")
Back to Journals » Journal of Inflammation Research » Volume 15
MBL Binding with AhR Controls Th17 Immunity in Silicosis-Associated Lung Inflammation and Fibrosis
Authors Liu Y, Zhao N , Xu Q, Deng F, Wang P, Dong L, Lu X, Xia L, Wang M, Chen Z, Zhou J, Zuo D
Received 17 January 2022
Accepted for publication 1 June 2022
Published 28 July 2022 Volume 2022:15 Pages 4315—4329
DOI https://doi.org/10.2147/JIR.S357453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam D Bachstetter
Yunzhi Liu,1,2,* Na Zhao,3,* Qishan Xu,1,2 Fan Deng,1 Ping Wang,1,2 Lijun Dong,1,2 Xiao Lu,2 Lihua Xia,3 Mingyong Wang,4 Zhengliang Chen,2 Jia Zhou,2 Daming Zuo1,5
1Department of Medical Laboratory, School of Laboratory Medicine and Biotechnology, Southern Medical University, Guangzhou, Guangdong, 510515, People’s Republic of China; 2Guangdong Province Key Laboratory of Proteomics, Department of Immunology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, Guangdong, 510515, People’s Republic of China; 3Department of Medical Laboratory, Guangdong Province Hospital for Occupational Disease Prevention and Treatment, Guangzhou, Guangdong, 510399, People’s Republic of China; 4Xinxiang Key Laboratory of Immunoregulation and Molecular Diagnostics, Xinxiang, 453003, People’s Republic of China; 5Microbiome Medicine Center, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, 510282, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jia Zhou, Guangdong Province Key Laboratory of Proteomics, Department of Immunology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, Guangdong, 510515, People’s Republic of China, Tel +86-20-61648220, Fax +86-20-61648221, Email [email protected] Daming Zuo, Department of Medical Laboratory, School of Laboratory Medicine and Biotechnology, Southern Medical University, Guangzhou, Guangdong, 510515, People’s Republic of China, Tel +86-20-61648552, Fax + 86-20-61648221, Email [email protected]
Objective: Mannan-binding lectin (MBL), a soluble pattern recognition molecule of the innate immune system, is primarily synthesized in the liver and secreted into the circulation. Low serum level of MBL has been reported to be related to an increased risk of lung diseases. Herein, we aimed to investigate the function of MBL in silicosis-associated pulmonary inflammation.
Methods: Serum collected from silicosis patients was tested for correlation between serum MBL levels and Th17 immunity. In vitro studies were performed to further demonstrated the effect of MBL on Th17 polarization. Silica was intratracheally injected in wild type (WT) or MBL-deficient (MBL–/–) mice to induce silicosis-associated lung inflammation and fibrosis. Th17 response was evaluated to explore the effect of MBL on silicosis in vivo.
Results: Silicosis patients with high serum MBL levels displayed ameliorative lung function. We demonstrated that serum MBL levels negatively correlated to Th17 cell frequency in silicosis patients. MBL protein markedly reduced expression of IL-17 but enhanced expression of Foxp3 in CD4+ T cells in vitro when subjected to Th17 or Treg polarizing conditions, respectively. The presence of MBL during Th17 cell polarization significantly limited aryl hydrocarbon receptor (AhR) expression and suppressed the signal transducer and activator of transcription 3 (STAT3) phosphorylation. Treatment with the AhR antagonist abolished the effect of MBL on Th17 response. Strikingly, MBL directly bound to AhR and affected its nuclear translocation. Furthermore, MBL–/– mice displayed elevated Th17 cell levels compared with WT mice in response to the silica challenge. The CD4+ T lymphocytes from silica-administrated MBL–/– mice exhibited more AhR expression than the wild-type counterparts.
Conclusion: Our study suggested that MBL limited the Th17 immunity via controlling the AhR/STAT3 pathway, thus providing new insight into silicosis and other inflammatory diseases in patients with MBL deficiency.
Keywords: silicosis, mannan-binding lectin, aryl hydrocarbon receptor, signal transducer and activator of transcription 3, Th17 cell
Introduction
Silica is the major component of respirable particles, and over time inhalation may cause silicosis, a fatal occupational lung disease characterized by lung inflammation and irreversible fibrosis.1,2 Silicosis has become one of the most important occupational diseases worldwide in past decades. Although lung transplantation may be offered to a minority of patients, there is still no specific treatment for silicosis to halt or reverse the disease progression.3 The Th17 cell is one of the subtypes of T helper (Th) cells and has been reported to take part in the progression of pulmonary fibrosis.4 Previous studies determined that Th17 cells contributed to the inflammatory and fibrotic responses in a hypersensitivity pneumonitis mouse model.5,6 Additionally, infiltrated Th17 cells have been detected in the bronchial submucosa of patients with cystic fibrosis (CF).7 Notably, Th17 response significantly raised during the inflammatory phase of silicosis.8 Besides, declined neutrophil infiltration and attenuated silica-induced lung inflammation and fibrosis progression were observed in a mice model of experimental silicosis when IL-17a was neutralized.9
The Aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor associated with cellular responses to endogenous and exogenous stimuli. Increasing evidence suggested that AhR acts as an essential modulator for Th17 cell differentiation. AhR is highly expressed in Th17 cells while slightly expressed in Treg cells and undetectable in Th1 or Th2 cells.10 It is worth mentioning that CD4+ T cells isolated from AhR−/− mice exhibited declined IL-17 production compared with WT counterparts under Th17 polarization conditions. Besides, tryptophan metabolite 6-formylindolo[3,2-b] carbazole (FICZ), a high-affinity AhR endogenous ligand, could enhance Th17 development and functions.11 Notably, FICZ aggravated the progression of experimental autoimmune encephalomyelitis and collagen-induced arthritis, characterized as Th17 cell-related autoimmune diseases.10,12 Similarly, PM2.5 promoted Th17 cell differentiation and aggravated asthma in an AhR-dependent manner.13 Jaligama et al observed that radical containing combustion-derived particulate matter induced Th17 response in the lung tissue through regulation of AhR.14
Mannan binding lectin (MBL) is a prototypic pattern recognition molecule mainly produced in the liver and can activate the lectin complement pathway.15 Although MBL has been considered to exert an essential effect on the lectin complement pathway for decades, it regulates the immune response in a complement-independent way. Dendritic cells (DCs) isolated from MBL-deficient cases exhibited boosted pro-inflammatory cytokine expression in the condition of microbial stimulation compared to those from healthy counterparts.16 Our previous study demonstrated that MBL regulated the inflammatory cytokine production and cytotoxicity of peripheral NK cells through direct interaction.17 Similarly, MBL inhibits the production of pro-inflammatory cytokines by monocytes during innate immune response.18,19 Interestingly, we demonstrated that serum MBL levels negatively correlated to the frequency of activated T cells in silicosis patients. In vitro studies proved that MBL interacted with T cells and suppressed T cells activation,20 which indicated the regulatory role of MBL in T cell function.
In this study, we purposed to study the function of MBL in the pathogenesis of silicosis and whether modulation of Th17 immunity is involved. It is noteworthy that we demonstrated that silicosis patients with high serum levels of MBL exhibited improved lung function compared with the patients with low serum MBL levels. In vitro study revealed that MBL inhibited the Th17 cell differentiation by suppressing the AhR/STAT3 axis. MBL-deficient mice generated elevated Th17 cells and reduced levels of Tregs compared with wild-type mice in response to the silica challenge. In summary, our findings provide evidence that MBL might have potential modulatory effects on Th17 immunity, which offers new insight into the disease mechanisms of silicosis and other Th17-related diseases, especially in patients with MBL deficiency.
Materials and Methods
Clinical Samples
Clinical data were collected at Guangdong Province Hospital for Occupational Disease. The diagnosis of silicosis was identified based on the Chinese National Diagnostic Criteria for pneumoconiosis (GBZ 70–2009). The study was approved by the Guangdong Province Hospital for Occupational Disease Prevention and Treatment (GDHOD MEC 2021001), and complied with the Declaration of Helsinki. Each participant was informed consent for taking part in the study before collecting the blood.
Animals
WT C57BL/6J mice were purchased from the Laboratory Animal Center of Southern Medical University (Guangzhou, China). MBL-deficient (MBL–/–) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Male mice were included and housed under a 12 h light/dark cycle condition at a constant temperature (19–23°C) and (55±10%) humidity, fed with commercial diet, and had free access to food and water. All animal experiments were approved by the Welfare and Ethical Committee for Experimental Animal Care of Southern Medical University (2016011). Animal work had taken place in Southern Medical University, department of immunology, according to the National Institute of Health Guideline for the Care and Use of Laboratory Animals. All mice were anaesthetized or euthanized with isoflurane.
Experimental Silicosis Mice Model
5 mg silica (S5631, Sigma-Aldrich, St. Louis, MO, USA) dissolved in 50μL sterile PBS were intratracheally injected in WT or MBL–/– mice. Mice were sacrificed 3 days after silica injection for the inflammation model and sacrificed at 35 days for the fibrosis model. Lung tissues were lavaged 3 times with 0.5 mL PBS for bronchoalveolar lavage fluid (BALF) collection. BALF was centrifugated at 450×g for 5 min at 4°C. Then, the supernatant was kept for total protein measurement, and the precipitants were collected for cell count detection and flow cytometry assay (FCM) assay. Hilar lymph nodes (HLN) were harvested for FCM assay.
Differentiation of Th17 Cell
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood obtained from healthy donors by Ficoll density gradient centrifugation. Naïve CD4+ T cells were purified from PBMCs by immunomagnetic selection using the naïve CD4+ T cell isolation kit (130–094-131, Miltenyi Biotec, Bergisch Gladbach, Germany). For Th17 cell differentiation assay, naïve CD4+ T cells were cultured in 96-well U-bottomed plates (2×105/well) coated with 1μg/mL anti-CD3 mAb (MA1-10175, Thermo Fisher Scientific, Inc) and cultured in 1μg/mL anti-CD28 mAb (MA1-10166, Thermo Fisher Scientific, Inc), 20ng/mL IL-6 (200–06, peprotech), 50ng/mL IL-23 (200–23, peprotech), 10ng/mL IL-1β (200–01B, peprotech), 1ng/mL TGF-β (100–21, peprotech), 5μg/mL anti-IFN-γ (16–7318-85, Thermo Fisher Scientific, Inc) and 5μg/mL anti-IL-4 (16–7048-85, Thermo Fisher Scientific, Inc) condition for 7 days. For Treg cell differentiation assay, naïve CD4+ T cells were cultured in 96-well U-bottomed plates (2×105/well) coated with 1μg/mL anti-CD3 mAb and cultured in 1μg/mL anti-CD28 mAb, 1ng/mL TGF-β, 5μg/mL anti-IFN-γ and 5μg/mL anti-IL-4 condition for 7 days. MBL protein was prepared as previously described.20 In some cases, cells were pretreated 5μg/mL MBL, 10μM Sc144 (S7124, Selleck Chemicals), or 1μM CH-223191 (S7711, Selleck Chemicals) for 2 h. For AhR activation assay, CD4+ T cells were treated with 200nM FICZ (SML1489, Sigma-Aldrich; Merck KGaA) for 6 h.
RT-qPCR Analysis
Cells were combined with 1 mL TRIzol® (Thermo Fisher Scientific, Inc), and total RNA was extracted based on the manufacturer’s instruction. Then, 1000 ng RNA was synthesized into cDNA at 50°C for 10 min and 85°C for 5 sec. SYBR Green Real-Time PCR Master Mix (cat. no. A46112; Applied Biosystems; Thermo Fisher Scientific, Inc) was used on an ABI Prism 7500 Sequence Detection System (Applied Biosystems; Thermo Fisher Scientific, Inc), based on the following conditions: Initial denaturation at 94°C for 30 sec, followed by 40 cycles of denaturation at 94°C for 5 sec, and extension at 60°C for 30 sec. The expression of the target genes was normalized to GAPDH gene expression and calculated using the 2−∆∆CT method. The primer sequences involved in this study are shown in Table 1.
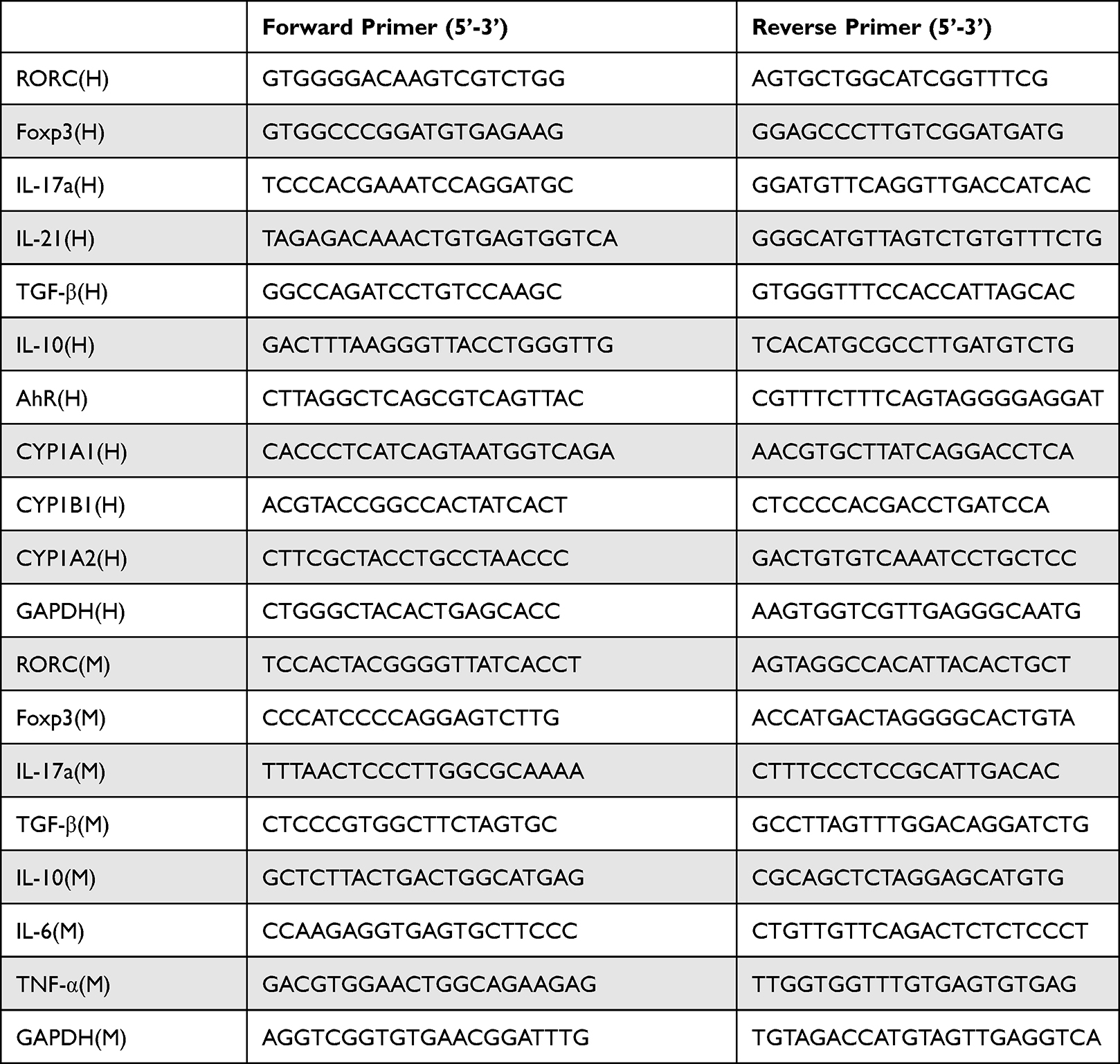 |
Table 1 List of Primer Sequences Used for RT-qPCR Analysis in This Study |
Flow Cytometry
Cells were harvested and stained with indicated antibodies at 4°C in the dark for 30 min. LIVE/DEAD Fixable Violet Dead Cell Stain Kit (L34966, Thermo Fisher Scientific, Inc) was used to identify live cells. For intracellular protein staining, cells were fixed and permeabilized by Fixation/Permeabilization kit (88–8824-00, Thermo Fisher Scientific, Inc). For transcription factor staining, cells were fixed and permeabilized by Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent kit (00–5521-00, Thermo Fisher Scientific, Inc). For phosphor-STAT3 staining, cells were fixed by ice methanol before staining. Cell suspensions from mice tissues were treated with Cell Stimulation Cocktail (plus protein transport inhibitors) (500×) (00–4975-93, Thermo Fisher Scientific, Inc) before intracellular cytokine staining. CD4+IL-17a+ cells were identified as Th17 cell in PBMCs and CD4+CD25+Foxp3+ cells were identified as Treg cell. Antibodies involved in this subsection were listed below: mouse anti-human/mouse CD4 monoclonal antibody (11–0049-42, Thermo Fisher Scientific, Inc), mouse anti-human IL-17a monoclonal antibody (12–7178-42, Thermo Fisher Scientific, Inc), mouse anti-human Foxp3 monoclonal antibody (17–4777-42, Thermo Fisher Scientific, Inc), mouse anti-human CD4/CD25 antibody cocktail (22–0425-71, Thermo Fisher Scientific, Inc), rat anti-human/mouse RORγ monoclonal antibody (12–6988-82, Thermo Fisher Scientific, Inc), rat anti-human/mouse IL-17a monoclonal antibody (12–7177-81, Thermo Fisher Scientific, Inc), rat anti-mouse CD25 monoclonal antibody (56–0251-82, Thermo Fisher Scientific, Inc), mouse- anti-human/mouse phosphor-STAT3 (TYR705)(48–9033-42, Thermo Fisher Scientific, Inc), mouse anti-human/mouse AhR (67785-1-Ig, Proteintech, Chicago, USA), Alexa Fluor® 488 goat anti-mouse IgG antibody (405319, Biolegend, San Diego, USA). Cells were analyzed using the FACS LSRFortessa™ flow cytometer (BD Biosciences) with BD FACSDiva™ software (version 8.0.1; BD Biosciences).
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum from silicosis patients or supernatants from culture media were collected. The cytokine levels were assessed using commercial ELISA kits purchased from Thermo Fisher Scientific, Inc. The ELISA kits involved in this report were listed as follows: Human MBL (EHMBL2X10), human IL-17a (BMS2017), human IL-21 (BMS2043), human TGF-β (BMS249-4), human IL-10 (BMS215-2), mouse IL-17a (BMS6001), mouse IL-6 (BMS603-2), mouse TNF-α (BMS607-3), mouse TGF-β (BMS608-4), mouse IL-10 (BMS614INST). Standard curves were established by recombinant cytokines included in ELISA kits.
Immunoprecipitation and Western Blot
Whole-cell lysates were incubated with 1μg antibodies at 4°C overnight and treated with protein A/G agarose (Santa Cruz Biotechnology) for another 2 h at 4°C. SDS-PAGE was used to detect the eluted immunoprecipitates. For Western blot analysis, protein samples were separated on SDS-PAGE and then transferred onto polyvinylidene fluoride membranes. The membranes were blocked by 5% BSA for 1 h at room temperature and then incubated with indicated primary antibodies at 4°C overnight. The membranes were stained with HRP-conjugated secondary antibody (goat anti-rabbit, S0001, Affinity Biosciences) at room temperature for another 1 h. Measurement of the target protein was conducted with ECL substrate (1705062, Bio-Rad Laboratories, Inc). For nuclear protein isolation, a Nuclear and Cytoplasmic Protein Extraction Kit was used (P0027, Beyotime Biotechnology, Shanghai, China). Antibodies involved in this subsection were listed below: mouse anti-human MBL monoclonal antibody (ab26277, Abcam, USA), AhR (67785-1-Ig, Proteintech, Chicago, USA), GAPDH (5174, Cell Signaling Technology, USA), rabbit anti-human Histone H3 polyclonal antibody (AF0863, Ancaster, ON, Canada).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 for another 5 min at room temperature. After blocking for 1 h, cells were stained with anti-AhR and anti-MBL (ab23457, Abcam, USA) antibody, respectively, overnight at 4°C. Subsequently, the slides were incubated with Alexa Fluor® 488 goat anti-mouse IgG antibody (405319, Biolegend, San Diego, USA) and PE Donkey anti-rabbit IgG (406421, Biolegend, San Diego, USA) antibodies for 1 h at room temperature.
Histology Analysis
Lung tissues were removed and collected in 1 mL 4% paraformaldehyde for 24 h at room temperature. The sections were stained with hematoxylin and eosin (H&E) to observe morphological changes. Hydroxyproline was detected using a hydroxyproline detection kit (MAK008-1KT, Sigma-Aldrich, Merck KGaA). The Ashcroft method21 was used to estimate lung fibrosis as follows: grade 0, normal lung; grade 1, minimal fibrous thickening of the alveolar or bronchiolar walls; grade 3, moderate thickening of the walls without apparent damage to lung architecture; grade 5, increased fibrosis with actual damage to the lung structure and the formation of fibrous bands or small fibrous masses; grade 7, severe distortion of the structure and large fibrous areas; grade 8, total fibrous obliteration of fields.
Statistical Analysis
Statistical analysis was performed using SPSS 12.0 (SPSS, Inc.). The data are presented as the mean ± SD. An unpaired Student’s t-test was used to compare between two groups. Pearson’s correlation test was used to estimate the correlation between two factors. P < 0.05 was considered to indicate a statistically significant difference.
Results
MBL Level Positively Correlated with Lung Function and Th17/Treg Cell Frequency in Silicosis Patients
To investigate the potential effect of MBL on silicosis, serum from silicosis patients was obtained and tested for MBL levels. Interestingly, serum MBL levels were positively correlated to DLCO%, TLC%, VC%, FEV1/FVC%, PEF%, FEF50%, FEF25%, and MVV% in silicosis patients (Figure 1A). Consistently, patients with ≥80% DLCO or TLC value exhibited high serum MBL levels than patients with <80% DLCO or TLC value. Similarly, patients with normal pulmonary ventilation and small airway function showed increased serum MBL levels compared to patients with abnormal pulmonary ventilation and small airway function (Figure 1B). Since Th17 cells contributed to the development of silicosis,22,23 we then tried to figure out whether MBL serum level was correlated to the frequency of Th17 cells. The results showed elevated Th17 cell frequency and RORγt expression in peripheral CD4+ T lymphocytes obtained from silicosis patients compared to those from healthy controls. Meanwhile, declined Treg cell frequency and Foxp3 expression were found in patients with silicosis compared with healthy individuals (Figure 1C and D). Besides, serum MBL levels were negatively correlated to Th17 cell frequency and RORγt expression, while Treg cell frequency and Foxp3 expression positively correlated to serum MBL levels (Figure 1E and F). Taken together, these data indicate that serum MBL levels positively correlated to the lung function of silicosis patients, and this phenomenon may involve the regulation of Th17 and Treg cells.
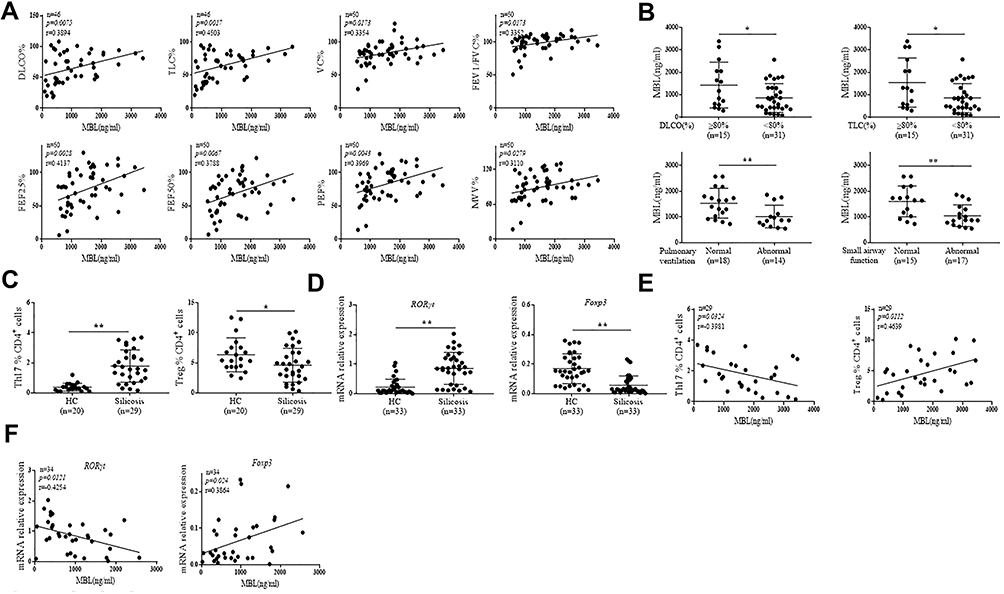 |
Figure 1 MBL level positively correlated with lung function and Th17/Treg cell balance in silicosis patients. (A and B) Serum MBL levels were detected by ELISA analysis. (A) The correlation between serum MBL levels and DLCO%, TLC%, VC%, FEV1/FVC%, PEF%, FEF75%, FEF50%, FEF25%, MVV% in silicosis patients was determined. (B) The correlation of serum MBL levels with different DLCO%, TLC% levels, pulmonary ventilation or small airway function condition was analyzed. (C) The frequency of Th17 and Treg in PBMCs from silicosis patients or healthy counterparts was determined by FCM assay. (D) RORγt and Foxp3 mRNA expression were measured by RT-qPCR analysis in CD4+ T cells isolated from silicosis patients and healthy controls. (E) The correlation between serum MBL levels and Th17 or Treg frequency in silicosis patients was analyzed. (F) The correlation between serum MBL levels and RORγt or Foxp3 mRNA expression in CD4+ T cells from silicosis patients. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. |
MBL Inhibits Th17 Cells Polarization in vitro
To determine whether MBL regulated Th17 and Treg cell polarization, naïve CD4+ T lymphocytes were sorted from human PBMCs and cultured in Th17 or Treg cell differentiation conditions, respectively. The flow cytometry assay demonstrated that MBL inhibited IL-17a and RORγt expression under the Th17 polarization condition (Figure 2A). Accordingly, lower IL-17a and IL-21 levels were detected in the culture supernatants from the Th17 culture condition in the presence of MBL protein than those from controls (Figure 2B). Moreover, MBL significantly suppressed RORγt, IL-17a, and IL-21 expression at the transcriptional level (Figure 2C). However, MBL treatment promoted Foxp3 expression in CD4+ T cells under Treg polarization (Figure 2D). In addition, elevated TGF-β and IL-10 production was found in the culture medium obtained from Treg-polarized cells treated with MBL protein compared to those from control cells (Figure 2E). Similarly, MBL exposure promoted Foxp3, TGF-β, and IL-10 mRNA expression during Treg cell differentiation (Figure 2F). Collectively, these data suggest that MBL inhibited Th17 polarization but promoted Treg differentiation in vitro.
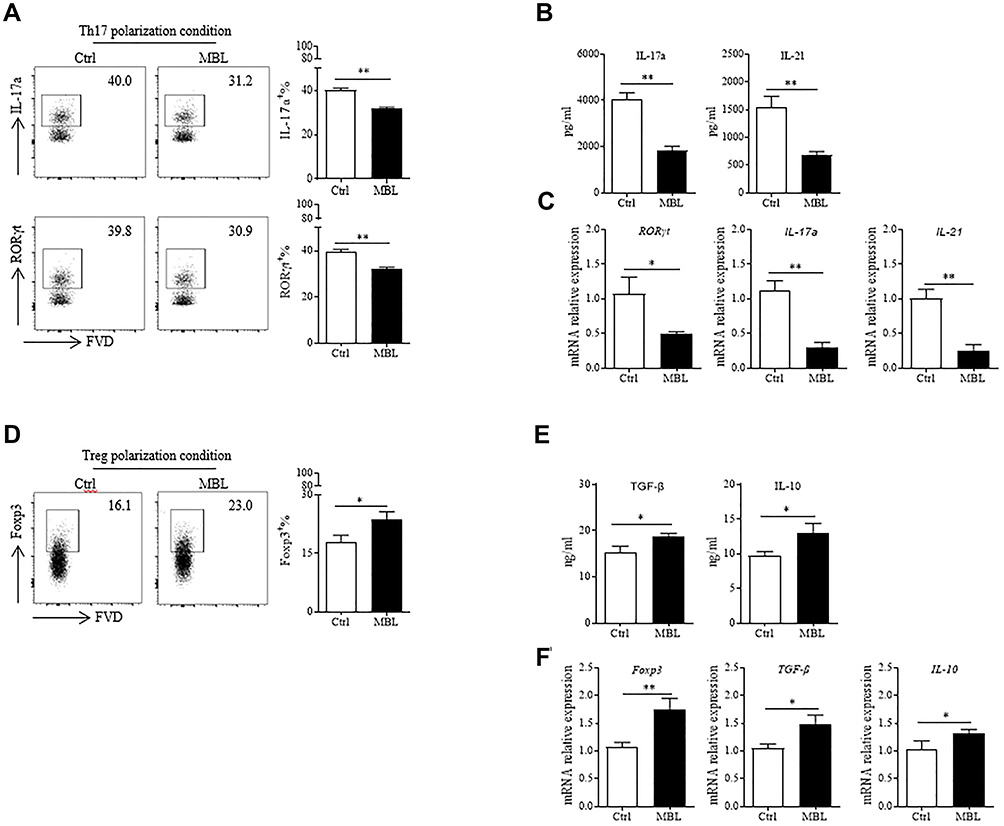 |
Figure 2 MBL inhibited Th17 cells polarization in vitro. (A and C) Naïve CD4+ T cells were cultured under Th17 differentiation conditions in the presence or absence of MBL protein (5μg/mL). (A) The expression of IL-17a and RORγt in CD4+ T cells were determined by FCM analysis. (B) The levels of IL-17a and IL-21 in the culture medium were evaluated by ELISA assay. (C) The mRNA levels of RORγt, IL-17a, and IL-21 were detected by RT-qPCR analysis. (D–F) Naïve CD4+ T cells were cultured under Treg differentiation conditions with or without MBL protein (5μg/mL). (D) The expression of Foxp3 in CD4+ T cells was determined by FCM analysis. (E) The levels of TGF-β and IL-10 in the culture medium were evaluated by ELISA assay. (F) The mRNA transcription levels of Foxp3, TGF-β, and IL-10 were detected by RT-qPCR analysis. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. |
MBL Limited AhR Expression and Translocation in CD4+ T Lymphocytes
It has been reported that AhR activation is essential for Th17 cell polarization.10,11 We, therefore, attempted to discover whether MBL affected AhR signaling during Th17 differentiation. The flow cytometry and Western blot assay displayed attenuated AhR expression in CD4+ T lymphocytes treated with MBL during Th17 polarization (Figure 3A and B). Notably, AhR expression is regulated by STAT3 signaling, and activated AhR may also induce STAT3 phosphorylation.24,25 Interestingly, the results showed that MBL treatment significantly limited STAT3 phosphorylation in CD4+ T lymphocytes during Th17 polarization (Figure 3C). Besides, inhibition of STAT3 signaling by a STAT3 inhibitor, Sc144, abolished the effect of MBL on AhR expression at both mRNA and protein levels (Figure 3D and E). FICZ, an AhR ligand, can promote the process of Th17 cell differentiation.10 AhR has been known to exert a transcription factor function after nuclear translocation. Surprisingly, MBL treatment increased AhR levels in cytosolic but decreased its nuclear level in the condition of Th17 polarization (Figure 3F), indicating that MBL limited the nuclear translocation of AhR. Transcriptional activation of CYP1A1, CYP1A2, and CYP1B1 is believed to be regulated by AhR.26 Herein, we observed that MBL significantly limited the mRNA expression of CYP1A1, CYP1A2, and CYP1B1 in CD4+ T cells upon FICZ stimulation (Figure 3G). Moreover, the mRNA levels of RORγt and IL-17a were also decreased in CD4+ T cells upon FICZ stimulation in the presence of MBL protein (Figure 3H). Together, these data suggest that MBL inhibits AhR activation in CD4+ T lymphocytes.
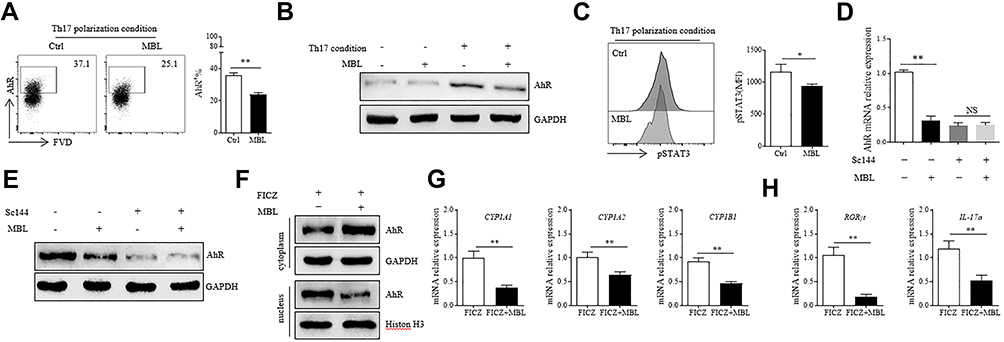 |
Figure 3 MBL limited AhR expression and its nuclear translocation in CD4+ T lymphocytes. (A and C) Naïve CD4+ T cells were cultured under Th17 differentiation conditions with or without MBL protein (5μg/mL). (A) The expression of AhR in CD4+ T cells was determined by FCM analysis. (B) The protein levels of AhR were determined by Western blot. (C) The level of phosphorylated STAT3 was assessed by FCM analysis. (D–E) Naïve CD4+ T cells were pretreated with 10μM Sc144 for 2h, then cultured under Th17 differentiation conditions with or without MBL protein (5μg/mL). (D) The mRNA expression of AhR in CD4+ T cells was determined by RT-qPCR analysis. (E) The protein level of AhR was assessed by Western blot analysis. (F–H) CD4+ T cells were treated with 200nM FICZ for 6h in the presence or absence of 5μg/mL MBL. (F) The fraction of cytoplasm and nucleus protein were separated, and the expression of AhR was determined by Western blot. (G) The mRNA levels of CYP1A1, CYP1A2, and CYP1B1 were detected by RT-qPCR analysis. (H) The mRNA levels of RORγt and IL-17 were detected by RT-qPCR analysis. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. Abbreviation: N.S, not significant. |
MBL Interacts with AhR and Regulates Th17 Cell Polarization Through AhR Signaling
Then we investigate whether there is a physical interaction between MBL and AhR. Surprisingly, the results showed that MBL interacted with AhR under FICZ stimulation (Figure 4A). The immunofluorescence staining validated the direct association of AhR with MBL in the T cells (Figure 4B). Furthermore, we cultured CD4+ naïve T cells under Th17-polarizing condition with MBL protein in the presence or absence of the AhR inhibitor, CH-223191. CH-223191 treatment significantly inhibited the Th17 differentiation. Interestingly, MBL-mediated inhibition of Th17 polarization was abolished when treated with CH-223191 (Figure 4C). Moreover, supernatants from MBL treated cells exhibited comparable IL-17a and IL-21 production levels with those from counterparts in the presence of CH-223191 (Figure 4D). Besides, no significant difference in IL-17a, IL-21, and RORγt mRNA levels was found between cells treated with or without MBL upon CH-223191 challenge (Figure 4E). These data suggested that MBL might regulate Th17 cell polarization through AhR signaling.
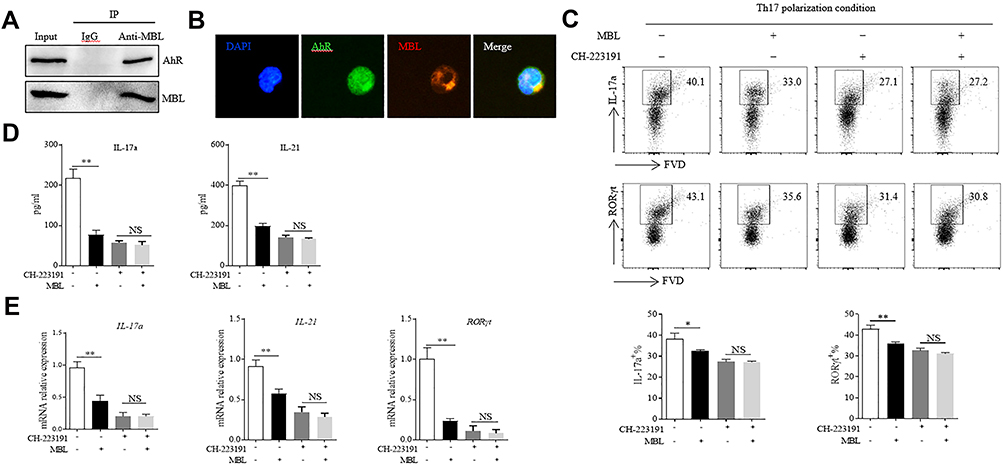 |
Figure 4 MBL interacted with AhR and regulated Th17 cell polarization through AhR signaling. (A and B) CD4+ T cells were stimulated with 200nM FICZ with 5μg/mL MBL for 6 h. (A) The association of AhR with MBL was analyzed by immunoprecipitation assay. (B) Colocalization of AhR and internalized MBL in T cells was determined by immunofluorescence analysis. (C–E) Naïve CD4+ T cells were cultured under Th17 differentiation conditions in the presence or absence of MBL protein (5μg/mL) combined with 1μM CH-223191 for 7d, t. (C) The expression of IL-17a and RORγt in CD4+ T cells was determined by FCM analysis. (D) The levels of IL-17a and IL-21 in the culture medium were evaluated by ELISA assay. (E) The mRNA levels of RORγt, IL-17a, and IL-21 were detected by RT-qPCR analysis. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. Abbreviation: N.S, not significant. |
MBL Deficiency Aggravates Silica-Induced Pulmonary Inflammation in Mice
To confirm the effect of MBL on silica-induced pulmonary inflammation. WT and MBL−/− mice were intratracheally injected with silica for 3 days. H&E staining revealed more inflammatory cell infiltration and severe abnormal follicular epithelial cell proliferation in lung tissues from MBL−/− mice than from WT mice (Figure 5A). Besides, elevated cell numbers and protein levels were observed in bronchoalveolar lavage fluid (BALF) from MBL−/− mice compared with WT counterparts, which indicated MBL deficiency aggravated pulmonary inflammation response (Figure 5B and C). Flow cytometry assay showed higher Th17 cell frequency and lower Treg cell frequency in lung tissues from MBL−/− mice than those from WT mice. Besides, samples of hilar lymph node (HLN) and BALF from the mice with silica-induced pulmonary inflammation exhibited a similar trend (Figure 5D). Consistently, increased levels of IL-17a, IL-6, and TNF-α were detected in BALF from silica-challenged MBL−/− mice compared to WT counterparts, while declined TGF-β and IL-10 levels were found in samples from MBL−/− silicosis mice (Figure 5E). Besides, we observed promoted IL-17a, IL-6, TNF-α, and RORγt mRNA levels in lung tissues from MBL−/− mice, while decreased TGF-β, IL-10, and Foxp3 transcription levels were measured compared to WT counterparts (Figure 5F). Besides, CD4+ T lymphocytes from lung tissues of silica-administred MBL−/− mice exhibited elevated AhR expression compared to WT controls (Figure 5G). Taken together, these data indicate that MBL deficiency aggravates silica-induced pulmonary inflammation in mice.
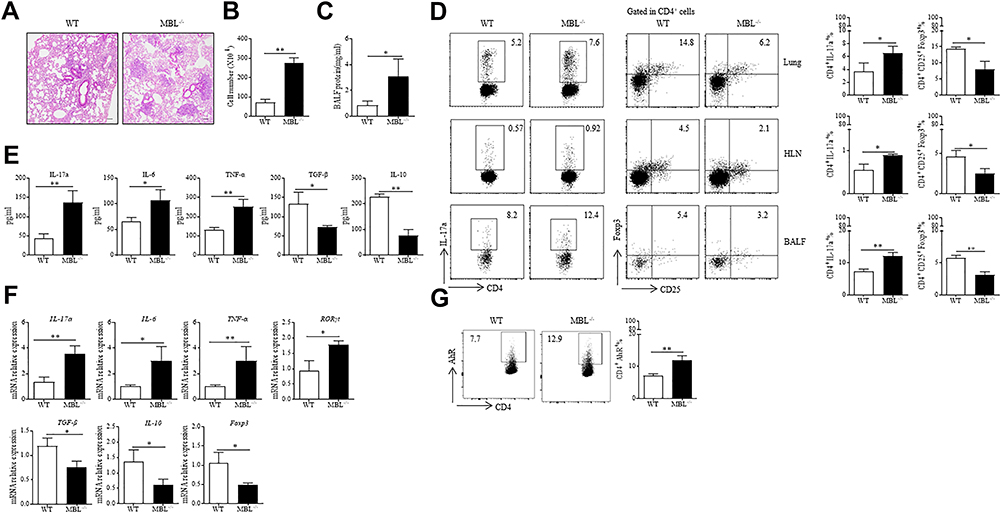 |
Figure 5 MBL deficiency aggravated silica-induced pulmonary inflammation in mice. (A–G) WT and MBL−/− mice were intratracheally injected with 50μL silica suspension containing 5mg silica for 3 d. The lung tissues and sera were collected at the end of the experiment. (A) Histological analysis of mouse lungs was determined by H&E staining. (B) Cell counts in BALF were calculated under the microscope. (C) Total protein level was detected in BALF by BCA analysis. (D) FCM experiments were performed to measure Th17 or Treg frequency in lung tissues, HLN, and BALF from silica-challenged mice. (E) The levels of IL-17a, IL-6, TNFα, TGF-β, and IL-10 levels in BALF were evaluated by ELISA analysis. (F) The mRNA expression levels of IL-17a, IL-6, TNFα, RORγt, TGF-β, IL-10, and Foxp3 in lung tissues were determined by RT-qPCR analysis. (G) AhR expression in CD4+ T cells was evaluated by FCM assay. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. |
MBL Deficiency Aggravates Silica-Induced Pulmonary Fibrosis in Mice
Furthermore, we investigate the function of MBL in silica-induced pulmonary fibrosis. Increased amounts of fibrous nodules and consolidation areas were observed in lung tissues from MBL−/− mice compared to those from WT mice after silica administration for 14 days (Figure 6A). Additionally, we found a higher Ashcroft score in MBL−/− mice lung tissues than WT mice (Figure 6B). Similarly, enhanced hydroxyproline levels were detected in lung homogenate from MBL−/− mice compared to WT counterparts, which indicated severe lung fibrosis in silica-challenged MBL−/− mice (Figure 6C). Flow cytometry analysis showed elevated Th17 cell frequency and declined Treg cell frequency in lung tissues, HLN, BALF from MBL−/− mice compared to those from WT mice (Figure 6D). Besides, the levels of IL-17a, IL-6, and TNF-α were increased in the BALF from silica-challenged MBL−/− mice compared to WT counterparts, while the levels TGF-β and IL-10 levels were reduced consistently (Figure 6E). Moreover, we found elevated IL-17a, IL-6, TNF-α, and RORγt mRNA levels in lung tissues acquired from MBL−/− mice, while declined TGF-β, IL-10, and Foxp3 transcription levels were observed compared to WT mice (Figure 6F). Furthermore, CD4+ T lymphocytes from MBL−/− mice lung tissues showed more AhR expression than those from WT mice (Figure 6G). These data suggest MBL deficiency exacerbates silica-induced pulmonary fibrosis in mice.
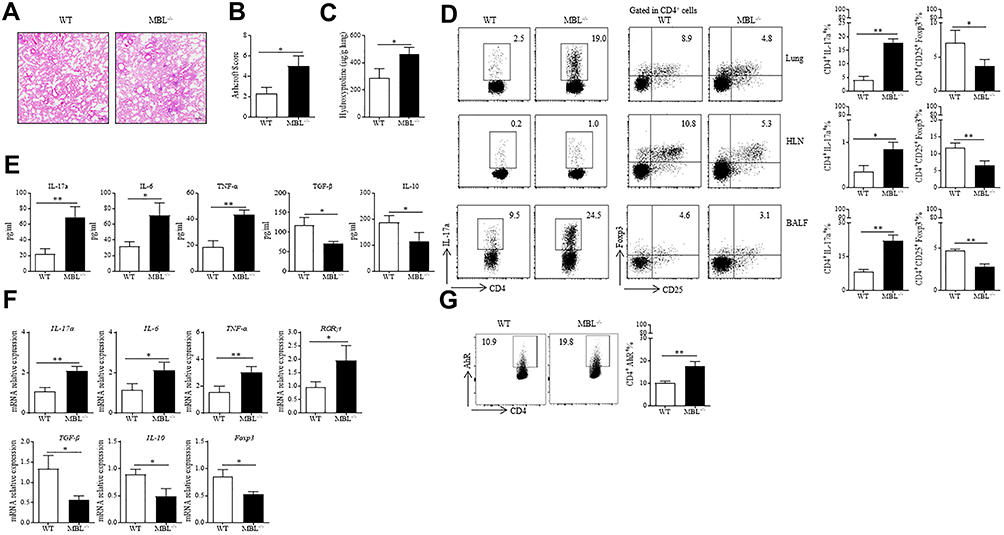 |
Figure 6 MBL-deficient mice exhibited aggravated silica-induced pulmonary fibrosis. (A–G) Mice were intratracheally injected with silica suspension (5mg silica in 50μL sterile PBS) for 35 d. The lung tissues and sera were collected at the end of the experiment. (A) Histological analysis of lung tissues was detected by H&E staining. (B) Ashcroft scores of lung tissues were determined under a microscope. (C) Hydroxyproline levels in lung tissues were tested. (D) Th17 or Treg frequency in lung tissues, HLN, and BALF were assessed by FCM analysis. (E) ELISA analysis was performed to evaluate IL-17a, IL-6, TNFα, TGF-β, and IL-10 levels in BALF collected from silica-treated mice at the last day. (F) The mRNA expression levels of IL-17a, IL-6, TNFα, RORγt, TGF-β, IL-10, and Foxp3 in lung tissues were determined by RT-qPCR analysis (G) Expression of AhR in CD4+ T cells was assessed by FCM assay. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. |
MBL Restoration Ameliorates Silica-Induced Pulmonary Fibrosis in Mice
To further confirm the effect of MBL on silica-induced pulmonary fibrosis in mice, MBL-expressed AAV was injected into MBL−/− mice for MBL restoration as we previously reported.27 As expected, MBL restoration ameliorated lung fibrosis in the silicosis model (Figure 7A). Consistently, declined hydroxyproline levels were shown in lung homogenate obtained from MBL−/− mice injected with MBL-expressed AAV compared to those injected with control AAV (Figure 7B). Furthermore, restoration of MBL expression limited Th17 differentiation, while promoted Treg differentiation (Figure 7C). Flow cytometry analysis showed decreased AhR expression in CD4+ T lymphocytes from pAAV-MBL injected MBL−/− mice lung tissues than those from control mice (Figure 7D). Above all, these data demonstrated that MBL restoration ameliorated silica-induced pulmonary fibrosis in mice.
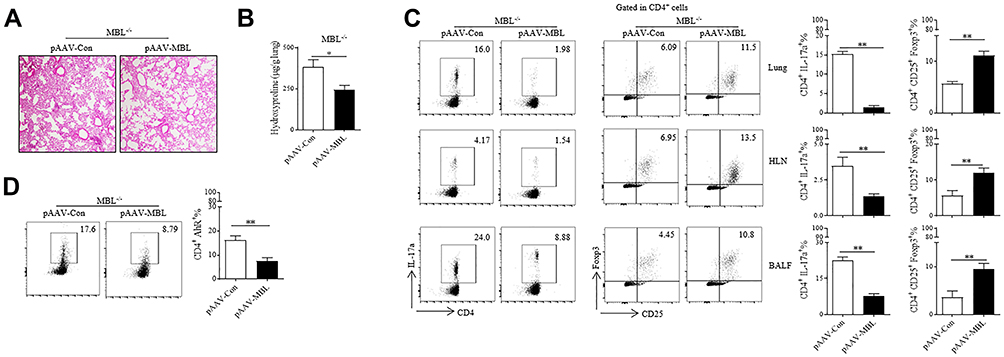 |
Figure 7 MBL restoration ameliorated silica-induced pulmonary fibrosis in mice. (A–D) MBL−/− mice were tail vein injected with MBL-expressed AAV or control AAV combined with intratracheally injected with silica suspension (5mg silica in 50μL sterile PBS) for 35 d. The lung tissues and sera were collected at the end of the experiment. (A) Histological analysis of lung tissues was determined by H&E staining. (B) Hydroxyproline levels in lung tissues were detected. (C) FCM analysis was performed to evaluate Th17 or Treg frequency in lung tissues, HLN, and BALF. (D) Expression of AhR in CD4+ T cells was detected by FCM assay. *p < 0.05, **p < 0.01. The data represent three independent experiments with similar results. |
Discussion
MBL is a circulating plasma protein of the innate immune system, which can modulate host inflammatory response. Our previous reports indicated that MBL might be correlated to the progression of silicosis, and MBL directly interacted with T cells and suppressed T cell activation.20 The present study demonstrated the negative correlation between serum MBL levels and disease progression in silicosis patients. Our results indicated that MBL inhibited the Th17 differentiation through limiting AhR expression and its nuclear translocation. Furthermore, we observed that MBL deficiency aggravates silica-induced pulmonary inflammation and fibrosis in mice, which was associated with the regulation of Th17 immunity (Figure 8).
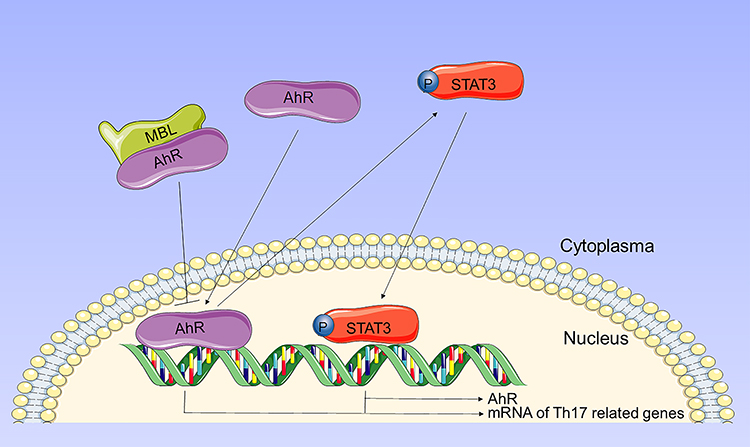 |
Figure 8 Schematic representation of the mechanism by which MBL regulates Th17 cell differentiation through AhR signaling. Upon activation, AhR translocates to the nucleus and induces the expression of Th17-related transcripts. Besides, activated AhR promotes phosphorylation of STAT3. Phosphorylated STAT3 not only induces expression of Th17-related transcripts, but also promotes AhR expression. MBL interacts with AhR and inhibits AhR translocation, thereby limiting Th17 cell differentiation. |
MBL deficiency is the most common complement deficiency in humans.28 Low MBL serum levels have been shown to enhance the risk for infection, and individuals with MBL deficiency may have an increased frequency of inflammatory autoimmune diseases. MBL is primarily a circulating protein that accumulates in the lung during inflammation.29 Clinical studies in humans have demonstrated that MBL deficiency predisposes to severe respiratory tract infection.30,31 MBL could bind with influenza virus and was ascribed a protective role of MBL in mice infected with influenza.32 A previous longitudinal study revealed that functional deficiency of MBL was associated with frequent exacerbations of chronic obstructive pulmonary disease (COPD), and increased serum MBL levels correlated with improved survival.31 Interestingly, the addition of MBL protein as a supplement significantly reduced smoking-related lung inflammation in mice.33 Garred et al reported that the presence of MBL variant alleles was correlated with poor prognosis and early death in patients with CF.34 Structural MBL-2 gene mutations could result in the impairment of FEV1 and reduced patient survival. Indeed, serum MBL levels were also positively correlated to the lung function determined by FEV1 and FVC in CF patients,35 which was similar to our observation that silicosis patients with higher serum MBL levels exhibited better lung function. Also, MBL deficiency was associated with steeper declines in FEV1 in patients with primary ciliary dyskinesia (PCD), a congenital lung disease.36 Therefore, we postulated that MBL might be critical in regulating lung function in the pathogenesis of acute and chronic lung diseases. Currently, the association of MBL with lung diseases was mainly evaluated by the longitudinal study of clinical parameters in patients. Further investigation is urgent to reveal the mechanisms through which MBL and related pathways modulate the pulmonary response with experimental animal studies and biological research.
Th17 cells are believed to be critical pro-inflammatory cells that contribute to the pathogenesis of diverse pulmonary diseases.4 COPD patients exhibited remarkable elevated Th17 cell frequency compared to healthy controls.37,38 Th17 lymphocytes secrete IL-17a that promotes the activation of bronchial fibroblasts, epithelial cells, and smooth muscle cells, inducing them to produce pro-inflammatory cytokines responsible for recruiting neutrophils and their local infiltration, thus aggravating the COPD symptomatology.6 Indeed, Th17/Treg cytokine imbalance was observed in the experimental model of COPD, and the imbalance cytokine profile plays a pivotal role in the exacerbated inflammatory response.39 Similarly, Th17 cells were found in airway submucosa in CF patients and have been thought to account for the inflammatory response.7 Silicosis sometimes complicates autoimmune diseases, implying that the autoreactive T-cell responses were involved in the progress of the disease. Our previous study demonstrated that the serum concentrations of MBL correlated negatively with in vivo T-cell activation status in patients with early-stage silicosis.20 Song et al, observed that the Th17 response could induce lung inflammation during the pathogenesis of silicosis by regulating the homeostasis of the Th immune responses and affecting the production of IL-22 and IL-1β.22 Treg cells controlled the inflammation against silica via suppression of inflammatory cells in the early stage.40 In examining Th17 cells in the mice challenged with silica, we found elevated levels of CD4+ T cells expressing IL-17 in the MBL−/− mice. Considering the reciprocal regulation of IL-17 and FoxP3 expression, we also examined the frequency of CD4+FoxP3+ T cells from silica-administrated mice. We confirmed that the enhanced IL-17 production by T cells from MBL-deficient mice is accompanied by reduced FoxP3 expression. In vitro study also showed that MBL protein limited IL-17 expression but promoted Foxp3 expression in CD4+ T cells, which is partially correlated with a previous study that showed MBL inhibit the differentiation of CD4+ T cells to Th17 cells.41 These results indicate that MBL shifts the program of regulation of IL-17 and FoxP3 gene expression. MBL circulates in complexes with MBL-associated serine proteases (MASPs). Upon binding of MBL to their targets, MASP becomes activated and initiates complement activation. The complement component, C5a, has been reported to induce Th17 cytokine expression from human T cells. Therefore, it is possible that MBL-mediated complement activation might be also involved in the regulation of Th17 responses in vivo, and further investigation is warranted. During Th17 cell differentiation, activated STAT3 induces the expression of RORγt and RORα, which are the master transcription factors driving the lineage commitment to Th17. MBL inhibited the IL-2-induced activation of STAT5 in NK cells, limiting inflammatory cytokine production.17 Indeed, genetic variants in the STAT3/STAT5 region are associated with immune-mediated diseases and inflammatory diseases.42 In this study, we demonstrated that MBL attenuated STAT3 activation under the Th17 polarization conditions. Indeed, MBL, like many other lectins, binds to a large number of different glycoproteins expressed on various cells. Further investigation about the effect of MBL on JAK-STAT activation could reveal the role of MBL in inflammatory responses. Notably, STAT3 expression and activity are regulated through transcriptional control, post-translational regulation, and intracellular localization.43 The cooperation of STAT3 with associated transcription factors appears to control the consequent inflammatory cytokine production.42 Activated AhR causes phosphorylation of Janus kinase 2 (Jak2) and subsequent STAT3 activation.44 Knockdown of AhR significantly suppressed the Jak2/STAT3 signaling. Alternatively, phosphorylated STAT3 binds to the promoter of AhR, thereby promoting AhR expression.45 We supposed MBL firstly inhibited AhR signal, leading to impaired STAT3 phosphorylation, which contributed to subsequent downregulation of AhR expression. Furthermore, these data indicated the potential role of MBL in STAT3 signalling regulation.
A previous study determined that MBL attenuated the expression of CYP1A1, CYP1A2, and CYP1B1, genes that are considered as biomarkers for AhR activation.46 The induction of CYPs could increase oxidative stress and inflammation that are intimately linked to promoting complications in people chronically exposed to incense smoke.47 Notably, CYP1A1, CYP1A2, and CYP1B1 have been reported to produce carcinogenic metabolites.48,49 Previous studies documented that MBL had been involved in the development of several cancer types,27,50 while the precise mechanisms are still limited. Further investigation of whether MBL modulates carcinogenic metabolite production through AhR may provide new insight into tumorigenesis. We detected interaction between AhR and MBL, while whether MBL coupled with AhR directly still needs more evidence. AhR forms complex with several chaperone proteins, such as HSP90, AIP, and p23 in the cytoplasm. Upon activation, AhR releases from the complex and translocates to the nucleus.51 We previously demonstrated that MBL coupled with calreticulin and functioned like a chaperone protein.20 Thus, we assumed that MBL interacted with the AhR complex and contributed to the stability of the complex, thereby regulating AhR translocation.
In summary, we have made the novel observation that MBL suppresses the differentiation and activation of Th17 cells. MBL treatment reduces IL-17 expression in CD4+ T cells associated with enhanced expression of FoxP3, which implies that the circulating MBL is crucial for maintaining Th17/Treg balance in vivo. Mechanistically, our study elucidates that MBL might inhibit the Th17 differentiation through AhR/STAT3 pathway. The finding that MBL modulates the Th17/Treg counterbalance suggests that mediators and mechanisms that regulate MBL expression or function may serve as targets for manipulating this balance, thereby regulating autoimmune and inflammatory disease outcomes.
Conclusion
Our studies indicated that MBL inhibited Th17 response, thereby ameliorating silicosis-associated lung inflammation and fibrosis. We demonstrated that MBL down-regulated AhR/STAT3 pathway through interacting with AhR and limiting AhR nucleus translocation. This report provided new insight into silicosis and other inflammatory diseases in patients with MBL deficiency.
Data Sharing Statement
All data presented or analyzed during this research are included in this published study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported in part by the National Natural Science Foundation of China (grant no.: 82071781, 81971550, 81873872, and 81903269), Science and Technology Planning Project of Guangzhou (grant no.: 202002030160 and 202102080005), Innovation team of chronic kidney disease with integrated traditional Chinese and Western Medicine (grant no.: 2019KCXTD014), Natural Science Foundation of Guangdong (grant no.:2021A1515011546, 2021A1515010081, 2021A1515012205) and Guangdong Provincial Key Laboratory of Occupational Disease Prevention and Treatment (grant no.:2017B030314152).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Leung CC, Yu IT, Chen W. Silicosis. Lancet. 2012;379(9830):2008–2018. PubMed PMID: 22534002. doi:10.1016/S0140-6736(12)60235-9
2. Latoche JD, Ufelle AC, Fazzi F, Ganguly K, Leikauf GD, Fattman CL. Secreted phosphoprotein 1 and sex-specific differences in silica-induced pulmonary fibrosis in mice. Environ Health Perspect. 2016;124(8):1199–1207. PubMed PMID: 26955063; PubMed Central PMCID: PMCPMC4977050. doi:10.1289/ehp.1510335
3. The Lancet Respiratory M. The world is failing on silicosis. Lancet Respir Med. 2019;7(4):283. PubMed PMID: 30872128. doi:10.1016/S2213-2600(19)30078-5
4. Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. PubMed PMID: 20148686. doi:10.1146/annurev-physiol-021909-135926
5. Simonian PL, Roark CL, Wehrmann F, et al. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182(1):657–665. PubMed PMID: 19109199; PubMed Central PMCID: PMCPMC2766086. doi:10.4049/jimmunol.182.1.657
6. Ponce-Gallegos MA, Ramirez-Venegas A, Falfan-Valencia R. Th17 profile in COPD exacerbations. Int J Chron Obstruct Pulmon Dis. 2017;12:1857–1865. PubMed PMID: 28694696; PubMed Central PMCID: PMCPMC5491572. doi:10.2147/COPD.S136592
7. Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. 2011;184(2):252–258. PubMed PMID: 21474644; PubMed Central PMCID: PMCPMC3381840. doi:10.1164/rccm.201102-0236OC
8. Song L, Weng D, Liu F, et al. Tregs promote the differentiation of Th17 cells in silica-induced lung fibrosis in mice. PLoS One. 2012;7(5):e37286. PubMed PMID: 22615967; PubMed Central PMCID: PMCPMC3352873. doi:10.1371/journal.pone.0037286
9. Chen Y, Li C, Weng D, et al. Neutralization of interleukin-17A delays progression of silica-induced lung inflammation and fibrosis in C57BL/6 mice. Toxicol Appl Pharmacol. 2014;275(1):62–72. PubMed PMID: 24291675. doi:10.1016/j.taap.2013.11.012
10. Veldhoen M, Hirota K, Westendorf AM, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453(7191):106–109. PubMed PMID: 18362914. doi:10.1038/nature06881
11. Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206(1):43–49. PubMed PMID: 19114668; PubMed Central PMCID: PMCPMC2626686. doi:10.1084/jem.20081438
12. Nakahama T, Kimura A, Nguyen NT, et al. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc Natl Acad Sci USA. 2011;108(34):14222–14227. PubMed PMID: 21825138; PubMed Central PMCID: PMCPMC3161527. doi:10.1073/pnas.1111786108
13. Sun L, Fu J, Lin SH, et al. Particulate matter of 2.5 μm or less in diameter disturbs the balance of TH17/regulatory T cells by targeting glutamate oxaloacetate transaminase 1 and hypoxia-inducible factor 1α in an asthma model. J Allergy Clin Immunol. 2020;145(1):402–414. PubMed PMID: 31647966. doi:10.1016/j.jaci.2019.10.008
14. Jaligama S, Patel VS, Wang P, et al. Radical containing combustion derived particulate matter enhance pulmonary Th17 inflammation via the aryl hydrocarbon receptor. Part Fibre Toxicol. 2018;15(1):20. PubMed PMID: 29724254; PubMed Central PMCID: PMCPMC5934866. doi:10.1186/s12989-018-0255-3
15. Garred P, Genster N, Pilely K, et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. 2016;274(1):74–97. PubMed PMID: 27782323. doi:10.1111/imr.12468
16. Dean MM, Flower RL, Eisen DP, Minchinton RM, Hart DN, Vuckovic S. Mannose-binding lectin deficiency influences innate and antigen-presenting functions of blood myeloid dendritic cells. Immunology. 2011;132(2):296–305. PubMed PMID: 21091907; PubMed Central PMCID: PMCPMC3050452. doi:10.1111/j.1365-2567.2010.03365.x
17. Zhou J, Hu MY, Li J, et al. Mannan-binding lectin regulates inflammatory cytokine production, proliferation, and cytotoxicity of human peripheral natural killer cells. Mediat Inflamm. 2019;2019:1–12. PubMed PMID: WOS: 000504247100001. doi:10.1155/2019/6738286
18. Tang Y, Ma D, Ming S, et al. Mannan-binding lectin reduces CpG DNA-induced inflammatory cytokine production by human monocytes. Microbiol Immunol. 2015;59(4):231–237. PMID: 25664598. doi:10.1111/1348-0421.12245
19. Liu H, Zhou J, Ma D, et al. Mannan binding lectin attenuates double-stranded RNA-mediated TLR3 activation and innate immunity. FEBS Lett. 2014;588(6):866–872. PubMed PMID: 24530528. doi:10.1016/j.febslet.2014.01.064
20. Zhao N, Wu J, Xiong S, et al. Mannan-binding lectin, a serum collectin, suppresses T-cell proliferation via direct interaction with cell surface calreticulin and inhibition of proximal T-cell receptor signaling. FASEB J. 2017;31(6):2405–2417. PubMed PMID: 28209773. doi:10.1096/fj.201601200RR
21. Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41(4):467–470. PMID: 3366935; PubMed Central PMCID: PMCPMC1141479. doi:10.1136/jcp.41.4.467
22. Song L, Weng D, Dai W, et al. Th17 can regulate silica-induced lung inflammation through an IL-1beta-dependent mechanism. J Cell Mol Med. 2014;18(9):1773–1784. PubMed PMID: 25091058; PubMed Central PMCID: PMCPMC4196653. doi:10.1111/jcmm.12341
23. Lee S, Hayashi H, Mastuzaki H, Kumagai-Takei N, Otsuki T. Silicosis and autoimmunity. Curr Opin Allergy Clin Immunol. 2017;17(2):78–84. PubMed PMID: 28177948. doi:10.1097/ACI.0000000000000350
24. Liu X, Hu H, Fan H, et al. The role of STAT3 and AhR in the differentiation of CD4+ T cells into Th17 and Treg cells. Medicine. 2017;96(17):e6615. PubMed PMID: 28445259; PubMed Central PMCID: PMCPMC5413224. doi:10.1097/MD.0000000000006615
25. Yuan X, Dou Y, Wu X, Wei Z, Dai Y. Tetrandrine, an agonist of aryl hydrocarbon receptor, reciprocally modulates the activities of STAT3 and STAT5 to suppress Th17 cell differentiation. J Cell Mol Med. 2017;21(9):2172–2183. PubMed PMID: 28332288; PubMed Central PMCID: PMCPMC5571555. doi:10.1111/jcmm.13141
26. Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279(23):23847–23850. PubMed PMID: 15028720. doi:10.1074/jbc.R400004200
27. Li J, Li H, Yu Y, et al. Mannan-binding lectin suppresses growth of hepatocellular carcinoma by regulating hepatic stellate cell activation via the ERK/COX-2/PGE2 pathway. Oncoimmunology. 2019;8(2):e1527650. PubMed PMID: 30713782; PubMed Central PMCID: PMCPMC6343806. doi:10.1080/2162402X.2018.1527650
28. Thiel S, Frederiksen PD, Jensenius JC. Clinical manifestations of mannan-binding lectin deficiency. Mol Immunol. 2006;43(1–2):86–96. PubMed PMID: 16023210; PubMed Central PMCID: PMCPMC7132399. doi:10.1016/j.molimm.2005.06.018
29. Chalmers JD, Fleming GB, Hill AT, Kilpatrick DC. Impact of mannose-binding lectin insufficiency on the course of cystic fibrosis: a review and meta-analysis. Glycobiology. 2011;21(3):271–282. PMID: 21045008. doi:10.1093/glycob/cwq161
30. Eisen DP. Mannose-binding lectin deficiency and respiratory tract infection. J Innate Immun. 2010;2(2):114–122. PubMed PMID: 20375630; PubMed Central PMCID: PMCPMC7179718. doi:10.1159/000228159
31. Mandal J, Malla B, Steffensen R, et al. Mannose-binding lectin protein and its association to clinical outcomes in COPD: a longitudinal study. Respir Res. 2015;16:150. PubMed PMID: 26684757; PubMed Central PMCID: PMCPMC4750539. doi:10.1186/s12931-015-0306-3
32. Chang WC, White MR, Moyo P, et al. Lack of the pattern recognition molecule mannose-binding lectin increases susceptibility to influenza A virus infection. BMC Immunol. 2010;11:64. PubMed PMID: 21182784; PubMed Central PMCID: PMCPMC3022599. doi:10.1186/1471-2172-11-64
33. Hodge S, Matthews G, Dean MM, et al. Therapeutic role for mannose-binding lectin in cigarette smoke-induced lung inflammation? Evidence from a murine model. Am J Respir Cell Mol Biol. 2010;42(2):235–242. PubMed PMID: 19411612. doi:10.1165/rcmb.2008-0486OC
34. Garred P, Pressler T, Madsen HO, et al. Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J Clin Invest. 1999;104(4):431–437. PubMed PMID: 10449435; PubMed Central PMCID: PMCPMC408526. doi:10.1172/JCI6861
35. Garred P, Pressler T, Lanng S, et al. Mannose-binding lectin (MBL) therapy in an MBL-deficient patient with severe cystic fibrosis lung disease. Pediatr Pulmonol. 2002;33(3):201–207. PubMed PMID: 11836800. doi:10.1002/ppul.10064
36. Videbaek K, Buchvald F, Holgersen MG, et al. The impact of mannose-binding lectin polymorphisms on lung function in primary ciliary dyskinesia. Pediatr Pulmonol. 2019;54(8):1182–1189. PubMed PMID: 31012247. doi:10.1002/ppul.24346
37. Zheng X, Zhang L, Chen J, Gu Y, Xu J, Ouyang Y. Dendritic cells and Th17/Treg ratio play critical roles in pathogenic process of chronic obstructive pulmonary disease. Biomed Pharmacother. 2018;108:1141–1151. PubMed PMID: 30372815. doi:10.1016/j.biopha.2018.09.113
38. Wang H, Ying H, Wang S, et al. Imbalance of peripheral blood Th17 and Treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J. 2015;9(3):330–341. PubMed PMID: 24720797. doi:10.1111/crj.12147
39. Cervilha DA, Ito JT, Lourenco JD, et al. The Th17/Treg cytokine imbalance in chronic obstructive pulmonary disease exacerbation in an animal model of cigarette smoke exposure and lipopolysaccharide challenge association. Sci Rep. 2019;9(1):1921. doi:10.1038/s41598-019-38600-z
40. Liu FW, Liu J, Weng D, et al. CD4+CD25+Foxp3+Regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS One. 2010;5(11):e15404. PubMed PMID: WOS: 000283780800027. doi:10.1371/journal.pone.0015404
41. Zhang W, Wang Y, Yang L, et al. Effects of mannan-binding lectin on differentiation of Th17 cells. Chin J Microbiol Immunol. 2017;15(12):666–673. PubMed PMID: 35300210; PubMed Central PMCID: PMCPMC8923702.
42. Wingelhofer B, Neubauer HA, Valent P, et al. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia. 2018;32(8):1713–1726. PubMed PMID: 29728695; PubMed Central PMCID: PMCPMC6087715. doi:10.1038/s41375-018-0117-x
43. Qi QR, Yang ZM. Regulation and function of signal transducer and activator of transcription 3. World J Biol Chem. 2014;5(2):231–239. PubMed PMID: 24921012; PubMed Central PMCID: PMCPMC4050116. doi:10.4331/wjbc.v5.i2.231
44. Xiong J, Zhang X, Zhang Y, et al. Aryl hydrocarbon receptor mediates Jak2/STAT3 signaling for non-small cell lung cancer stem cell maintenance. Exp Cell Res. 2020;396(1):112288. PubMed PMID: 32941808. doi:10.1016/j.yexcr.2020.112288
45. Stobbe-Maicherski N, Wolff S, Wolff C, et al. The interleukin-6-type cytokine oncostatin M induces aryl hydrocarbon receptor expression in a STAT3-dependent manner in human HepG2 hepatoma cells. FEBS J. 2013;280(24):6681–6690. PubMed PMID: 24127753. doi:10.1111/febs.12571
46. Wincent E, Amini N, Luecke S, et al. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J Biol Chem. 2009;284(5):2690–2696. PubMed PMID: 19054769. doi:10.1074/jbc.M808321200
47. Hussain T, Al-Attas OS, Al-Daghri NM, et al. Induction of CYP1A1, CYP1A2, CYP1B1, increased oxidative stress and inflammation in the lung and liver tissues of rats exposed to incense smoke. Mol Cell Biochem. 2014;391(1–2):127–136. PubMed PMID: WOS: 000335570400014. doi:10.1007/s11010-014-1995-5
48. Chang TK, Chen J, Yang G, Yeung EY. Inhibition of procarcinogen-bioactivating human CYP1A1, CYP1A2 and CYP1B1 enzymes by melatonin. J Pineal Res. 2010;48(1):55–64. PubMed PMID: 19919601. doi:10.1111/j.1600-079X.2009.00724.x
49. Mescher M, Haarmann-Stemmann T. Modulation of CYP1A1 metabolism: from adverse health effects to chemoprevention and therapeutic options. Pharmacol Ther. 2018;187:71–87. PubMed PMID: 29458109. doi:10.1016/j.pharmthera.2018.02.012
50. Swierzko AS, Kilpatrick DC, Cedzynski M. Mannan-binding lectin in malignancy. Mol Immunol. 2013;55(1):16–21. PubMed PMID: 23062612. doi:10.1016/j.molimm.2012.09.005
51. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. 2014;32:403–432. PubMed PMID: 24655296. doi:10.1146/annurev-immunol-032713-120245
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.