")
Back to Journals » Vascular Health and Risk Management » Volume 10
Macitentan for the treatment of pulmonary arterial hypertension
Authors Kholdani C, Fares W, Trow T
Received 1 August 2014
Accepted for publication 23 September 2014
Published 25 November 2014 Volume 2014:10 Pages 665—673
DOI https://doi.org/10.2147/VHRM.S33904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Daniel Duprez
Cyrus A Kholdani,1 Wassim H Fares,2 Terence K Trow2
1Section of Pulmonary, Critical Care and Sleep Medicine, 2Yale Pulmonary Vascular Disease Program, Section of Pulmonary, Critical Care and Sleep Medicine, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT, USA
Abstract: Macitentan is the most recently approved dual endothelin-receptor antagonist (ERA) for the treatment of symptomatic pulmonary arterial hypertension. Compared to other available ERAs, it demonstrates superior receptor-binding properties, with consequently improved tissue penetration, and a longer duration of action allowing for once-daily dosing. It has a favorable adverse-effect profile, with notably no demonstrable increase in the risk of hepatotoxicity or peripheral edema, but like other ERAs, it is potentially limited by significant anemia. Phase I data have demonstrated a favorable drug–drug interaction profile and no need for dose adjustment with hepatic and renal impairment. In the pivotal SERAPHIN study, treatment of symptomatic pulmonary arterial hypertension patients with macitentan led to statistically significant improvements in functional class, exercise tolerance, and hemodynamic parameters, in addition to a reduction in morbidity in an event-driven long-term trial.
Keywords: endothelin, endothelin receptor antagonists, macitentan, pulmonary arterial hypertension
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a progressive and lethal disease characterized by remodeling of the pulmonary arterioles with consequent increase of the pulmonary vascular resistance (PVR) with eventual right ventricular failure and ultimately death. Furthermore, despite targeted treatments, the disease remains fatal.1 The natural history of PAH and the survival rates for patients suffering from this disease were initially understood through analysis of the first registry of patients through the National Institutes of Health, with a reported median survival with supportive care of 2.8 years after diagnosis.2 Subsequent to this, several other registries have been established around the world that have demonstrated improved survival in PAH in the era of PAH-specific therapies. More recently an analysis of a cohort of patients enrolled in the REVEAL (Registry to EValuate EArly and Long-term PAH disease management) trial between March 2006 and December 2009 at 55 different centers within the US demonstrated 1-, 3-, 5-, and 7-year survival rates of 85%, 68%, 57%, and 49%, respectively.1
The endothelins and the endothelin receptors
The endothelins (ETs) are composed of three isoforms – ET-1, ET-2, and ET-3 – with the first considered to be the most clinically significant and physiologically active of the compounds. ET-1, a 21-amino acid peptide generated by the vascular endothelium, is a smooth-muscle mitogen, and is among the most potent and longest-acting vasoactive compounds.3 Its release from endothelial cells occurs through both constitutive and inducible pathways.4,5 While its production can be stimulated by hypoxia, shear stress, and thrombin, its biosynthesis can be inhibited by nitric oxide (NO).6 Based on rat models, the lungs are believed to be the most significant source of ET.7 ET is released as an inactive precursor molecule that is ultimately acted upon by the ET-converting enzyme (ECE) within the lung to generate its active form.8
There are two ET receptors, ETRA and ETRB, which are distributed among nearly all cell types, with the notable exception of the absence of ETRA on endothelial cells.9 These receptors are G-protein coupled, 7-transmembrane domain receptors. The binding of ET-1 to the ETRA results in the activation of phospholipase C, with a consequent intracellular rise in inositol triphosphate and ionized calcium and subsequent vasoconstriction.10 Conversely, it has been shown that binding of ET-1 to ETRB results in the release of both NO and prostacyclin, as well as inhibition of apoptosis.11,12 The antagonism of the ETRB results in a reduction of NO-mediated vasodilation, and deficiency of the receptor results in a hypertensive phenotype in murine models.13,14 In rat models, both the ETRA and ETRB contribute to ET-1-mediated pulmonary arterial vasoconstriction and dual blockade resulted in maximal reduction of sensitivity to ET-1.15 The biology of ETR function is highly complex and remains incompletely understood, with evidence of significant cross talk between the two receptors.16,17
Targeting the endothelin pathway
With the accumulation of evidence that ET-1 has a role to play in the pathogenesis of PAH, multiple therapies targeting this axis were researched.18 Despite aberrant levels in a multitude of cardiovascular diseases, the ET-1-axis has only been successfully targeted in the treatment of PAH.19 Trials of treatments for patients with World Health Organization (WHO) functional class III and IV PAH with the dual-receptor antagonist bosentan were encouraging, with significant increases in 6-minute walk distance (6MWD) and improvement in both pulmonary hemodynamics and functional class.20,21 Improvement in exercise capacity and hemodynamic parameters were also noted in subsequent trials evaluating the efficacy of the ETRA-specific antagonist sitaxsentan and the ETRA-specific ambrisentan.22,23 During the evaluation of bosentan for the treatment of PAH, however, there was evidence of significant elevations in aminotransferase levels, with approximately 14% of patients showing elevations three times the upper limit of normal (ULN). This adverse event was also prominent in the evaluation of sitaxsentan, and in one instance resulted in death from liver failure.24 Sitaxsentan has subsequently been removed from the market.
Macitentan, a novel ERA: metabolism and drug interactions
Macitentan is a dual ET-receptor antagonist (ERA) developed in the setting of extensive efforts to maximize blockade of the ET axis while generating compounds with improved adverse-effect profiles when compared to prior compounds in the class. Unlike bosentan and sitaxsentan, macitentan is not characterized by a sulfonamide structure, but belongs to the sulfamide class of compounds.25 The presence of a bromine atom in the parent compound of macitentan allowed for straightforward determination of its metabolites.26 Macitentan is metabolized via oxidative depropylation into ACT-132577 and via oxidative cleavage into ACT-373898 (Figure 1).27 Of the two aforementioned metabolites, only the former, ACT-132577, has been shown to be metabolically active, and like its parent compound, also demonstrates dual-receptor antagonist properties, albeit with less potency.25,28 The metabolites are the result of a reaction catalyzed by the cytochrome P450 (CYP), system with enzyme kinetic data showing ACT-132577 primarily formed by CYP3A4 with negligible contribution from CYP2C19.29 Coadministration of macitentan with the maximum approved dose of ketoconazole, one of the prototypical inhibitors of CYP3A4, resulted in an increase in the concentration of the parent drug, with an area under the curve comparable to the 30 mg dose of macitentan administered during its Phase I evaluation.29 Nonetheless, it is recommended by the manufacturers that its use be avoided with strong inhibitors of CYP3A4.
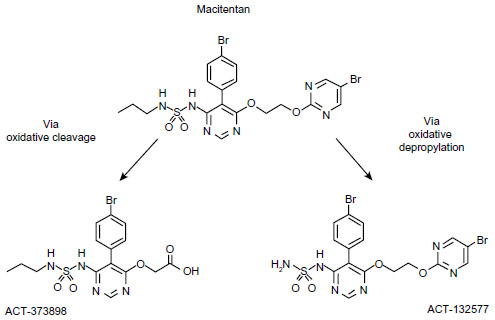 | Figure 1 Macitentan and its two metabolites. |
At a cellular level, macitentan demonstrates slower dissociation kinetics compared to all other available ERAs, in addition to insurmountable antagonism in functional pulmonary artery smooth-muscle assays.30 There are multiple lines of evidence showing that heterodimerization of the ETRA and ETRB receptors is necessary for activation, as with other G-coupled proteins, and upon binding macitentan interferes with this heterodimerization, blocking both receptor pathways (Figure 2).17 It has been demonstrated that macitentan’s functional inhibition constant (KB) ratio of 6.3 is higher than that of both bosentan and ambrisentan (0.7 and 1.0, respectively), indicating significantly more potent antagonism (Table 1).30 Both macitentan and its metabolites are highly bound to plasma proteins (>99%), and are primarily excreted via the hepatic and renal systems (combined hepatic and renal excretion of 49.7%±3.9%), with fecal elimination representing 23.9%±4.8% of total excretion.27,31 Pharmacokinetic evaluation of the 10 mg dose of macitentan has been shown to be safely tolerated in cirrhosis patients with Child–Pugh classes A, B, and C, with the expected caveat, given its metabolism, of decreased exposure to ACT-132577 in patients with hepatic impairment.32 Severe renal impairment, defined as a creatinine clearance of 15–29 mL/min, did not have any bearing on the safety profile of macitentan either.32
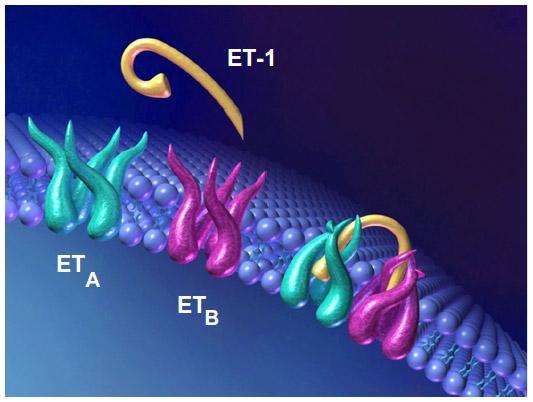 | Figure 2 Heterodimerization of G-coupled protein ERAA and ERAB receptors required to initiate downstream second messaging. |
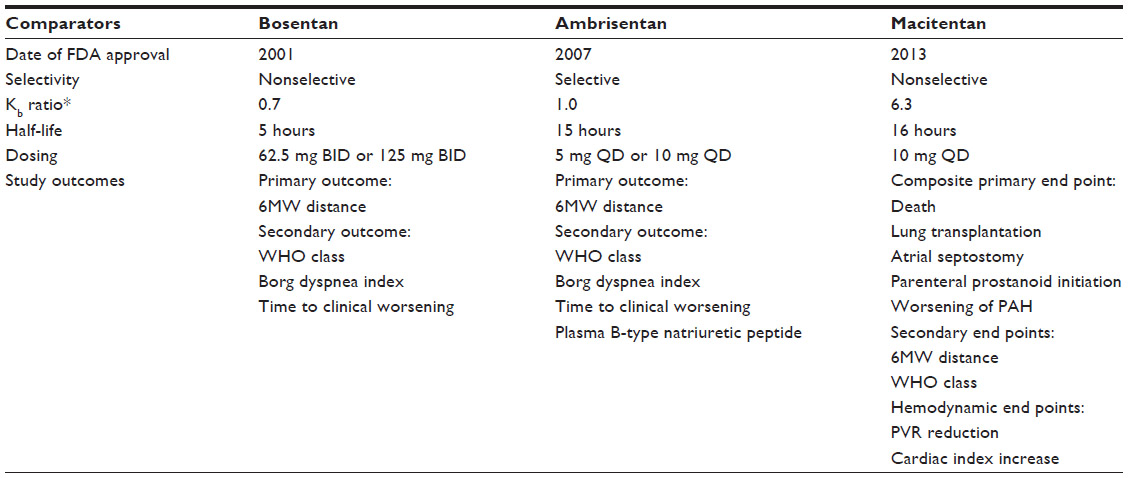 | Table 1 Comparative features for FDA-approved ERAs |
Evaluation of macitentan’s pharmacokinetics has been extensively studied with single doses ranging from 0.2 mg to 600 mg tested in healthy subjects. Macitentan is absorbed slowly, with a time to reach maximal concentration ranging from 8 hours to 30 hours.26 Reliable assessment of terminal half-life has been achieved with the higher non-US Food and Drug Administration (FDA)-approved doses, and is approximately 16 hours. The half-life of its main metabolites ranges from 40.2 hours to 65.6 hours. The plasma concentration of ET-1 demonstrated a dose-dependent rise, but was only noted to be statistically significant with doses of 25 mg and higher, which are higher than any dose used in trials evaluating macitentan’s clinical efficacy.33 The medication appears not to have proarrhythmic potential, as demonstrated in one randomized, double-blind, crossover, controlled study of 64 healthy subjects.33
Macitentan has demonstrated a favorable profile of drug interactions. With respect to combination therapy, a recent study of 12 healthy subjects demonstrated that the pharmacokinetics of macitentan were unaffected by concomitant sildenafil use, although there was a slight decrease in the concentration of ACT-132577.34 Cyclosporine, considered a weaker inhibitor of CYP3A4 than ketoconazole, has also been coadministered with macitentan with no clinically significant changes in the pharmacokinetic profile of the drugs.35 Rifampin, which is an inducer of CYP3A4, has also been coadministered with macitentan with preserved steady-state exposure to ACT-132577; however, this coadministration resulted in a nearly fourfold reduction in levels of macitentan.35 As such, the manufacturers advise against coadministration with strong CYP3A4 inducers, such as rifampin. In a randomized, open-label, crossover study assessing the effects of multiple doses of macitentan on the pharmacokinetics and pharmacodynamics of a single 25 mg dose of warfarin, there was no change in either the INR or factor VII activity when compared to controls.36 As with other ERAs, macitentan has the potential for teratogenicity, and it is therefore contraindicated in pregnancy. The manufacturers advise the use of two forms of birth control, one of which should be a barrier method, for women with PAH taking macitentan. It has not been studied in patients with pediatric pulmonary hypertension and is not approved for use in this population.
ERAs and significant adverse events
Hepatotoxicity
The clinical trial evaluating the efficacy of bosentan in PAH21 was marred by the presence of a significant amount of hepatotoxicity, with several patients developing an asymptomatic rise in serum aminotransferases that resolved with the cessation of the therapy. While rises in serum aminotransferases signal the presence of potential hepatotoxicity, they provide no insight into the mechanism. None of the patients in the study underwent a liver biopsy to help clarify the mechanism. Of the numerous potential mechanisms for bosentan-induced hepatotoxicity, the possibility of cholestatic or “mixed hepatitis” was explored by Fattinger et al, in part due to the fact that bosentan and its metabolites are primarily excreted in the bile.37 Several medications, including the insulin secretagogue glyburide, have been previously shown to result in cholestatic liver injury via inhibition of the hepatocyte canalicular bile salt-export pump (BSEP).38 In this scenario, the intracellular accumulation of cytotoxic cholephilic compounds, or bile salts, results in hepatocyte damage. Intracellular accumulation of bile salts has been previously established as a cause of hepatotoxicity via interference with normal mitochondrial function.39 The BSEP mediates the adenosine triphosphate-dependent secretion of bile salts across the canalicular membrane of hepatocytes. Mutations resulting in a defective BSEP at the canalicular membrane are a known cause of progressive familial intrahepatic cholestasis.40
The data from the initial Phase II study of bosentan for essential systemic hypertension were suggestive of a dose-dependent hepatotoxicity.41 Although there was no significant difference in serum bilirubin levels between dosage groups, the level of serum bile salts was higher among those patients receiving the higher doses. Administration of bosentan results in a dose-dependent increase in plasma bile-salt concentration in a rat model, and the degree to which it occurs is exacerbated by the concurrent administration of glyburide.37 Administration of bosentan along with its three known metabolites results in inhibition of adenosine triphosphate-dependent taurocholate transport based on data from uptake experiments performed on rat liver canalicular plasma-membrane vesicles and BSEP-expressing cells. The conclusion based on these data is that the hepatotoxic profile of bosentan and its metabolites results from inhibition of the canalicular BSEP. Whereas intravenous administration of bosentan resulted in an increase in plasma bile salts in rats, this effect was not demonstrated in the case of macitentan.26 The withdrawal of sitaxsentan from the market secondary to its hepatotoxicity is an example of the ongoing concern about this class of drugs and their adverse side-effect profile. An ongoing evaluation of the long-term hepatic safety profile of macitentan was mandated by the FDA42 (Table 2).
 | Table 2 Adverse-effect profile for FDA-approved ERAs |
Peripheral edema
Peripheral edema is also a notable side effect of the ERA class, and the mechanism is also unknown. The observation that peripheral edema is less common in patients treated with dual-receptor antagonists, such as bosentan, as opposed to ETRA-specific medications, such as ambrisentan, supports the hypothesis that the effect is mediated through circulating ET-1 and its activation of the ETRB (Table 2).17,43
Macitentan in animal models
Macitentan’s effect on hemodynamics was assessed using the monocrotaline model of pulmonary hypertension in rats.44 Four weeks of oral administration of macitentan at a maximally efficacious dose of 30 mg/kg/day prevented the development of both PAH and right ventricular hypertrophy. Rat models of both systemic and pulmonary hypertension were also used to compare the potency of blockade by macitentan when compared to bosentan.44 In this rat model of PAH, the addition of macitentan to a regimen of bosentan caused a reduction in mean pulmonary arterial pressure by 4 mmHg, whereas the addition of bosentan to a background regimen of macitentan resulted in no change to mean pulmonary arterial pressure (P<0.01). This supports a more potent and comprehensive blockade of the receptors by macitentan.
Human trials of macitentan in PAH
The Phase I trials evaluating macitentan in humans consisted of drug–drug interaction trials and pharmacokinetic studies discussed earlier.29,32,33,35,36 The Phase II trial was an unpublished study evaluating the dosing range for macitentan in patients with essential systemic hypertension. In this multicenter, double-blind, placebo-controlled study of 466 patients, a total of 337 were deemed evaluable at the end of the two-phased protocol. After an initial 2- to 4-week period of placebo treatment, patients were randomized to one of four doses of macitentan (0.3, 1, 3, or 10 mg daily), enalapril 20 mg, or placebo. After 8 weeks of treatment, macitentan 10 mg was associated with a reduction in systemic diastolic blood pressure when compared to placebo, with a similar overall frequency of adverse events. There were no cases of peripheral edema associated with macitentan, and most instances of a rise in aminotransferases to a value >3× ULN were attributed to other, more compelling causes. Of note, there was evidence of a modest decrease in mean hemoglobin level, which was deemed to be possibly dose-related.45
The findings of the Phase III double-blind, randomized, controlled SERAPHIN (Study with an ET Receptor Antagonist in Pulmonary arterial Hypertension to Improve cliNical outcome) trial published in 2013 revealed clinically significant findings using a novel composite primary end point.46 A total of 742 patients were enrolled to receive placebo therapy or either the 3 mg or 10 mg dose of macitentan. While no patient who enrolled was on an ERA, the use of other background PAH-specific therapy, including oral or inhaled (but not infused) prostanoid therapy was allowed. Previous trials exploring the efficacy of PAH-specific therapies used short-term changes in exercise capacity as a primary end point. In light of more recent guidelines arguing for the use of morbidity and mortality as the primary end point for Phase III trials in PAH, the SERAPHIN investigators designed their trial to that end.47 The SERAPHIN investigators used an “event-driven” composite primary end point looking at the time to worsening of PAH, the need for initiation of either intravenous or subcutaneous prostanoid therapy, lung transplantation, atrial septostomy, or death. Worsening of PAH was defined as a decrease of 15% 6MWD from baseline, the need for use of an additional PAH agent, and worsening symptoms of PAH manifested as either a worsening of functional class or clinical right heart failure that was not responsive to oral diuretic therapy. Of the 14 enrolled patients with WHO functional class IV symptoms, a lack of improvement in functional class was considered synonymous with clinical worsening for purposes of the analysis. The mean durations of treatment were 85.3 weeks, 99.5 weeks, and 103.9 weeks for the placebo, 3 mg, and 10 mg groups, respectively. A total of 742 patients were enrolled, with 287 patients having a primary end-point event after a median period of 115 weeks. Within the placebo group, 116 patients (46.4%) met the primary end point, whereas 95 patients (38.0%) and 76 patients (31.4%) met the end point in the 3 mg and 10 mg treatment groups, respectively. The most frequent primary end point was worsening of PAH, which occurred in 224 patients. Data from the 94 patients who did not continue with therapy without having a primary end point were censored from the analysis. While this represents a limitation of the study, this did not appear to impact significantly on the conclusions of the study.
The SERAPHIN investigators prespecified a change in baseline 6MWD at month 6, an improvement in WHO functional class at month 6, and hospitalization for a PAH-related reason or any occurrence of death during and up to the end of the study as secondary end points. With respect to exercise capacity, the mean decrease in 6MWD at 6 months in the placebo group was 9.4 meters, whereas there was a mean increase of 7.4 meters and 12.5 meters in the 3 mg and 10 mg treatment groups, respectively. Improvement in functional class occurred in 13% of patients in the placebo group by month 6 compared to the statistically significant 20% and 22% improvements, respectively, in the 3 mg and 10 mg treatment groups. It is worth noting that among the patients enrolled in the placebo group, 154 of the 250 were already receiving background therapy for their PAH, which likely contributed to their clinical improvement during this interval of the study. In fact, approximately two-thirds of all enrolled patients were on background therapy without any suggestion of untoward effects from combination therapy.
A subset of 145 patients participated in a hemodynamic assessment, with right-heart catheterization performed at baseline and at month 6. The baseline mean PVR in the 50 patients of the placebo group was 886 dyn·sec/cm5 compared to 945 dyn · sec/cm5 and 907 dyn · sec/cm5 in the 3 mg and 10 mg treatment groups, respectively. After a 6-month treatment period, the PVR had increased to a mean value of 1,042 dyn · sec/cm5 in the placebo group, whereas it decreased to 736 dyn · sec/cm5 and 680 dyn · sec/cm5 in the 3 mg and 10 mg treatment groups, respectively. Furthermore, while there was a decrease in cardiac index from a baseline of 2.54 L/min/m2 to 2.21 L/min/m2 after 6 months in the placebo group, the index increased from 2.34 L/min/m2 to 2.69 L/min/m2 and 2.63 L/min/m2 to 2.93 L/min/m2 in the 3 mg and 10 mg treatment groups, respectively.
The duration of the trial allowed for a comprehensive assessment of the adverse-effect profile of macitentan. Given macitentan’s structural relationship to bosentan, there was great interest in its effects on aminotransferase levels. Compared to the baseline levels, aminotransferases were assessed at months 3 and 6 and every 6 months thereafter until the end of the study, with an elevation >3× ULN seen in eleven patients (4.5%) in the placebo group compared to nine (3.6%) and eight (3.4%) in the 3 mg and 10 mg treatment groups, respectively. An elevation >3× ULN in combination with a doubling of the ULN bilirubin level was even more infrequent and comparable between all three groups. There was additionally no significant difference in the appearance of peripheral edema, which has been a concern when studying the adverse-effect profile of other ERAs.23,48 As stated before, investigators have hypothesized that dual-receptor antagonists, such as macitentan and bosentan before it, are less likely to generate this effect, which may well be a receptor phenomenon and not an effect for all ERAs accross the board. These findings are quite favorable when compared to the subset of patients who received 250 mg of bosentan twice daily in the original BREATHE-1 (Bosentan: Randomized trial of ET receptor Antagonist THErapy) trial, in which ten of 70 patients (14%) developed elevations of their aminotransferases.21 In the STRIDE-1 (Sitaxsentan To Relieve ImpaireD Exercise) trial, the subset of 63 patients who received the 300 mg dose of sitaxsentan had six cases of reversible elevations of aminotransferases to >3× the ULN during the initial 12-week period of observation.22 Highlighting the importance of more prolonged observation, however, was the increasing frequency of increased aminotransferase levels when assessments were carried out at 58 weeks, with the incidence increasing to 21% with the 300 mg dose.
More significant is the issue of anemia with use of macitentan. The SERAPHIN data show that only one of the 237 patients in the placebo arm developed a reduction in hemoglobin to a level ≤8 g/dL, whereas this occurred in four of 241 (1.7%) and ten of 230 (4.3%) patients in the 3 mg and 10 mg treatment arms, respectively. In fact, the mean decrease in hemoglobin was approximately 1 g/dL. As was concluded from the Phase II data, the effect seems to be dose-dependent and resolves with discontinuation of the drug. Anemia secondary to ERA use was also noted in the cases of the landmark bosentan and ambrisentan trials.20,23 The mechanism behind the development of anemia is unknown, although some theories have been postulated. For example, experimental data support the concept that stimulation of the ETRB may inhibit eryptosis, or suicidal erythrocyte death.49 Consequently, blockade of the ETRB, which results in decreased stimulation, may lead to unopposed eryptosis via the ET pathway.
Given the fact that PAH is a chronic and life-threatening condition, studies that are geared to assess the long-term consequences of a therapy have significant clinical utility. For example, since death as a result of PAH is likely to be preceded by hospitalization resulting from the disease, assessing the effect of macitentan on hospitalization rates provided clinically meaningful results. Of the 180 hospitalizations that were documented in the study,46 79 (31.6%) were from the placebo group, whereas 56 (22.4%) and 45 (18.6%) were in the 3 mg and 10 mg treatment groups, respectively.
Potential role of macitentan in other disorders
Idiopathic pulmonary fibrosis
Macitentan has also been investigated for its potential role in the management of idiopathic pulmonary fibrosis (IPF). IPF is a chronic fibrotic pulmonary disease that is progressive and fatal. ET-1 has been implicated in the pathogenesis of IPF, and its receptor blockade has theoretical antifibrotic consequences.50 Previous trials evaluating the role of bosentan and ambrisentan either did not meet their primary end points or were terminated early.51–53 The combination of macitentan and ACT-132577 has been shown to reduce the amount of profibrotic α-smooth-muscle actin in the lesional skin fibroblasts taken from patients with systemic sclerosis.54 Additional promising data regarding antifibrotic properties of macitentan were obtained from a rat model of pulmonary fibrosis.55 Consequently, the randomized, double-blind, placebo-controlled MUSIC trial was undertaken in several centers to investigate the effects of macitentan on the progression of IPF using change in forced vital capacity as the primary outcome and surrogate for clinical response.56 A total of 178 subjects were enrolled, with 119 randomized to receive macitentan. After 12 months of treatment with 10 mg of macitentan, the treatment group demonstrated no significant difference in forced vital capacity, forced expiratory volume in 1 second, or corrected diffusion capacity of carbon monoxide. While the reasons for the lack of efficacy are not known, the authors postulated that the complex and multifaceted pathways leading to pulmonary fibrosis make achieving significant clinical improvement through solely the ET axis unlikely. Of note, and not dissimilar from findings in the SERAPHIN trial, was a rise in aminotransferase levels to ≥3× ULN in 3.4% of the macitentan arm compared to 5.1% of the placebo group.
Chronic thromboembolic pulmonary hypertension
Chronic thromboembolic pulmonary hypertension (CTEPH), or WHO group IV pulmonary hypertension, is the only form of pulmonary hypertension that can undergo definitive curative therapy via surgical pulmonary endarterectomy. Nonetheless, a significant subset of patients with CTEPH have an inoperable form of the condition, owing to a predominantly distal distribution of disease in the pulmonary vascular bed or because of prohibitive comorbidities. At the moment, there is only one FDA-approved medical therapy for inoperable CTEPH in the form of riociguat, a stimulator of soluble guanylate cyclase, which demonstrated both improved exercise capacity and PVR after 16 weeks of therapy.57 Prior to this, however, there were promising preliminary data on treatment of the ET axis to improve hemodynamics in patients with inoperable CTEPH.58,59 In light of this, the Phase II placebo-controlled MERIT-1 (MacitEntan in the tReatment of Inoperable chronic Thromboembolic pulmonary hypertension) trial is under way, with the primary end point being the change in PVR after 16 weeks of therapy when compared to baseline using the 10 mg dose in the treatment group.60
Summary
Macitentan is the latest ERA with highly efficacious and enduring binding properties to both ETRA and ETRB. In part due to the properties of its sole active metabolite, it is available for once-daily use. Extensive Phase I data have demonstrated a favorable drug–drug interaction profile, with no dosage adjustments necessary in the face of chronic hepatic or renal impairment. Unlike some prior ERAs, it has demonstrated no increased risk for hepatotoxicity, with its only notable significant adverse event being anemia. The first long-term trial designed to evaluate a PAH-specific medicine’s effect on morbidity and mortality demonstrated both favorable hemodynamic effects and significant reduction in morbidity in both treatment-naïve patients with PAH and those already on background therapies.
Disclosure
The authors report no conflicts of interest in this work.
References
Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448–456. | |
D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. | |
Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. | |
Russell FD, Skepper JN, Davenport AP. Evidence using immunoelectron microscopy for regulated and constitutive pathways in the transport and release of endothelin. J Cardiovasc Pharmacol. 1998;31(3):424–430. | |
Russell FD, Davenport AP. Secretory pathways in endothelin synthesis. Br J Pharmacol. 1999;126(2):391–398. | |
Boulanger C, Luscher TF. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J Clin Invest. 1990;85(2):587–590. | |
Firth JD, Ratcliffe PJ. Organ distribution of the three rat endothelin messenger RNAs and the effects of ischemia on renal gene expression. J Clin Invest. 1992;90(3):1023–1031. | |
Takahashi M, Matsushita Y, Iijima Y, Tanzawa K. Purification and characterization of endothelin-converting enzyme from rat lung. J Biol Chem. 1993;268(28):21394–21398. | |
Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102(19):2434–2440. | |
Pollock DM, Keith TL, Highsmith RF. Endothelin receptors and calcium signaling. FASEB J. 1995;9(12):1196–1204. | |
Hirata Y, Emori T, Eguchi S, et al. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest. 1993;91(4):1367–1373. | |
Shichiri M, Kato H, Marumo F, Hirata Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension. 1997;30(5):1198–1203. | |
Verhaar MC, Strachan FE, Newby DE, et al. Endothelin-A receptor antagonist-mediated vasodilatation is attenuated by inhibition of nitric oxide synthesis and by endothelin-B receptor blockade. Circulation. 1998;97(8):752–756. | |
Ohuchi T, Kuwaki T, Ling GY, et al. Elevation of blood pressure by genetic and pharmacological disruption of the ETB receptor in mice. Am J Physiol. 1999;276(4 Pt 2):R1071–R1077. | |
Sauvageau S, Thorin E, Caron A, Dupuis J. Endothelin-1-induced pulmonary vasoreactivity is regulated by ET(A) and ET(B) receptor interactions. J Vasc Res. 2007;44(5):375–381. | |
Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur Respir J. 2008;31(2):407–415. | |
Trow TK, Taichman DB. Endothelin receptor blockade in the management of pulmonary arterial hypertension: selective and dual antagonism. Respir Med. 2009;103(7):951–962. | |
MacLean MR. Endothelin-1: a mediator of pulmonary hypertension? Pulm Pharmacol Ther. 1998;11(2–3):125–132. | |
Kirkby NS, Hadoke PW, Bagnall AJ, Webb DJ. The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? Br J Pharmacol. 2008;153(6):1105–1119. | |
Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001; 358(9288):1119–1123. | |
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. | |
Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169(4):441–447. | |
Galie N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(3):529–535. | |
Barst RJ, Rich S, Widlitz A, Horn EM, McLaughlin V, McFarlin J. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot study. Chest. 2002;121(6):1860–1868. | |
Dingemanse J, Sidharta PN, Maddrey WC, Rubin LJ, Mickail H. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf. 2014;13(3):391–405. | |
Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67(10):977–984. | |
Bruderer S, Hopfgartner G, Seiberling M, et al. Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans. Xenobiotica. 2012;42(9):901–910. | |
Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327(3):736–745. | |
Atsmon J, Dingemanse J, Shaikevich D, Volokhov I, Sidharta PN. Investigation of the effects of ketoconazole on the pharmacokinetics of macitentan, a novel dual endothelin receptor antagonist, in healthy subjects. Clin Pharmacokinet. 2013;52(8):685–692. | |
Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One. 2012;7(10):e47662. | |
Hong IS, Coe HV, Catanzaro LM. Macitentan for the treatment of pulmonary arterial hypertension. Ann Pharmacother. 2014;48(4):538–547. | |
Sidharta PN, Lindegger N, Ulc I, Dingemanse J. Pharmacokinetics of the novel dual endothelin receptor antagonist macitentan in subjects with hepatic or renal impairment. J Clin Pharmacol. Epub October 3, 2013. | |
Sidharta PN, van Giersbergen PL, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol. 2013;53(11):1131–1138. | |
Sidharta PN, van Giersbergen PL, Woltz N, Dingemanse J. Investigation of mutual pharmacokinetic interactions between macitentan, a novel endothelin receptor antagonist, and sildenafil in healthy subjects. Br J Clin Pharmacol. Epub June 24, 2014. | |
Bruderer S, Aänismaa P, Homery MC, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 2012;14(1):68–78. | |
Sidharta PN, Dietrich H, Dingemanse J. Investigation of the effect of macitentan on the pharmacokinetics and pharmacodynamics of warfarin in healthy male subjects. Clin Drug Investig. 2014;34(8):545–552. | |
Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69(4):223–231. | |
Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000; 118(2):422–430. | |
Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest. 1993;92(1):17–24. | |
Jansen PL, Strautnieks SS, Jacquemin E, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117(6):1370–1379. | |
Krum H, Viskoper RJ, Lacourciere Y, Budde M, Charlon V. The effect of an endothelin-receptor antagonist, bosentan, on blood pressure in patients with essential hypertension. Bosentan Hypertension Investigators. N Engl J Med. 1998;338(12):784–790. | |
Actelion. OPsumit USers Registry (OPUS). Available from: NLM identifier: NCT02126943. http://clinicaltrials.gov/ct2/show/NCT02126943?term=macitentan&rank=13. Accessed July 29, 2014. | |
Comellas AP, Briva A, Dada LA, et al. Endothelin-1 impairs alveolar epithelial function via endothelial ETB receptor. Am J Respir Crit Care Med. 2009;179(2):113–122. | |
Iglarz M, Bossu A, Wanner D, et al. Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension. Life Sci. Epub February 26, 2014. | |
Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutics Review, application number: 204410Orig1s000. 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/ 2013/204410Orig1s000ClinPharmR.pdf. Accessed July 29, 2014. | |
Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. | |
Galie N, Simonneau G, Barst RJ, Badesch D, Rubin L. Clinical worsening in trials of pulmonary arterial hypertension: results and implications. Curr Opin Pulm Med. 2010;16 Suppl 1:S11–S19. | |
Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24(3):353–359. | |
Föller M, Mahmud H, Qadri SM, et al. Endothelin B receptor stimulation inhibits suicidal erythrocyte death. FASEB J. 2010;24(9):3351–3359. | |
Fonseca C, Abraham D, Renzoni EA. Endothelin in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;44(1):1–10. | |
King TE Jr, Behr J, Brown KK, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81. | |
King TE Jr, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–99. | |
Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. | |
Corallo C, Pecetti G, Iglarz M, et al. Macitentan slows down the dermal fibrotic process in systemic sclerosis: in vitro findings. J Biol Regul Homeost Agents. 2013;27(2):455–462. | |
Iglarz M, Landskroner K, Rey M, Wanner D, Hess P, Clozel M. Optimization of tissue targeting properties of macitentan, a new dual endothelin receptor antagonist, improves its efficacy in a rat model of pulmonary fibrosis associated with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;183:A6445. | |
Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J, Group MS. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622–1632. | |
Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319–329. | |
Bonderman D, Nowotny R, Skoro-Sajer N, et al. Bosentan therapy for inoperable chronic thromboembolic pulmonary hypertension. Chest. 2005;128(4):2599–2603. | |
Hoeper MM, Kramm T, Wilkens H, et al. Bosentan therapy for inoperable chronic thromboembolic pulmonary hypertension. Chest. 2005;128(4):2363–2367. | |
Actelion. MERIT-1: macitentan in the treatment of inoperable chronic thromboembolic pulmonary hypertension. Available from: http://clinicaltrials.gov/show/NCT02021292. NLM identifier: NCT02021292. Accessed July 29, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.