are factors in airway remodeling. If IL-6 secretion via OGR1 activation is involved in neutrophilic airway inflammation in OA, then our results might support the usefulness of ICS in the pharmacotherapy of OA caused by some metals. In future, it would be important to investigate whether DEX inhibits OGR1-mediated IL-6 secretion in BSMCs isolated from patients with steroid-resistant asthma or OA, because the BSMCs of patients with severe asthma seem resistant to GCs.70–72 Since the inhibitory effect of GCs on cytokine production in BSMCs is not specific to OGR1-mediated cytokine production, further elucidation of the mechanism by which GCs inhibit cytokine production at the cellular level is necessary.
Conclusions
We demonstrate for the first time that Co and Ni induce IL-6 secretion by human BSMCs through OGR1-mediated cellular activation, and that DEX inhibits OGR1-mediated IL-6 secretion by BSMCs originated from non-diseased individuals. These results suggest that Co and Ni not only induce inflammation as sensitizers in the airway but may also be involved in airway inflammation as ligands for OGR1. Additionally, it is suggested that GCs may be effective in the treatment of airway inflammation induced by Co and Ni.
Acknowledgments
The authors are grateful to Ayumi Suzuki and Emi Takeuchi for technical assistance and to Kazuyo Takeuchi for manuscript preparation. We would like to thank Editage for English language editing.
Funding
This work was supported by a Grant-in-Aid for scientific research from the Japan Society for the Promotion of Science from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, 20K08538 and 21K08174 (M. Kadowaki and T. Ishizuka).
Disclosure
The authors have no conflicts of interest to declare for this work.
References
1. Hirst SJ. Airway smooth muscle as a target in asthma. Clin Exp Allergy. 2000;30(Suppl 1):54–59. doi:10.1046/j.1365-2222.2000.00099.x
2. Ludwig MG, Vanek M, Guerini D, et al. Proton-sensing G-protein-coupled receptors. Nature. 2003;425:93–98. doi:10.1038/nature01905
3. Saxena H, Deshpande DA, Tiegs BC, et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br J Pharmacol. 2012;166:981–990. doi:10.1111/j.1476-5381.2011.01807.x
4. Nayak AP, Penn RB. The proton-sensing receptor ovarian cancer G-protein coupled receptor 1 (OGR1) in airway physiology and disease. Curr Opin Pharmacol. 2020;51:1–10. doi:10.1016/j.coph.2020.03.004
5. Huang XP, Karpiak J, Kroeze WK, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015;527:477–483. doi:10.1038/nature15699
6. Pera T, Deshpande DA, Ippolito M, et al. Biased signaling of the proton-sensing receptor OGR1 by benzodiazepines. FASEB J. 2018;32:862–874. doi:10.1096/fj.201700555R
7. Nayak AP, Pera T, Deshpande DA, et al. Regulation of ovarian cancer G protein-coupled receptor-1 expression and signaling. Am J Physiol Lung Cell Mol Physiol. 2019;316:L894–L902. doi:10.1152/ajplung.00426.2018
8. Ichimonji I, Tomura H, Mogi C, et al. Extracellular acidification stimulates IL-6 production and Ca2+ mobilization through proton-sensing OGR1 receptors in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2010;299:L567–577. doi:10.1152/ajplung.00415.2009
9. Kadowaki M, Yamada H, Sato K, et al. Extracellular acidification-induced CXCL8 production through a proton-sensing receptor OGR1 in human airway smooth muscle cells: a response inhibited by dexamethasone. J Inflamm (Lond). 2019;16:4. doi:10.1186/s12950-019-0207-1
10. Matsuzaki S, Ishizuka T, Yamada H, et al. Extracellular acidification induces connective tissue growth factor production through proton-sensing receptor OGR1 in human airway smooth muscle cells. Biochem Biophys Res Commun. 2011;413:499–503. doi:10.1016/j.bbrc.2011.08.087
11. Nakajima M, Kawaguchi M, Ota K, et al. IL-17F induces IL-6 via TAK1-NFkappaB pathway in airway smooth muscle cells. Immun Inflamm Dis. 2017;5:124–131. doi:10.1002/iid3.149
12. Ge Q, Moir LM, Trian T, et al. The phosphoinositide 3ʹ-kinase p110delta modulates contractile protein production and IL-6 release in human airway smooth muscle. J Cell Physiol. 2012;227:3044–3052. doi:10.1002/jcp.23046
13. Shan L, Redhu NS, Saleh A, et al. Thymic stromal lymphopoietin receptor-mediated IL-6 and CC/CXC chemokines expression in human airway smooth muscle cells: role of MAPKs (ERK1/2, p38, and JNK) and STAT3 pathways. J Immunol. 2010;184:7134–7143. doi:10.4049/jimmunol.0902515
14. Henness S, Johnson CK, Ge Q, et al. IL-17A augments TNF-alpha-induced IL-6 expression in airway smooth muscle by enhancing mRNA stability. J Allergy Clin Immunol. 2004;114:958–964. doi:10.1016/j.jaci.2004.06.023
15. Oltmanns U, Chung KF, Walters M, et al. Cigarette smoke induces IL-8, but inhibits eotaxin and RANTES release from airway smooth muscle. Respir Res. 2005;6:74. doi:10.1186/1465-9921-6-74
16. Pang L, Knox AJ. Bradykinin stimulates IL-8 production in cultured human airway smooth muscle cells: role of cyclooxygenase products. J Immunol. 1998;161:2509–2515.
17. Wuyts WA, Vanaudenaerde BM, Dupont LJ, et al. Interleukin-17–induced interleukin-8 release in human airway smooth muscle cells: role for mitogen-activated kinases and nuclear factor-kappaB. J Heart Lung Transplant. 2005;24:875–881. doi:10.1016/j.healun.2004.05.003
18. McWhinnie R, Pechkovsky DV, Zhou D, et al. Endothelin-1 induces hypertrophy and inhibits apoptosis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L278–286. doi:10.1152/ajplung.00111.2006
19. Zhong H, Belardinelli L, Maa T, et al. A 2B adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:118–125. doi:10.1165/rcmb.2003-0118OC
20. Lin CC, Lee IT, Yang YL, et al. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: role of TLR4-dependent NADPH oxidase activation. Free Radic Biol Med. 2010;48:240–254. doi:10.1016/j.freeradbiomed.2009.10.047
21. Hollins F, Kaur D, Yang W, et al. Human airway smooth muscle promotes human lung mast cell survival, proliferation, and constitutive activation: cooperative roles for CADM1, stem cell factor, and IL-6. J Immunol. 2008;181:2772–2780. doi:10.4049/jimmunol.181.4.2772
22. Dodge IL, Carr MW, Cernadas M, et al. IL-6 production by pulmonary dendritic cells impedes Th1 immune responses. J Immunol. 2003;170:4457–4464. doi:10.4049/jimmunol.170.9.4457
23. Doganci A, Sauer K, Karwot R, et al. Pathological role of IL-6 in the experimental allergic bronchial asthma in mice. Clin Rev Allergy Immunol. 2005;28:257–270. doi:10.1385/CRIAI:28:3:257
24. Neveu WA, Allard JB, Dienz O, et al. IL-6 is required for airway mucus production induced by inhaled fungal allergens. J Immunol. 2009;183:1732–1738. doi:10.4049/jimmunol.0802923
25. Yokoyama A, Kohno N, Fujino S, et al. Circulating interleukin-6 levels in patients with bronchial asthma. Am J Respir Crit Care Med. 1995;151:1354–1358. doi:10.1164/ajrccm.151.5.7735584
26. Neveu WA, Allard JL, Raymond DM, et al. Elevation of IL-6 in the allergic asthmatic airway is independent of inflammation but associates with loss of central airway function. Respir Res. 2010;11:28. doi:10.1186/1465-9921-11-28
27. Zhang JG, Chen XJ, Liu T, et al. FOXP3(+) associated with the pro-inflammatory regulatory T and T helper 17 effector cells in asthma patients. Exp Ther Med. 2016;12:2753–2758. doi:10.3892/etm.2016.3662
28. Tillie-Leblond I, Pugin J, Marquette CH, et al. Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. Am J Respir Crit Care Med. 1999;159:487–494. doi:10.1164/ajrccm.159.2.9805115
29. Dixon AE, Shade DM, Cohen RI, et al. Effect of obesity on clinical presentation and response to treatment in asthma. J Asthma. 2006;43:553–558. doi:10.1080/02770900600859123
30. Virchow JC, Kroegel C, Walker C, et al. Inflammatory determinants of asthma severity: mediator and cellular changes in bronchoalveolar lavage fluid of patients with severe asthma. J Allergy Clin Immunol. 1996;98(5 Pt 2):
31. Bruckner HC. Extrinsic asthma in a tungsten carbide worker. J Occup Med. 1967;9:518–519. doi:10.1097/00043764-196710000-00006
32. Weissman DN, Lewis DM. Is specific antibody determination diagnostic for asthma attributable to low-molecular-weight agents? Occup Med. 2000;15:385–398.
33. Walters GI, Robertson AS, Moore VC, et al. Cobalt asthma in metalworkers from an automotive engine valve manufacturer. Occup Med (Lond). 2014;64:358–364. doi:10.1093/occmed/kqu043
34. Shirakawa T, Kusaka Y, Fujimura N, et al. Occupational asthma from cobalt sensitivity in workers exposed to hard metal dust. Chest. 1989;95:29–37. doi:10.1378/chest.95.1.29
35. Kusaka Y, Yokoyama K, Sera Y, et al. Respiratory diseases in hard metal workers: an occupational hygiene study in a factor. Br J Ind Med. 1986;43:474–485. doi:10.1136/oem.43.7.474
36. Shirakawa T, Kusaka Y, Fujimura N, et al. The existence of specific antibodies to cobalt in hard metal asthma. Clin Allergy. 1988;18:451–460. doi:10.1111/j.1365-2222.1988.tb02895.x
37. Shirakawa T, Kusaka Y, Morimoto K. Specific IgE antibodies to nickel in workers with known reactivity to cobalt. Clin Exp Allergy. 1992;22:213–218. doi:10.1111/j.1365-2222.1992.tb03075.x
38. Jones MG, Floyd A, Nouri-Aria KT, et al. Is occupational asthma to diisocyanates a non-IgE-mediated disease? J Allergy Clin Immunol. 2006;117:663–669. doi:10.1016/j.jaci.2005.09.053
39. Kusaka Y, Nakano Y, Shirakawa T, et al. Lymphocyte transformation test with cobalt in hard metal asthma. Ind Health. 1989;27:155–163. doi:10.2486/indhealth.27.155
40. Kusaka Y, Nakano Y, Shirakawa T, et al. Lymphocyte transformation test with nickel in hard metal asthma: another sensitizing component of hard metal. Ind Health. 1991;29:153–160. doi:10.2486/indhealth.29.153
41. Anees W, Huggins V, Pavord ID, et al. Occupational asthma due to low molecular weight agents: eosinophilic and non-eosinophilic variants. Thorax. 2002;57:231–236. doi:10.1136/thorax.57.3.231
42. Abe-Ohya R, Ishikawa T, Shiozawa H, et al. Identification of metals from osteoblastic ST-2 cell supernatants as novel OGR1 agonists. J Recept Signal Transduct Res. 2015;35:485–492. doi:10.3109/10799893.2015.1015736
43. Mochimaru Y, Negishi J, Murakami S, et al. Metals differentially activate ovarian cancer G protein-coupled receptor 1 in various species. Zoolog Sci. 2018;35:109–114. doi:10.2108/zs170145
44. Negishi J, Omori Y, Shindo M, et al. Manganese and cobalt activate zebrafish ovarian cancer G-protein-coupled receptor 1 but not GPR4. J Recept Signal Transduct Res. 2017;37:401–408. doi:10.1080/10799893.2017.1298130
45. Okajima F, Sho K, Kondo Y. Inhibition by islet-activating protein, pertussis toxin, of P2-purinergic receptor-mediated iodide efflux and phosphoinositide turnover in FRTL-5 cells. Endocrinology. 1988;123:1035–1043. doi:10.1210/endo-123-2-1035
46. Kotake M, Sato K, Mogi C, et al. Acidic pH increases cGMP accumulation through the OGR1/phospholipase C/Ca2+/neuronal NOS pathway in N1E-115 neuronal cells. Cell Signal. 2014;26:2326–2332. doi:10.1016/j.cellsig.2014.07.010
47. Sato K, Mogi C, Mighell AJ, et al. A missense mutation of Leu74Pro of OGR1 found in familial amelogenesis imperfecta actually causes the loss of the pH-sensing mechanism. Biochem Biophys Res Commun. 2020;526:920–926. doi:10.1016/j.bbrc.2020.04.005
48. Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi:10.1038/sj.onc.1209937
49. Tomura H, Mogi C, Sato K, et al. Proton-sensing and lysolipid-sensitive G-protein-coupled receptors: a novel type of multi-functional receptors. Cell Signal. 2005;17:1466–1476. doi:10.1016/j.cellsig.2005.06.002
50. Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal. 2013;25:2263–2271. doi:10.1016/j.cellsig.2013.07.022
51. Aoki H, Mogi C, Okajima F. Ionotropic and metabotropic proton-sensing receptors involved in airway inflammation in allergic asthma. Mediat Inflamm. 2014;2014:712962. doi:10.1155/2014/712962
52. Aoki H, Mogi C, Hisada T, et al. Proton-sensing ovarian cancer G protein-coupled receptor 1 on dendritic cells is required for airway responses in a murine asthma model. PLoS One. 2013;8:e79985. doi:10.1371/journal.pone.0079985
53. Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. 2011;163:29–43. doi:10.1111/j.1476-5381.2010.01199.x
54. Hinks TSC, Levine SJ, Brusselle GG. Treatment options in type-2 low asthma. Eur Respir J. 2021;57:2000528. doi:10.1183/13993003.00528-2020
55. Damera G, Tliba O, Panettieri RA. Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther. 2009;22:353–359. doi:10.1016/j.pupt.2008.12.006
56. Howarth PH, Knox AJ, Amrani Y, et al. Synthetic responses in airway smooth muscle. J Allergy Clin Immunol. 2004;114(2 Suppl):S32–S50. doi:10.1016/j.jaci.2004.04.041
57. Tliba O, Panettieri RA. Noncontractile functions of airway smooth muscle cells in asthma. Annu Rev Physiol. 2009;71:509–535. doi:10.1146/annurev.physiol.010908.163227
58. Xia YC, Redhu NS, Moir LM, et al. Pro-inflammatory and immunomodulatory functions of airway smooth muscle: emerging concepts. Pulm Pharmacol Ther. 2013;26:64–74. doi:10.1016/j.pupt.2012.05.006
59. Oliver BG, Johnston SL, Baraket M, et al. Increased proinflammatory responses from asthmatic human airway smooth muscle cells in response to rhinovirus infection. Respir Res. 2006;7:71. doi:10.1186/1465-9921-7-71
60. Gosens R, Rieks D, Meurs H, et al. Muscarinic M3 receptor stimulation increases cigarette smoke-induced CXCL8 secretion by human airway smooth muscle cells. Eur Respir J. 2009;34:1436–1443. doi:10.1183/09031936.00045209
61. Iwata S, Ito S, Iwaki M, et al. Regulation of endothelin-1-induced interleukin-6 production by Ca2+ influx in human airway smooth muscle cells. Eur J Pharmacol. 2009;605:15–22. doi:10.1016/j.ejphar.2008.12.045
62. Berair R, Hollins F, Brightling C. Airway smooth muscle hypercontractility in asthma. J Allergy (Cairo). 2013;2013:185971. doi:10.1155/2013/185971
63. Hudey SN, Ledford DK, Cardet JC. Mechanisms of non-type 2 asthma. Curr Opin Immunol. 2020;66:123–128. doi:10.1016/j.coi.2020.10.002
64. Ferreira MA, Matheson MC, Duffy DL, et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet. 2011;378:1006–1014. doi:10.1016/S0140-6736(11)60874-X
65. Ullah MA, Revez JA, Loh Z, et al. Allergen-induced IL-6 trans-signaling activates γδ T cells to promote type 2 and type 17 airway inflammation. J Allergy Clin Immunol. 2015;136:1065–1073. doi:10.1016/j.jaci.2015.02.032
66. Chu DK, Al-Garawi A, Llop-Guevara A, et al. Therapeutic potential of anti-IL-6 therapies for granulocytic airway inflammation in asthma. Allergy Asthma Clin Immunol. 2015;11:14. doi:10.1186/s13223-015-0081-1
67. Peters MC, Mauger D, Ross KR, et al. Evidence for exacerbation-prone asthma and predictive biomarkers of exacerbation frequency. Am J Respir Crit Care Med. 2020;202:973–982. doi:10.1164/rccm.201909-1813OC
68. Jevnikar Z, Ostling J, Ax E, et al. Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol. 2019;143:577–590. doi:10.1016/j.jaci.2018.05.026
69. Holgate ST, Peters-Golden M, Panettieri RA, et al. Roles of cysteinyl leukotrienes in airway inflammation, smooth muscle function, and remodeling. J Allergy Clin Immunol. 2003;111:
70. Chang PJ, Bhavsar PK, Michaeloudes C, et al. Corticosteroid insensitivity of chemokine expression in airway smooth muscle of patients with severe asthma. J Allergy Clin Immunol. 2012;130:877–885.e5. doi:10.1016/j.jaci.2012.07.017
71. Chang PJ, Michaeloudes C, Zhu J, et al. Impaired nuclear translocation of the glucocorticoid receptor in corticosteroid-insensitive airway smooth muscle in severe asthma. Am J Respir Crit Care Med. 2015;191:54–62. doi:10.1164/rccm.201402-0314OC
72. Chachi L, Abbasian M, Gavrila A, et al. Protein phosphatase 5 mediates corticosteroid insensitivity in airway smooth muscle in patients with severe asthma. Allergy. 2017;72:126–136. doi:10.1111/all.13003
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.