Back to Journals » International Journal of General Medicine » Volume 17
Long-Term Follow-Up of Patients Undergoing Thalidomide Therapy for Transfusion-Dependent β-Thalassaemia: A Single-Center Experience
Authors Zhu W ![]() , He Y, Huang M
, He Y, Huang M ![]() , Fu S, Liu Z, Wang X, Li Z, Li X, Chen J, Li Y
, Fu S, Liu Z, Wang X, Li Z, Li X, Chen J, Li Y ![]()
Received 12 March 2024
Accepted for publication 24 April 2024
Published 30 April 2024 Volume 2024:17 Pages 1729—1738
DOI https://doi.org/10.2147/IJGM.S462991
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Weijian Zhu,1 Ying He,1 Mufang Huang,1 Shezhu Fu,1 Ziyi Liu,1 Xiaoqi Wang,1 Zhixin Li,1 Xiaoliang Li,1 Jiangming Chen,2 Yangqiu Li3
1Department of Hematology, Zhuhai Clinical Medical College of Jinan University (Zhuhai People’s Hospital), Zhuahai, 519050, People’s Republic of China; 2Department of Haematology, Wuzhou Gongren Hospital, Wuzhou, 543001, People’s Republic of China; 3Institute of Hematology, School of Medicine, Jinan University, Guangzhou, 10632, People’s Republic of China
Correspondence: Yangqiu Li, Institute of Hematology, School of Medicine, Jinan University, Guangzhou, 510632, People’s Republic of China, Email [email protected]
Objective: We evaluated the long-term safety and efficacy of thalidomide in the treatment of transfusion-dependent β-thalassemia (TDT).
Methods: Fifty patients with TDT were treated with thalidomide and followed-up for 5 years. Thalidomide at a 50 mg dose was administered once a day after dinner. The dose was increased to 150 mg/d after 3 d if well tolerated. After 1 year of treatment, the hemoglobin (Hb) level was stabilized at its maximum, and thalidomide was gradually reduced and maintained at the minimum dose. The hematological response, transfusion dependence, and haemolytic indicators were assessed.
Results: At 9 month of follow-up, 38 (76%) patients achieved an excellent response, 1 (2%) a good response, 4(8%) a minor response, and 7(14%) did not show a response. The overall response rate was 86%. At 9 months, the Hb level increased from 79.0 ± 13.2 g/L at baseline to 99.0 ± 13.7g/L (P< 0.001). Patients who achieved excellent response continued to show an increase in Hb levels during follow-up. At 48 months, the mean Hb level was 98.99 ± 10.3g/L; 21 patients (84.0%) became transfusion independent. Thalidomide was reduced and maintained to 25 mg/d in three of these patients. Moreover, five patients completed 60 months of follow-up, and with a mean Hb level of 99.8 ± 6.7g/L. During follow-up, grade 1– 2 adverse drug reactions were noted; however, no grade 3 or higher adverse event was reported. However, no decrease in hemolytic indicators was observed.
Conclusion: Thalidomide was well tolerated in the long term, while it significantly improved Hb levels and reduced the transfusion burden.
Keywords: β-thalassemia, transfusion-dependent, thalassemia, thalidomide, foetal hemoglobin
Introduction
Beta-thalassemia is one of the most common single gene inherited diseases. The World Health Organization reported that approximately 150 million people worldwide carry the hemoglobinopathy gene, representing a substantial global health burden. The incidence of hemolytic anemia is mainly concentrated in tropical and subtropical areas, primarily on the Mediterranean coast, African America, North Africa, Southeast Asia, the Indian subcontinent, and Melanesia in the Pacific Islands.1 In these areas, the high incidence of hemolytic anemia caused by hemoglobinopathies is a serious public health problem.2,3 Beta-thalassemia is mainly caused by point mutation of the β-globin gene or loss or insertion of individual nucleotides. This results in accumulation of excess, unbound α-globin chains that precipitate in erythroid precursors in the bone marrow and mature erythrocytes, leading to ineffective erythropoiesis and peripheral hemolysis.4 Beta-thalassemia was classified clinically as transfusion dependent or non-transfusion dependent.5 Patients with transfusion-dependent β-thalassemia need regular red blood cell transfusions and iron chelation therapy to long-term survival; however, blood transfusion is associated with iron overload, which causes end-organ damage, thus, increasing morbidity and mortality. Currently, curative options for these diseases include allogeneic bone marrow transplantation and gene therapy or gene editing;6–8 however these methods are costly, and safety challenges limit their use worldwide, especially in developing countries.
Fetal hemoglobin (HbF) inducers activate γ-globin genes that are largely turned off in patients with β-thalassaemia. The synthesized γ-chain can replace the defective β-chain, and the HbF formed by the relative excess of α-chain can be compensated by an increase in the production of the β-like globin molecule, the γ-globin. Reducing the number of unbound α-globin chains in red blood cells may ameliorate its detrimental effects and increase total (Hb) levels.9 Hydroxyurea is the most widely use HbF inducer used in sickle cell disease; however, it is less effective for patients with β-thalassemia.10 Other inducers such as 5-azacytidine and decitabine have not been widely use in clinical practice due to their poor efficacy and potential carcinogenic effects.
Thalidomide, an immunomodulatory drug and strong HbF inducer, reduces dependence on transfusions by inducing γ-globin gene expression and significantly increasing in Hb concentration.11–14 In a Phase 2, multicenter, randomized, double-blind clinical trial, thalidomide effectively improved Hb levels in patients with transfusion-dependent β-thalassaemia (TDT) with few adverse events (AEs).15 All the clinical trials were short-term, and the long-term efficacy of thalidomide in patients with TDT remains unknown. Therefore, we aimed to evaluate the long-term safety and clinical response to thalidomide and its relationship with hemolytic markers, such as bilirubin.
Methods
Patients
This prospective study enrolled patients aged ≥14 years who were diagnosed with TDT from May 2018 to December 2020. Transfusion dependence was defined when as the need of at least eight transfusions or 100 mL per kilogram of body weight of leukoreduced packed RBCs per year, or frequent transfusion to maintain Hb > 70 g/L in the 2 years before enrolment. The inclusion and exclusion criteria is available in Supplementary Material 1. This study recruited patients from Guangdong Province, South China. All patients provided written informed consent prior to enrolment. This consent was obtained from the parents or guardians of the adolescent participants. The protocol and consent form were approved by the independent ethics committee of the Zhuhai People’s Hospital (ethics number: 2018(4)/ZY[2019](28)), and the study was conducted in accordance with the tenets of the Declaration of Helsinki.
Treatment
Patients received thalidomide at a dose of 50mg/d, administered after dinner. For those who could tolerate the drug, the dose was increased to 150 mg/d after 3 d. If necessary, the dose could be adjusted according to the adverse reactions. All patients were advised to take folic acid (5 mg/d) orally, and aspirin (100 mg/d) was prescribed to prevent thrombosis. Patients with aspirin contraindications (G6PD deficiency) were administered clopidogrel as prophylaxis. Transfusion was recommended to maintain the Hb level at > 90.0 g/L, based on local blood transfusion standards, during treatment.
Considering that thalidomide has the potential risk of teratogenicity and lethality to fetuses, pregnancy was forbidden during the period of taking thalidomide administration. Fertile females must took a medically supervised pregnancy test before starting the trial and during each clinic visit, and were engaged to take reliable contraceptive measures during treatment. The dosage of thalidomide is tapered after 12 months based on the hemoglobin level and maintained to keep the hemoglobin at an appropriate level.
Treatment Response Criteria
Response to thalidomide was defined as follows: excellent response, free of blood transfusion for at least 6 weeks, reaching a final total Hb level of 90 g/L, or an elevation in total Hb level of ≥ 20 g/L; good response, a reduction in the transfusion burden of at least 50% from baseline (the 3-month period before the first of thalidomide); minor response, reduction of at least 33% and <50% from baseline; and no response, reduction in transfusion burden of <33%.16,17
Patients were followed up every 3 months for 1 year and every 6 months thereafter. Follow-up included monitoring for thrombosis, nervous system toxicity, and other possible AEs as well as routine blood, serum alanine transaminase (ALT), aspartate transferase (AST), total bilirubin (TBIL), indirect bilirubin (DBIL), indirect bilirubin (IBIL), blood urea nitrogen(BUN), creatinine(Cr), and lactate dehydrogenase (LDH) levels. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.18
Statistical methods
Statistical analysis was performed using SPSS (version 24.0; SPSS Institute, Cary, NC), with a P-value ≤ 0.05 indicating statistical significance. Continuous variables that approximate a normal distribution are represented by mean ± standard deviation. Median (interquartile range) was used for continuous variables that did not follow a normal distribution. Paired t-tests were used for pre- and post-treatment comparisons. Analysis of the measurement indicators at each monitoring time point was performed using a repeated-measures analysis of variance. Pearson’s correlation analysis was used for correlation analysis.
Safety data were used to describe the occurrence of blood or non-blood toxic reactions in detail, calculate the incidence rates of different events, and determine the composition ratios of the severity of each event.
Results
Fifty patients were included in the study and followed-up for at least 9 months until October 2023. Mean follow-up was 4.2 years (standard deviation, 0.74) and the median value was 4.21 years. The longest follow-up time was 5.5 years. Of these patients, 29 were enrolled in the randomized controlled trial15 and were followed up after the extension study (Figure 1). Moreover, 24 men and 26 women, with a median age of 16.5 years (range, 14–30 years) were included. Nineteen patients underwent splenectomy. Eleven (22%) patients had β0/β0, 26 (52.0%) β0/β+, including a case of a merger - SEA/α-gene, and 13 (26.0%) β+/β+ genotype (Supplementary Table 1). The baseline patient characteristics are shown in Table 1.
 |
Table 1 Baseline Demographic Characteristics of Patients |
 |
Figure 1 Treatment, and follow-up. Twenty-nine patients were enrolled in this randomized controlled trial. The placebo-controlled period included data from month 0 (baseline) to 12. Additional 21 patients who meet the inclusion criteria were enrolled in this open-label study. The study population comprised of 50 patients who received at least one dose of thalidomide. (allo-HSCT: allogeneic hematopoietic stem cell transplantation). |
Primary Efficacy Analysis
Fifty patients were administered thalidomide, and its efficacy was evaluated at 3, 6, 9, 12, 18, 24, 36, 48, and 60 months. The primary endpoints were changes in Hb levels and transfusion volume in patients who received thalidomide. After 3 months of treatment, 24 (48.0%), 11 (22.0%), 11 (22.0%) and 4 (8.0%) patients had excellent, good, minor, and no response respectively. The overall response rate (ORR) was 92%, with excellent, good and minor rates accounting for 48.0%,22.0% and 22.0%, respectively. At 6 months, 35 patients (70.0%) showed an excellent response. The effectiveness rate was higher than in the third month. Four (8.0%) patients showed a good response, four (8.0%) a minor response, and seven (14.0%) no response. The ORR was 86.0%. At 9 months, 38 (76.0%), 1 (2.0%), 4 (8.0%), and 7(14.0%) patients had excellent, good, minor, no responses, respectively. The ORR was 86.0%.
A significant increase was observed in Hb levels after 3 months. The Hb levels increased from 79. 0±13.2 g/L at baseline to 92.5 ± 14.4g/L (P<0.001) after thalidomide therapy. At month 6, Hb level increased to 97.2 ± 15.7g/L (P<0.001), presenting a significant increase of 18.2 ± 20.1g/L. At month 9, Hb increased to 99.0 ± 13.7g/L (P<0.001), with a rise of 20.1 ± 19.8 g/L from baseline, After 12 months, thalidomide was discontinued in four patients who failed to respond, and a total of 46 patients completed 12 months of follow-up, with an average Hb level of 101.4 ± 13.7g/L. After 12 months, the minimum dose was maintained according to Hb levels. A total of 42 patients were followed up for 24 months; 7 patients discontinued treatment due to ineffectiveness, and 1 patient due to nephrotic syndrome. The mean Hb level was 102.6 ± 10.2g/L, which persisted throughout the follow-up period (Table 2, and Figure 2).
 |
Table 2 Change in Haemoglobin Levels |
 |
Figure 2 The mean change in haemoglobin levels from baseline to 24 months after thalidomide treatment. The effective group includes excellent responder, good and minor responders. The level of Hb was expressed as mean ± standard deviation (SD) and P- values were determined by Student’s t-test. |
Analysis of Long-Term Efficacy After Drug Reduction
Thalidomide dose was reduced after 12 months and maintained to keep the hemoglobin at an appropriate level at 30 months (Hb level remained stable at ≥90g/L). A total of 38 patients receiving oral thalidomide for 36 months had a mean Hb level of 100.8 ± 8.8 g/L, of whom 31 (81.5%) had an excellent response (free of blood transfusion and maintaining Hb level at >90g/L), 3 (7.9%) had a good response, and 3 (7.9%) had a minor response. The patient who achieved a reduction in transfusion burden of ≤33% continued to receive thalidomide. Two patients with excellent responses were proposed to receive gene therapy; one patient stopped using thalidomide after 2 years and the other at 42 months. One patient discontinued treatment at 42 months due to receive allogeneic hematopoietic stem cell transplantation. A total of 25 patients received oral thalidomide for 48 months. The mean Hb level was 98.99 ± 10.3 g/L, 21 (84.0%),1 (4.0%), and 3 (12.0%) patients had an excellent, good, and minor responses respectively. From the 21 patients who had an excellent response, in 3 the dose was reduced and maintained to 25 mg/d, whereas the dose was 50 mg/d, 75 mg/d, 100 mg/d, 125 mg/d, and 150mg/d in 1,2,9,3 and 3 patients respectively. Of the 21 patients with and excellent response, 15 (71.4%) had a −28 (HBB:c-78A>G) locus mutation, suggesting that this mutation may be associated with a high thalidomide response. However, Hb level did not correlate with HbF levels at month 48 (r=−0.364, P>0.05; Table 3). Furthermore, five patients were followed for 60 months, with a mean Hb level of 99.8 ± 6.7g/L.
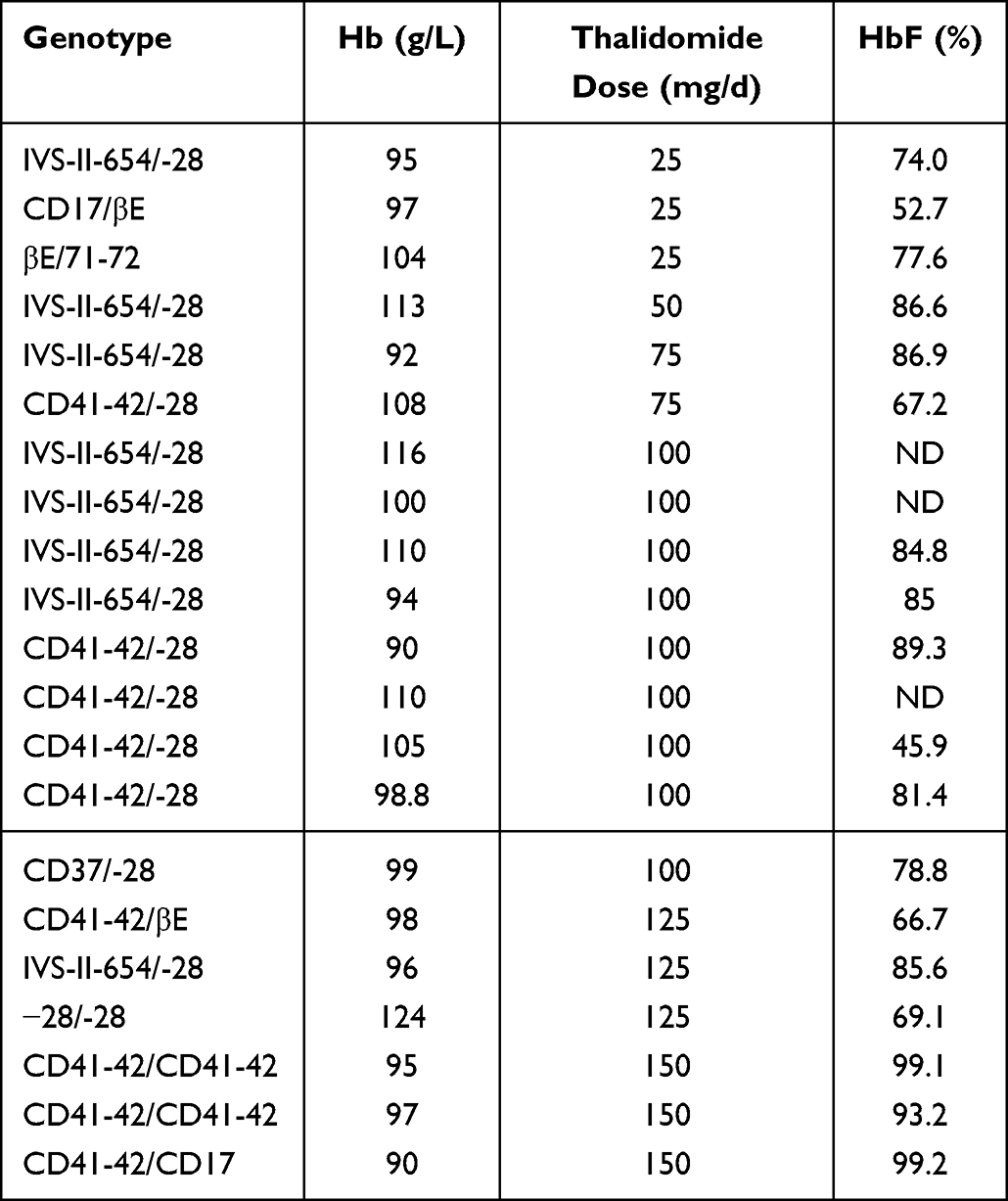 |
Table 3 Genotype of Patients Who Had an Excellent Response and Thalidomide Maintenance Dose |
Secondary Study Endpoints
Laboratory assessments were performed to evaluate changes in hemolysis indices (TBIL, DBIL, IBIL, LDH, reticulocyte count, and peripheral blood nucleated red cell count), iron load, liver and kidney function, and HbF level. At 3 months of treatment, HbF significantly increased from 16.09± 12.79% to 51.72± 25.59% (P < 0.001). Serum ferritin levels presented a reduction of 4046.42± 4098.12 ng/mL from baseline (P>0.05). At 4 years, HbF increased to 74.40± 20.10%. Serum ferritin levels were reduced to 1651.45± 1247.60 ng/mL (P < 0.05). Thirty patients completed the 4-year follow-up period, and complete data were obtained from 20 patients. Univariate repeated measurement ANOVA was performed for WBC, PLT, TBIL, IBIL, LDH, reticulocyte count, liver and kidney function and other indicators at 0, 12, 24, 36 and 48 months: WBC (F[2.24,20.20]) = 0.585, P=0.585>0.05), PLT (F[2.10,18.86] = 0.152 P=0.538>0.05), reticulocyte percentage (F[1.81,16.31] = 0.548, P=0.572>0.05), TBIL (F[2.49,47.24]=0.40, P=0.714>0.05), IBIL (F[2.32,44.11]=0.29, P=0.781>0.05), LDH (F[1.63,13.05]=0.915, P=0.406>0.05), Cr (F[5,60]= 0.725, P=0.725>0.05). No significant differences were observed any time point. ALT (F [2.85,56.90] = 4.54, P=0.007 P < 0.05), and AST (F[2.36,39.91]=3.14, P=0.045< 0.05) levels decreased significantly at each time point (Figure 3).
 |
Figure 3 (A)Changes in liver function from baseline to 48 months. (B) Changes in LDH and CR from baseline to 48 months. (C) Changes in RET and WBC from baseline to 48 months. (D) Changes in PLT from baseline to 48 months. |
Safety
Thalidomide was administered for a median of 46 months. During follow-up, all AEs were grade 1–2, and no grade 3 or higher AE was noted. The incidence of AEs is shown in Table 4. The most common AE was drowsiness, with an incidence rate of 42%, all of which were grade 1. Drowsiness occurred more often after starting medication and gradually decreased or becomes tolerable after approximately 1 week. Constipation (26%) was also commonly observed but generally mild. Rashes and allergies were noted in 12% and 8% of the patients, respectively, and manifested as maculopapular lesions, rubella, or blushing. They mostly occurred within 1 week of drug administration and were tolerated without increasing or decreasing the dose. The incidence rate of edema was 10%, and manifesting as edema of the foot, ankle, or face, and relieved by the short-term use of diuretics. Other common AEs included dizziness (2%), fatigue (6%), and limb numbness (2%), while the AEs of grade 2 were relieved after temporary drug discontinuation or drug dose reduction. Abdominal pain or thrombosis was not observed. One patient was free of transfusion 1 year after taking the drug, while the Hb level remained above 90g/L. This patient was diagnosed with nephrotic syndrome; the pathology of kidney biopsy showed stage III membranous nephropathy, accompanied by decreased Hb, and was withdrawn from the study to receive symptomatic treatment.
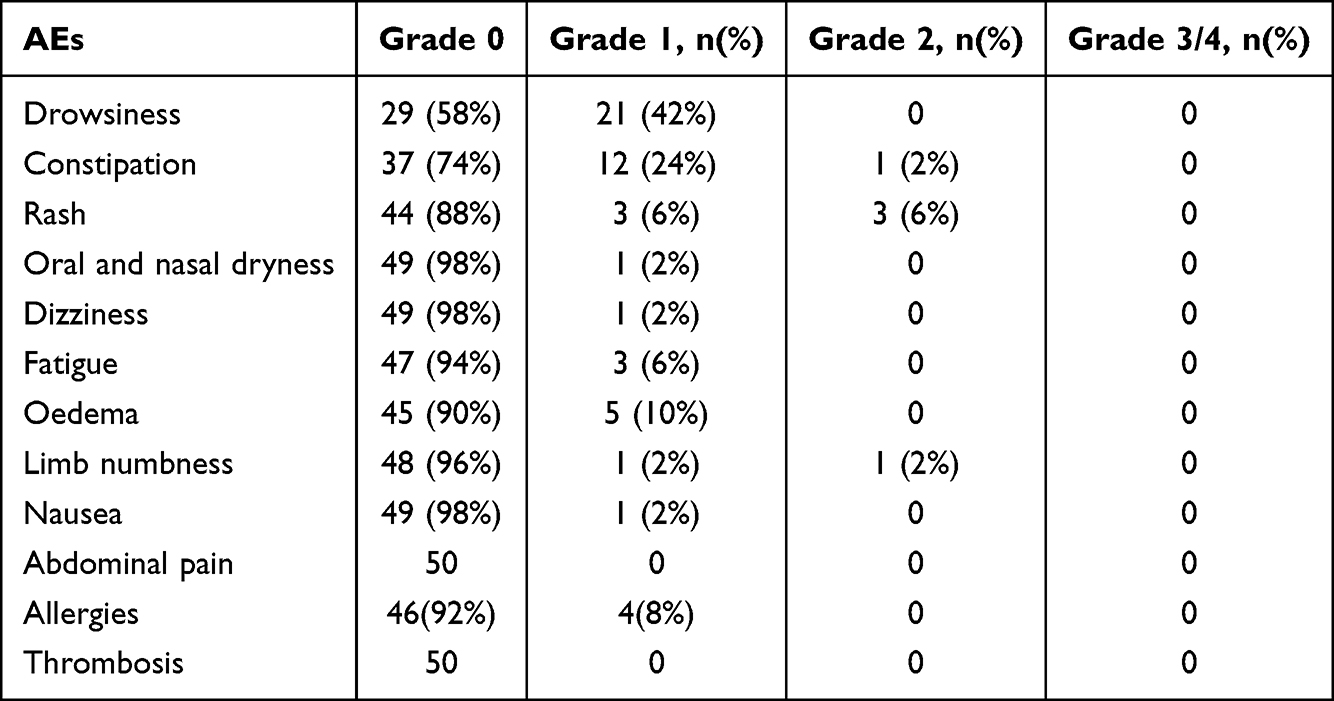 |
Table 4 Incidence of Adverse Events (AEs) |
Discussion
This long-term follow-up study investigated the efficacy and safety of thalidomide for TDT. In this clinical trial, 50 patients with TDT showed a high response rate to thalidomide, with sustained increases in Hb levels, while more than 70% of the patients with TDT did not require blood transfusion. Thalidomide was well tolerated and did not cause serious AEs in 5 years of follow-up. However, in this study, no decrease in hemolysis-related indices (TBIL, DBIL, IBIL, LDH, and reticulocyte count) was noted, despite the improvement of anemia.
Previous studies showed dramatic erythroid response to thalidomide in transfusions dependent β-thalassemia. In an Indian report including 21 patients with transfusion dependent E-β thalassemia, who have failed a trial of hydroxyurea, 15 (71.4%) attained transfusion independence (complete responders) and 1 (4.7%) attained partial response (50% decrease in transfusion requirement), while 5 (23.9%) were non-responders.19 In a multicenter study in a southern Chinese population, among 23 patients with TDT, transfusions were terminated in 43.5% (10/23) of the patients and decreased by more than 50% in 52.2% (12/23), whereas the average Hb level increased.20 In our study, at 9 months, 38 (76.0%), 1(2.0%), 4(8.0%), and 7(14.0%) patients had excellent, good, minor and no responses, respectively. The ORR was 86.0%. The most previous studies on thalidomide the patients were followed-up for 1–2 years. In the current study, the response was sustained over a longer duration of approximately 5 years (range: 35–65 months). Moreover, a significant decrease in serum ferritin levels was observed in the responders. These levels were reduced to 1651.45± 1247.60 ng/mL from baseline after 4 years of treatment.
Recently, luspatercept (a erythroid maturation agent) was studied in the double-blind Phase III BELIEVE trial (NCT02604433) and was associated with a significant reductions in the RBC transfusion burden in adults with TDT. The percentage of patients who achieved the primary endpoint of ≥33% reduction from baseline in transfusion burden during weeks 13–24 plus a reduction of ≥2 RBC units during the same period was 21.4%.16 In our study, thalidomide was more effective in reducing the transfusion burden and increasing Hb levels. Regarding the degree of transfusion independence as the primary endpoint, the volume of transfusion requirement was significantly decreased in most enrolled patients, the majority of which were from Guangdong Province. However, the transfusion criteria were inconsistent across different regions. The previous blood transfusion volume at the same interval was used as baseline, which may be clinically feasible for all patients and reflects real-world clinical practice.
The unbalanced synthesis of α- and β-globin chains is the core mechanism of the pathogenesis of β-thalassemia, while thalidomide acts as an anti-angiogenic and immunomodulatory agent with an unknown mechanism of action in the treatment of β-thalassemia. Previous studies confirmed that thalidomide promotes erythroid differentiation and induces γ-globin and HbF production. In the current trial, we also observed a sustained increase in HbF levers after 4 years of treatment. HbF increased to 73.49± 21.0%; however, it was not associated with elevated Hb levels. Simultaneously, the most frequent β-globin gene mutation in our patients was −28 (−78A>G) locus mutation, which may have contributed to the high rates of thalidomide response. We also analyzed the levels of several hemolytic markers (TBIL, DBIL, IBIL, LDH, and reticulocyte counts); however, we did not observe a decline after treatment. This may be due to the fact that most of the patients in this group came from Guangdong Province and had adequate medical follow-up. Most patients had received adequate blood transfusions before admission, and bone marrow hematopoiesis was suppressed. Thalidomide corrects some of the imbalance in α- and β-globin chain synthesis and inhibits ineffective hematopoiesis. In the absence of transfusion, the destruction of immature red blood cells provokes a compensatory hyperplasia of the erythroid marrow.
The optimal dose of thalidomide for ß-thalassemia has not yet been determined. We found that efficacy was dose-dependent. In the current study, the patients received a dose of 150 mg/d for 1 year and achieved a maximum stable Hb level. AEs associated with thalidomide were mild in this patient group. During follow-up, grade 1–2 AEs occurred; no grade 3 or higher AEs were observed, and there was no clinically evident incidence of thrombosis or secondary malignancies in any patient. The risk of AEs is related to the daily dose and duration of treatment.21 As patients need to take thalidomide for a long time to maintain its efficacy, and considering the safety of the drug, we reduced the dose and administered maintenance therapy at a relatively low dose, accompanied by an acceptable decrease in Hb levels. In the subsequent follow-up, no greater than Grade 1 AEs occurred. During the treatment, the women were not pregnant; however, the teratogenic effects of the drug remain a concern, and although there were no pregnancy-related problems during follow-up, it is necessary to increase awareness and medication guidance in case of pregnancy in patients receiving thalidomide.
However, the limitations of this study were that the sample size was relatively small and we used serum ferritin instead of liver R2* magnetic resonance imaging for to assess iron overload. As thalidomide is a lifelong drug for patients with thalassemia, its AEs remain a matter of concern, and further clinical trials with extended follow-up periods are required to comprehensively drug efficacy and safety.
Conclusions
Thalidomide can effectively improve Hb levels and reduce the need for blood transfusions in patients undergoing TDT. This drug causes mild adverse reactions, while it is a convenient, effective, and affordable treatment for patients with thalassemia who cannot undergo allogeneic stem cell transplantation. Thalidomide can also act as a bridge between transplantation and gene therapy, thereby reducing the need for blood transfusions.
No studies have been conducted to determine the optimal therapeutic dose of thalidomide for TDT. An oral thalidomide dose of 150 mg/d was used in this trial, and a preliminary exploration was conducted to determine the optimal therapeutic dose. However, to further validate these finding, designing randomized controlled studies using different treatment doses is necessary.
Data Sharing Statement
The original data sets are also available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank the patients with transfusion-dependent β-thalassaemia for their participation and cooperation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (82260026), the Zhuhai Medical Health Science and Technology Plan project (ZH2202200041HJL), and the Guangxi Natural Science Foundation (2020GXNSFAA159097).
Disclosure
The authors have no competing interests to declare that are relevant to the content of this article.
References
1. Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M. IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLoS One. 2014;9(7):e103020. doi:10.1371/journal.pone.0103020
2. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–4336. doi:10.1182/blood-2010-01-251348
3. Aydinok Y. Thalassemia. Hematology. 2012;17(sup1):s28–s31. doi:10.1179/102453312X13336169155295
4. Shah FT, Sayani F, Trompeter S, Drasar E, Piga A. Challenges of blood transfusions in β-thalassemia. Blood Rev. 2019;37:100588. doi:10.1016/j.blre.2019.100588
5. Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. doi:10.1016/S0140-6736(17)31822-6
6. Persons DA. Hematopoietic stem cell gene transfer for the treatment of hemoglobin disorders. ASH Education Program Book. 2009;2009(1):690–697. doi:10.1182/asheducation-2009.1.690
7. Michlitsch JG, Walters MC. Recent advances in bone marrow transplantation in hemoglobinopathies. Curr Mole Med. 2008;8(7):675–689. doi:10.2174/156652408786241393
8. Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479–1493. doi:10.1056/NEJMoa1705342
9. Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood. 2012;119(2):364–367. doi:10.1182/blood-2011-09-382408
10. Ansari SH, Shamsi TS, Ashraf M, et al. Efficacy of hydroxyurea in providing transfusion Independence in β-thalassemia. J Pediatr Hematol. 2011;33(5):339–343.
11. Chen J, Zhu W, Cai N, Bu S, Li J, Huang L. Thalidomide induces haematologic responses in patients with β‐thalassaemia. Eur J Haematol. 2017;99(5):437–441. doi:10.1111/ejh.12955
12. Li X, Hu S, Liu Y, et al. Efficacy of thalidomide treatment in children with transfusion dependent β-thalassemia: a retrospective clinical study. Front Pharmacol. 2021;12:722502. doi:10.3389/fphar.2021.722502
13. Jain M, Chakrabarti P, Dolai TK, et al. Comparison of efficacy and safety of thalidomide vs hydroxyurea in patients with Hb E-β thalassemia-a pilot study from a tertiary care Centre of India. Blood Cells. 2021;88:102544. doi:10.1016/j.bcmd.2021.102544
14. Li Y, Ren Q, Zhou Y, Li P, Lin W, Yin X. Thalidomide has a significant effect in patients with thalassemia intermedia. Hematology. 2018;23(1):50–54. doi:10.1080/10245332.2017.1354427
15. Chen J-M, Zhu W-J, Liu J, et al. Safety and efficacy of thalidomide in patients with transfusion-dependent β-thalassemia: a randomized clinical trial. Sig Transduct Target Ther. 2021;6(1):1–7. doi:10.1038/s41392-021-00811-0
16. Cappellini MD, Viprakasit V, Taher AT, et al. A Phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2020;382(13):1219–1231. doi:10.1056/NEJMoa1910182
17. Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood. 2013;121(12):2199–2212. doi:10.1182/blood-2012-10-408021
18. Services UDoHH. Common terminology criteria for adverse events (CTCAE) version 4.0. Nat Instit Health. 2009;4(03):1.
19. Nag A, Radhakrishnan VS, Kumar J, et al. Thalidomide in patients with transfusion-dependent E-beta thalassemia refractory to hydroxyurea: a single-center experience. Indian J Hematol Blood Transfusion. 2020;36(2):399–402. doi:10.1007/s12288-020-01263-2
20. Yang K, Wu Y, Zhou Y, et al. Thalidomide for patients with thalassemia intermedia: a retrospective multicenter clinical study. Mediterr J Hematol Infect Dis. 2020;12(1):e2020021–e2020021. doi:10.4084/mjhid.2020.021
21. Bastuji-Garin S, Ochonisky S, Bouche P, et al. Incidence and risk factors for thalidomide neuropathy: a prospective study of 135 dermatologic patients. J Invest Dermatol. 2002;119(5):1020–1026. doi:10.1046/j.1523-1747.2002.19502.x
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.