Back to Journals » Journal of Inflammation Research » Volume 14
Lung Inflammatory Response to Environmental Dust Exposure in Mice Suggests a Link to Regional Respiratory Disease Risk
Authors Burr AC, Velazquez JV, Ulu A, Kamath R, Kim SY, Bilg AK, Najera A, Sultan I, Botthoff JK, Aronson E, Nair MG ![]() , Nordgren TM
, Nordgren TM ![]()
Received 20 May 2021
Accepted for publication 6 August 2021
Published 21 August 2021 Volume 2021:14 Pages 4035—4052
DOI https://doi.org/10.2147/JIR.S320096
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Abigail C Burr,1,* Jalene V Velazquez,1,* Arzu Ulu,1 Rohan Kamath,1 Sang Yong Kim,1 Amanpreet K Bilg,1 Aileen Najera,1 Iman Sultan,1 Jon K Botthoff,2 Emma Aronson,3 Meera G Nair,1 Tara M Nordgren1
1Division of Biomedical Sciences, University of California Riverside, Riverside, CA, 92521, USA; 2Center for Conservation Biology, University of California Riverside, Riverside, CA, 92521, USA; 3Department of Plant Pathology and Microbiology, University of California Riverside, Riverside, CA, 92521, USA
*These authors contributed equally to this work
Correspondence: Tara M Nordgren
Division of Biomedical Sciences, University of California Riverside, 92521 University Avenue, Riverside, CA, 92521, USA
Tel +1 951 827 3148
Email [email protected]
Purpose: The Salton Sea, California’s largest lake, is designated as an agricultural drainage reservoir. In recent years, the lake has experienced shrinkage due to reduced water sources, increasing levels of aerosolized dusts in surrounding regions. Communities surrounding the Salton Sea have increased asthma prevalence versus the rest of California; however, a connection between dust inhalation and lung health impacts has not been defined.
Methods: We used an established intranasal dust exposure murine model to study the lung inflammatory response following single or repetitive (7-day) exposure to extracts of dusts collected in regions surrounding the Salton Sea (SSDE), complemented with in vitro investigations assessing SSDE impacts on the airway epithelium.
Results: In these investigations, single or repetitive SSDE exposure induced significant lung inflammatory cytokine release concomitant with neutrophil influx. Repetitive SSDE exposure led to significant lung eosinophil recruitment and altered expression of genes associated with allergen-mediated immune response, including Clec4e. SSDE treatment of human bronchial epithelial cells (BEAS-2B) induced inflammatory cytokine production at 5- and 24-hours post-treatment. When BEAS-2B were exposed to protease activity-depleted SSDE (PDSSDE) or treated with SSDE in the context of protease-activated receptor-1 and − 2 antagonism, inflammatory cytokine release was decreased. Furthermore, repetitive exposure to PDSSDE led to decreased neutrophil and eosinophilic influx and IL-6 release in mice compared to SSDE-challenged mice.
Conclusion: These investigations demonstrate potent lung inflammatory responses and tissue remodeling in response to SSDE, in part due to environmental proteases found within the dusts. These studies provide the first evidence supporting a link between environmental dust exposure, protease-mediated immune activation, and respiratory disease in the Salton Sea region.
Keywords: Salton Sea, dust exposure, lung inflammation, asthma, proteases, protease-activated receptors
Introduction
The Salton Sea is situated within the northern part of the Imperial Valley, found in the southeastern border region of California. The “sea” is California’s largest lake, a 345 mile2 shallow body of water, with no natural outlets. In the 1900s, the lake was inadvertently created when water from the Colorado River accidently flooded a dry salt bed. Located within the Imperial Valley, known for its strong agricultural industry, the lake is framed by local farming communities. Since its inception, the lake has been sustained by runoff from surrounding agricultural operations. Historically, the Salton Sea has served as a repository for this irrigation wastewater.1 Agricultural run-off has been shown to bring pesticides, such as organophosphorus insecticides, chlorpyrifos, as well as industrial contaminants into the sea.2
Combined with the natural desert climate, the shallowness of the lake has historically contributed to its gradual shrinkage.3 However, the Salton Sea now experiences increased shrinkage and is predicted to shrink by 100 mile2 by 2030 due to limitations in the water resources that feed it.4 As the water recedes, lakebed is exposed (known as playa) which can then be transformed to aerosolized dusts by strong winds. This increase in aerosolized playa dust could contribute to airborne particulate matter (PM) mass and raise health concerns, as increases in PM mass have been linked to cardiovascular disease, respiratory disease, and mortality.5–7 In addition to increased mass loadings, the composition of PM emissions could increase particulate toxicity.8,9 Dust events represent a significant exposure to children whose respiratory and immune systems are still developing. Currently, the community surrounding the Salton Sea is disproportionately affected by pediatric asthma with 20% of its pediatric population diagnosed with asthma compared to 8% nationally.10
Research has identified toxic metals within the lake sediments, including cadmium, arsenic, and selenium,9,11 and recent studies have found that the soil collected from the now exposed lakebed is enriched in Selenium (Se).8,9 Selenium is an essential nutrient; however, it is toxic depending on its oxidation state. Through microbial transformation in the soil, Se can be removed and expelled into the atmosphere as Dimethyl selenide (DMSe), and secondary organic aerosols derived from DMSe oxidation were recently found to increase IL-6 release and upregulate HOX-1, TNF-α, and IL-6 in the BEAS-2B bronchial epithelial cell line.12 Previous studies have also confirmed the presence of diatoms (a major group of microalgae), cyanobacteria, and fungi in the Salton Sea. These biological species can typically be found in marine settings and are all capable of releasing a variety of proteins, including proteases.13–15 Proteases are necessary for metabolism and function as enzymes that cleave proteins and catalyze reactions by breaking down active sites.16 A subset of environmental proteins inhaled by humans can trigger an allergic response, and numerous studies suggest that this ability is due to active proteolytic activity in these airborne particles.17 Environmental proteases can elicit an immune response through epithelial cell activation via protease-activated receptors (PARs), leading to increased release of proinflammatory cytokines and chemokines.16–19 However, a connection between inhalation of the aerosolized Salton Sea dusts and lung health impacts has not been defined.
In this study, we hypothesized that exposure to aqueous extracts of dust collected from areas surrounding the Salton Sea (SSDE) would cause lung inflammation and deleterious activation of the immune response. To test this hypothesis, mice were challenged with an acute (1 time) or repetitive (7 days) exposure to SSDE and assessed for lung inflammatory impacts and alterations in immune-related gene expression. We also assessed inflammatory cytokine production in BEAS-2B following 5- and 24-hour treatments with SSDE. Through these investigations, we identified a gene activation signature consistent with the activation of pro-allergic environmental protease-mediated signaling. Exploring these findings further, we identified active proteases within SSDE, and depletion of protease activity led to alterations in the inflammatory response to SSDE both in vitro and in vivo. Through these studies, we identified that SSDE elicits potent inflammatory responses both in vitro and in vivo that were in part mediated through proteases found in the dusts. These results identify immune-mediated and tissue remodeling cellular and molecular mechanisms that are directly triggered by Salton Sea dusts and may contribute to exacerbated respiratory diseases such as asthma.
Materials and Methods
Mice
C57BL/6 mice were purchased from Jackson Laboratories. Mice were age- and sex-matched and housed five per cage under ambient temperature with 12-hour light/dark cycles. Studies were performed in mice aged 8–12 weeks. All protocols for animal use and euthanasia were approved by the University of California Riverside Institutional Animal Care and Use Committee and were in accordance with National Institutes of Health guidelines, the Animal Welfare Act, and Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Salton Sea Dust Extract (SSDE) Preparation
Dust samples were collected as described previously.20 Briefly, airborne particles in regions adjacent to the Salton Sea were collected using passive collectors. These collectors consisted of Teflon-coated round pans (25.4 cm in diameter) filled with quartz marbles that were suspended from the bottom of the pan with Teflon mesh. Collectors were mounted on 2.4 m tall wooden posts in open areas to minimize local contributions of wind-suspended material (such as those from nearby trees). The collectors were continuously deployed from December 2015 to March 2017 at the Dos Palmas Preserve at 33○29ʹ22.1” N 115○50ʹ06.3” W. Dusts that had settled into the collectors were recovered first by washing the marbles with >18.2MΩcm water and allowing dust suspensions to collect in the Teflon-coated pan bottom. These dust suspensions were then transferred to pre-cleaned 1 L LDPE Nalgene bottles. The samples were frozen and stored until use in −20°C fridge. To prepare the extracts, dust suspensions were thawed and aliquoted into 50-mL conical tubes and lyophilized (Labconco, model 2.5, Kansas City, MO). The resulting dust was reconstituted in sterile PBS to a final protein concentration of 40 μg/mL. Dust samples from regions surrounding the Salton Sea, collected during a similar time period and by the same methods, have been previously characterized for major compositional components, including elements and soluble anions.9 Concentrations/doses of SSDE used in in vitro and in vivo studies were chosen based on initial pilot studies to confirm inflammatory potential while preventing overt cytotoxic effects (data not shown).
Protease Activity Inhibition/Depletion of Salton Sea Dust Extract
Protease activity was inhibited/depleted from undiluted SSDE similarly as previously described.21 Briefly, Halt™ Protease Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA) was diluted from 10× to 1× in SSDE or PBS. The protease inhibitors’ working concentrations were as follows: AEBSF 1 mM, Aprotinin 800 nM, Bestatin 50 µM, E64 15 µM, Leupeptin 20 µM and Pepstatin A 10 µM. Mixtures were incubated at 37°C with agitation for 2 hours and maintained at 4°C until use within 24 hours or stored at −20°C if not used within 24 hours. This resulted in protease activity-depleted Salton Sea dust extract (PDSSDE). The PBS incubated with protease inhibitor was used as a vehicle control for studies (PI), diluted as appropriate to replicate PDSSDE for in vitro studies.
In vivo SSDE Exposure Model
Murine studies were performed using a standardized model of dust exposure.22 For acute (one-time) and repetitive (once daily for 7 days) exposure, mice were given an intranasal (i.n.) instillation consisting of 50 μL of sterile saline, 100% SSDE, PI vehicle, or 100% PDSSDE. At 5 hours after final SSDE instillation, mice were euthanized, and tissues were collected for subsequent analyses.
Sample Collection, Processing, and Cell Differentials
Murine left lungs were ligated at the primary bronchi, removed, flash frozen in liquid nitrogen, and stored at −80°C for future analyses. Bronchoalveolar lavage fluid (BALF) was collected from remaining right lungs in three washes of 400 μL of ice-cold PBS. BALF was stored on ice until centrifugation at 1200 rpm and 4°C for 5 minutes. Supernates from the first collected wash were aliquoted and stored for cytokine analyses. The remaining pellets from the first wash were combined with the cell pellets of washes 2 and 3. Recovered cells were visualized and counted via hemocytometer. Cytospins were prepared using a Shandon Cytospin 4 cytocentrifuge (Thermo Fisher Scientific, Waltham, MA) at 600 rpm for 5 minutes. Resulting slides were dried overnight before staining with Siemens Diff-Quik Stain Set (Newark, DE). Differential cell counts were then quantified under light microscopy (Revolve Microscope, Echo Laboratories, San Diego, CA).
Flow Cytometry
BALF cells collected from mice receiving repetitive SSDE were analyzed via flow cytometry. Recovered cells from the BALF were resuspended in FACS buffer and counted by a hematocytometer, and 10,000 cells were blocked with 0.6 μg rat IgG and anti-CD16/32 (2.4G2). Blocked cells were stained for 30 min with antibodies for CD4 (RM4-5) (all from BD Biosciences, San Jose, CA); CD11b (M1/70), CD11c (N418), Ly6C (HK 1.4), MHCII (AF6-120.1), SiglecF (E50-2440) (all from eBioscience, Affymetrix; Santa Clara, CA); Ly6G (1A8) (BioLegend, San Diego, CA) and F4/80 (CI:A3-1, Bio-Rad Laboratories, Hercules, CA). After staining, cells were washed with FACS buffer and analyzed on an LSRII instrument (BD Biosciences, San Jose, CA), followed by data analysis using FlowJO v10 (Tree Star Inc.; Ashland, OR).
Cytokine Quantification
Murine TNF-α, IL-6, and chemokine C-X-C motif ligand 1 (CXCL1) levels, along with human IL-8 and IL-6 were quantitated using DuoSet enzyme-linked immunosorbent assays according to manufacturer’s instructions (R&D Systems, Minneapolis, MI).
Histology and Histopathology
For one-time and 7-day SSDE studies, right lungs were inflated with 400 μL one part 10% formalin/30% sucrose and one part OCT embedding medium (Fisher Scientific; Hampton NH) and stored overnight in formalin/30% sucrose at 4°C. The following day right lungs were transferred to new solutions of 30% sucrose in PBS and stored overnight at 4°C. Right lungs were blocked in OCT and sectioned at 12 μm. In in vivo protease depletion studies, the right lung was slowly inflated with 10% formalin at 20 cm pressure overnight. The following day formalin-fixed lungs were moved into 70% ethanol and stored at 4°C until they were sent to the University of California Irvine Research Services Core Facility for paraffin-embedding. For pathology, lung sections were stained with hematoxylin and eosin and visualized using an Echo Revolve Microscope (Echo Laboratories, San Diego, CA, USA). Histopathological evaluations for perivascular inflammation and granulomatous tissues were conducted on formalin-fixed paraffin-embedded (FFPE) right lung sections from in vivo PDSSDE studies, similarly as previously described.23 For each sample, all left lung lobes present were assessed, in their entirety, for perivascular, peribronchial and the presence of immune cell aggregate/granulomatous inflammation. For perivascular inflammation, a protocol was adapted from methods previously described.23 Samples were given a single score based on the following criteria: 0, no inflammation; 1, occasional inflammatory cells; 2, greater and more frequent accumulations of inflammatory cells on vessels; 3, multifocal and even greater inflammation around vessels; 4, severe accumulations of inflammatory cells around multiple vessels present. For immune cell aggregate/granulomatous inflammation, tissue sections were assessed individually. Scores were assigned individually to samples based on the following: 0, 1 or no granulomatous inflammation present; 1, 2–9 small, granulomatous lesions present; 2, 10–20 granulomatous lesions present; 3, 21–29 well-defined lesions; 4, >30 pervasive and well-organized granulomatous lesions present. Peribronchial inflammation was assessed by quantification of inflammatory cells as previously described.24 A value of 0 was assigned when no inflammation was detected; a value of 1 was given when occasional inflammatory cells were present; a value of 2 indicated that most bronchi were surrounded by a thin layer (1–5 cells) of inflammatory cells; and a value of 3 was assigned when a majority of bronchi were surrounded by a thick layer (>5 cells) of inflammatory cells.
Gene Expression Analysis
An overview of gene expression was achieved using the nCounter Analysis System (Nanostring Technologies, Seattle, WA, USA) run according to manufacturer’s instructions. We analyzed gene expression profiles from a total of 12 mice lungs (3 females, 3 males in each group) harvested on different dates. No differences were identified in gene expression between males and females within treatment groups based on principal component analyses (Supplemental Figure 1). Briefly, RNA was isolated from mouse lungs using an RNA isolation kit (Invitrogen, 12183555). RNA samples were quantified using NanoDrop ND-100 (NanoDrop Technologies, Inc, Wilmington, DE), and the integrity of the RNA samples was determined using RIN (RNA integrity numbers) and 28S-to-18S rRNA as control obtained with Agilent 2100 Bioanalyzer (UC Riverside Core Facilities, Agilent Technologies, Santa Clara, CA) and samples were run on a NanoString mouse Inflammation Panel following an overnight hybridization step. Analysis of the data was performed with nSolver software (version 4.0, NanoString Technologies, Seattle, WA, USA, http://www.NanoString.com/products/nSolver). Housekeeping gene normalization as well as background subtraction (mean ± 2 standard deviations of negative controls) were applied to all samples to control for variation in RNA isolation, hybridization and overall run efficiency. Those genes with less than 100 counts were excluded from further analysis. Normalized data were contrasted to positive controls (SSDE-exposed mouse lung RNA) to detect any differences in gene expression. To further explore the protein–protein interactions among differentially regulated proteins, we used STRING database (https://string-db.org). Raw and normalized NanoString data are deposited to https://www.ncbi.nlm.nih.gov/geo/info/spreadsheet.html.
Immunofluorescent Staining and Microscopy
Lung tissues were OCT-embedded and sectioned on a cryostat (Leica Biosystems, Illinois, USA) with each tissue slice being 12 mm. Tissues were then stained with rabbit a-mouse LYVE-1 primary antibody diluted 1:50 (NOVUS Biologicals, Colorado, USA, Cat. #NB600-1008). For immunofluorescence at 647 nm, tissues were stained with goat a-rabbit IgG secondary antibody diluted 1:500 (Thermo Fisher Scientific, Waltham, MA, USA, Cat. #A21244). Images of the stained lung tissues were taken under fluorescent microscopy on a Revolve microscope at 10× magnification (Echo Laboratories, San Diego, CA, USA). For the analysis of LYVE-1 expression in the whole lung, each lung was divided into four sections and an image was taken in each section for five control lungs and five SSDE exposed lungs, for a total of 20 images per treatment group. The images were analyzed, and integrated density/area was calculated on ImageJ (software downloaded from nih.gov). The subsequent data were statistically analyzed using GraphPad Prism software (La Jolla, CA, USA).
In vitro Bronchial Epithelial Cell Exposures
For in vitro studies, human bronchial epithelial cells (BEAS-2B; obtained from the American Type Culture Collection, Manassas, VA) were cultured similarly as previously described.25 The cells were grown in T-75 flasks coated with type 1 collagen (Sigma-Aldrich, St. Louis, MO) and maintained in LHC9 medium (Thermo Fisher Scientific, Waltham, MA) at 37°C. The LHC9 medium was changed every 2–3 days. After reaching 60–70% of confluence, cells were plated in 24-well flat bottom plates. For exposure studies, cell cultures at 85–90% of confluence were treated with either control (LHC9 medium only), 1% protease inhibitor (PI), 5% PI, 1% SSDE, 5% SSDE, 1% PDSSDE, or 5% PDSSDE for 5 hours or 24 hours. After each treatment period (5 hours or 24 hours), supernatant fractions were collected to be used for cytokine quantification. For protease-activated receptor antagonism investigations, bronchial epithelial cells were pre-treated with media control, 0.75 nM PAR-1 antagonist SCH-79797 (Tocris, Bristol UK) or 45 nM PAR-2 antagonist FSLLRY-NH2 (Cayman Chemical, Michigan, USA) for 1 hour before being exposed to either media control or SSDE for 24 hours. After 24 hours, supernates were collected to be used for cytokine quantification.
Assessment of Cytotoxicity and Proliferation in vitro
To assess for SSDE-induced bronchial epithelial cell cytotoxicity, lactate dehydrogenase activity (LDH) assays (Pierce LDH Cytotoxicity Assay Kit, Thermo Scientific, IL, USA) were performed according to manufacturer’s protocol using supernatant fractions of BEAS-2B exposed to 0%, 1%, or 5% SSDE for 5 or 24 hours. To assess the impact of SSDE on proliferation/metabolic activity, a tetrazolium (MTT) assay was performed, as previously described.26 Briefly, cells were plated in a 96-well plate and treated for 24 or 48 hours with 0%, 1%, or 5% SSDE. At 2 hours prior to the endpoint of the assay, 25 µL of 5 mg/mL MTT was added to each well. At endpoints, media were decanted, and dimethyl sulfoxide added to lyse cells. Absorbance at 590 nm was read using a Varioskan Lux plate reader with 620 nm used as a reference wavelength (Thermo Fisher Scientific, Waltham, MA). Wavelengths were corrected from blank well readouts (no cells) and data were calculated from absorbances as percentages of control (0% SSDE).
Gel Zymography
To assess the presence of proteolytic activity in the dust extracts, gel zymography was performed similarly as previously described.27 SSDE concentrations of 100% SSDE and 50% SSDE were made using PBS. Similarly, PDSSDE concentrations of 100% PDSSDE and 50% PDSSDE were made using PBS. Each sample (50 μL) was loaded onto an 8% SDS polyacrylamide gel copolymerized with 0.1% gelatin. Following electrophoresis, proteins were renatured in zymogram renaturing buffer (Bio-Rad Laboratories, Hercules, CA) for 30 minutes, developed in developing buffer (Bio-Rad Laboratories, Hercules, CA) overnight, and visualized by staining with 0.5% Coomassie Brilliant Blue R-250 (Bio-Rad Laboratories, Hercules, CA). After de-staining, proteolytic activity was visible as clear bands against the field of undigested substrate.
Statistical Analysis
Mouse studies included a minimum of 3 animals per treatment per experiment and each experiment was repeated a minimum of 2 times. All statistical analyses were performed using GraphPad Prism software (La Jolla, California), and data were expressed as the mean ± standard error of the mean. A Grubb’s test was performed to identify data outliers. Student’s t test, one-way analysis of variance or two-way analysis of variance with Tukey’s method for post hoc comparisons were used, as appropriate. A P value less than or equal to 0.05 was considered statistically significant.
Results
Acute and Repetitive Salton Sea Dust Extract Exposure Drives Rapid Chemokine Production, Inflammatory Cell Influx to the Airways and Lung Tissue Remodeling in vivo
To assess the lung inflammatory potential of Salton Sea dust exposure, mice were challenged with SSDE (100%) or saline via intranasal inhalation given once (acute exposure), or daily for 7 days (repetitive exposure). As shown in Figure 1A, at 5 hours following the single or final repetitive SSDE challenge, SSDE-treated mice exhibited a significant increase in total cell influx as evidenced in their BALF. Differential cell counts identified that this influx was largely attributable to neutrophil influx (Figure 1B) in both acute and repetitive exposure models, while mice exposed to SSDE for 7 days also exhibited significant increases in eosinophils and lymphocytes (Figure 1C and D). While no statistically significant alterations in total monocyte/macrophage populations were identified via differential cell counts (Figure 1E), using flow cytometry (Figure 1F–K) we identified a significant increase in infiltrating monocytes (Figure 1H) in mice receiving repetitive SSDE exposure, while alveolar macrophage populations remained the same as compared to saline controls (Figure 1G). Similar to the findings by differential cell counting, flow cytometric analysis also identified significant increases in neutrophils (Figure 1I), CD4+ T cells (Figure 1J) and eosinophils (Figure 1K), suggesting that repetitive SSDE exposure drives innate and adaptive immune cell recruitment to the airways.
 |
Figure 1 Effects of acute and repetitive SSDE exposure on lung cellular influx in vivo. (A) Total cell counts; (B) neutrophil; (C) eosinophil; (D) lymphocyte; and (E) macrophage recruitment in BALF following single and repetitive exposure to saline or SSDE based on differential cell counting. (F) Flow diagram of positive and negative (Neg) gating strategy to determine frequencies of CD11c+F4/80+ alveolar macs; SiglecF+ eos; Ly6G+ neutrophils; Ly6C+Ly6G- monocytes; CD4+ T cells. BALF population counts for alveolar macrophages (G); monocytes (H); neutrophils (I); CD4+ T cells (J); and eosinophils (K) following repetitive exposure to SSDE compared to saline controls. Error bars are standard error of mean (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). |
To evaluate the chemokines and inflammatory cytokines that may be responsible for this cell influx, we measured CXCL1, TNFα and IL-6 in the BALF. Consistent with our findings of a rapid immune response with neutrophil influx following an acute exposure, we identified that mice challenged with a single SSDE exposure exhibited significant elevations in BALF TNFα and CXCL1 (Figure 2A and B). Meanwhile, coincident with the increased monocyte influx following 7 days of SSDE exposure, repetitively exposed mice exhibited significant increases in IL-6 release (Figure 2C). Furthermore, repetitively exposed animals exhibited evidence of perivascular inflammation, while both acutely and repetitively SSDE-exposed mice had evidence of lung granulocyte influx (Figure 2D–G). Because angiogenesis, endothelial dysfunction, and changes in vascular density constitute distinct aspects of remodeled airways in respiratory diseases,28 we next assessed the effects of the observed increased inflammatory cytokine secretion and immune cell influx on the lymphatic vasculature of the lung. Here, we stained lung sections with anti-LYVE-1 (lymphatic vessel hyaluronan receptor) antibody. LYVE-1 functions as a hyaluronic acid (HA) receptor on the surface of lymphatic endothelial cells and promotes lymphangiogenesis through intracellular signaling,29 and previous studies have shown that TNFα and TNFβ downregulate LYVE-1 gene expression and lead to the receptor’s internalization and degradation via lysosomes.30 Consistent with the early increase in TNFα observed in our model, quantification of LYVE-1 expression in the mouse lungs revealed a decrease in LYVE-1 expression in repetitively SSDE-exposed lung images versus saline controls (Figure 2H–J).
 |
Figure 2 SSDE-induced alterations in inflammatory mediators and lung histopathology in vivo. (A–D) Cytokine release in BALF for (A) TNF-α; (B) IL-6; and (C) CXCL1 following single and repetitive exposure to SSDE. (D–G) Representative histology of lungs following single and repetitive exposure to saline and SSDE. (D) Single exposure, saline; (E) single exposure, SSDE; (F) 7-day exposure, saline; and (G) 7-day exposure, SSDE. Green arrow indicates perivascular inflammation. Yellow arrows indicate representative infiltrating granulocytes. Scale bar in D is 120 µm. Images taken at 20× with 150% optical zoom. (H–J) Representative images of lung immunofluorescence of LYVE-1 are shown. (H) Seven-day exposure, saline; (I) 7-day exposure, SSDE; (J) quantification of fluorescent intensity of LYVE-1 in saline- and SSDE-exposed mice. Quantification data are mean ± SEM, four lung sections were imaged per mouse, with five mice each in saline and control groups (total of 20 images per group). Error bars are standard error of mean (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). |
Effects of Salton Sea Dust Extract Treatment on Inflammatory Cytokine Production by Bronchial Epithelial Cells in vitro
Bronchial epithelial cells are among the first cells exposed to inhaled irritants, and they can initiate the innate immune response to these exposures through the release of pro-inflammatory cytokines including TNFα and CXCL1.31 To further explore the impact of SSDE on the airway epithelial inflammatory responses and assess the translational relevance of our in vivo findings using human cells, we treated a human bronchial epithelial cell line (BEAS-2B) with 0% (control), 1%, or 5% SSDE and assessed inflammatory cytokine release at 5 hours or 24 hours post-treatment. As shown in Figure 3, treatment of BEAS-2B with SSDE led to a significant induction of IL-6 and IL-8 release at both 5 hours (Figure 3A and B) and 24 hours (Figure 3D and E). Importantly, this induction was not associated with any significant alterations in cytotoxicity, as assessed via measuring lactate dehydrogenase (LDH) activity at 5 and 24 hours following SSDE treatment (Figure 3C and F), but was associated with a significant reduction in metabolic activity/proliferation with the 5% SSDE at both 24 and 48 hours of treatment, as indicated by MTT assay (Figure 3G and H).
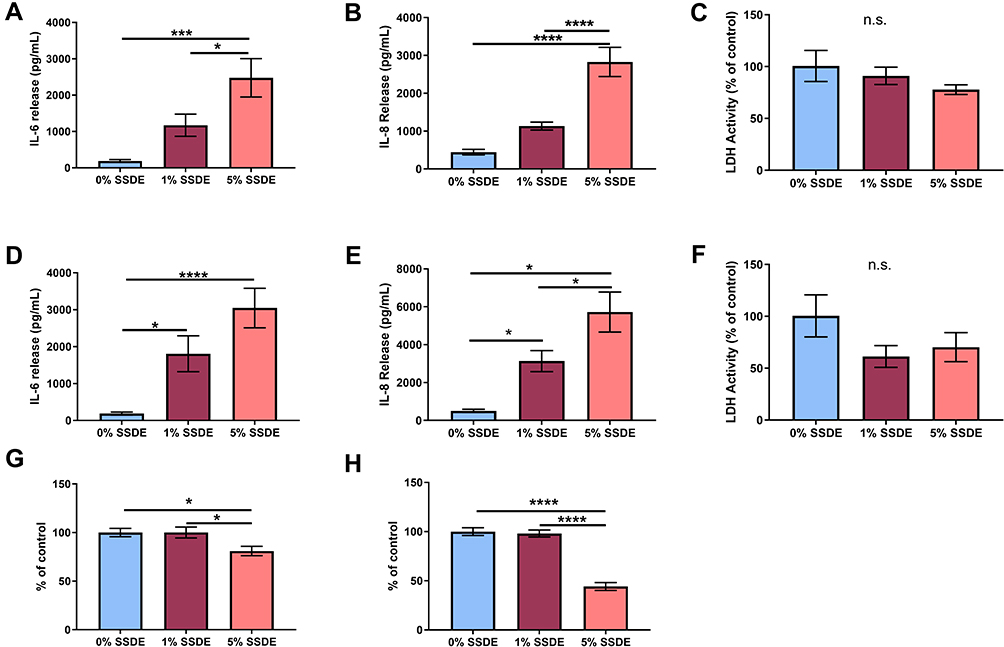 |
Figure 3 Effects of SSDE on bronchial epithelial cell inflammatory response following 5- and 24-hours treatments. Supernatant fractions from BEAS2-B treated with 1 or 5% SSDE were assayed at 5 or 24 hours post-treatment for IL-6 ((A), 24 hours; (C), 48 hours), IL-8 ((B), 24 hours; (D), 48 hours), and lactate dehydrogenase ((E), 24 hours; (F), 48 hours). Cells treated with 1 or 5% SSDE were also assessed for proliferative/metabolic activity via MTT assay at 24 (G) or 48 (H) hours. Error bars are standard error of mean (*p ≤ 0.05; ***p ≤ 0.001; ****p ≤ 0.0001). Abbreviation: ns, not significant. |
Repetitive Salton Sea Dust Extract Exposure Leads to Significant Alterations in Immune Pathways Associated with Protease-Related Activation and Allergic Response
To further explore the molecular mechanisms underlying the lung immune response to repetitive SSDE exposure, we next performed NanoString gene expression profiling using a panel that assesses 560 genes related to immune response pathways. When evaluating overarching changes in immune-related pathways, we identified clear SSDE-associated gene expression signatures in pathways associated with immune response (Figure 4A), chemokine activity (Figure 4B), and cell–cell signaling (Figure 4C). In addition, we also found alterations in calcium-mediated signaling (Figure 4D), calcium ion binding (Figure 4E), and activation of mitogen-activated protein kinase (MAPK) signaling (Figure 4F). Considering specific gene expression alterations between SSDE-treated animals and saline controls, gene-by-gene expression analyses identified 84 significantly altered genes among SSDE-treated vs saline-treated mice, where 73 were upregulated and 11 were downregulated in SSDE vs control samples. The 11 downregulated genes in SSDE- versus saline-challenged mice are shown in Figure 4G and include Il16, Gzma, Cd55, Cd19, Cfd, Ikzf3, Pax5, Mr1, Adal, Dpp4, and Igf2r. Both Cfd and Cd55 are regulators of the complement pathway, with Cfd (complement factor D) promoting activation of the alternative pathway of complement activation, and Cd55 (complement decay-accelerating factor) serving as a negative regulator of complement activation and formation of the membrane attack complex.32,33 Meanwhile, Gzma, Cd19, Il16, Ikzf3, Pax5, Mr1, and Dpp4 are all involved in the recruitment, differentiation, or functioning of lymphocytes.34–40
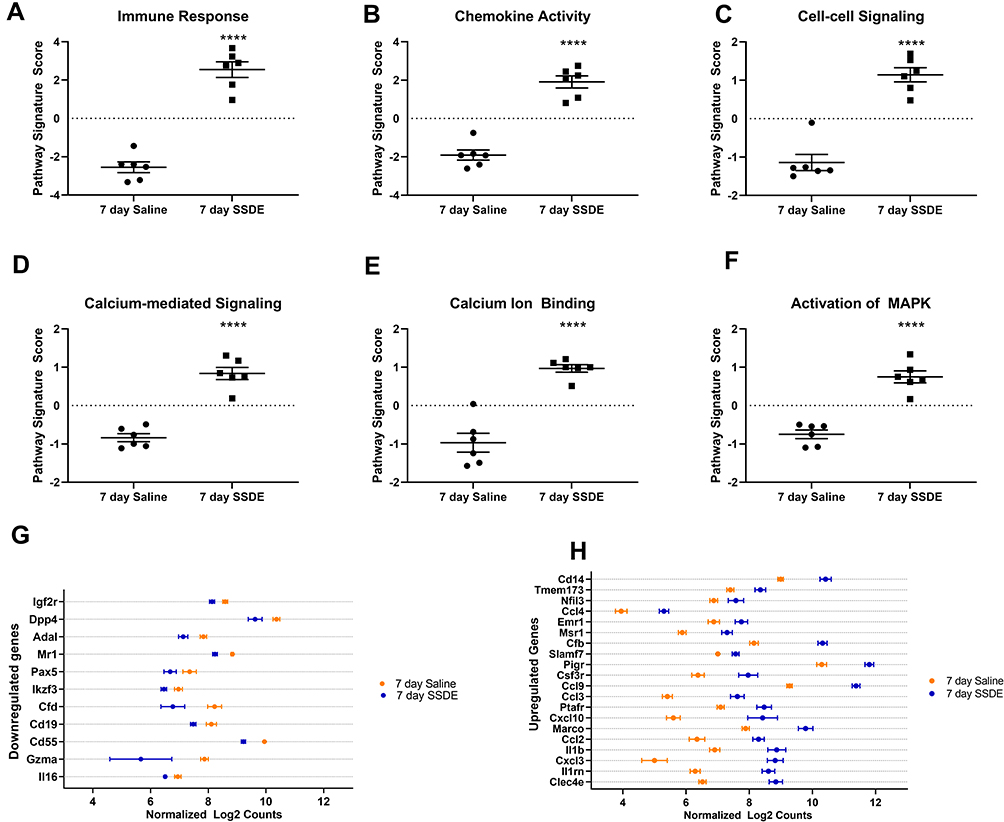 |
Figure 4 Pathways and transcripts altered after repetitive SSDE exposure in vivo. Pathway signature scores for (A) immune response; (B) chemokine activity; (C) cell-cell signaling; (D) calcium-mediated signaling; (E) calcium ion binding; and (F) activation of MAPK pathways. (G) Genes found to be significantly (P < 0.05) downregulated in SSDE- versus saline-instilled mice in a gene-by-gene analysis. (H) Top 20 significantly (P < 0.05) upregulated genes in SSDE- versus saline-instilled mice in a gene-by-gene analysis. N=6 mice per group. ****p < 0.0001. |
Of the upregulated genes, the top 20 significantly altered (based on P value) are listed in Figure 4H and include Clec4e, IL1rn, Cxcl3, Il1b, Ccl2, Marco, Cxcl10, Ptafr, Ccl3, Ccl9, Csf3r, Pigr, Slamf7, Cfb, Msr1, Emr1, Ccl4, Nfil3, Tmem173, and Cd14. The genes Cxcl3, Ccl2, Marco, Ccl3, Ccl9, Csf3r, Msr1, Emr1, Ccl4, and Cd14 all play important roles particularly in macrophages and/or other cells of the myeloid lineage, regulating differentiation, recruitment, activation, and/or functions.41–48 Furthermore, the most significantly altered gene (P = 1.37E-06), Clec4e, recognizes a variety of fungal-derived allergens that play a critical role in the sensitization, persistence, and severity of asthma,49,50 and has also been shown to play a novel and nonredundant role in the initiation of a proinflammatory response to C. albicans – a known sensitizer to asthma.51 A recent study has also shown that granulocyte colony stimulating factor receptor (encoded by Csf3r) signaling is important for modulating infection-dependent asthma at the cellular and molecular level,52 while CCL2 secretion has been shown to be increased following downstream signaling of protease-activated receptor (PAR)-1.53 The significant upregulation of these genes associated with fungal allergen/protease response also correlated with what is known regarding PAR signaling and our pathway activation results; when cleaved by serine proteases, PARs induce calcium mobilization and induce calcium-mediated signaling along with MAPK activation.54 The upregulation of these pathways, along with the increased eosinophilia, reduced LYVE-1 expression, and upregulation of environmental protease-responsive genes in SSDE-exposed lungs provide support for SSDE induction of an allergic-type response that may be partially mediated by fungal allergen/protease activity.
Impacts of Protease Activity on Salton Sea Dust Extract Inflammatory Potential in vitro and in vivo
Environmental proteases are known to induce lung inflammatory and allergic responses.17,18,21,55–57 These proteases interact with the immune system by cleaving and irreversibly activating host PARs, which are recognized contributors to chronic inflammatory diseases.54 Based on the NanoString gene expression signatures suggestive of protease-mediated immune activation, we next determined whether SSDE exhibited protease activity. Using gel zymography, we identified distinct bands of proteolytic activity in SSDE (Figure 5A) and this protease activity could be limited via treatment with a cocktail of protease inhibitors (Figure 5B; PDSSDE: protease activity-depleted SSDE). Treatment of BEAS-2B with 5% PDSSDE resulted in a significant decrease in IL-6 and IL-8 release at 5 hours following treatment as compared to cells treated with 5% SSDE (Figure 5C and D). Interestingly, this effect was no longer evident by 24 hours, whereby SSDE and PDSSDE yielded similar increases in IL-6 and IL-8 release compared to untreated cells or cells treated with vehicle only (Figure 5E and F). Furthermore, when BEAS-2B were pretreated with antagonists to the protease-activated receptors (PAR) PAR-1 and PAR-2, the SSDE-induced release of IL-6 and IL-8 at 24 hours was significantly reduced (Figure 5G and H).
 |
Figure 5 Effects of proteases on the SSDE inflammatory potential in bronchial epithelial cells. Gel zymography was performed on complete SSDE (A) or PDSSDE (B) to assess protease activity in the extracts. Solid rectangular boxes highlight main protease activity, evidenced by clearing (white banding) in gels. Dashed rectangular boxes identify gel lanes. BEAS-2B were treated with 1% and 5% SSDE or PDSSDE and IL-6 and IL-8 release were assessed at 5 hours following treatment (C and D) or 24 hours following treatment (E and F). BEAS-2B were pretreated for 1 hour with media control, PAR-1 antagonist SCH-79797 or PAR-2 antagonist FSLLRY-NH2 before being exposed to either medium control or SSDE for 24 hours. After 24 hours, supernatant was collected and IL-6 (G) and IL-8 (H) release were quantified. Error bars are standard error of mean (δ, p <0.05 compared to all other groups; β, p <0.05 versus control; λ, p <0.05 versus control, 1% PI, and 5% PI groups; ε, p <0.05 versus all groups except 5% PDSSDE; φ, p <0.05 versus all groups except 5% SSDE). |
Due to the impacts of protease inhibition or PAR1/PAR2 antagonism in the human bronchial epithelial responses to SSDE stimulation, we next assessed the impacts of SSDE protease depletion in vivo. Mice challenged with a single PDSSDE exposure exhibited similar total cellular influx (Figure 6A), neutrophilia (Figure 6C), and IL-6, CXCL1, and TNFα release (Figure 6F–H) as compared to SSDE-exposed mice, and no differences were identified in macrophage influx (Figure 6B). Interestingly, eosinophil influx in single SSDE-exposed mice, but not PDSSDE-exposed mice, reached statistical significance (Figure 6E), whereas exposure to PDSSDE led to a significant increase in lymphocyte influx into the airways, while SSDE exposure did not (Figure 6D). When mice were challenged with the repetitive (7-day) exposure, we found that protease depletion of SSDE (PDSSDE) led to a significant reduction in total cellular influx in the lavage fluid compared to SSDE-treated mice (Figure 6I). Macrophage and lymphocyte influx were similarly increased in both SSDE- and PDSDE-treated mice (Figure 6J and L). Interestingly, repetitive exposure to PDSSDE resulted in a significant reduction of eosinophils compared to SSDE exposure (Figure 6M). The overall reduction of cellular influx was further supported by a significant reduction in neutrophils in PDSSDE-treated animals compared to SSDE-treated animals (Figure 6K). In addition, PDSSDE-treated mice also exhibited a significant decrease in IL-6 release following 7 days of exposure, as compared to SSDE-treated mice (Figure 6O), while both PDSSDE- and SSDE-treated mice exhibited similar elevations in CXCL1 release (Figure 6N). However, when assessing the lung histopathology (Figure 6P–Y), including areas surrounding large airways (Figure 6P–S) and small airways (Figure 6T–W), both perivascular inflammation (Figure 6X) and immune cell aggregate/granulomatous tissue (Figure 6Y) yielded no significant differences between SSDE- or PDSSDE-exposed tissues.
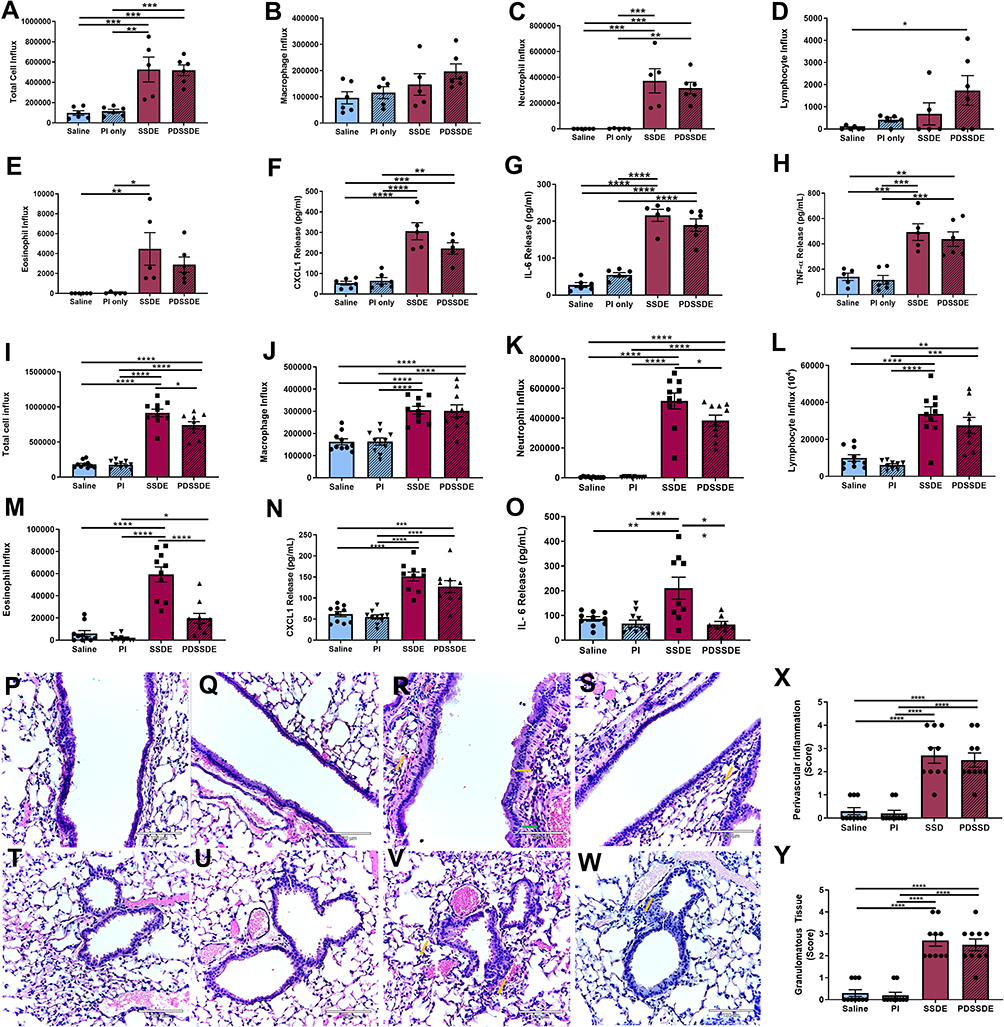 |
Figure 6 Effects of protease activity depletion on SSDE-induced lung immune response in vivo. (A–H) Cellular influx and inflammatory mediators in BALF at 5 hours following an acute SSDE or PDSSDE exposure. (A) Total cell counts; (B) macrophages; (C) neutrophils; (D) lymphocytes; (E) eosinophils; (F) CXCL1; (G) IL-6; (H) TNFα. (I–O) Cellular influx and inflammatory mediators in BALF following a 7-day repetitive exposure to SSDE or PDSSDE. (I) Total cell counts; (J) macrophages; (K) neutrophils; (L) lymphocytes; (M) eosinophils; (N) CXCL1; (O) IL-6. (P–W) Representative images of lung histopathology following 7 days of daily instillations to saline (P), saline treated with protease inhibitor (Q and U), SSDE (R and V), or PDSSDE (S and W). Large airways are represented in P-S; small airways represented in T-W. Green arrow indicates perivascular inflammation. Yellow arrows indicate representative infiltrating leukocytes. Blinded scoring for perivascular inflammation (X) and granulomatous/immune aggregate tissue (Y) were performed. Images were taken at 20x with 150% optical zoom, scale bar is 120 µm. Error bars are standard error of mean (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). |
Discussion
The Imperial/Coachella Valley has been implicated as the dustiest region among the southern California deserts.58 Here, we have identified that single and repetitive exposure to extracts of dusts collected from regions surrounding the Salton Sea induce a potent inflammatory response within the lungs of mice. A single intranasal exposure to SSDE resulted in significant increases in inflammatory cell influx and cytokine release in BALF. Repeated exposures to SSDE also elicited a potent inflammatory response, with increases in lymphocyte and eosinophil populations as compared to the single exposure, in addition to continued neutrophil influx dominating the lung cellular response. These changes in immune response were also mirrored in altered cytokine release between acute and repeated exposure models, changes in gene expression associated with environmental allergen/protease response, chemokine activity, cell–cell, calcium-mediated signaling and MAPK signaling. Overall, our findings indicate that aerosolized dusts in regions surrounding the Salton Sea possess proteases capable of eliciting potent lung inflammatory responses that could progress to allergic or inflammatory lung disease.
It has been shown that the salt-based crusts of exposed playa have a largely variable contribution to dust emissions.59 However, with increased drying of the Salton Sea, these contributions to PM10 are expected to increase.8 Wind-blown dusts not only contribute to PM10 and PM2.5 but also can carry biological materials such as bacteria, pollen, fungi, and pollutants including pesticides and heavy metals.9,60 Additionally, dust events worldwide have been implicated with increased respiratory symptoms and hospital visits in children.61–63 A study in Greece suggested that desert dust from the Sahara led to elevated PM10 levels, which resulted in a 2.54% increase in hospital admissions for pediatric asthma.6 Furthermore, researchers in Japan found increased hospitalizations for asthma in children during heavy dust events.64
Our in vivo findings corroborate the inflammatory potential of Salton Sea dusts; mice receiving a single or repetitive intranasal exposure to SSDE experienced significantly increased inflammatory cell influx to the lung. Following a single exposure, neutrophils were significantly elevated within the lung. After repetitive exposure, neutrophils as well as eosinophils and lymphocytes were increased. Recent studies have shown that increased neutrophil influx was associated with decreased lung function and steroid insensitivity.65,66 The significant elevation in eosinophils between single and repetitive exposures to SSDE is also suggestive of a developing allergic-type immune response. Eosinophils are known to be elevated in the immune response to parasitic infections and allergic reactions, such as anaphylaxis, making eosinophilia a key characteristic of allergic asthma.67 The development of a CD4-dominated immune response is also known to play a role in allergic asthma pathogenesis;68 flow cytometry revealed a significant increase in the CD4+ T-cell population in BALF following repetitive exposure, and these data together with the increased eosinophilia warrant studies to further assess the allergic potential of Salton Sea dust exposure. It is interesting to note that we identified a gene signature consistent with a protease/allergen-mediated inflammatory response in the lungs of mice exposed to SSDE for 7 days, including significant transcriptional upregulation of the fungal allergen-binding protein Clec4e as well as genes for scavenger receptors including Msr1 and Marco. Marco is expressed on alveolar macrophages and dendritic cells and binds environmental particles; its upregulation has been previously identified to occur during allergic airway inflammation.43,69 Changes in cell–cell, calcium ion binding, MAPK activation, and calcium-mediated signaling were also identified in SSDE-exposed mice, which have significant roles in modulating airway responsiveness and in responding to PAR activation.70 Consistent with airway remodeling, calcium-binding epidermal growth factors have been shown to contribute to lymphatics during development.54,71 Previously, a mouse model of allergic airway disease demonstrated decreased lymphatic activity through Th2 cell-dependent down regulation of pro-lymphatic genes.72 Given this finding, the changes we observed in LYVE-1 expression may be a consequence of a developing allergen-mediated immune response. While pro-inflammatory cytokines and chemokines contribute to changes in lymphangiogenesis, the underlying mechanisms affecting lymphatic vessels and lymphatic endothelial cells in respiratory diseases are largely unknown.72 For example, while asthmatic patients have decreased lymphangiogenesis, patients with COPD exhibit increased lymphatic density evidenced by increased LYVE-1 and D2-40 staining in lung tissue.73 The decrease in LYVE-1 we identified suggests reduced lymphangiogenesis during SSDE-induced lung inflammation, which is similar to what has been observed in asthmatic patients.74 Furthermore, impaired lymphatics during an inflammatory response can lead to hypoxia and edema, both of which exacerbate asthmatic symptoms.75 Overall, the changes in inflammatory cytokines, eosinophil influx, the lung gene signature of SSDE exposure, and the changes in lymphatic vessel density are consistent with an allergen-mediated immune response following repetitive SSDE exposure.
Previous studies have identified environmental proteases as having a pathogenic role as human allergens.76–78 Indeed, active proteases within allergens are crucial in initiating Th2-dependent allergic inflammation, including via induction of allergic airway responses through epithelial cell activation.79–81 The bronchial epithelium is a first-line defense against inhaled toxicants and is thus critical to inflammation initiation in the setting of environmental dust exposures including through the release of pro-inflammatory cytokines such as TNFα and CXCL1.31 Furthermore, proteases found in agricultural dusts were found to be potent potentiators of lung inflammation.21 Given the role of environmental proteases in promoting allergic-type responses in the lung,17,18,79,82 our results corroborate that proteases are contributing to the lung immune response, including via actions on epithelial cell activation in SSDE-induced inflammation. It is notable that protease depletion of SSDE had the most profound effect in the early response of the bronchial epithelial cells (5 hours) but did not affect overall IL-6 and IL-8 accumulation at 24 hours. This may suggest that additional components of the dust are contributing to the overall pro-inflammatory response of these cells to SSDE in a temporal manner. Interestingly, blocking PAR-1 or PAR-2 pharmacologically did significantly impact 24-hour IL-6 and IL-8 release, suggesting that endogenously derived PAR signaling (as opposed to just exogenously derived from the dust proteases) resulting from bronchial epithelial cell activation could still be driving their overall inflammatory response. This finding has potentially important implications when considering future therapeutic strategies for PAR inhibition. Additionally, the lack of changes in LDH levels between SSDE-treated cells and saline controls suggest that the response of the bronchial epithelial cells is independent of epithelial cell cytotoxicity. In our investigations, depletion of protease activity from SSDE led to significant decreases in pro-inflammatory cytokine release from bronchial epithelium in vitro and following exposure in vivo. Allergen-derived proteases can disrupt tight junctions in lung epithelium by cleaving occludin and other tight junction proteins. This leads to impaired barrier function in airway epithelium, a common characteristic of asthma.83–87 The protease-mediated epithelial degradation leads to easier facilitation of antigen presentation in sub-epithelial tissues due to increased permeability, further triggering innate immunity. They can also initiate airway remodeling starting with the recruitment of inflammatory cells by activating epithelial cell receptors.17 The reduced total cell, neutrophil, and eosinophil influx following the repetitive exposure to PDSSDE versus SSDE suggests that proteases are involved with the recruitment of immune cells in response to SSDE inhalation. Our data also suggest that proteases are acting through PAR-1 and PAR-2 activation in bronchial epithelial cells. PARs are membrane-bound receptors on a variety of cells and are activated by proteases through proteolytic cleavage at the amino terminus.19,88,89 PAR-1 and PAR-2 are both activated by serine proteases and are among the most activated PARs in lung pathogenesis. The activation of PARs stimulates the activation of nuclear factor-kB, calcium-mediated signaling, and MAPK, leading to the secretion of various pro-inflammatory cytokines and chemokines.54 The antagonism of these two receptors in bronchial epithelial cells exposed to SSDE in vitro led to decreased secretion of IL-6 and IL-8, linking PAR-1 and -2 activation to SSDE-induced inflammation. PAR-2 is especially associated with antigen-induced asthma, including increasing release of IL-6, IL-8, GM-CSF and promoting eosinophil infiltration.16,88 Together, these findings provide evidence concerning the role of proteases derived from aerosolized dust in the pathogenesis of lung disease near the Salton Sea.
Our study does include numerous limitations. We have utilized a mouse model for Salton Sea dust exposure that relies on the preparation of an aqueous extract of dust that is intranasally instilled to mice, given as a single exposure challenge or daily for 7 days. While this is a well-established and robust model for dust exposures,21,22,25,90–96 it cannot completely replicate the chronic inhaled exposure experienced by individuals living near the Salton Sea. Future studies using a chamber inhaled exposure method and for longer time durations are warranted to ascertain the lung impacts of more chronic exposures to dusts found in the Salton Sea region. Our investigations also utilized dust samples from a single site near the Salton Sea; previous studies have explored dust samples from other regions surrounding the Salton Sea, collected during a similar time period and by the same methods, and have characterized their major compositional components, including elements and soluble anions.9 It is notable, however, that our data are among the first studies to characterize the impacts of dusts associated with salt water lake evaporation on lung health, and they will be important for environmental and health policy considerations for the Inland Empire of California as well as other regions in the world experiencing similar conditions.
Furthermore, while our data identify a role for proteases in the lung inflammatory response to SSDE, the use of a protease cocktail inhibitor limits the capacity for identifying the specific protease(s) or classes of protease(s) involved in eliciting this response. Thus, future studies are needed to better characterize the proteases found in these environmental dusts. In addition, while we have designed the SSDE protease activity depletion in such a way to minimize impacts on endogenous protease inhibition, we cannot rule out the possibility that some endogenous proteases may also be inhibited, and this is a recognized limitation of this method.21 We have also used commercially available inhibitors for studying the impact of PAR inhibition on the airway epithelial response to SSDE; however, alternative methods could have been utilized including siRNA as we have previously published.21 Nonetheless, equivalent outcomes were found when using the pharmacologic inhibitors versus siRNA strategies, justifying our use of the inhibitors herein.
Conclusion
Given the high incidence of respiratory disease in the Imperial Valley,10 our results suggest that the high concentration of PM, including dusts from regions surrounding the Salton Sea, could be contributing to lung inflammation and disease risk in these individuals. Furthermore, our findings indicate that this dust-induced inflammation may be in part protease-mediated, which can potentially serve as a therapeutic target. In pre-clinical studies, serine protease inhibitors have been proven to be effective in reducing asthma-induced inflammation and cytokine release in mice.97,98 Additionally, the inhibition of PAR-2 in a mouse model for allergen-induced asthma led to decreased airway hyperresponsiveness and eosinophilia.99 Although there have been clinical trials for PAR antagonism in the treatment of cardiovascular and gastrointestinal disease,54 there have yet to be any accepted clinical trials for PAR-1 or PAR-2 antagonism in humans for the treatment of lung disease. Our findings warrant further investigations to ascertain the health risks associated with dust inhalation and other environmental exposures associated with the Salton Sea in order to identify suitable preventive or therapeutic options for exposed individuals living in this and other similar ecological regions.
Acknowledgments
This work was supported in part by the National Institute of Environmental Health Sciences (R00ES025819 to TMN) and the National Institute of Allergy and Infectious Diseases (R01AI153195 to MGN and TMN). Abigail C Burr and Jalene V Velazquez are co-first authors for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hart CM, Gonzalez MR, Simpson EP, Hurlbert SH. Salinity and fish effects on Salton Sea microecosystems: zooplankton and nekton. Hydrobiologia. 1998;381(1–3):129–152.
2. Tompson AFB. Born from a flood: the Salton Sea and its story of survival. J Earth Sci. 2016;27(1):89–97. doi:10.1007/s12583-016-0630-7
3. Barnum DA, Bradley T, Cohen M, Wilcox B, Yanega G State of the Salton Sea–A science and monitoring meeting of scientists for the Salton Sea: U.S. Geological Survey Open-File Report 2017-1005. 2017.
4. Johnston JE, Razafy M, Lugo H, Olmedo L, Farzan SF. The disappearing Salton Sea: a critical reflection on the emerging environmental threat of disappearing saline lakes and potential impacts on children’s health. Sci Total Environ. 2019;663:804–817. doi:10.1016/j.scitotenv.2019.01.365
5. Franklin BA, Brook R, Arden Pope C. Air pollution and cardiovascular disease. Curr Probl Cardiol. 2015;40(5):207–238. doi:10.1016/j.cpcardiol.2015.01.003
6. Samoli E, Nastos P, Paliatsos A, Katsouyanni K, Priftis K. Acute effects of air pollution on pediatric asthma exacerbation: evidence of association and effect modification. Environ Res. 2011;111(3):418–424. doi:10.1016/j.envres.2011.01.014
7. Forastiere F, Stafoggia M, Tasco C, et al. Socioeconomic status, particulate air pollution, and daily mortality: differential exposure or differential susceptibility. Am J Ind Med. 2007;50(3):208–216. doi:10.1002/ajim.20368
8. Frie AL, Dingle JH, Ying SC, Bahreini R. The effect of a receding saline lake (the Salton Sea) on airborne particulate matter composition. Environ Sci Technol. 2017;51(15):8283–8292. doi:10.1021/acs.est.7b01773
9. Frie A, Garrison A, Schaefer M, et al. Dust sources in the Salton Sea basin: a clear case of an anthropogenically impacted dust budget. Environ Sci Technol. 2019;53(16):9378–9388. doi:10.1021/acs.est.9b02137
10. Marshall JR. Why emergency physicians should care about the Salton Sea. West J Emerg Med. 2017;18(6):1008–1009. doi:10.5811/westjem.2017.8.36034
11. Vogl RA, Henry RN. Characteristics and contaminants of the Salton Sea sediments. Feature. Hydrobiologia. 2002;473(1–3):47. doi:10.1023/A:1016509113214
12. Ahmed CMS, Cui Y, Frie AL, et al. Exposure to dimethyl selenide (DMSe)-derived secondary organic aerosol alters transcriptomic profiles in human airway epithelial cells. Environ Sci Technol. 2019;53(24):14660–14669. doi:10.1021/acs.est.9b04376
13. Anastasiou C. Fungi from Salt Lakes. I. A new species of Clavariopsis. Mycologia. 1961;53(1):11–16. doi:10.1080/00275514.1961.12017929
14. Carmichael WW, Li R. Cyanobacteria toxins in the Salton Sea. Saline Syst. 2006;2:5. doi:10.1186/1746-1448-2-5
15. Paul C, Pohnert G. Induction of protease release of the resistant diatom chaetoceros didymus in response to lytic enzymes from an algicidal bacterium. PLoS One. 2013;8(3):e57577. doi:10.1371/journal.pone.0057577
16. Matsumura Y. Role of allergen source-derived proteases in sensitization via airway epithelial cells. J Allergy. 2012;2012(2012):903659. doi:10.1155/2012/903659
17. Jacquet A. Interactions of airway epithelium with protease allergens in the allergic response. Clin Exp Allergy. 2011;41(3):305–311. doi:10.1111/j.1365-2222.2010.03661.x
18. Kauffman HF, Tomee JF, van de Riet MA, Timmerman AJ, Borger P. Protease-dependent activation of epithelial cells by fungal allergens leads to morphologic changes and cytokine production. J Allergy Clin Immunol. 2000;105(6 Pt 1):1185–1193. doi:10.1067/mai.2000.106210
19. Reed C, Kita H. The role of protease activation of inflammation in allergic respiratory diseases. J Allergy Clin Immunol. 2004;114(5):997–1008. doi:10.1016/j.jaci.2004.07.060
20. Peng X, Maltz MR, Botthoff JK, et al. Establishment and characterization of a multi-purpose large animal exposure chamber for investigating health effects. Rev Sci Instrum. 2019;90(3):035115. doi:10.1063/1.5042097
21. Romberger DJ, Heires AJ, Nordgren TM, et al. Proteases in agricultural dust induce lung inflammation through PAR-1 and PAR-2 activation. Am J Physiol Lung Cell Mol Physiol. 2015;309(4):L388–99. doi:10.1152/ajplung.00025.2015
22. Poole JA, Wyatt TA, Oldenburg PJ, et al. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296(6):L1085–95. doi:10.1152/ajplung.90622.2008
23. Raftis E, Delday M, Cowie P, et al. Bifidobacterium breve MRx0004 protects against airway inflammation in a severe asthma model by suppressing both neutrophil and eosinophil lung infiltration. Sci Rep. 2018;8(1):1–13. doi:10.1038/s41598-018-30448-z
24. Gueders M, Balbin M, Rocks N, et al. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175(4):2589–2597. doi:10.4049/jimmunol.175.4.2589
25. Nordgren TM, Friemel TD, Heires AJ, Poole JA, Wyatt TA, Romberger DJ. The omega-3 Fatty Acid docosahexaenoic Acid attenuates organic dust-induced airway inflammation. Nutrients. 2014;6(12):5434–5452. doi:10.3390/nu6125434
26. Nordgren TM, Bailey KL, Heires AJ, Katafiasz D, Romberger DJ. Effects of agricultural organic dusts on human lung-resident mesenchymal stem (stromal) cell function. Toxicol Sci. 2018;162(2):635–644. doi:10.1093/toxsci/kfx286
27. Lantz MS, Ciborowski P. Zymographic techniques for detection and characterization of microbial proteases. Methods Enzymol. 1994;235:563–594.
28. Kan S, Hariyadi DM, Grainge C, Knight DA, Bartlett NW, Liang M. Airway epithelial-targeted nanoparticles for asthma therapy. Am J Physiol Lung Cell Mol Physiol. 2020;318(3):L500–l509. doi:10.1152/ajplung.00237.2019
29. Wu M, Du Y, Liu Y, et al. Low molecular weight hyaluronan induces lymphangiogenesis through LYVE-1-mediated signaling pathways. PLoS One. 2014;3:556.
30. Johnson L, Prevo R, Clasper S, Jackson D. Inflammation-induced uptake and degradation of the lymphatic endothelial hyaluronan receptor LYVE-1. J Biol Chem. 2007;282(46):33671–33680. doi:10.1074/jbc.M702889200
31. Takizawa H. Airway epithelial cells as regulators of airway inflammation (Review). Int J Mol Med. 1998;1(2):367–378.
32. Giang J, Seelen M, van Doorn M, Rissmann R, Prens E, Damman J. Complement activation in inflammatory skin diseases. Front Immunol. 2018;9:639. doi:10.3389/fimmu.2018.00639
33. Volanakis J, Narayana S. Complement factor D, a novel serine protease. Protein Sci. 1996;5(4):553–564. doi:10.1002/pro.5560050401
34. Russell J, Ley T. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi:10.1146/annurev.immunol.20.100201.131730
35. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131(4):959–971. doi:10.1016/j.jaci.2013.01.046
36. Center D, Kornfeld H, Cruikshank W. Interleukin-16. Int J Biochem Cell Biol. 1997;29(11):757–766. doi:10.1016/s1357-2725(97)00053-8
37. Schmitt C, Tonnelle C, Dalloul A, Chabannon C, Debré P, Rebollo A. Aiolos and Ikaros: regulators of lymphocyte development, homeostasis and lymphoproliferation. Apoptosis. 2002;7(3):277–284. doi:10.1023/a:1015372322419
38. Medvedovic J, Ebert A, Tagoh H, Busslinger M. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol. 2011;111:179–206. doi:10.1016/B978-0-12-385991-4.00005-2
39. McWilliam H, Villadangos J. MR1: a multi-faceted metabolite sensor for T cell activation. Curr Opin Immunol. 2020;64:124–129. doi:10.1016/j.coi.2020.05.006
40. Klemann C, Wagner L, Stephan M, von Hörsten S. Cut to the chase: a review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin Exp Immunol. 2016;185(1):1–21. doi:10.1111/cei.12781
41. Gulati K, Gangele K, Agarwal N, Jamsandekar M, Kumar D, Poluri K. Molecular cloning and biophysical characterization of CXCL3 chemokine. Int J Biol Macromol. 2018;107(PtA):575–584. doi:10.1016/j.ijbiomac.2017.09.032
42. Larsson K. Inflammatory markers in COPD. Clin Respir J. 2008;2(Suppl 1):84–87. doi:10.1111/j.1752-699X.2008.00089.x
43. Palecanda A, Paulauskis J, Al-Mutairi E, et al. Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J Exp Med. 1999;189(9):1497–1506. doi:10.1084/jem.189.9.1497
44. Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol. 2004;36(10):1882–1886. doi:10.1016/j.biocel.2003.10.019
45. Tsuji K, Ebihara Y. Expression of G-CSF receptor on myeloid progenitors. Leuk Lymphoma. 2001;42(6):1351–1357. doi:10.3109/10428190109097763
46. Gordon S. Macrophage-restricted molecules: role in differentiation and activation. Immunol Lett. 1999;65(1–2):5–8. doi:10.1016/s0165-2478(98)00116-3
47. Lin H, Stacey M, Stein-Streilein J, Gordon S. F4/80: the macrophage-specific adhesion-GPCR and its role in immunoregulation. Adv Exp Med Biol. 2010;706:149–156. doi:10.1007/978-1-4419-7913-1_13
48. Wu Z, Zhang Z, Lei Z, Lei P. CD14: biology and role in the pathogenesis of disease. Cytokine Growth Factor Rev. 2019;48:24–31. doi:10.1016/j.cytogfr.2019.06.003
49. Hadebe S, Brombacher F, Brown G. C-Type lectin receptors in asthma. Front Immunol. 2018;9:733. doi:10.3389/fimmu.2018.00733
50. Negi S, Pahari S, Bashir H, Agrewala J. Gut Microbiota regulates mincle mediated activation of lung dendritic cells to protect against mycobacterium tuberculosis. Front Immunol. 2019;10:1142. doi:10.3389/fimmu.2019.01142
51. Wells C, Salvage-Jones J, Li X, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol. 2008;180(11):7404–7413. doi:10.4049/jimmunol.180.11.7404
52. Wang H, FitzPatrick M, Wilson N, et al. CSF3R/CD114 mediates infection-dependent transition to severe asthma. J Allergy Clin Immunol. 2019;143(2):785–788.e6. doi:10.1016/j.jaci.2018.10.001
53. Deng X, Mercer P, Scotton C, Gilchrist A, Chambers R. Thrombin induces fibroblast CCL2/JE production and release via coupling of PAR 1 to Gαq and cooperation between ERK1/2 and Rho kinase signaling pathways. Mol Biol Cell. 2008;19:2520–2533. doi:10.1091/mbc.e07-07-0720
54. Heuberger D, Schuepbach R. Protease-activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J. 2019;17:4. doi:10.1186/s12959-019-0194-8
55. Asokananthan N, Graham PT, Stewart DJ, et al. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immunol. 2002;169(8):4572–4578. doi:10.4049/jimmunol.169.8.4572
56. Bhat RK, Page K, Tan A, Hershenson MB. German cockroach extract increases bronchial epithelial cell interleukin-8 expression. Clin Exp Allergy. 2003;33(1):35–42. doi:10.1046/j.1365-2222.2002.01481.x
57. Boitano S, Flynn AN, Sherwood CL, et al. Alternaria alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR2. Am J Physiol Lung Cell Mol Physiol. 2011;300(4):L605–14. doi:10.1152/ajplung.00359.2010
58. Bach AJ, Brazel AJ, Lancaster N. Temporal and spatial aspects of blowing dust in the Mojave and Colorado deserts of Southern California, 1973–1994. Analytic Serial. Physical Geography. 1996;17(4):329–353. doi:10.1080/02723646.1996.10642589
59. King J, Etyemezian V, Sweeney M, Buck BJ, Nikolich G. Dust emission variability at the Salton Sea, California, USA. Aeolian Res. 2011;3(1):67–79. doi:10.1016/j.aeolia.2011.03.005
60. Goudie AS. Desert dust and human health disorders. Environ Int. 2014;63:101–113. doi:10.1016/j.envint.2013.10.011
61. Kunii O, Hashizume M, Chiba M, et al. Respiratory symptoms and pulmonary function among school-age children in the Aral Sea region. Arch Environ Health. 2003;58(11):676–682. doi:10.3200/AEOH.58.11.676-682
62. Hong YC, Pan XC, Kim SY, et al. Asian Dust Storm and pulmonary function of school children in Seoul. Sci Total Environ. 2010;408(4):754–759. doi:10.1016/j.scitotenv.2009.11.015
63. Chien LC, Yang CH, Yu HL. Estimated effects of Asian dust storms on spatiotemporal distributions of clinic visits for respiratory diseases in Taipei children (Taiwan). Environ Health Perspect. 2012;120(8):1215–1220. doi:10.1289/ehp.1104417
64. Kanatani KT, Ito I, Al-Delaimy WK, et al. Desert dust exposure is associated with increased risk of asthma hospitalization in children. Am J Respir Crit Care Med. 2010;182(12):1475–1481. doi:10.1164/rccm.201002-0296OC
65. Chen S, Yao L, Huang P, et al. Blockade of the NLRP3/caspase-1 axis ameliorates airway neutrophilic inflammation in a toluene diisocyanate-induced murine asthma model. Toxicol Sci. 2019;170(2):462–475. doi:10.1093/toxsci/kfz099
66. Moore WC, Hastie AT, Li X, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. 2014;133(6):1557–63.e5. doi:10.1016/j.jaci.2013.10.011
67. Davoine F, Lacy P. Eosinophil cytokines, chemokines, and growth factors: emerging roles in immunity. Front Immunol. 2014;5:570. doi:10.3389/fimmu.2014.00570
68. Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 2012;8(9):1281–1290. doi:10.7150/ijbs.4874
69. Arredouani MS, Franco F, Imrich A, et al. Scavenger receptors SR-AI/II and MARCO limit pulmonary dendritic cell migration and allergic airway inflammation. J Immunol. 2007;178(9):5912–5920. doi:10.4049/jimmunol.178.9.5912
70. Sakai H, Suto W, Kai Y, Chiba Y. Mechanisms underlying the pathogenesis of hyper-contractility of bronchial smooth muscle in allergic asthma. J Smooth Muscle Res. 2017;53:37–47. doi:10.1540/jsmr.53.37
71. Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438(7070):946–953. doi:10.1038/nature04480
72. Stump B, Cui Y, Kidambi P, Lamattina A, El-Chemaly S. Lymphatic changes in respiratory diseases: more than just remodeling of the lung? Am J Respir Cell Mol Biol. 2017;57(3):272–279. doi:10.1165/rcmb.2016-0290TR
73. Hardavella G, Tzortzaki EG, Siozopoulou V, et al. Lymphangiogenesis in COPD: another link in the pathogenesis of the disease. Respir Med. 2012;106(5):687–693. doi:10.1016/j.rmed.2011.11.011
74. Ebina M. Remodeling of airway walls in fatal asthmatics decreases lymphatic distribution; beyond thickening of airway smooth muscle layers. Allergol Int. 2008;57(2):165–174. doi:10.2332/allergolint.O-07-497
75. El-Chemaly S, Levine S, Moss J. Lymphatics in lung disease. Ann N Y Acad Sci. 2008;1131:195–202. doi:10.1196/annals.1413.017
76. Hewitt CR, Brown AP, Hart BJ, Pritchard DI. A major house dust mite allergen disrupts the immunoglobulin E network by selectively cleaving CD23: innate protection by antiproteases. J Exp Med. 1995;182(5):1537–1544. doi:10.1084/jem.182.5.1537
77. Schulz O, Sewell HF, Shakib F. Proteolytic cleavage of CD25, the alpha subunit of the human T cell interleukin 2 receptor, by Der p 1, a major mite allergen with cysteine protease activity. J Exp Med. 1998;187(2):271–275. doi:10.1084/jem.187.2.271
78. Kolattukudy PE, Lee JD, Rogers LM, et al. Evidence for possible involvement of an elastolytic serine protease in aspergillosis. Infect Immun. 1993;61(6):2357–2368. doi:10.1128/iai.61.6.2357-2368.1993
79. Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, Corry DB. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol. 2002;169(10):5904–5911. doi:10.4049/jimmunol.169.10.5904
80. Kale S, Agrawal K, Gaur S, Arora N. Cockroach protease allergen induces allergic airway inflammation via epithelial cell activation. Sci Rep. 2017;7:42341. doi:10.1038/srep42341
81. Cayrol C, Duval A, Schmitt P, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. 2018;19(4):375–385. doi:10.1038/s41590-018-0067-5
82. Page K. Role of cockroach proteases in allergic disease. Curr Allergy Asthma Rep. 2012;12(5):448–455. doi:10.1007/s11882-012-0276-1
83. Holgate S. The airway epithelium is central to the pathogenesis of asthma. Allergol Int. 2008;57(1):1–10. doi:10.2332/allergolint.R-07-154
84. Holgate ST. Epithelial damage and response. Clin Exp Allergy. 2000;30(Suppl 1):37–41. doi:10.1046/j.1365-2222.2000.00095.x
85. Holgate S. Epithelium dysfunction in asthma. J Allergy Clin Immunol. 2007;120(6):1233–1244. doi:10.1016/j.jaci.2007.10.025
86. Gon Y, Hashimoto S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int. 2018;67(1):12–17. doi:10.1016/j.alit.2017.08.011
87. Bartemes K, Kita H. Dynamic role of epithelium-derived cytokines in asthma. Clin Immunol. 2012;143(3):222–235. doi:10.1016/j.clim.2012.03.001
88. Schmidlin F, Amadesi S, Dabbagh K, et al. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol. 2002;169(9):5315–5321. doi:10.4049/jimmunol.169.9.5315
89. Reed C. Inflammatory effect of environmental proteases on airway mucosa. Curr Allergy Asthma Rep. 2007;7(5):368–374. doi:10.1007/s11882-007-0056-5
90. Nordgren TM, Bauer CD, Heires AJ, et al. Maresin-1 reduces airway inflammation associated with acute and repetitive exposures to organic dust. Translational Res. 2015;166(1):57–69. doi:10.1016/j.trsl.2015.01.001
91. Nordgren TM, Heires AJ, Bailey KL, et al. Docosahexaenoic acid enhances amphiregulin-mediated bronchial epithelial cell repair processes following organic dust exposure. Am J Physiol Lung Cell Mol Physiol. 2018;314(3):L421–L431. doi:10.1152/ajplung.00273.2017
92. Nordgren TM, Heires AJ, Zempleni J, Swanson BJ, Wichman C, Romberger DJ. Bovine milk-derived extracellular vesicles enhance inflammation and promote M1 polarization following agricultural dust exposure in mice. J Nutr Biochem. 2019;64:110–120. doi:10.1016/j.jnutbio.2018.10.017
93. Poole JA, Gleason AM, Bauer C, et al. CD11c +/CD11b+cells are critical for organic dust–elicited murine lung inflammation. Am J Respir Cell Mol Biol. 2012;47(5):652–659. doi:10.1165/rcmb.2012-0095OC
94. Poole JA, Wyatt TA, Romberger DJ, et al. MyD88 in lung resident cells governs airway inflammatory and pulmonary function responses to organic dust treatment. Respir Res. 2015;16:111. doi:10.1186/s12931-015-0272-9
95. Poole JA, Nordgren TM, Heires AJ, et al. Amphiregulin modulates murine lung recovery and fibroblast function following exposure to agriculture organic dust. Am J Physiol Lung Cell Mol Physiol. 2020;318(1):L180–l191. doi:10.1152/ajplung.00039.2019
96. Romberger DJ, Heires AJ, Nordgren TM, et al. Beta2-adrenergic agonists attenuate organic dust-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2016;311(1):L101–10. doi:10.1152/ajplung.00125.2016
97. Lin C, Lin L, Wang S, et al. The effect of serine protease inhibitors on airway inflammation in a chronic allergen-induced asthma mouse model. Mediators Inflamm. 2014;2014(2014):879326. doi:10.1155/2014/879326
98. Oh S, Pae C, Lee D, et al. Tryptase inhibition blocks airway inflammation in a mouse asthma model. J Immunol. 2002;168(4):1992–2000. doi:10.4049/jimmunol.168.4.1992
99. Asaduzzaman M, Nadeem A, Arizmendi N, et al. Functional inhibition of PAR2 alleviates allergen-induced airway hyperresponsiveness and inflammation. Clin Exp Allergy. 2015;45(12):1844–1855. doi:10.1111/cea.12628
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.