Back to Journals » Journal of Inflammation Research » Volume 14
Melatonin Attenuates Neuroinflammation by Down-Regulating NLRP3 Inflammasome via a SIRT1-Dependent Pathway in MPTP-Induced Models of Parkinson’s Disease
Authors Zheng R, Ruan Y, Yan Y, Lin Z, Xue N, Yan Y, Tian J, Yin X, Pu J, Zhang B
Received 28 April 2021
Accepted for publication 25 June 2021
Published 8 July 2021 Volume 2021:14 Pages 3063—3075
DOI https://doi.org/10.2147/JIR.S317672
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Ran Zheng, Yang Ruan, Yiqun Yan, Zhihao Lin, Naijia Xue, Yaping Yan, Jun Tian, Xinzhen Yin, Jiali Pu, Baorong Zhang
Department of Neurology, the Second Affiliated Hospital, School of Medicine, Zhejiang University, Zhejiang, 310009, People’s Republic of China
Correspondence: Baorong Zhang; Jiali Pu
Department of Neurology, the Second Affiliated Hospital, School of Medicine, Zhejiang University, Zhejiang, People’s Republic of China
Fax +86-571-87784752
Email [email protected]; [email protected]
Background: Inflammasome-induced neuroinflammation is a key contributor to the pathology of Parkinson’s disease (PD). NLR family pyrin domain-containing 3 (NLRP3) inflammasome activation has been implicated in PD in postmortem human PD brains, indicating it as a potential target for PD treatment. Melatonin, a multitasking molecule, has been found to have anti-inflammatory activities, mediated by silence information regulator 1 (SIRT1). However, whether and how melatonin is involved in inflammasome-induced neuroinflammation in PD pathogenesis remains unclear.
Methods: We investigated the potential anti-inflammatory effects of melatonin in vitro and in vivo, using 1-methyl-4-phenylpyridinium (MPP+)-simulated BV2 and primary microglia cell models, and a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced murine PD model, with or without melatonin treatment. Rotarod, grip strength, and open-field tests were performed to measure the effects of melatonin on MPTP-induced motor disorders. Degeneration of dopaminergic neurons was evaluated by immunofluorescence. Changes in microglia were examined by immunofluorescence and Western blotting, and the expression levels of the involved signaling molecules were assessed by Western blotting and enzyme-linked immunosorbent assay (ELISA). Intracellular reactive oxygen species (ROS) was detected using fluorescent probes via flow cytometry.
Results: We found that melatonin significantly alleviated motor dysfunction and prevented MPTP-induced neurotoxicity in dopaminergic neurons. Additionally, melatonin reduced MPTP-induced microglial activation and suppressed NLRP3 inflammasome activity, and also inhibited IL-1β secretion. Moreover, in MPP+-primed BV2 cells, melatonin markedly restored the downregulation of SIRT1 and attenuated the activation of the NLRP3 inflammasome. This was reversed by SIRT1 inhibitor treatment.
Conclusion: In conclusion, our data demonstrated that melatonin attenuates neuroinflammation by negatively regulating NLRP3 inflammasome activation via a SIRT1-dependent pathway in MPTP-induced PD models. These findings provide novel insights into the mechanism underlying the anti-inflammatory effects of melatonin in PD.
Keywords: inflammasomes, melatonin, NLRP3, Parkinson disease, sirtuin 1
Introduction
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders,1 characterized by the degeneration of dopaminergic neurons and the accumulation of α-synuclein-rich Lewy bodies.2 However, its pathogenesis is still not fully understood, which makes it difficult to develop disease-modifying treatments. Several etiological theories of PD have been proposed. Among these, neuroinflammation, microglial activation, and the inflammasome have been implicated in PD pathogenesis.3,4 Inflammasomes are intracellular proinflammatory pattern recognition receptors (PRRs) that induce proinflammatory cytokines and amplify the inflammatory response.5 Several factors contribute to the stimulation of inflammasome, such as reactive oxygen species (ROS), microbial or damage-associated stimuli, aggregated proteins and metabolic abnormalities.6 As a large multi-protein complex, inflammasomes recruit pro-inflammatory cysteinyl aspartate-specific proteinases (caspases) via the apoptosis-associated speck-like protein containing a CARD (ASC) and then proceeds to cleave the proinflammatory cytokine precursors, into their mature forms.7 Upon assembly of inflammasomes, ASC is mobilized to form a large singular paranuclear structure termed the ASC speck, which is essential for the recruitment of caspase-1 and its inflammatory activity.8 In particular, the NLR family pyrin domain-containing 3 (NLRP3) inflammasome is increasingly implicated in PD, based on postmortems of human PD brains, and in PD progression.9–11 Therefore, inhibition of NLRP3 inflammasome activation may be a promising therapeutic target for PD.
Melatonin, a neurohormone produced by the pineal gland,12 has been reported to exert a wide variety of biological activities, such as anti-inflammatory, anti-oxidative, anti-apoptotic, and immunomodulatory activities.13–15 Preclinical and clinical studies have shown that melatonin decreases neuroinflammation in PD,12 but the mechanism underlying this effect remains to be explored. Recent studies on mouse brain injury and major depressive disorder models showed that the anti-inflammatory effect of melatonin was partially attributed to the suppression of NLRP3 inflammasome activity, where melatonin reduced the expression of NLRP3 inflammasome components and the protein levels of IL-1β.16–18 However, the inhibitory effect of melatonin on NLRP3 inflammasome activation in PD per se has not been studied.
Numerous studies have reported that the anti-inflammatory effects of melatonin can be mediated by silence information regulator 1 (SIRT1).19 After melatonin treatment, increased SIRT1 activity has been found in a variety of cells and animal models, particularly in the context of inflammation or aging.14 Interestingly, it has been reported that SIRT1 activity is decreased in patients with PD and in the midbrains of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated rats.20–22 Upregulation of SIRT1 suppressed NLRP3 inflammasome activation in many conditions,23–25 suggesting that SIRT1 may participate in the progression of PD by regulating neuroinflammation.
Therefore, our study aimed to investigate whether melatonin inhibits activation of the NLRP3 inflammasome in PD through a SIRT1-dependent pathway. We demonstrated that melatonin reduced motor defects and NLRP3 inflammasome activation in an MPTP-induced mouse model of PD. Additionally, melatonin inhibited the activation of the NLRP3 inflammasome in microglia through a SIRT1-dependent pathway.
Materials and Methods
Chemicals and Reagents
Melatonin (M5250), MPTP-HCL (M0896), 1-methyl-4-phenylpyridinium (MPP+, D048), adenosine 5-triphosphate disodium hydrate (ATP, A6419) were obtained from Sigma‒Aldrich (St Louis, MO), and nigericin was purchased from InvivoGen (San Diego, CA). Selisistat (EX-527) was purchased from MedChemExpress (Monmouth Junction, NJ). Anti-NLRP3 (AG-20B-0014), anti-caspase1 (AG-20B-0042), and anti-ASC (AG-25B-0006) antibodies were purchased from AdipoGen Life Sciences (San Diego, CA). Anti-IBA-1 (ab5076) antibody and anti-NF-κB p65 (ab16502) antibody were purchased from Abcam (Cambridge, MA). Anti-tyrosine hydroxylase (TH) (AB152) was purchased from Merck Millipore (Billerica, MA). Anti-SIRT1 (No.8469) antibody and anti-phospho-NF-κB p65 (No.3033) were purchased from Cell Signaling Technology (Danvers, MA). Anti-IL18 (No. A1115) antibody was purchased from ABclonal (Woburn, MA). Alexa fluor-488 conjugated anti-rabbit and Alexa fluor-546 conjugated anti-goat antibodies were purchased from Invitrogen (Carlsbad, CA). The mouse IL-1β ELISA kit (EK0394) was purchased from Boster Bio (Pleasanton, CA). The 2ʹ,7ʹ-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe was purchased from NJJCBIO (Nanjing, China).
Ethics Statement
All procedures were performed in accordance with the animal ethics guidelines of the Second Affiliated Hospital of Zhejiang University, and were approved by the Animal Research Ethics Committees of the Second Affiliated Hospital of Zhejiang University.
Animals and Experimental Design
Eight-week-old adult male C57BL/6 mice (weight 22–25 g) were obtained from SLAC Laboratory Animal Co., Ltd. (Shanghai, China). Animals were housed under a 12-h light/dark cycle with free drinking and eating. After 1 week of acclimation and 5 days of behavioral test pre-training, mice were randomly divided into three groups (n = 8/group): (1) the control group; (2) MPTP model group; (3) pretreated melatonin + MPTP group (mice were pretreated with 10 mg/kg melatonin 1 h before each MPTP injection). Mice received intraperitoneal injections of MPTP (30 mg/kg) daily for 5 consecutive days to establish a subacute PD model. Control animals were injected with an equal volume of 0.9% saline. MPTP was dissolved in 0.9% saline. Melatonin was dissolved in 1% EtOH/0 and 0.9% saline (v/v). The animals were subjected to behavioral experiments and were sacrificed by decapitation.
Behavioral Tests
The rotarod test, open field test, and grip strength test were performed to evaluate the behavioral defects of MPTP-induced mice.
For the rotarod test, the mice were assessed using a rotarod apparatus (Panlab, Barcelona, Spain; LE8205). The mice were placed on the rotating rod which is divided into five compartments, and the rotation speed was progressively increased from 4 to 40 rpm within a 5 -min period. The latency to falling (the duration that mice remained on the rod until their first drop, or a maximum cutoff time of 120 s) was recorded. The measurements were averaged across three trials per day, at least 1 h apart. Before modeling, the mice were adapted to the test environment and received pre-training. The tests were performed on the 7th‒10th days after the 1 week of acclimation and the 5 days of training according to the time schedule (Figure 1A).
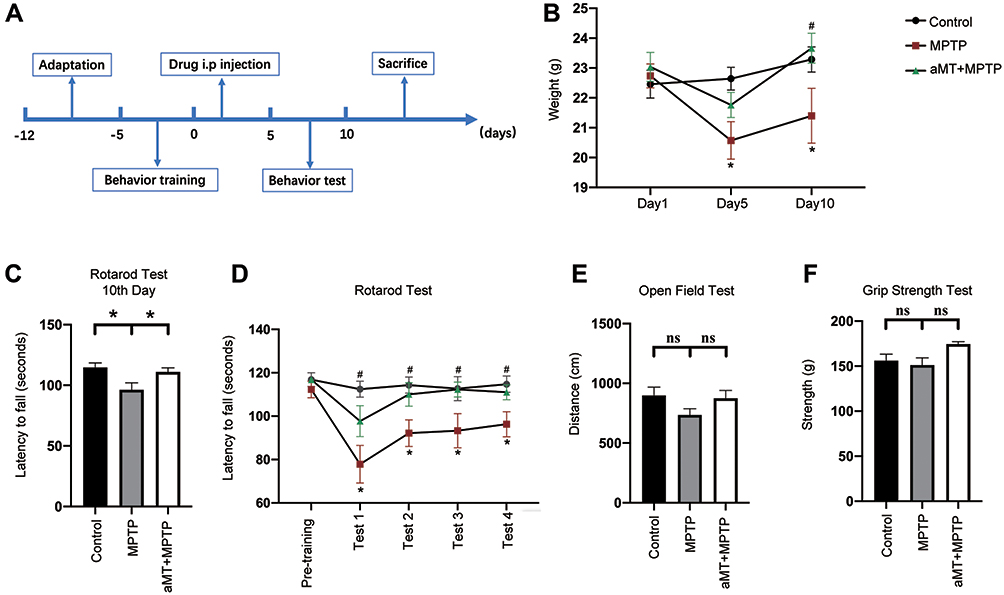 |
Figure 1 Melatonin attenuates weight loss and behavior disorder. (A) Schematic representation illustrating the experimental design (timeline). (B) Melatonin restored body weight in MPTP-treated mice. (C‒F) Behavior changes in the indicated mice were analyzed. The grip strength (C), total travelled distance in the open field test (D), and the latency to falling in the rotarod test on the 10th day (E) were recorded and analyzed. (F) The latency to falling during the rotarod test on the accelerated rotarod was recorded for four consecutive trials. aMT, melatonin. *P < 0.05 vs control group; #P < 0.05 vs MPTP group; ns, no significant differences. Data are presented as mean ± SEM, n = 7. |
For the open field test, animals were transported to the testing room and left undisturbed for 30 min before the test. The mouse was placed in the middle of a cubic box under bright illumination, and its movements were recorded over the course of 5 min as it moved around and explored the environment. The total distance traveled during this 5-min period was measured by SMART video tracking software (Smart 3.0).
For the grip strength test, a grip strength meter (Ugo Basile, Cat. No. 47200; Gemonio, Italy) was used to measure the grip strength (peak force and time resistance) of the forelimbs. The mouse was lowered to just above the grid and the torso was kept in line with the grid. Once the mouse clasped the grid, it was gently pulled back by its tail, and the maximal value of grip strength was recorded. This procedure was repeated twice and the values averaged for data analysis.
Immunofluorescence of Brain Slices
After decapitation, animals were perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). The brains were dissected out, fixed in 4% PFA solution overnight at 4°C, and then dehydrated in 30% sucrose in PBS solution until the brains sunk to the bottom of the container. Brains were embedded in OCT compound (Sakura, Osaka, Japan) and were serially sectioned (thickness: 30 μm) using a cryostat microtome (Leica CM1950; Wetzlar, Germany). For blocking and permeabilization, brain slices were incubated with PBS containing 5% bovine serum albumin (BSA) and 0.3% Triton X-100 for 1 h at room temperature (RT). After a brief rinse in carrier solution (PBS with 1% BSA and 0.3% Triton X-100), the slices were incubated with primary antibodies diluted in carrier solution and then incubated at 4°C overnight. Next, the slices were washed with the carrier solution and incubated with fluorescent secondary antibodies for 2 h at RT. After washing with the carrier solution, anti-fading agents containing 4',6-diamidino-2-phenylindole (DAPI) was added before mounted. Sections were screened using laser scanning confocal microscopy (Leica TCS SP8) and VS120 virtual slide microscope (Olympus, Shinjuku City, Japan). Images were analyzed using ImageJ software (NIH, Bethesda, MD). The number of TH-positive cells in substantia nigra compacta (SNc) was obtained stereologically as previously described with modifications.26 Briefly, every sixth section covering the entire extent of the SNc was included in the counting procedure (7–8 sections per animal). A 2x lens was used to outlined the SNc on the sections, while 40x lens was used for counting.
Primary Microglia Isolation
Primary microglial cells were isolated as previously described with modifications.27 Briefly, the newborn Sprague Dawley rats (postpartum day 0–2) were decapitated and the brains harvested. The meninges were carefully removed, and the cerebral cortices were collected and trypsinized in 0.25% trypsin at 37°C for 15 min. After centrifugation at 500 × g for 5 min at 4°C, the pellets were suspended in 5 mL warm Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Waltham, MA) with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin‒streptomycin) and filtered through a 70-μm-pore-size filter. Cells from two cortices were seeded in a poly-D-lysine-coated T-75 culture flask containing 15 mL culture media and were incubated in 5% CO2 at 37°C. After 24 h, the entire culture medium was replaced, and half of the medium was replaced with fresh culture medium every 3–5 days. On days 12–14, microglia were isolated from the mixed glial culture by shaking the flask at 200 rpm for 1.5 h at 37°C. The collected microglia were seeded in 12-well plates for further treatment. The purity of microglia was > 90%, as determined by Iba-1 immunostaining.
Cell Culture and Treatments
Mouse BV2 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Beijing, China). Cells were maintained in DMEM supplemented with 10% FBS in 5% CO2 at 37°C. Following stimulation with MPP+ (250 μM or 500 μM, 6 h), with or without melatonin, or a specific SIRT1 inhibitor, Selisistat (EX527), BV2 cells were treated with ATP (2.5 mM, 30 min) or nigericin (10 µM, 45 min).
Primary microglial cells were treated with 100 μM MPP+ for 6 h in the presence and absence of melatonin. Then, treatment with ATP (2 mM, 30 min) was conducted.
Western Blot Analysis
After treatment, proteins were extracted from cell lysates of BV2 cells using RIPA lysis buffer containing 1% protease inhibitor cocktail (Halt, Thermo Scientific, Waltham, MA). For mouse brain tissue, the striatum and substantia nigra of the midbrain were isolated as previously described.28 The tissue was minced and homogenized using a tissue grinder. The supernatant was collected after centrifugation at 12,000 × g for 20 min at 4°C. The extracted protein was added to 5 × loading buffer by volume, denatured at 95°C for 5 min. SDS-PAGE was used to resolve proteins isolated from cell lysate and brain tissue along with a molecular weight marker, and then transferred to polyvinylidene fluoride membranes (Merck Millipore). Blocking was performed with 5% skim milk in 0.1% Tween-20/ Tris-buffered saline (TBS-T) for 1 h at RT. Then, the membranes were incubated with the primary antibodies overnight at 4°C. Following the incubation of horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at RT, the protein bands were visualized by chemiluminescence detection using an enhanced chemiluminescent reagent (WBKLS0500, Millipore) and quantitated using a densitometer (Bio-Rad Imaging System, Hercules, CA). Band density was analyzed with ImageJ and normalized to GAPDH.
Enzyme-Linked Immunosorbent Assay (ELISA)
After stimulation, supernatant samples of cultured cell were collected. The levels of IL-1β were determined by ELISA according to the manufacturer’s instructions. The absorbance was then measured at 450 nm using a multimode plate reader (PerkinElmer EnVision; Waltham, MA).
Detection of ASC Specks
Primary microglial cells were seeded at a density of 1×106 cells per well in a six-well plate. After treatment, cells were fixed with 4% PFA at RT for 30 min and washed with PBS three times. PBS containing 10% BSA and 0.5% Triton X‐100 was used for blocking and permeabilization at 37°C for 30 min. Following incubation with anti-ASC (1:500) and anti-IBA-1 primary antibody (1:500) overnight at 4 °C, cells were incubated with Alexa fluor-488 conjugated anti-rabbit antibody (1:500) and Alexa fluor-546 conjugated anti-goat antibody (1:500) for 2 h at RT. ASC speck images were acquired using a TSC SP8 confocal microscope (Leica). ASC speck-positive microglial cells were counted using ImageJ software.
Detection of Reactive Oxygen Species (ROS)
Intercellular ROS was detected using DCFH-DA fluorescent probe according to manufacturer’s protocol. In brief, the BV2 cells were incubated in the dark with 8μM DCFH-DA at 37°C for 30min after treatments and washed with PBS. The fluorescence intensity was measured using flow cytometry (CytoFLEX, Beckman) and analyzed with FlowJo.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA). All data were analyzed using one-way ANOVA test, followed by Tukey’s multiple comparison test. Data are presented as the mean ± SEM with at least 3 independent experiments. Statistical significance was set at p < 0.05.
Results
Melatonin Ameliorates Weight Loss and Motor Dysfunction
To evaluate the neuroprotective effects of melatonin on the MPTP-induced mouse model of PD, we first recorded the changes in body weight and measured motor function using the rotarod test, open field test, and grip strength test (Figure 1A). Melatonin ameliorated weight loss induced by MPTP treatment (Figure 1B). In the rotarod test, melatonin significantly restored the decreased latency to falling on an accelerated rotarod that was induced by MPTP treatment in four consecutive trials (Figure 1C and D; Supplementary Figure 1A‒D). In the open field test and grip strength test, however, no statistically significant correlation was observed between the different groups (Figure 1E and F; Supplementary Figure 1E). Collectively, these findings indicate that melatonin partially reversed the adverse effects of MPTP on motor function.
Melatonin Attenuates Dopaminergic Neuron Loss and Microglial Activation
We next performed tyrosine hydroxylase (TH) immunostaining and Western blotting to examine the protective effect of melatonin on dopaminergic neurons. The results indicated that compared with the control group, MPTP-treated mice exhibited severe loss of TH-positive neurons, and melatonin treatment partially alleviated this situation (Figure 2A and B). Moreover, melatonin also upregulated TH expression in the brain striatum and substantia nigra (SN) of MPTP-treated mice (Figure 2C–E). Furthermore, while IBA-1 positive cells in the striatum were significantly increased in the MPTP-treated mice, melatonin reduced IBA-1 expression in this region (Figure 2F and G). Consistent with this result, while MPTP significantly increase the expression levels of IBA-1 in the striatum and SN region, melatonin downregulated the level of IBA-1 in the striatum of the mouse brain, indicating that melatonin partially reduced microglial activation in response to MPTP (Figure 2H–J).
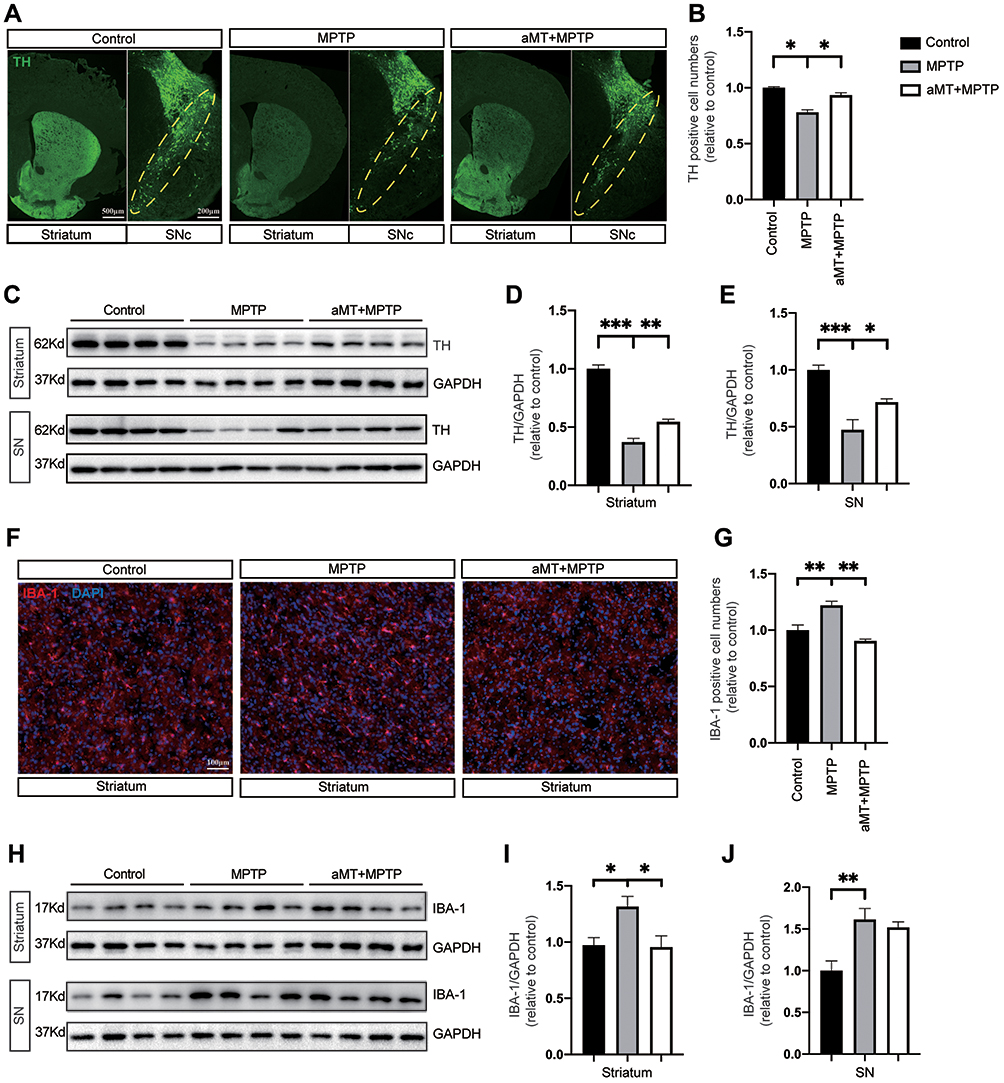 |
Figure 2 Protective effect of melatonin on dopaminergic neurons and microglia. (A) Representative immunofluorescence staining of neurons in the striatum and substantia nigra compacta (SNc) of mouse brain for TH. Yellow dotted circles indicate the SNc. Scale bars are indicated. (B) Quantification of relative TH-positive cells in SNc (n = 4). (C‒E) Western blotting was performed to determine the expression of TH in striatum and SN of PD mice (n = 4). Data are shown as representative plots (C) and bands quantified by densitometric analysis (D and E). (F) Representative immunofluorescence image of the microglia in the brain striatum stained with anti-IBA1 antibody (red). DAPI represents nuclear staining (blue). Scale bars = 100 μm. (G) Quantification of relative IBA1-positive cells per DAPI (n = 4). (H–J) Western blotting was performed to determine the expression of IBA-1 in the striatum and SN of PD mice (n = 4). Data are shown as representative plots (H) and bands quantified by densitometric analysis (I and J). aMT, melatonin; TH, tyrosine hydroxylase; SN, Substantia nigra, PD, Parkinson disease. *P < 0.05, **P < 0.01, ***P<0.001. Data are expressed as means ± SEM. |
Melatonin Inhibits NLRP3 Inflammasome Activation in vivo
We then examined whether melatonin-mediated inhibition of neuroinflammation was associated with the NLRP3 inflammasome signaling pathway by Western blotting. We found that, compared with the control group, the expression of NLRP3, cleaved-caspase 1 and mature IL-1β in the striatum and SN tissues of MPTP-treated mice was significantly increased, suggesting the activation of the NLRP3 inflammasome. However, melatonin markedly inhibited the activation of the NLRP3 inflammasome by decreasing the levels of NLRP3 and cleaved-caspase 1 in the SN and the striatum (Figure 3A and B, E and F, G and H, K and L). In addition, we also investigated the influence of melatonin on NLRP3 inflammasome-related priming effector NF-κB and activation effector pro-inflammatory cytokine IL-1β. Our data demonstrated that melatonin reduced IL-1β secretion and phosphorylation of NF-κB induced by MPTP in the striatum and SN, although it is not significant in some results (Figure 3C and D, I and J). These results suggest that melatonin prevents MPTP-induced NLRP3 inflammasome activation in vivo.
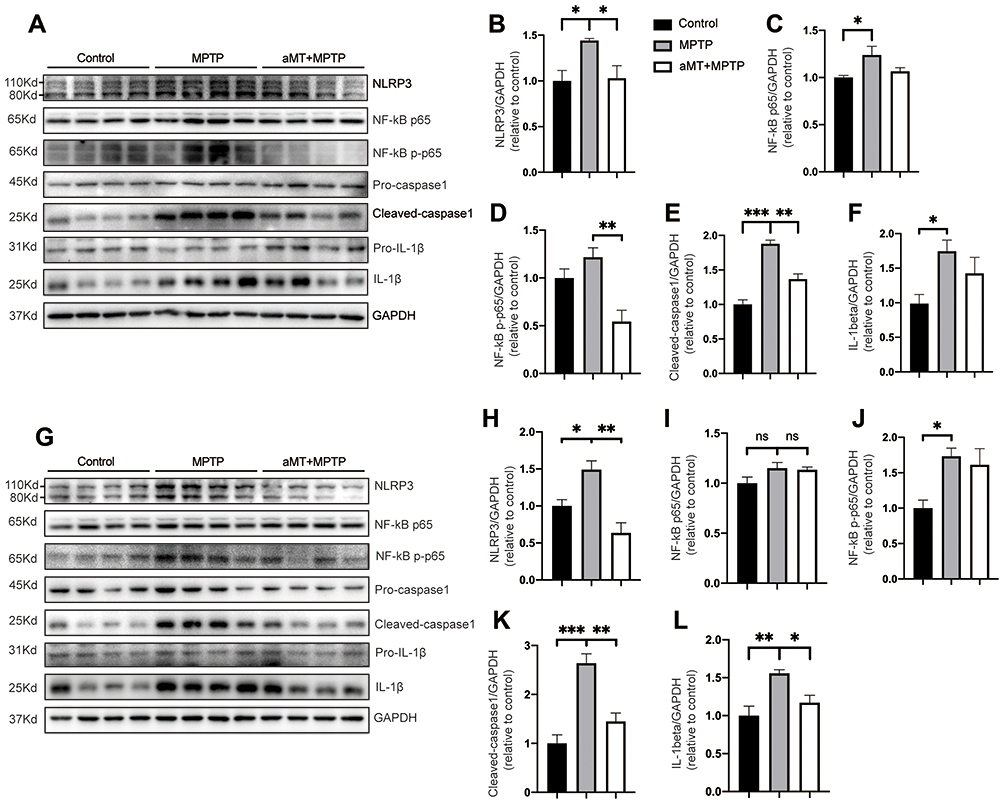 |
Figure 3 Melatonin inhibits NLRP3 inflammasome activation in vivo. (A–F) Immune blotting analysis of NLRP3 inflammasome signals in the striatum from mouse brains (n = 4). Representative blots are shown in (A). Relative expression of NLRP3, NF-κB p65, phospho-NF-κB, cleaved-caspase 1, IL-1β were quantified by densitometric analysis in (B–F). (G–L) Immune blotting analysis of NLRP3 inflammasome signals in the SN region of the brains (n = 4). Data are shown as representative plots (G) and quantified immunoblotting bands (H–L). aMT, melatonin. *P < 0.05, **P < 0.01, ***P<0.001; ns, no significant differences. Data are expressed as the mean ± SEM. |
Melatonin Suppresses Activation of NLRP3 and the Assembly of ASC Specks in vitro
Next, to investigate the effect of melatonin on NLRP3 inflammasome activation further in vitro, we evaluated the constitutive protein levels of the NLRP3 inflammasome after treatment. When BV2 cells were primed with MPP+ alone, we found that NLRP3 inflammasome activation failed to elicit. While MPP+ priming, followed by ATP or nigericin treatment, increased the protein levels of NLRP3 and cleaved-caspase 1, as well as the release of IL-1β, melatonin pretreatment almost completely abrogated activation of the NLRP3 inflammasome (Figure 4A‒E). In addition, we measured the intracellular production of ROS in BV2 cells to evaluate whether melatonin regulated the oxidative stress, which is an important contributor in the activation of NLRP3 inflammasome. While intracellular ROS levels significantly increased in MPP+-induced cells, pretreatment with melatonin reduced production of intracellular ROS (Figure 4F and G). Consistent with the findings in BV2 cells, melatonin treatment also ameliorated the formation of ASC specks induced by MPP+ and ATP in primary microglia (Figure 4H and I). Taken together, we demonstrated that melatonin treatment prevented MPP+-induced inflammasome activation as well as ASC speck formation in vitro.
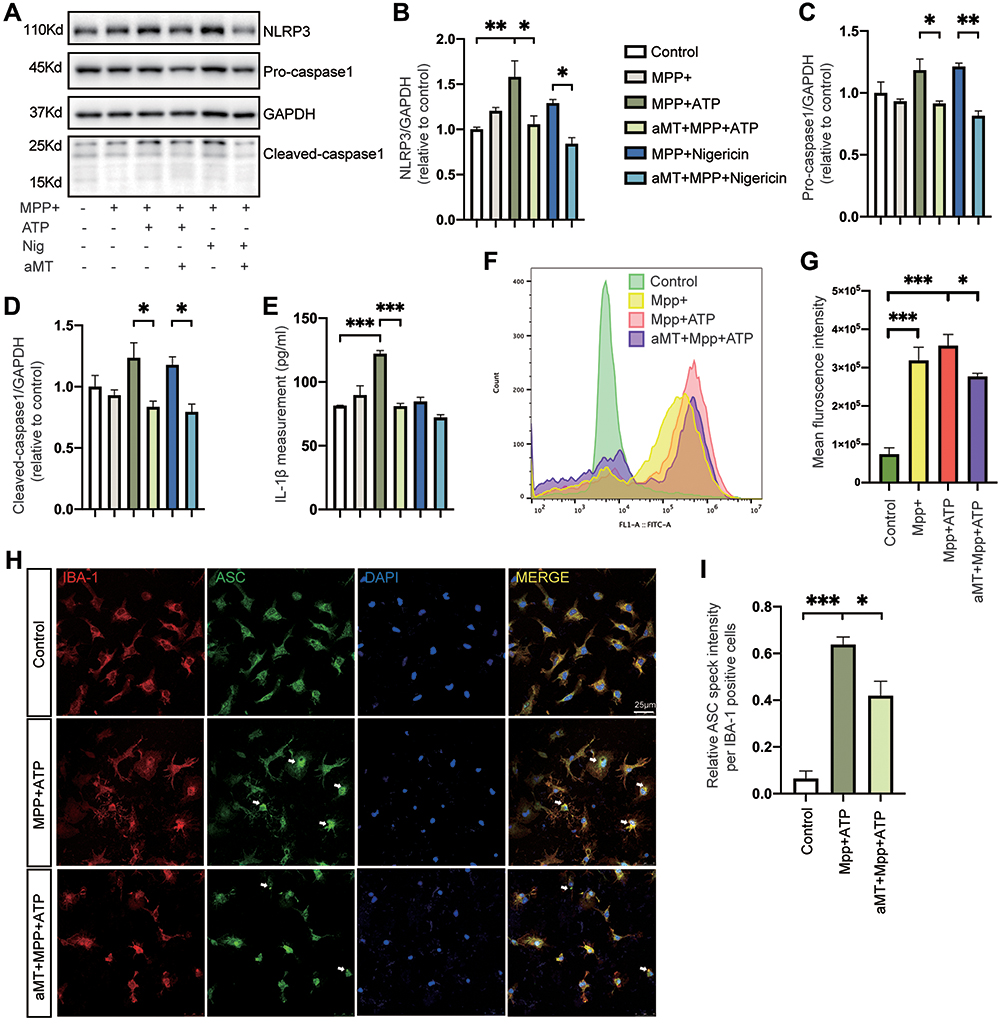 |
Figure 4 Melatonin suppresses MPP+-induced NLRP3 inflammasome activation in vitro. (A‒D) Immunoblot analysis of NLRP3, caspase 1 (full-length and cleaved forms) from mouse BV2 cells primed with MPP+ (500 μM, 6 h), followed by ATP (2.5 mM, 30 min) or nigericin (10 μM, 45 min); or MPP+ (500 μM, 6 h) + melatonin (100 μM, 6 h) followed by ATP (2.5 mM, 30 min) or nigericin (10 μM, 45 min). (E) Quantification of IL-1β in the BV2 cell culture supernatants. (F and G) Intracellular levels of ROS were detected in MPP+-treated (500 μM, 6 h) BV2 cells followed by ATP (2.5 mM, 30 min), with or without melatonin (100 μM, 6 h) pretreatment. Mean fluorescence intensity was quantified by FlowJo in (G). (H) Representative immunofluorescence image of primary microglia treated with PBS or MPP+ followed by ATP, with or without melatonin pretreatment, stained with anti-ASC antibody (green) and anti-IBA1 antibody (red). DAPI represents the nuclear signal (blue). Scale bars = 25 μm. White arrows indicate ASC specks. (I) Quantification of relative ASC speck intensity per IBA1-positive cells. aMT, melatonin; Nig, Nigericin. *P < 0.05, **P < 0.01, ***P<0.001. Data are expressed as the mean ± SEM, all experiments were repeated at least 3 times. |
Melatonin Negatively Regulates the NLRP3 Inflammasome via Its Effects on SIRT1 Expression
To evaluate the role of SIRT1 in regulating the effect of melatonin on NLRP3 inflammasome activation, we first examined SIRT1 protein expression levels in the presence or absence of melatonin, within a given time frame. We found that melatonin significantly increased the expression of SIRT1 over time, but high concentrations of melatonin decreased SIRT1 expression levels, indicating that melatonin exerts its effects within a particular concentration range (Figure 5A and B). Next, we investigated whether MPP+-induced inflammasome activation could affect the expression of SIRT1 in BV2 cells. We found that when the concentration of MPP+ stimulation increased, the expression of SIRT1 showed a downward trend and declined significantly under subsequent stimulation with ATP (Figure 5C and D). Then, we evaluated the regulatory effect of melatonin on SIRT1 expression and NLRP3 inflammasome suppression. Not surprisingly, we found the expression of SIRT1 increased while the protein levels of NF-κB, and cleaved-caspase 1 decreased as the concentration of melatonin increased (Figure 5E and F). Finally, we investigated whether SIRT1 suppression affects the inhibitory effect of melatonin on NLRP3 inflammasome activation using selisistat, a specific SIRT1 inhibitor. As expected, selisistat suppressed the expression of SIRT1 and reversed the protective effect of melatonin on MPP+- and ATP-treated cells by increasing the protein levels of NLRP3 and cleaved-caspase 1, which is consistent with the results we found in primary microglia except for changes in SIRT1 expression (Supplementary Figure 2A–D). This suggested that melatonin negatively regulates the NLRP3 inflammasome through its effects on SIRT1 expression (Figure 5G and H).
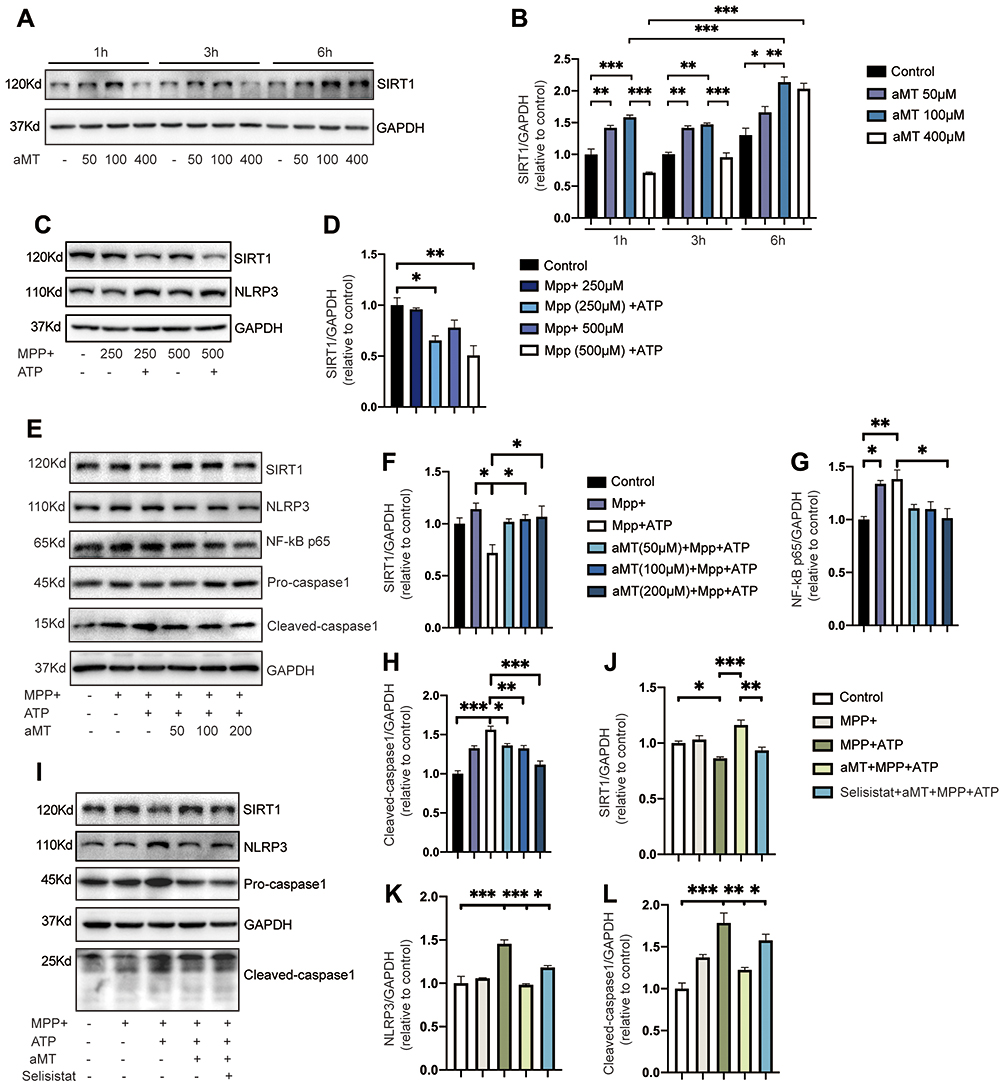 |
Figure 5 Inhibition of SIRT1 reverses the suppressive effect of melatonin on NLRP3 inflammasome activation. (A and B) BV2 cells were treated with melatonin (50 μM, 100 μM, 400 μM) for 0–6 h. Representative Western blotting of SIRT1 expression is shown as plots (A) and quantified bands (B). (C and D) Immunoblot analysis of SIRT1 expression from MPP+-treated BV2 cells (250 μM or 500 μM, 6 h), with or without ATP (2.5 mM, 30 min). Representative blots are shown in (C). Bands were quantified by densitometric analysis in (D). (E–H) Immunoblot analysis of NLRP3 inflammasome signals and SIRT1 expression from BV2 cells primed with MPP+ (500 μM, 6 h), followed by ATP (2.5 mM, 30 min), with or without melatonin (50 μM, 100 μM, 200 μM, 6 h) pretreatment. Data are shown as representative plots (E) and quantified bands (F–H). (I–L) Immunoblot analysis of SIRT1, NLRP3, and caspase 1 from mouse BV2 cells primed with MPP+ (500 μM, 6 h), followed by ATP (2.5 mM, 30 min); or MPP+ (500 μM, 6 h) + melatonin (100 μM, 6 h) pretreatment, with or without selisistat (50 μM, 6 h), followed by ATP (2.5 mM, 30 min). Data are shown as representative plots (I) and quantified bands (J–L). aMT, melatonin. *P < 0.05, **P < 0.01, ***P<0.001. Data are expressed as the mean ± SEM, all experiments were repeated at least 3 times. |
Discussion
The current study revealed that melatonin suppresses MPTP-induced dopaminergic neuron degeneration, microglial activation, and motor dysfunction by inhibiting NLRP3 inflammasome activation via a SIRT1-dependent pathway. Specifically, MPTP treatment induced NLRP3 inflammasome activation in vivo, but only acted as a priming signal in vitro. Melatonin ameliorated neuroinflammation in PD models by inhibiting NLRP3 inflammasome activation in vitro and in vivo. Melatonin prevents MPP+-induced production of ROS. Melatonin inhibited NLRP3 inflammasome activation through a SIRT1-dependent pathway (Figure 6). Collectively, our results demonstrated a previously unrecognized mechanism through which melatonin suppresses inflammasome-induced neuroinflammation in PD.
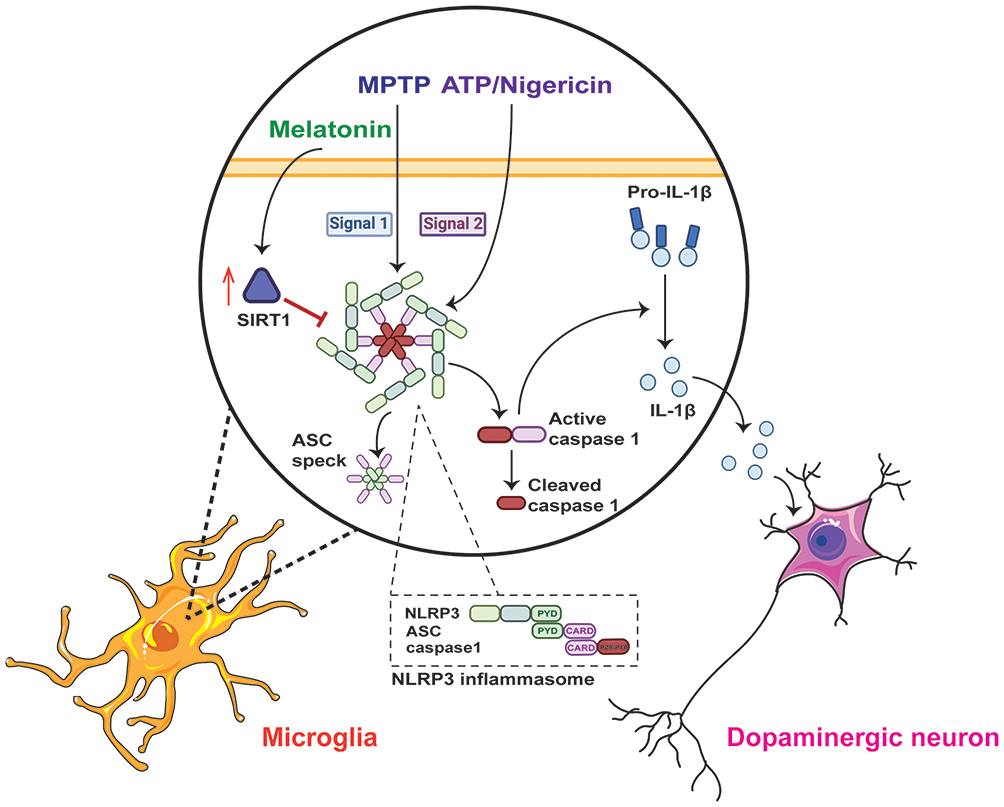 |
Figure 6 The underlying mechanism of the anti-inflammatory effect of melatonin in Parkinson’s disease. The NLRP3 inflammasome is assembled and activated in microglia when MPTP acts as the priming signal (signal 1), and ATP or nigericin acts as the activation signal (signal 2). Once the NLRP3 inflammasome is activated, it cleaves pro-IL-1β into mature IL-1β and promotes the release of ASC specks. However, melatonin can negatively regulate NLRP3 inflammasome activation via the SIRT1-dependent pathway and protect dopaminergic neurons in Parkinson’s disease. |
Although the precise pathogenesis of PD remains elusive, accumulating evidence indicates that inflammasome-induced neuroinflammation is an important component of PD etiopathogenesis.29,30 In particular, the NLRP3 inflammasome, including NLRP3, ASC, and caspase 1, is the most studied inflammasome in PD.9 To activate the NLRP3 inflammasome successfully, two signals have been proposed: (1) Signal 1 (priming). Priming signals are required for the transcription of NLRP3 and pro-IL-1β. (2) Signal 2 (activation). Activation signals are responsible for assembly and activation of the NLRP3 inflammasome.4 A recent study reported that priming with MPTP or MPP+ alone failed to activate inflammasomes in primary mixed glial and mouse bone marrow-derived macrophages.11 Similar to this result, our study demonstrated that MPP+ treatment without ATP or nigericin failed to elicit NLRP3 inflammasome activation in BV2 cells, suggesting that MPP+ only plays a priming role in in vitro studies. As previously reported, expression of the proinflammatory cytokine IL-1β, which is mainly regulated by the NLRP3 inflammasome, was notably increased in PD patients.31–33 Recent studies have shown that the NLRP3 inflammasome is implicated in postmortem human PD brains and is significantly activated in MPTP and α-synuclein preformed fibril (PFF)-induced PD mouse models.10,11 Consistent with this finding, our results showed that activation of NLRP3 inflammasome and levels of IL-1β were significantly increased in the MPTP-treated mice.
Emerging evidence suggests that melatonin treatment protects dopaminergic neurons in PD by decreasing neuroinflammation.13 However, no previous study has evaluated the effect of melatonin on inflammasome activation in PD. Here, we demonstrated that melatonin almost completely counteracted MPTP-induced NLRP3 inflammasome activation both in vivo and in vitro. The anti-inflammatory effects of melatonin are known to be mediated by SIRT1 in some contexts.14 Recent studies have revealed that SIRT1 inhibits the transcriptional activity of nuclear factor-kappa B and NLRP3 inflammasome activation.18,34 Interestingly, the enzymatic activity of SIRT1 is disturbed in patients with PD,22 suggesting that SIRT1 may participate in PD progression by regulating neuroinflammation. Indeed, in our study, we found that the expression of SIRT1 significantly decreased upon treatment with MPP+ and ATP, indicating a correlation between SIRT1 and NLRP3 inflammasome activation. However, whether there is a direct link between SIRT1 and the inhibitory effect of melatonin on NLRP3 inflammasome activation in PD has not been reported. Thus, to determine whether the inhibitory effect of melatonin on NLRP3 inflammasome depends on the SIRT1 signaling pathway, we first evaluated the effect of melatonin on SIRT1 expression using melatonin pretreatment within a concentration gradient. As expected, we found that the expression of SIRT1 increased accompanied by the inhibition of NLRP3 inflammasome as the concentration of melatonin increased. Then, we used a SIRT1-specific inhibitor selisistat and found that SIRT1 suppression partly abolished the inhibitory effect of melatonin on NLRP3 inflammasome in vitro, demonstrating a previously unrecognized mechanism through which melatonin suppresses inflammasome-induced neuroinflammation in PD.
There were some limitations to our study. First, the results in Figure 5 suggest that melatonin regulates SIRT1 expression in a dose-and time-dependent manner. Additional studies are required to validate the different doses and routes of administration both in vitro and in vivo. Second, we only investigated the involvement of SIRT1 in NLRP3 inflammasome activation in vitro, but we did not find significant differences in the expression of SIRT1 between different groups in the in vivo study (Supplementary Figure 3A-D). Its role in vivo prompted further research. Moreover, species differences should be considered when translating bench-side findings into human studies.
Conclusion
In conclusion, our study showed that melatonin treatment ameliorated motor deficits and dopaminergic neuron loss, and inhibited microglial activation in an MPTP-induced PD model. Melatonin also diminished NLRP3 inflammasome activation both in vivo and in vitro. Furthermore, we concluded that the SIRT1 signaling pathway is involved in the inhibitory effects of melatonin on NLRP3 inflammasome activation in microglia. The present study shed light on the mechanism underlying the preventative effect of melatonin on NLRP3 inflammasome activation in PD.
Data Sharing Statement
All datasets generated for this study are included in the article or supplementary material.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No.81771216 and No.81520108010), the Natural Science Foundation of Zhejiang Province (No.LY18H090003) and the Key Research and Development Program of Zhejiang Province (No.2020C03020).
Thanks to our colleagues Zhong-Xuan Wang, Yi Fang, Chong-Yao Jin, Xiao-Li Si for their suggestions and help in this study. ZXW, XLS contributed to the data curation. YF, CYJ contributed to writing the manuscript.
Disclosure
Dr Jiali Pu reports grants from National Natural Science Foundation of China, grants from Natural Science Foundation of Zhejiang Province, grants from Key Research and Development Program of Zhejiang Province, during the conduct of the study. The authors declare that there are no conflicts of interest.
References
1. Li G, Ma J, Cui S, et al. Parkinson’s disease in China: a forty-year growing track of bedside work. Transl Neurodegener. 2019;8:22. doi:10.1186/s40035-019-0162-z
2. Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi:10.1038/nrdp.2017.13
3. Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–397. doi:10.1016/S1474-4422(09)70062-6
4. Haque ME, Akther M, Jakaria M, Kim IS, Azam S, Choi DK. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov Disord. 2020;35:20–33. doi:10.1002/mds.27874
5. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748. doi:10.1038/ni.2919
6. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi:10.1038/nm.3893
7. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: emerging Mechanisms and Effector Functions. Cell. 2016;165:792–800. doi:10.1016/j.cell.2016.03.046
8. Lu A, Magupalli VG, Ruan J, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell. 2014;156:1193–1206. doi:10.1016/j.cell.2014.02.008
9. de Araujo FM, Cuenca-Bermejo L, Fernandez-Villalba E, Costa SL, Silva VDA, Herrero MT. Role of Microgliosis and NLRP3 Inflammasome in Parkinson’s Disease Pathogenesis and Therapy. Cell Mol Neurobiol. 2021. doi:10.1007/s10571-020-01027-6
10. Gordon R, Albornoz EA, Christie DC. et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med;2018. 10. doi: 10.1126/scitranslmed.aah4066
11. Lee E, Hwang I, Park S, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2018.
12. Tamtaji OR, Reiter RJ, Alipoor R, Dadgostar E, Kouchaki E, Asemi Z. Melatonin and Parkinson Disease: current Status and Future Perspectives for Molecular Mechanisms. Cell Mol Neurobiol. 2020;40:15–23. doi:10.1007/s10571-019-00720-5
13. Alghamdi BS. The neuroprotective role of melatonin in neurological disorders. J Neurosci Res. 2018;96:1136–1149. doi:10.1002/jnr.24220
14. Hardeland R. Melatonin and inflammation-Story of a double-edged blade. J Pineal Res. 2018;65:e12525.
15. Ashrafizadeh M, Najafi M, Kavyiani N, Mohammadinejad R, Farkhondeh T, Samarghandian S. Anti-Inflammatory Activity of Melatonin: a Focus on the Role of NLRP3 Inflammasome. Inflammation. 2021. doi:10.1007/s10753-021-01428-9
16. Xu G, Shi D, Zhi Z, Ao R, Yu B. Melatonin ameliorates spinal cord injury by suppressing the activation of inflammasomes in rats. J Cell Biochem. 2019;120:5183–5192. doi:10.1002/jcb.27794
17. Dong Y, Fan C, Hu W, et al. Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J Pineal Res. 2016;60:253–262. doi:10.1111/jpi.12300
18. Arioz BI, Tastan B, Tarakcioglu E, et al. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation Through the SIRT1/Nrf2 Pathway. Front Immunol. 2019;10:1511. doi:10.3389/fimmu.2019.01511
19. Mayo JC, Sainz RM, Gonzalez Menendez P, Cepas V, Tan DX, Reiter RJ. Melatonin and sirtuins: a “not-so unexpected” relationship. J Pineal Res. 2017;62.
20. Li X, Feng Y, Wang XX, Truong D, Wu YC. The Critical Role of SIRT1 in Parkinson’s Disease: mechanism and Therapeutic Considerations. Aging Dis. 2020;11:1608–1622. doi:10.14336/AD.2020.0216
21. Pallas M, Pizarro JG, Gutierrez-Cuesta J, et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience. 2008;154:1388–1397. doi:10.1016/j.neuroscience.2008.04.065
22. Singh P, Hanson PS, Morris CM. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 2017;18:46. doi:10.1186/s12868-017-0364-1
23. Han Y, Sun W, Ren D, et al. SIRT1 agonism modulates cardiac NLRP3 inflammasome through pyruvate dehydrogenase during ischemia and reperfusion. Redox Biol. 2020;34:101538. doi:10.1016/j.redox.2020.101538
24. Park S, Shin J, Bae J, et al. SIRT1 Alleviates LPS-Induced IL-1beta Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells. 2020;9.
25. Tu Y, Song E, Wang Z, et al. Melatonin attenuates oxidative stress and inflammation of Muller cells in diabetic retinopathy via activating the Sirt1 pathway. Biomed Pharmacother. 2021;137:111274. doi:10.1016/j.biopha.2021.111274
26. Jimenez-Ferrer I, Backstrom F, Duenas-Rey A, et al. The MHC class II transactivator modulates seeded alpha-synuclein pathology and dopaminergic neurodegeneration in an in vivo rat model of Parkinson’s disease. Brain Behav Immun. 2021;91:369–382. doi:10.1016/j.bbi.2020.10.017
27. Lian H, Roy E, Zheng H. Protocol for Primary Microglial Culture Preparation. Bio Protoc. 2016;6.
28. Spijker S. Dissection of Rodent Brain Regions. Neuroproteomics. 2011;13–26.
29. Soderbom G, Zeng BY. The NLRP3 inflammasome as a bridge between neuro-inflammation in metabolic and neurodegenerative diseases. Int Rev Neurobiol. 2020;154:345–391.
30. Chatterjee K, Roy A, Banerjee R, et al. Inflammasome and alpha-synuclein in Parkinson’s disease: a cross-sectional study. J Neuroimmunol. 2020;338:577089. doi:10.1016/j.jneuroim.2019.577089
31. Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: a Systematic Review and Meta-Analysis. Front Immunol. 2018;9:2122. doi:10.3389/fimmu.2018.02122
32. Hanamsagar R, Torres V, Kielian T. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem. 2011;119:736–748. doi:10.1111/j.1471-4159.2011.07481.x
33. Fan Z, Pan YT, Zhang ZY, et al. Systemic activation of NLRP3 inflammasome and plasma alpha-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation. 2020;17:11. doi:10.1186/s12974-019-1670-6
34. Ye J, Liu Z, Wei J, et al. Protective effect of SIRT1 on toxicity of microglial-derived factors induced by LPS to PC12 cells via the p53-caspase-3-dependent apoptotic pathway. Neurosci Lett. 2013;553:72–77. doi:10.1016/j.neulet.2013.08.020
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.