Back to Journals » Journal of Inflammation Research » Volume 17
Macrophage-Related Gene Signatures for Predicting Prognosis and Immunotherapy of Lung Adenocarcinoma by Machine Learning and Bioinformatics
Authors Xiang Y, Wang G, Liu B, Zheng H, Liu Q, Ma G, Du J ![]()
Received 5 October 2023
Accepted for publication 30 January 2024
Published 7 February 2024 Volume 2024:17 Pages 737—754
DOI https://doi.org/10.2147/JIR.S443240
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tara Strutt
Yunzhi Xiang,* Guanghui Wang,* Baoliang Liu, Haotian Zheng, Qiang Liu, Guoyuan Ma, Jiajun Du
Institute of Oncology, Shandong Provincial Hospital, Shandong University, Jinan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiajun Du, Shandong Provincial Hospital, No. 324 Jingwu Road, Huaiyin District, Jinan, 250000, People’s Republic of China, Email [email protected]
Background: In recent years, the immunotherapy of lung adenocarcinoma has developed rapidly, but the good therapeutic effect only exists in some patients, and most of the current predictors cannot predict it very well. Tumor-infiltrating macrophages have been reported to play a crucial role in lung adenocarcinoma (LUAD). Thus, we want to build novel molecular markers based on macrophages.
Methods: By non-negative matrix factorization (NMF) algorithm and Cox regression analysis, we constructed macrophage-related subtypes of LUAD patients and built a novel gene signature consisting of 12 differentially expressed genes between two subtypes. The gene signature was further validated in Gene-Expression Omnibus (GEO) datasets. Its predictive effect on prognosis and immunotherapy outcome was further evaluated with rounded analyses. We finally explore the role of TRIM28 in LUAD with a series of in vitro experiments.
Results: Our research indicated that a higher LMS score was significantly correlated with tumor staging, pathological grade, tumor node metastasis stage, and survival. LMS was identified as an independent risk factor for OS in LUAD patients and verified in GEO datasets. Clinical response to immunotherapy was better in patients with low LMS score compared to those with high LMS score. TRIM28, a key gene in the gene signature, was shown to promote the proliferation, invasion and migration of LUAD cell.
Conclusion: Our study highlights the significant role of gene signature in predicting the prognosis and immunotherapy efficacy of LUAD patients, and identifies TRIM28 as a potential biomarker for the treatment of LUAD.
Keywords: lung adenocarcinoma, macrophage, TRIM28, prognosis, immunotherapy
Introduction
Lung cancer is the second most common cancer in the world and one of the leading causes of cancer-related deaths globally.1–3 Nearly 90% of lung cancer cases are non-small cell lung cancer, the most common histologic subtype of which is lung adenocarcinoma (LUAD).4–7 Although much progress has been made in the arena of surgical treatment, prognosis has generally remained poor. The International Association for the Study of Lung Cancer (IASLC) staging project showed a decrease in overall 5-year survival after surgical resection from 73% in stage IA to 9% in stage IIIB of LUAD.7
Immune checkpoint inhibition (ICI) therapies such as programmed cell death protein 1 (PD1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA4) have demonstrated significant efficacy in the treatment of LUAD.8 However, these improvements in therapeutic response have remain limited to a subset of LUAD patients with specific molecular characteristics.9 A percentage of LUAD patients are resistant to immunotherapies, resulting in cancer recurrence.10 The same treatment regimen may lead to different responses and clinical outcomes among different LUAD patients.8 This may be due to the exceptionally complex genomes of lung cancer, as demonstrated by in-depth sequencing studies, which support its extensive heterogeneity at the cellular and tumor microenvironment (TME) level.4,11 Therefore, to enhance risk stratification, survival prediction, and treatment management of LUAD, identification of molecular abnormalities and prognostic markers is crucial.12
There is growing evidence that the immune phenotypes of cancer can be determined by components in the TME, which can affect patient prognosis.13–15 As important components of the immune microenvironment, macrophages play an irreplaceable role in innate and acquired immunity.16 Tumor-infiltrating macrophages, as the most abundant infiltrating immune cells in the TME, perform a wide repertoire of functions in LUAD through diverse phenotypes.17 Macrophages are typically divided into different subsets, including M1 (classically activated macrophages) and M2 (alternatively activated macrophages).18,19 In the early stages of tumor development, macrophages in their dormant state (M0) are mainly polarized to the M1 phenotype, which is pro-inflammatory and mediates immune responses that inhibit tumor growth. As tumors develop, M0 macrophages are gradually transformed into the M2 functional phenotype, which, in turn, contributes to immune suppression and tumor angiogenesis.20,21 Accumulating evidence has demonstrated that dynamic changes in macrophage phenotypes can affect tumorigenesis, progression, and metastasis.22 Therefore, evaluation of macrophages may be an effective way to predict the prognosis and therapeutic response of individual patients. However, direct use of macrophages is relatively difficult. In this case, an intuitive and effective tool may be required.
Our study evaluated the expression of macrophage-related differentiated expressed genes (DEGs) with a non-negative matrix factorization (NMF) algorithm. Based on these genes, 458 patients with LUAD from The Cancer Genome Atlas (TCGA) were clustered using COX risk assessment model, and eventually a gene signature was constructed for predicting prognosis and immunotherapy efficacy. Four Gene-Expression Omnibus (GEO) cohorts including 496 patients with LUAD were used to validate the predictive performance of the signature. Two independent immunotherapy cohorts were identified to validate the predictive performance for immunotherapy efficacy. Finally, we investigated the regulation of the key gene TRIM28 on the biological behavior of LUAD cells using in vitro experiments.
Materials and Methods
Public Data Collection
A total of 953 LUAD patients from five public LUAD cohorts (TCGA-LUAD cohort, n=457; GSE3141 cohort, n=58; GSE31210 cohort, n=226; GSE32019 cohort, n=85; GSE50081 cohort, n=127) were included in this study as training and validation cohorts. Inclusion criteria were as follows: (1) LUAD patients and normal controls must be included in the profiles, (2) The discovery profile should have at least ten samples to ensure accuracy, and (3) complete clinical information must be included. The mRNA expression, clinical information, copy number variation, and somatic mutation data of TCGA-LUAD were downloaded from https://portal.gdc.cancer.gov/. The data of the GSE3141, GSE31210, GSE32019 and GSE50081 cohorts were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). In addition, two immunotherapy cohorts (GSE135222 and IMvigor210 downloaded from http://research-pub.gene.com/IMvigor210CoreBiologies) were obtained for subsequent analysis.
Clinical Sample Collection
We selected 30 patients diagnosed with lung adenocarcinoma who were treated in Shandong Provincial Hospital as samples to perform differential expression analysis of TRIM28. The inclusion criteria were: (1) They were diagnosed with lung adenocarcinoma; (2) Complete clinical data were included; (3) They were informed and signed informed consent. We collected basic patient information and tumor and normal tissues were collected and stored at −80°C. This study was approved by the Biomedical Research Ethics Committee of Shandong Provincial Hospital and conducted in accordance with the Declaration of Helsinki. All subjects were required to provide written informed consent.
Analysis of Tumor-Infiltrating Macrophages
MCPCOUNTER is an algorithm that estimates the content of different cell types in tumor tissue through a linear regression model based on the gene expression characteristics of immune and non-immune cell types. By “ggpubr” R package and MCPCOUNTER algorithm, we analyzed macrophage infiltration between normal and tumor tissues of LUAD patients. Then, we introduced COATES_MACROPHAGE_M1_VS_M2 gene sets and adopted “GSVA” and “ggpubr” packages into R to assess the infiltration of M1 and M2 macrophages in tumor vs normal samples.23 Lastly, we conducted Kaplan-Meier analysis of macrophages by “survival” and “survminer” R packages.
Construction of Cell Subtypes
The NMF algorithm is a matrix decomposition method which can decompose a large non-negative matrix into two small non-negative matrices and can continue to decompose it. With NMF algorithm, patients were categorized into distinct molecular subtypes based on expression of macrophages. We generated the NMF rank survey, and then selected the smallest rank value at which the cophenetic correlation coefficient began to decline as the number of groups for the final grouping.
Relationship Between Cell Subtypes and Prognosis of LUAD
To explore the clinical value of the two subtypes identified by NMF algorithm, we used Kaplan–Meier curves to assess the differences in overall survival (OS) between them, generated by “survival” and “survminer” R packages. Principal Component Analysis (PCA) is an unsupervised machine learning method that extracts the largest individual differences displayed by the principal components through dimensionality reduction, so that each group can be clustered. Single sample Gene Set Enrichment Analysis (ssGSEA) algorithm is commonly used to analyze immune cell infiltration by comparing the gene expression data of each sample to a specific gene set to estimate the relative enrichment of that gene set in that sample. Thus, they were used to validate the subsets. Later, gene set variation analysis (GSVA) was performed to investigate the differences between two subtypes in biological processes, with the hallmark gene set (c2.cp.kegg. v7.4) derived from the MSigDB database.
Construction of the Gene Signature
“limma” package uses linear models to estimate the mean and variance of gene expression in different groups to perform differential analysis. Using the “limma” package in R, we identified differentially expressed genes (DEGs) between two subtypes (cutoff value was | log2FC| ≥ 1 and FDR < 0.05). These DEGs were then subjected to univariate Cox regression analysis (coxPfilter=0.05), least absolute shrinkage and selection operator (LASSO) regression analysis, and multivariate Cox regression analysis to identify genes which were linked to LUAD prognosis. Finally, twelve genes were selected to construct a prognostic-related risk score, termed LUAD macrophage gene signature (LMS). According to the results of the multivariate Cox regression analysis, the LMS score was constructed as follows:
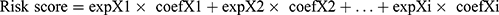 , where, coefXi represents the synergetic coefficient and expXi represents the relative expression of genes.
, where, coefXi represents the synergetic coefficient and expXi represents the relative expression of genes.
All patients were divided into low-risk (LMS score< median value) and high-risk (LMS score> median value) groups based on the median risk score. We conducted Kaplan–Meier survival analysis to explore the prediction direction of LMS on the prognosis of LUAD patients, and made the ROC curve to verify the prediction effect of LMS on the prognosis of LUAD patients. ROC curve is an indicator for evaluating the performance of a binary classification model, generally passing through the upper left of the coordinate axis. The area under the curve is called AUC, and the larger the value, the better the performance, generally between 0.5–1. ROC curve and AUC possess an advantage that they will not be affected by the imbalance in the number of positive and negative samples and can stably reflect the quality of the model, so they were used to predict the predictive ability of LMS.
Prognostic and Clinical Correlation Analysis of LMS
With the “ssGSEA” R package, we calculated the infiltration degree of most immune cells between the two risk groups. Then, using “pheatmap” package in R, risk curve, risk heat maps, and survival maps were constructed to explore the relationship between patient prognosis and LMS. Furthermore, through univariate Cox and multivariate Cox regression analysis, LMS was proved to be an independent prognostic factor. A heatmap was used to investigate the relationship between LMS and clinical features (age, gender, tumor stage, and tumor–node–metastasis (TNM) stage). We also performed clinical subgroup survival analysis to exclude the impact of clinical stage itself. COATES_MACROPHAGE_M1_VS_M2 was used to assess the differential degrees of infiltration by M1 and M2 macrophages between the high and low-risk groups. With “ggpubr” and “reshape2” R packages, we calculated and compared tumor mutation burden (TMB) of two risk groups.
Establishment and Validation of a Nomogram Scoring System
We developed a predictive nomogram combining clinical characteristics and LMS using the “rms” package in R. In the nomogram scoring system, each variable matched a score, and the total score was obtained by adding the scores across all variables of each sample. Calibration plots were utilized to illustrate the performance of the nomogram in predicting 1-, 3-, and 5-year survival probabilities by comparing the predicted outcomes with the observed outcomes. Decision Curve Analysis (DCA) and ROC curve were then used to compare the ability in predicting prognosis between nomogram and other features, including age, gender, stage and LMS.
Validation of External Cohorts
We obtained four LUAD cohorts (GSE3141, GSE31210, GSE32019 and GSE50081) from GEO databases. Kaplan-Meier survival analysis, ROC curve, risk curve, and survival map were then performed on them respectively.
Immune Correlation Analysis of the LMS
Using the R package “Mcpcounter”, we quantified the abundance of 10 infiltrating immune cells in all samples to explore the association between immune cell and LMS. We employed Gene Set Enrichment Analysis (GSEA) with the hallmark gene set (c2.cp.kegg. v7.4) from the MSigDB database to investigate immune-related pathways enriched in the high and low risk groups. In addition, we utilized the ssGSEA algorithm and the “GSEABase” R package to identify differences in immune functions between the high and low risk groups. The association between immune checkpoints and LMS was then investigated by the “limma” package in R. In the end, with the Estimation of Stromal and Immune cells in Malignant Tumors using Expression data (ESTIMATE) algorithm, we calculated the immune and stromal scores to predict the level of infiltrating immune and stromal cells between the two groups, which formed the basis to infer tumor purity.
Identification of Potential Predictive Value of Immunotherapy Efficacy
We calculated the tumor immune dysfunction and exclusion (TIDE) score and immunophenotype score (IPS) for each LUAD patient in two groups. The TIDE algorithm, developed by Jiang et al, was employed to model different tumor immune evasion mechanisms.24 A higher TIDE score usually suggests a greater possibility of inducing immune escape, thus indicating a lower response rate to ICI treatment. IPS is a superior predictor of response to anti-CTLA4 and anti-PD1 regimens, quantifying the determinants of tumor immunogenicity and describing the intratumoral immune landscapes and cancer antigenomes.25 IMvigor210, an immunotherapy cohort, was adopted to explore the prediction of LMS for immunotherapy. We also acquired the GSE135222 cohort from GEO database for prediction of immunotherapy.
Drug Susceptibility Analysis
To quantify the comparative efficacy of chemotherapeutic drugs in the two groups, we calculated the semi-inhibitory concentration (IC50) values of chemotherapeutic drugs which were commonly used to treat LUAD using the “pRRophetic” package.
RNA Extracting and Real-Time PCR
The method is based on previous report.26 First, LUAD tissues and cells were lysed with Triddle reagent, in which RNA was extracted with trichloroacetic acid, isopropanol and absolute ethanol. The RNA was then reverse-transcribed using the reverse transcription machine. Evo M-MLVRT Master Mix kit (Accurate Biotechnology (Hunan) Co., Ltd China) were then used to synthesize cDNA. Finally, SYBR Premix Ex Tap kit (Accurate Biotechnology (Hunan)Co., Ltd China) were used to measure the expression of related genes on the qRT-PCR machine. The primers are listed in Table S1.
Cell Culture
Human A549 and PC9 cell lines were purchased from Procell Life Science & Technology Co., Ltd. A549 cells were cultured in F12K medium (Gibco) supplemented with 10% FBS and PC9 cells were cultured in RPMI 1640 medium (Gibco). These cell lines were cultured at 37 °C in a humidified incubator containing 5% CO2.
Proliferation Assay
First, A549 and PC9 cells were seeded in several 96-well plates with about 4000 cells per well. After the cells adhered to the wall, one of the 96-well plates was fixed with trichloroacetic acid and one plate was treated every 12 or 24 hours until the cells were all overgrown. Then all 96-well plates were washed with trichloroacetic acid, dried and stained with Sulforhodamine B sodium salt (SRB) (Sigma, USA). After the SRB was washed with acetic acid and dried, 150 µL of 10 mmol/L Tris was added to the 96-well plates, and the absorbance was measured with the microplate reader (Thermo Fisher, USA) at 562 nm. The method above is based on previous report.26
Transwell Assay
Migration chambers were placed in a 12-well plate. About 2.5% FBS F-12K or RPMI 1640 medium and 40,000 A549 or PC9 cells were added to the upper chambers. About 10% FBS medium was added to the lower chambers. After 48 hours, cells were fixed and stained with crystal violet dye for 20 minutes. After chambers were washed and dried, the migration of cells was observed under the microscope and photographed. We counted them with ImageJ software. The above steps are derived from previous literature report.26
Wound Healing Assay
This experiment drew on previous report.26 A549 and PC9 cells were seeded in a 12-well plate. After the cells grew to 100% density, a sharp instrument was used to draw four mutually perpendicular lines in each well to separate the cells and wells were photographed under the microscope as 0 hour. After waiting for 48 hours, we took pictures of the migration of each well, and calculated the area migrated between two time points with ImageJ software.
Statistical Analyses
All statistical analyzes were performed in R version 4.2.0 and GraphPad Prism8.0 software. Student’s t-test and Wilcoxon rank sum test were used to analyze the statistical significance between two groups, which test whether one of the two independent samples is significantly larger or smaller. Kruskal–Wallis test and one-way analysis were used to compare the statistical significance between more than two groups. Among them, student’s t-test and one-way analysis are suitable for normally distributed data, while Wilcoxon rank sum test and Kruskal–Wallis test are suitable for non-normally distributed variables. A P<0.05 was considered statistically significant.
Results
The Immune Landscape of Macrophages in LUAD
We began by analyzing the degree of macrophage infiltration in lung adenocarcinoma. The results showed that there was a significant difference in the degree of macrophage infiltration between normal tissues and tumor tissues (Figure S1A). Subsequently, through the introduction of the MI_VS_M2_UP gene set for GSVA analysis of normal tissues and tumor tissues, we found that the degree of M2 macrophage infiltration was higher than that of M1 macrophage infiltration in tumor tissues (Figure S1B). Furthermore, through Kaplan-Meier analysis, we found that patients with a higher degree of M2 macrophage infiltration tended to have poorer prognosis (Figure S1C). Therefore, we concluded that macrophages are closely related to the prognosis of lung adenocarcinoma, which is consistent with previous reports.27
Identification and Verification of Macrophage Subtypes of LUAD
To further explore the expression characteristics of macrophages in LUAD, we used a NMF algorithm to classify LUAD patients according to the expression levels of three kinds of macrophages (Figure 1A). Our results indicated k = 2 to be an optimal selection for sorting the entire cohort into subtypes A (n = 163) and B (n = 294). The Kaplan–Meier curves showed a longer OS in patients of subtype C2 than that in patients of subtype C1 in Figure 1B (Log rank test, p = 0.036). PCA analysis revealed significant differences in the expression of macrophages between the two subtypes (Figure 1C). Immune cell enrichment analysis also showed that there was a significant difference in the degree of macrophage infiltration between the two subtypes (Figure 1D). GSVA enrichment analysis shown that the C2 subtype was significantly enriched in various immune-related pathways, including antigen processing and presentation, primary immunodeficiency, and immune diseases (Figure 1E). So, we can see that the macrophage classification can well distinguish lung adenocarcinoma, and subtype C2 will survive better.
 |
Figure 1 The construction of macrophage-related LUAD subtypes in the TCGA cohort and clinicopathological and biological characteristics of two subtypes. (A) Two macrophage-related molecular subtypes were generated by NMF algorithm and heatmap defining two clusters and their correlation area. (B) Survival difference between patients in two distinct subtypes was compared using the Kaplan-Meier method. (C) A remarkable difference in transcriptomes between the two subtypes was shown by PCA analysis. (D) The fraction of tumor-infiltrating immune cells in two subtypes using the ssGSEA algorithm (E) GSVA of biological pathways between two distinct subtypes, in which red and blue represent activated and inhibited pathways, respectively. *P < 0.05; **P < 0.01; ***P < 0.001. Abeviations: LUAD, lung adenocarcinoma; TCGA, the cancer genome atlas database; NMF, non-negative matrix factorization; PCA, principal component analysis; ssGSEA, single sample Gene Set Enrichment Analysis; GSVA, gene set variation analysis. |
Construction of Gene Subtypes Based on DEGs
To explore the potential biological behavior of macrophage subtypes, we identified 1027 DEGs between two subtypes and performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis (Figures 2A and B and S1D and E). These genes were significantly enriched in biological processes correlated with immunity, especially antigen recognition and presentation (Figure 2A and B). KEGG analysis indicated the enrichment of DEGs in immune-related disease (Figure S1D and E). The results above suggested that macrophage subtypes have a strong correlation with immunity.
 |
Figure 2 The construction of LMS risk model and its prediction of OS. (A) The bubble diagram of the GO pathways enriched for 1027 DEGs between two macrophage subtypes. (B) The bar plot of the GO pathways enriched for 1027 DEGs between two macrophage subtypes. (C) The Kaplan-Meier survival analysis between high- and low-risk group. (D) ROC analysis for OS prediction of LMS signature including 1, 3, and 5 years. (E) TMB in two risk groups. (F) The risk score distribution, survival status distribution and survival heatmap of LMS signature. (G) Independent prognostic analysis by univariate Cox and multivariate Cox regression analysis. (H) Heatmap for the LMS signature based on clinicopathological manifestation. (I) Kaplan-Meier survival analysis between high- and low-risk group in different clinical subgroups. (J) Score of COATES_MACROPHAGE_M1_VS_M2 genesets between high- and low-risk group. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: LMS, LUAD macrophage-related signature; OS, overall survival; GO, gene ontology; DEG, differentially expressed genes; TMB, tumor mutation burden; ROC, receiver operating characteristic. |
Subsequently, we conducted univariate Cox regression analysis to DEGs and screened out 219 genes related to prognosis (p < 0.05). Applying LASSO regression analysis and multivariate Cox regression analysis to 219 genes, we screened out 12 key DEGs associated with LUAD prognosis (KSR1, MNDA, CDC42BPB, CC2D1A, ANGPTL4, PCBP4, CALCA, C1QTNF6, SHC1, CEBPB, CIB2, and TRIM28). Survival analysis was conducted on each of them (Figure S2), and each was found to be related to OS (P<0.05). Based on these 12 genes, we constructed a new prognostic signature, LUAD macrophage signature (LMS). The formula for constructing LMS is listed in the Methods. The synergetic coefficient of each gene is listed in Table S2.
Patients with LMS lower than the median risk score were categorized into the low-risk group (n = 229), whereas those with LMS greater than the median risk score were placed in the high-risk group (n = 228). The Kaplan–Meier curve predicted different OS between the high-risk and low-risk groups (Figure 2C). The areas under the ROC curve (AUCs) were 0.750, 0.747, and 0.72 for 1-, 3-, and 5-year survival times, respectively, demonstrating that this gene signature has good performance in predicting the survival of LUAD (Figure 2D). Furthermore, we found that TMB was higher in the high-risk group than in the low-risk group (Figure 2E). The distribution plot of the risk of LMS revealed that survival times decreased while recurrence rates increased with an increase in LMS (Figure 2F).
We then performed independent prognostic analysis of LMS and different clinical features. This analysis demonstrated that only LMS and tumor stage were independent prognostic factors (Figure 2G). The relationships between LMS and the distribution of clinicopathological parameters, including age (≤65 or >65 years), gender (male or female), tumor stage (I, II or III, IV) and TNM stage are shown in Figure 2H. We found obvious correlation between LMS and clinical stage and TNM stage of LUAD patients. Compared to the high-risk group, patients in the low-risk group had lower TNM stage and tumor stage. Considering that clinical stage itself has a certain impact on prognosis, we performed Kaplan-Meier analysis on the patients at different stages in the TCGA and GEO cohorts, respectively (Figures 2I and S3A–D). Results showed considerable difference in OS no matter early or late clinical stage, suggesting that LMS was truly independent of clinical stage. The degree of M1 vs M2 macrophage infiltration in two groups (Figure 2J) indicated that the low-risk group exhibited more M1 macrophage infiltration, whereas the high-risk group possessed more M2 macrophage infiltration. Finally, we also analyzed the relationship between 11 somatic gene mutations and LMS, considering the importance of oncogene mutations in LUAD. We found that the LMS scores of the KRAS and NTRK3 gene mutation groups were higher than those of the wild-type group (Figure S3E).
Verification by External Queues
We introduced four cohorts (GSE3141, GSE31210, GSE32019, and GSE50081) from the GEO as external validation cohorts to further assess the robustness and reliability of the prognostic signature (Figure 3A–D). We first classified patients in the GEO cohorts by the same median risk score we had used in LMS and then conducted Kaplan–Meier analysis and ROC curve. The survival outcome of the low-risk group was all around better (p < 0.001) compared to high-risk group. The AUCs were 0.91, 0.893, and 0.846 for 1-, 3-, and 5-year survival times respectively in GSE3141 cohort. Other cohorts also had good performance by AUC (Figure 3). These results demonstrate that this prognostic signature possesses good performance in predicting the survival of LUAD. Then we constructed risk curves and survival maps of the four cohorts. Within each cohort, survival times decreased while recurrence rates increased with an increase in LMS.
 |
Figure 3 Validation of LMS model by external cohorts. Kaplan-Meier survival analysis, ROC analysis including 1, 3, and 5 years, risk score distribution and survival status distribution in GEO cohorts (A) GSE3141 (B) GSE31210 (C) GSE32019 (D) GSE50081. Abbreviations: LMS, LUAD macrophage-related signature; ROC, receiver operating characteristic; GEO, gene expression omnibus database. |
Development of a Better Predictor Combined with Clinical Features
Considering the oneness of LMS in predicting OS in patients with LUAD, we established a nomogram incorporating the LMS and clinicopathological parameters to predict 1-, 3-, and 5-year OS rates (Figure 4A). Predictors included LMS, gender, age, and tumor stage. The subsequent calibration plots suggested that the proposed nomogram had close-to-ideal performance (Figure 4B). AUCs showed that the nomogram model had the highest accuracy, LMS came in second, and the accuracy of tumor stage was the lowest of these three (Figure 4C), suggesting that the nomogram exhibited superior survival predictive ability compared to the two, which was demonstrated in the decision curve analysis (Figure 4D). However, the nomogram without LMS showed relatively poor prediction performance, even worse than the predictive ability of one single clinical feature, suggesting the additional value of LMS to clinical parameters on prognosis (Figure S3F and G).
 |
Figure 4 Establishment and assessment of the nomogram for survival prediction. (A) The nomogram combining LMS risk score and other clinicopathological parameters was developed to predict 1-, 3-, and 5-year survival. (B) Calibration curves showing the predictions of the nomogram that we established for 1-, 3-, and 5-year overall survival. (C) ROC analysis for OS prediction of nomogram, LMS score, age, gender and tumor stage. (D) The decision curve analysis of the 1-year overall survival in the TCGA dataset. *P < 0.05; ***P < 0.001. Abbreviations: LMS, LUAD macrophage-related signature; ROC, receiver operating characteristic; OS, overall survival; TCGA, the cancer genome atlas database. |
LMS Was Significantly Related to Immunity
To investigate the correlation between our gene signature and immune cells, we assessed the association between LMS and the abundance of immune cells through MCPCOUNTER. As shown in Figure 5A, the LMS was positively correlated with fibroblasts and negatively correlated with myeloid dendritic cells, neutrophils, endothelial cells, and T cells. We then analyzed the two groups using the ESTIMATE algorithm, which confirmed that LMS was significantly correlated with the immune composition of the TME (Figure 5B).
 |
Figure 5 Evaluation of immune correlation of LMS score. (A) Correlations between twelve genes, LMS score and the abundance of immune cells in the LMS model. (B) Correlations between twelve genes, LMS model and both immune and stromal scores. (C) Enrichment plots from GSEA in the high-risk groups and low-risk groups according to LMS model. (D) Correlations between expression of KEGG signaling pathways and twelve genes in the LMS model. (E) The boxplot illustrating the difference in immune-related functions between the high and low-risk groups. (F) Kaplan-Meier survival analysis of three immune-related functions between two risk groups. (G) Correlations between twelve genes, LMS score and expression of immune checkpoints in the LMS model. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: LMS, LUAD macrophage-related signature; GSEA, gene set enrichment analysis; KEGG, Kyoto encyclopedia of genes and genomes. |
Subsequently, we utilized GSEA to investigate KEGG pathways enriched between the two groups. We found that the high-risk group was associated with cell cycle, DNA replication, the proteasome, and extracellular matrix receptor interaction, while the low-risk group was associated with allograft rejection, hematopoietic cell lineage, intestinal immune network for immunoglobulin A production, primary immunodeficiency, and systemic lupus erythematosus (Figure 5C). We also assessed the relationship between the twelve genes of LMS and the KEGG pathways and found that most of immune pathways were significantly correlated with the twelve genes (Figure 5D).
In addition, ssGSEA was used to analyze differences in immune-related functions between two groups (Figure 5E). We observed that various immune-related functions had significant difference in OS between two groups, including T cell co-inhibition, type II interferon response, and immune checkpoint (Figure 5F). We also investigated the associations between immune checkpoints and our signature (Figure 5G). Results showed that most immune checkpoints were differentially expressed in the two groups, including PD-L1 and CTLA4.
The Role of LMS in Predicting Immunotherapy
We wanted to investigate the ability of our signature to predict immunotherapy. Thus, we conducted immunotherapy analysis of anti-CTLA4 and anti-PD1 between two groups (Figure 6A–C). Results showed that, no matter what kind of treatment was utilized, the low-risk group fared better than the high-risk group. Furthermore, we evaluated the TIDE score between the two groups (Figure S1F), which indicated a significant difference between the high and low risk groups. In addition, through two immunotherapy cohorts, IMvigor210 and GSE135222, we found that better responses of immunotherapy both existed in low-risk groups (Figure 6D and E). Last, we analyzed chemotherapeutic drug sensitivity (Figure 6F). The high-risk group was sensitive to more than half of drugs assessed.
 |
Figure 6 Prediction of immunotherapy and chemotherapy of LMS model. (A) CTLA4+_PD1-, (B) CTLA4+_PD1+, and (C) CTLA4-_PD1+. The comparison of IPS between two risk group. (D and E) Immunotherapeutic response of IMvigor210 and GSE135222 cohorts between two risk groups. (F) Sensitivity analysis of chemotherapeutic drugs between two risk groups. **P < 0.01; ***P < 0.001. Abbreviations: LMS, LUAD macrophage-related signature; IPS, immunophenotype score. |
TRIM28 Affects the Biological Behaviors of LUAD Cells in vitro
We then figured out the mechanism of accurate prediction of LMS by evaluating 12 genes included in the signature. Combined with Kaplan–Meier analysis and differential expression analysis between normal and tumor tissues, we revealed that three genes, TRIM28, SHC1 and C1QTNF6, were significantly overexpressed in tumor tissues and negatively correlated to prognosis of LUAD patients (Figure 7A–C and Figure S4A–F). Enrichment analysis of KEGG pathway by GSEA suggested that only TRIM28 was enriched in the pathway related to malignant phenotype of tumor (Figures 7D and S4G and H). Pan-cancer analysis showed that TRIM28 was significantly different in most cancers and closely associated with prognosis in some cancers (Figures 7E and S4I–N). We realized that TRIM28 may play a promoting role in the occurrence and progression of LUAD and performed a series of experiments to elucidate it. We detected the expression of TRIM28 in 30 LUAD specimens and 11 normal specimens, and found that the expression of TRIM28 was significantly higher in the LUAD specimens compared to the normal specimens (Figure 7F). We further designed siRNA to knock out TRIM28 and transfected siRNA into A549 and PC9 cells. The knockout effect was detected using qRT-PCR (Figures 7G and S5A). We performed SRB assay to test cell proliferation, and the knockdown of TRIM28 significantly inhibited the proliferation of A549 and PC9 cells (Figures 7H and S5B). In addition, compared with NT group, TRIM28 knockout could reduce the rate of EDU proliferation positive cells in LUAD cells (Figures 7I and S5C). To investigate cell invasion, we performed transwell assay in cells with TRIM28 knockdown, and found that TRIM28 knockdown significantly decreased the invasion of A549 and PC9 cells (Figures 7J and S5D). The knockdown of TRIM28 also weakened the migration of A549 and PC9 cells in wound healing assay (Figures 7K and S5E). In short, knocking down TRIM28 significantly inhibited proliferation, migration and invasion of LUAD cells.
 |
Figure 7 TRIM28 affects the biological behaviors of LUAD cells in vitro. (A) Expression of TRIM28 in normal and tumor tissues of TCGA LUAD cohorts. (B) Paired difference analysis of TRIM28 in normal and tumor tissues of TCGA LUAD cohorts. (C) Kaplan-Meier survival analysis between low and high TRIM28 groups in LUAD cohorts. (D) KEGG pathway enrichment of GSEA analysis between low and high TRIM28 groups in LUAD cohorts. (E) Expression of TRIM28 in normal and tumor tissues of LUAD cohorts in 33 kinds of cancers. (F) The expression of TRIM28 in 30 tumor specimens and 11 normal specimens. (G) QRT-PCR was performed to detect the efficiency of TRIM28-siRNA transfection and expression of PD-L1 in A549 cells. (H) SRB assay showed growth curves of A549 cells treated with TRIM28. (I) EDU assay was used to detect the effect of TRIM28 downregulation on proliferation in A549 cell lines. (J) The invasion of A549 cells with treatment of TRIM28 knockdown was detected by transwell assay. (K) Wound healing assay was used to detect cell migration of TRIM28 knockdown in A549 cells. The data were expressed as means ± standard deviation (SD). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: LUAD, lung adenocarcinoma; TCGA, the cancer genome atlas database; KEGG, Kyoto encyclopedia of genes and genomes; GSEA, gene set enrichment analysis; QRT-PCR, real-time quantitative PCR; SRB, Sulforhodamine B; EDU, 5-ethynyl-2-deoxyuridine. |
Discussion
LUAD is a leading cause of cancer-related mortality worldwide.28 While pathological staging is currently the primary determinant of LUAD prognosis, patients with the same stage often exhibit varying clinical outcomes, highlighting the inadequacy of current predictive systems and the heterogeneity of the disease.29,30 Although significant advances have been made in lung cancer immunotherapy, a subset of LUAD patients remains resistant to these treatments, resulting in cancer recurrence.10,31 Therefore, we need methods that can accurately predict prognosis and immunotherapeutic response.
In our study, we developed a risk score based on the differentially expressed genes between M1 and M2 macrophage subtypes to predict these parameters in LUAD patients. We quantified the macrophages in the TCGA LUAD cohort and utilized an unsupervised NMF algorithm to divide this into two clusters. We observed that the prognosis of cluster 2 was significantly better than that of cluster 1. Cluster 2 was positively correlated with various immune pathways, while cluster 1 was positively correlated with multiple metabolic pathways. These evidences suggest that grouping tumors by their macrophage composition could effectively predict prognosis and therapeutic response.
However, when applied clinically, the quantification of macrophages is not a simple task. It is relatively easier to quantify genes. Therefore, in order to better predict prognosis, we identified 1027 DEGs between the two clusters. Enrichment analysis revealed that these genes are enriched in various aspects of antigen presentation pathways and are associated with a range of immune-related diseases. We used regression analysis to screen out 12 genes and used them to establish a risk score to quantify the prognostic risk to LUAD patients and provide prognostic biomarkers to predict responses to a variety of chemotherapies and immunotherapies. We termed this risk score the LUAD macrophage-related signature (LMS).
Our LMS model provides a new method for prognostic prediction in LUAD patients, as demonstrated by both Kaplan–Meier analysis and risk curves. Meanwhile, we found that LMS score of the KRAS mutation group was higher than that of the wild-type group. According to report, the activation of KRAS mutations can promote polarization of M1 to M2 macrophages.32 Combined with the result of more M2 macrophages in patients in the high-risk group, this may explain the poor prognosis of patients in the high-risk group and suggest a potential link between oncogene somatic mutations and LMS score.
Multivariate Cox regression analysis, which took patient age, gender, and tumor stage into account, confirmed the robustness of the model as an independent prognostic biomarker for evaluating patient outcomes. Additionally, more M1 macrophages in the low-risk group and more M2 macrophages in the high-risk group indicated the rationality of signature. Our model was robustly validated in four independent datasets. Time-dependent AUC showed that the signature had high accuracy in predicting OS in four GEO datasets. We also built a nomogram showing perfect consistency between the observed and predicted rates for 1-, 3-, and 5-year OS. Finally, the observed prognostic rates with 1-, 3-, and 5-year operating conditions show excellent agreement. Hence, our LMS model was quite accurate.
The immune microenvironment of tumors plays a crucial role in the development and progression of various types of cancers. We speculate that the accurate prediction of the prognosis of patients with lung adenocarcinoma by LMS model may be related to the tumor microenvironment. Through the correlation analysis of various immune cells in the microenvironment, we found that the LMS score was positively correlated with the degree of fibroblast infiltration, and negatively correlated with the infiltration of myeloid dendritic cells, endothelial cells, neutrophils and T cells. Reports indicate that cancer-related fibroblasts have been shown to promote tumor progression by various means, including the enhancement of malignant cells and fibrosis within the interstitial TME.33 In contrast, dendritic cells act as antigen-presenting cells and play a crucial role in initiating and controlling the immune response, serving as a bridge between the innate and acquired immune systems.34 Endothelial cells also play an active role in innate and acquired immune responses, performing many functions, such as cytokine secretion, antigen presentation, and anti-inflammation.35 This is consistent with our speculation. In addition, we found that immune pathways were more activated in low-risk patients. Activation of the immune pathway can enhance immune surveillance and immune defense, thereby improving the survival status of patients. To some extent, these results explain why the gene signature has a better prediction effect.
In clinical, ICIs that target programmed cell death 1 (PD-1) and PD-L1 have shown potent and durable anti-tumor activity in patients with LUAD.36 However, the overall response rate of ICI treatment is relatively low, and only a subset of LUAD patients can benefit from it.37 To date, a series of biomarkers, such as TMB, PD-L1 expression level and neoantigens, have been verified to be predictive of the efficacy of ICI treatment.38 However, the prediction of these indicators cannot achieve good results. So, we want to explore the role of LMS model in predicting immunotherapy. Because the information on ICI treatment was not available in the TCGA LUAD dataset, we used IPS as a surrogate for ICI treatment efficacy. The development of IPS was primarily based on TCGA RNAseq profiles, which enables the quantitative prediction of patient responses to anti-PD-1/PD-L1 and anti-CTLA4 therapies.25 We found that whether anti-PD1 or anti-CTLA4 therapy was used alone or in combination, the IPS values were significantly higher in the low-risk group than another group, which indicated that our signature might be useful for patient selection before ICI treatment. In addition, we introduced the IMvigor210 and GSE135222 cohort of patients with LUAD to test the predictive value of the signature for immunotherapeutic effect. Patients receiving anti-PD-L1 immunotherapy in the IMvigor210 cohort were assigned high or low ICI scores. Our observations showed that the low-risk LMS group had a higher proportion of patients achieving complete or partial remission after immunotherapy. We observed the same result in another cohort.
An increasing number of studies have employed the TIDE prediction score, which is a validated computational framework developed for predicting immunotherapeutic outcomes.24,39 In our study, the prediction of the TIDE algorithm suggested that responses to immunotherapy varied widely between the high- and low-risk group, thereby corroborating the validity of our signature. Additional analysis of drug sensitivity revealed that the gene signature could be helpful in chemotherapeutic applications. Most drugs responded differently between the low and high-risk group. Some drugs, such as gemcitabine, cisplatin, docetaxel, paclitaxel, and erlotinib, responded better in the high-risk group than the low-risk group. Thus, the LMS model may provide clinicians with a tool by which to suggest appropriate chemotherapies to patients for whom ICI is unlikely to be effective.
When further exploring internal mechanism of LMS score, we discovered a significant difference of TRIM28 expression between normal and tumor tissues of LUAD and higher expression of TRIM28 predicted a worse prognosis. As a member of a family of tripartite motif-containing (TRIM) proteins, TRIM28 has been reported to join in various aspects of cell biology to promote cell proliferation and regulate the malignant progression of various tumors through different signal pathways.40 In hepatocellular carcinoma, TRIM28 interacts with UBE2S in nucleus to accelerate the cell cycle through the ubiquitination of p27, thus promoting the development of cancer.41 In breast cancer, TRIM28 can accelerate cancer metastasis by stabilizing TWIST1 protein.42 In addition, TRIM28 facilitates the growth of cervical cancer via mTOR signal pathway.43 However, there are few studies on the role of TRIM28 in LUAD. In this study, we found that TRIM28 was closely associated with malignant progression of LUAD, promoting proliferation, invasion and migration of LUAD cells. GSEA suggested that TRIM28 is enriched into signal pathways such as cell cycle, DNA replication and nucleotide damage repair in LUAD, which may explain phenomena above. Interestingly, down-regulation of TRIM28 gene transcriptional levels in A549 cells leaded to decreasing transcriptional level of PD-L1. It reminds us of the possibility that TRIM28 upregulates expression of PD-L1 in tumor cells to promote immune escape and thus the malignant progression of tumor. Our research did not explore the mechanism further, which will be solved in future research.
Our study still has some limitations. First, we only used datasets from the online databases TCGA and GEO for the analysis. More patient data from different regions are needed for proper validation. Furthermore, public databases and samples used in our study were obtained retrospectively, which may contain some bias. Some data quality problems, such as technical noise, batch effects, etc., may exist. At the same time, in spite of large samples in these databases, differences between individuals are inevitable. These may have an impact on the universality and causing the prediction results to deviate from reality. Hence, large-scale prospective studies and additional in vivo and in vitro experimental studies are needed to confirm our findings.
Conclusions
We identified macrophage-related genes in LUAD patients. Furthermore, we established and validated the LMS model to predict the OS of LUAD patients, and it showed good predictive ability. We also assessed the differences in immunotherapeutic response and chemotherapeutic drug sensitivity between LMS risk groups. Finally, TRIM28, a key gene in the siganture was shown to promote the malignant progression of LUAD cells (Figure 8). The above results may help to advance our understanding of the features of macrophage infiltration and offer the possibility of translating gene signatures into clinical practice. In the future, genetic testing can be performed on clinical subjects and the results of LMS scores can be calculated to further stratify risk and provide personalized recommendations for different risk groups. Anyway, we still need to integrate various clinical and environmental factors, verify the effect of the score through in vivo experiments, develop targeted drugs, and conduct clinical trials to truly apply our findings to patient care.
 |
Figure 8 Article framework and workflow. Abbreviations: TCGA, the cancer genome atlas database; LUAD, lung adenocarcinoma; NMF, non-negative matrix factorization; GEO, gene expression omnibus database; DEG, differentially expressed genes; LASSO, least absolute shrinkage and selection operator; OS, overall survival; LMS, LUAD macrophage-related signature. |
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The present study was supported by Shandong Province Natural Science Foundation (grant no. ZR2021MH192 and ZR2020QH215).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi:10.3322/caac.21660
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
3. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi:10.3322/caac.21262
4. Testa U, Castelli G, Pelosi E. Lung cancers: molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers. 2018;10:1.
5. Little AG, Gay EG, Gaspar LE, Stewart AK. National survey of non-small cell lung cancer in the United States: epidemiology, pathology and patterns of care. Lung Cancer. 2007;57:253–260. doi:10.1016/j.lungcan.2007.03.012
6. Chang JT, Lee YM, Huang RS. The impact of the cancer genome atlas on lung cancer. Transl Res. 2015;166:568–585. doi:10.1016/j.trsl.2015.08.001
7. Travis, William D, Brambilla, Elisabeth, Noguchi, Masayuki et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–85.
8. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi:10.1056/NEJMoa1200690
9. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–454. doi:10.1038/nature25183
10. Zhang Y, Du H, Li Y, Yuan Y, Chen B, Sun S. Elevated TRIM23 expression predicts cisplatin resistance in lung adenocarcinoma. Cancer Sci. 2020;111:637–646. doi:10.1111/cas.14226
11. He D, Wang D, Lu P, et al. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene. 2021;40:355–368. doi:10.1038/s41388-020-01528-0
12. Zhao J, Guo C, Ma Z, Liu H, Yang C, Li S. Identification of a novel gene expression signature associated with overall survival in patients with lung adenocarcinoma: a comprehensive analysis based on TCGA and GEO databases. Lung Cancer. 2020;149:90–96. doi:10.1016/j.lungcan.2020.09.014
13. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi:10.1038/s41591-018-0014-x
14. Choi H, Na KJ. Integrative analysis of imaging and transcriptomic data of the immune landscape associated with tumor metabolism in lung adenocarcinoma: clinical and prognostic implications. Theranostics. 2018;8(7):1956–1965. doi:10.7150/thno.23767
15. Giraldo NA, Becht E, Vano Y, et al. Tumor-infiltrating and peripheral blood T cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin Cancer Res. 2017;23:4416–4428. doi:10.1158/1078-0432.CCR-16-2848
16. Heymann F, Tacke F. Immunology in the liver--from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110. doi:10.1038/nrgastro.2015.200
17. Wang T, Dai L, Shen S, et al. Comprehensive molecular analyses of a macrophage-related gene signature with regard to prognosis, immune features, and biomarkers for immunotherapy in hepatocellular carcinoma based on WGCNA and the LASSO algorithm. Front Immunol. 2022;13:843408. doi:10.3389/fimmu.2022.843408
18. Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79:541–566. doi:10.1146/annurev-physiol-022516-034339
19. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154:186–195. doi:10.1111/imm.12910
20. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. doi:10.1038/ni.1937
21. Wildes TJ, Dyson KA, Francis C, et al. Immune escape after adoptive T-cell therapy for malignant gliomas. Clin Cancer Res. 2020;26(21):5689–5700. doi:10.1158/1078-0432.CCR-20-1065
22. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi:10.1016/j.cell.2010.03.014
23. Coates PJ, Rundle JK, Lorimore SA, Wright EG. Indirect macrophage responses to ionizing radiation: implications for genotype-dependent bystander signaling. Cancer Res. 2008;68(2):450–456. doi:10.1158/0008-5472.CAN-07-3050
24. Jiang P, Gu S, Pan D, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–1558. doi:10.1038/s41591-018-0136-1
25. Charoentong P, Finotello F, Angelova M, et al. Pan-cancer immunogenomic analyses reveals genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2016;18:248–262. doi:10.1016/j.celrep.2016.12.019
26. Liu J, Shen H, Gu W, et al. Prediction of prognosis, immunogenicity and efficacy of immunotherapy based on glutamine metabolism in lung adenocarcinoma. Front Immunol. 2022;13:960738. doi:10.3389/fimmu.2022.960738
27. Chen J, Zhang K, Zhi Y, et al. Tumor-derived exosomal miR-19b-3p facilitates M2 macrophage polarization and exosomal LINC00273 secretion to promote lung adenocarcinoma metastasis via Hippo pathway. Clin Transl Med. 2021;11(9):e478. doi:10.1002/ctm2.478
28. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
29. Jurisic V, Obradovic J, Pavlovic S, Djordjevic N. Epidermal growth factor receptor gene in non-small-cell lung cancer: the importance of promoter polymorphism investigation. Anal Cell Pathol. 2018;2018:6192187. doi:10.1155/2018/6192187
30. Rafei H, El-Bahesh E, Finianos A, Nassereddine S, Tabbara I. Immune-based therapies for non-small cell lung cancer. Anticancer Res. 2017;37:377–387. doi:10.21873/anticanres.11330
31. Li F, Huang Q, Luster TA, et al. In vivo epigenetic CRISPR screen identifies asf1a as an immunotherapeutic target in kras-mutant lung adenocarcinoma. Cancer Discov. 2020;10:270–287. doi:10.1158/2159-8290.CD-19-0780
32. Salman RP, Vamsidhar V, Benjamin GN, Kwok-Kin W. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. 2022;19:637–655. doi:10.1038/s41571-022-00671-9
33. Boesch M, Baty F, Rumpold H, Sopper S, Wolf D, Brutsche MH. Fibroblasts in cancer: defining target structures for therapeutic intervention. Biochim Biophys Acta Rev Cancer. 2019;1872:111–121. doi:10.1016/j.bbcan.2019.06.003
34. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154:3–20. doi:10.1111/imm.12888
35. Shao Y, Saredy J, Yang WY, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol. 2020;40:e138–e152. doi:10.1161/ATVBAHA.120.314330
36. Forde PM, Chaft JE, Smith KN, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–1986. doi:10.1056/NEJMoa1716078
37. Li X, Shao C, Shi Y, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol Oncol. 2018;11:31. doi:10.1186/s13045-018-0578-4
38. Yi M, Jiao D, Xu H, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129. doi:10.1186/s12943-018-0864-3
39. Xu F, Huang X, Li Y, Chen Y, Lin L. m(6)A-related lncRNAs are potential biomarkers for predicting prognoses and immune responses in patients with LUAD. Mol Ther Nucleic Acids. 2021;24:780–791. doi:10.1016/j.omtn.2021.04.003
40. Czerwinska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. J Biomed Sci. 2017;24:63. doi:10.1186/s12929-017-0374-4
41. Zhang RY, Liu ZK, Wei D, et al. UBE2S interacting with TRIM28 in the nucleus accelerates cell cycle by ubiquitination of p27 to promote hepatocellular carcinoma development. Signal Transduct Target Ther. 2021;6:64. doi:10.1038/s41392-020-00432-z
42. Wei C, Cheng J, Zhou B, et al. Tripartite motif containing 28 (TRIM28) promotes breast cancer metastasis by stabilizing TWIST1 protein. Sci Rep. 2016;6:29822. doi:10.1038/srep29822
43. Li F, Wang Z, Lu G. TRIM28 promotes cervical cancer growth through the mTOR signaling pathway. Oncol Rep. 2018;39:1860–1866. doi:10.3892/or.2018.6235
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Platelet-Related Molecular Subtype to Predict Prognosis in Hepatocellular Carcinoma
Zhang G
Journal of Hepatocellular Carcinoma 2022, 9:423-436
Published Date: 19 May 2022
Molecular Characteristics and the Effect of KRAS Mutation on the Prognosis of Immunotherapy in Non-Small Cell Lung Cancer in Xinjiang, China
Gu G, Yu B, Wan H, Lu S, Zhu X, Zhao Y, Fuxi Y, Liu C
OncoTargets and Therapy 2022, 15:1021-1032
Published Date: 21 September 2022
Identification and Validation of Ferroptosis-Related Subtypes and a Predictive Signature in Hepatocellular Carcinoma
Zheng C, Peng Y, Wang H, Wang Y, Liu L, Zhao Q
Pharmacogenomics and Personalized Medicine 2023, 16:39-58
Published Date: 26 January 2023
Development and Validation of a Novel Nomogram Integrated with Hypoxic and Lactate Metabolic Characteristics for Prognosis Prediction in Hepatocellular Carcinoma
Qiu X, Dong L, Wang K, Zhong X, Xu H, Xu S, Guo H, Wei X, Chen W, Xu X
Journal of Hepatocellular Carcinoma 2024, 11:241-255
Published Date: 2 February 2024
Diagnosis, Prognosis, and Treatment of Triple-Negative Breast Cancer: A Review
Jie H, Ma W, Huang C
Breast Cancer: Targets and Therapy 2025, 17:265-274
Published Date: 17 March 2025