Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Long noncoding RNAs: from identification to functions and mechanisms
Authors Marín-Béjar O, Huarte M
Received 29 January 2015
Accepted for publication 11 April 2015
Published 2 July 2015 Volume 2015:5 Pages 257—274
DOI https://doi.org/10.2147/AGG.S61842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr John Martignetti
Oskar Marín-Béjar, Maite Huarte
Center for Applied Medical Research (CIMA), Department of Gene Therapy and Regulation of Gene Expression, University of Navarra, IDISNA, Navarra Institute of Health Research, Pamplona, Spain
Abstract: The complex universe of RNA transcribed by mammalian cells includes thousands of large RNA molecules that do not encode for proteins. Despite their lack of coding capacity and relatively low conservation, many large noncoding RNA molecules (lncRNAs) have been shown to be functional, adding a new level of complexity on the structural organization, function, and evolution of the genome. Here, we summarize the current knowledge of lncRNAs, starting from the methodologies that led to their identification to their genomic and evolutive features. We discuss the diverse mechanisms by which lncRNAs exert their functions, and the technical approaches for their study. Finally, we discuss the future directions of lncRNA research and the challenges involved. The advances in lncRNA research are definitively changing our conception of gene regulation.
Keywords: lncRNA, gene regulation, lncRNA function
Introduction
The central dogma of molecular biology that Francis Crick introduced in 1958 was able to answer the longstanding question of how cells are able to store and transfer information to produce the adequate functional output: the genetic information is encoded in DNA, transcribed to form messenger (m)RNA molecules, which then serve as a template to produce unique amino acid sequences that constitute individual proteins (ie, one gene, mRNA intermediate, one protein).1 However, as early as the 1960s, this view began to be challenged by the discovery of noncoding (nc)RNAs, which do not code for proteins, but rather function as RNA molecules – ie, ribosomal (r)RNA, transfer RNA, and small nucleolar RNAs.2 Another milestone in our understanding of gene function came about 15 years ago. The simple addition of double-stranded RNA control in an antisense experiment triggered a revolution in our comprehension of RNA function.3 Several new classes of small ncRNAs have burgeoned in the last years including small interfering RNAs (si)RNAs that deplete RNA transcript levels, Piwi-associated RNAs involved in germ cell differentiation, or micro (mi)RNAs. MiRNAs are now known to regulate multiple steps in gene expression, including RNA stability, mRNA translation, and transcriptional silencing.4 All these findings contributed to understand the enormous complexity and the extent of the regulatory potential of RNA.5
Presumably, the most significant shift in our conception of the role of RNA in gene regulation has emerged from the recent advances in genomic technologies, which have made it possible to survey the transcriptomes of many organisms to an unprecedented degree. By utilizing these technologies, several studies have revealed that the genomes of mammals, as well as other organisms, produce thousands of long transcripts that have no significant protein-coding capacity and are thus referred to as long (or large) noncoding (lnc)RNAs.6–12 It is now estimated that while 70% of the mammalian genome is actively transcribed, only 1%–2% of the transcriptionally active regions correspond to protein-coding genes.6–15 lncRNAs are transcripts longer than 200 nt that lack protein-coding capacity. Depending on their genomic localization relative to their neighboring protein-coding genes, lncRNAs can be classified into 1) long intergenic noncoding (linc)RNAs, when they do not overlap with any other gene; 2) intronic lncRNAs, when localized inside the intron of a gene; or 3) antisense lncRNAs, when transcribed from the opposite DNA strand overlapping with exons of a protein-coding gene. Many of the mammalian lncRNAs are transcribed by RNA polymerase II, and they are therefore strikingly similar to mRNAs: they are capped, spliced, and polyadenylated, while others are produced by RNA polymerase III or RNA polymerase I.16,17
Despite the fact that Xist – a functional lncRNA that mediates X chromosome inactivation in mammalian female cells – was already discovered in the early 1990s, the prevailing view until recently was that long noncoding transcripts are rare, and only a handful of functional lncRNAs are represented in the genome.18–20 However, research in the past several years has documented important functions for many lncRNAs, reporting roles in normal organism development and disease (examples of lncRNAs associated with pathologies are summarized in Table 1). Some of the varied processes where lncRNAs have been shown to intervene include transcriptional regulation, dosage compensation, genomic imprinting, cellular trafficking and nuclear organization, and compartmentalization.16,17,21–23
 | Table 1 Representative lncRNAs with role in disease |
In this review, we summarize the current knowledge of lncRNAs, starting from the methodologies that have led to their identification, their genomic and evolutive features, and their functions and mechanisms.
Identification of lncRNAs in the genomic context
The methodologies applied for the detection and identification of lncRNAs have evolved hand in hand with the genomic technologies (Figure 1). One of the first approaches to the genome-wide identification of lncRNAs was the Functional Annotation of the Mammalian genome project (FANTOM),24 where >21,000 complementary DNA clones were annotated, of which 3,000 did not keep any homology with protein-coding genes and were therefore annotated as lncRNAs. In the following FANTOM projects, the annotation of lncRNAs was improved by adding the identification of transcription start sites) detected by Cap Analysis of Gene Expression.6,25
 | Figure 1 Prediction of lncRNA gene organization by different NGS methodologies. |
A different method used for the identification of lncRNAs was based on the indirect determination of the transcriptional active regions of the genome. For instance, the detection of RNA polymerase II occupancy by ChIP-seq analysis has been applied as an indication of the presence of a transcriptional unit.25–27 However, this approach has some limitations, as it cannot discriminate between random RNA polymerase II binding events and the specific association with a transcribed gene, and it does not detect the regions transcribed by other RNA polymerases different to polymerase II.
Alternatively, the analysis of chromatin states has provided information about transcriptional activity genome-wide. There is a direct relationship between the transcriptional activity of RNA polymerase II and the pattern of histone post-translational modifications across the mammalian genome.28 It is known that H3K36me3 is linked to transcriptional elongation and it is strongly enriched across the transcribed regions of active genes, while the H3K4me3 signal is associated to the promoter of the gene, identifying the transcription start site.29 Analysis of these chromatin marks combined with gene expression data derived from tiling DNA microarrays have been applied to identify novel lncRNAs. Based on this principle, K4-K36 domains outside known protein-coding genes defined genomic regions encoding long lincRNAs.7
More recently, the development of next-generation sequencing applied to RNA has allowed for the direct detection and assembly of entire transcriptomes.30 RNA sequencing (RNA-seq) offers a more reliable and high-resolution method for measuring gene expression than previous techniques. Furthermore, thanks to computational methods for transcriptome reconstruction, novel transcribed regions, as well as alternative spliced forms of annotated genes, can be identified.31–34 Based on RNA-seq analyses of multiple cell types, the ENCODE project has released one of the most complete and updated lncRNA catalogs, including more than 16,000 lncRNAs (GENCODE version 21).35
The approaches described have allowed for the extension of the repertoire of lncRNAs present in the mammalian genome to more than 20,000. However, the lack of a long open reading frame is not a sufficient criterion for the classification of a transcript as noncoding. In order to predict whether the transcript may or not encode for a protein, some computational methods can be applied. Several features of bona fide protein-coding genes can be used as criteria to distinguish coding from ncRNAs. Coding regions 1) tend to have much longer open reading frames than expected by chance; and 2) they present with the conservation of amino acid sequence from multiple species alignments. Finally, 3) nucleotide substitution occurs frequently at the third position of the codon when compared to orthologous sequences.36–38 Methodologies such as Codon substitution frequency, PhyloCSF, and the Coding-Potential Assessment Tool are based on these principles that are used to calculate the probability of a transcript being coded.38–40 Still, a clear binary separation between coding and noncoding transcripts can be a hard task. Indeed, the ability of lncRNAs to encode peptides has also been experimentally tested using ribosome profiling and mass spectrometry.41–43 These studies have led to the surprising finding that some of the putative lncRNAs may encode small peptides with a low degree of conservation whose functions are still undefined. Independently of whether they may or may not encode for small peptides, it still remains to be shown how many of the thousands of lncRNAs identified are functional.
Evolution and conservation of lncRNAs
For decades, only the highly conserved genomic regions encoding for proteins were connected with basic cell functions. The evolutionary history of lncRNAs can therefore provide insights into their functionality. While the nucleotide sequence of protein-coding exons is highly conserved across species, noncoding regions are overall poorly conserved and, if they have some pattern of conservation, it is intermittent.44 Indeed, although exceptionally highly conserved lncRNAs exist, such as the transcribed ultraconserved regions, lncRNAs in general are under more selective pressure than ancestral repeat sequences with neutral selection, and about one-third of lncRNAs seem to have arisen within the primate lineage.45,46 Furthermore, it has been shown that 81% of lncRNA families are primate specific, which is consistent with previous studies that propose that lncRNA transcription can evolve extremely rapidly, even between closely related mammals.47,48 Interestingly, the number of lncRNAs encoded by the genome has increased during animal evolution, suggesting that the presence of lncRNAs could be linked to organismal complexity (Figure 2).47
 | Figure 2 Increase in the number of lncRNA along animal evolution. |
The growing number of lncRNAs along the evolutionary process is evident, but it is not so much the origin of this rise. During genomic evolution, it has been observed that 45%–65% of the genome was conquered by the “parasite genome”, transposable element (TE) insertions.49 The majority of lncRNAs (83%) contains at least one TE, and out of all the TE families, human endogenous retrovirus (ERV) is the most overrepresented in lncRNAs.50,51 It has been proposed that TE may have played a major role in the generation of alternative promoters, and thus of novel lncRNAs.50,52,53 The contribution of TE insertions to lncRNA function is known, for instance, in the case of Xist, the lncRNA responsible for X chromosome inactivation and dosage compensation in mammals. The Xist sequence contains several repeat domains. Among them are Rep-A, originated by an ERVB5 insertion, and Rep-C and Rep-F, originated by ERVB4 and DNA transposon insertions, respectively.54–56 While Rep-A interacts with the polycomb repressive complex 2 (PRC2) and acts as bait for X chromosome heterochromatin formation, Rep-C is responsible for specific binding to the transcription factor YY1, which tethers Xist to the inactive X nucleation center, and Rep-F is responsible for Jarid2 binding and subsequent PRC2 recruitment.51,56–58 Therefore, TE not only may have provided a mechanism for the origin of new transcripts, but they also constitute functional elements of lncRNAs.
Structural features of lncRNAs
The evolutionary conservation patterns of lncRNAs have been studied in sequence terms, but it is plausible that lncRNAs are conserved at a structural level, as the structure is most likely a key determinant for lncRNA function. However, the knowledge of the structure and folding of large molecules of RNA is nearly inexistent, and probably represents one of the major limitations when studying lncRNA biology. Computational methods have been developed to predict the secondary structure of RNAs, where the most frequently used are based in free energy, phylogenetic and machine-learning approaches, or various combinations of these three.59–61 However, as of today, it is nearly impossible to predict the structure of long RNAs. For example, a lncRNA of 2,200 nt in length has approximately 10888 possible predicted secondary structures.62 Experimental structural analyses are therefore more reliable than predictions, although they are quite challenging when dealing with long RNA molecules containing flexible and lowly structured domains. Nowadays, there are less than a dozen RNAs over 200 nt in size, whose structures have been solved in a crystallographic manner.63 For instance, thermodynamic algorithms combined with probing have been able to unveil the 16S rRNA structure.64 In the case of small molecules of RNA (<500 nt), the methodology of choice is selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE)-seq, which produces measurements of local nucleotide flexibility and, with subsequent data analysis, predicts RNA secondary structures.65 However, when dealing with larger molecules (>500 nt), the SHAPE profile often corresponds to a large number of different folds, complicating its interpretation. For example, approximately 45 RNA structure predictions are available for the steroid receptor RNA activator lncRNA, which has a length of 874 nt.66,67 An adapted experimental approach has been developed to solve the structure of longer RNAs.68 This approach combines the conventional SHAPE analysis of the full-length RNA with overlapping smaller fragments of the same RNA, defining the RNA in subdomains, followed by studies using in-line probing, dimethyl sulfate probing, or RNase V1 digestion.62 These structural analyses are, without a doubt, a crucial step to establishing the connection between lncRNA structure and function.
Experimental approaches for the functional study of lncRNAs
The elucidation of the role of lncRNAs represents a major challenge; as in most cases, it is unclear in what direction the investigation should focus. One approach to predict the putative function of lncRNAs uses “guilt-by-association”.7 This method associates lncRNAs with biological processes based on a common expression pattern across cell types and tissues of protein-coding genes and lncRNAs. In line with this observation, it has been shown that the promoters of lncRNAs, similarly to those of protein-coding genes, are subjected to regulation by transcription factors, which are the upstream controllers of biological pathways. In some cases, a coordinated expression between lncRNAs and neighbor protein-coding genes has also been observed. Although this correlation could indicate an activity in cis of the lncRNA, it is not higher than the correlation found for pairs of neighbor protein-coding genes.10
The results of this computational approach can lead to the conclusion that the vast majority of lncRNAs are associated with specific biological functions, an observation that could be used as a starting point for hypothesis generation that can then be confirmed by experimental analyses. For this, several methodologies have been developed and adapted for the study of lncRNAs, as will be explained.
Loss-of-function studies
Loss-of-function studies are crucial in order to address the role of lncRNAs. Among all strategies used for the inhibition of RNA levels in cells, post-transcriptional silencing with small interfering RNAs is the most widely used. This approach can be applied for the depletion of lncRNAs, although in many cases it has been shown to be inefficient.100 It has been argued that the predominantly nuclear localization of some lncRNAs impedes their targeting by RNA interference (RNAi) machinery, which is mainly localized to the cytoplasm of the cell. Alternative approaches can be used, such as antisense oligonucleotides (ASOs) or gapmers (Figure 3), which bind to the target RNA, forming a DNA/RNA hybrid and promote RNA cleavage by RNase H, which is present in the nucleus. Other RNase H-independent antisense DNA molecules that are used to knockdown spliced lncRNAs are the morpholinos or locked nucleic acid ASOs, which target the splice sites, blocking RNA processing into the functional spliced form.69,70 However, all these oligonucleotide-based techniques share limitations with RNAi technology (Figure 3); knockdown is incomplete, it diverges between experiments and laboratories, and it has unpredictable off-target effects. Furthermore, these techniques only provide temporary inhibition, which limits the loss-of-function analysis to a transient system.71,72
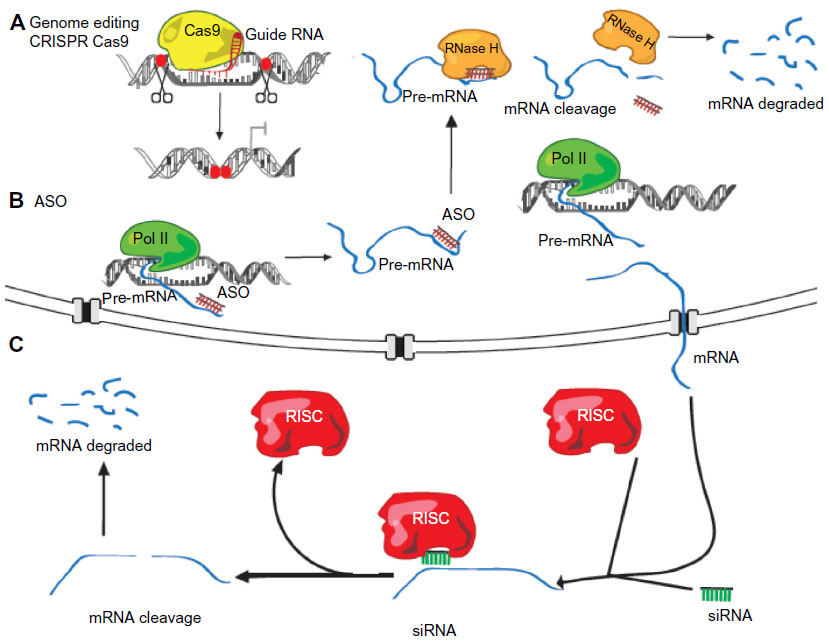 | Figure 3 lncRNA loss-of-function strategies. |
In order to avoid these limitations, gene-targeted knockout technology provides a powerful tool for elucidating the function of lncRNA in vivo. It has been applied for the generation of 18 lincRNA knockout mouse strains by removing the entire genomic loci containing the lncRNAs and replacing them with a lacZ reporter cassette.73,74 Beyond classical gene knockout approximations, genome editing can nowadays be performed in a fast and efficient way thanks to the development of strategies that allow for the targeting of nucleases to specific gene loci. Such techniques include programmable site-specific zinc-finger nucleases, transcription activator-like effector nucleases (TALENs), and clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated endonuclease (Cas9) (Figure 3).75–77 The design of the strategy for lncRNA targeting should take into account whether the transcription of the lncRNA requires silencing, or if the entire lncRNA gene has to be deleted. The silencing can be achieved through different strategies, including the deletion of the promoter region, or the targeted interruption between the promoter and the RNA sequence through the insertion of a polyadenylation signal or by an engineered inversion of the promoter. The latter may not always be successful, as reported in the case of bidirectional promoters where the inversion does not abolish the transcription, and promoter deletion may also affect the expression of the neighbor protein-coding gene. In all these scenarios, the possible reorganization of regulatory elements in the DNA has to be taken into account to be minimized.78
Gain-of-function studies
When a lncRNA exerts its functions in trans – ie, it acts distantly from the lncRNA genomic locus – the ectopic expression of the RNA may be able to mimic the function of the endogenous RNA. LncRNA overexpression (either transient or stable) requires the cloning of its sequence into an appropriate vector. For this approach it is important to take into account the architecture of the vector of choice, and to be aware of the possible additional sequences added to the lncRNA that may affect its structural features and result in experimental artifacts.
As an alternative to the use of expression vectors, lncRNA can be overexpressed from its endogenous locus, an approach that could be useful for both cis- and trans-acting lncRNAs. For instance, the TALEN methodology has been employed to insert a strong promoter upstream of the lncRNA CCAT1-L, which is able to drive its overexpression from the endogenous locus.79
Visualization of lncRNAs
To study lncRNA localization at the subcellular level by fluorescent in situ hybridization can provide important information regarding the cellular role of lncRNAs, indicating the number of RNA molecules present in different subcellular compartments. Currently, the most frequently used methods can be classified in two categories.80
Direct detection by tiling the sequence with multiple antisense labeled probes
This method employs a set of short single-stranded DNA oligonucleotides complementary to various regions of the target RNA, each labeled with one or more fluorescent moieties.80 This technique is able to simultaneously detect multiple RNA molecules in single cells, even in small cells such as the yeast Saccharomyces cerevisiae, using optical super-resolution microscopy and combinatorial labeling with the use of spectrally distinguishable fluorescent moieties.81
Detection by signal amplification
In order to detect lowly expressed or short RNA molecules, it may be necessary to amplify the signal. To this end, DNA polymerase and circular templates are used to locally create long, repetitive, single-stranded DNA tracts in situ, which can be targeted with short oligonucleotide probes.82 While amplification methods are capable of detecting short and low abundant RNAs, they are limited to only one fluorophore and cannot be directly applied for lncRNA quantification.
Determination of lncRNA molecular interactions
Both cis- and trans-acting lncRNAs have been shown to work in cooperation with proteins.83 Similarly, the interplay between chromatin and other RNA molecules is required for the function of some lncRNAs.84–86 The identification of the factors with which lncRNAs establish specific interactions is therefore crucial to understand the mechanism of lncRNAs. In recent years, new methodologies have been developed in order to obtain information about the binding of lncRNAs to gene loci and proteins. Several of these methods are based on the precipitation of the endogenous RNA by DNA biotinylated capture-oligonucleotides coupled with chromatin purification and high-throughput DNA sequencing. Such techniques include chromatin islolation by RNA purification (ChIRP), capture hybridization analysis of RNA targets (CHART), and RNA purification (RAP).87–89 In each of these methodologies, different data processing is used to discriminate between specific trans-DNA binding sites and trivial events.87–89 More recently, a variation of the RAP protocol named RAP-RNA has been implemented to identify RNA–RNA complexes through in vivo cross-linking, RNA capture with antisense biotinylated DNA oligonucleotides, and massive scale RNA-seq.90 The most commonly used methods, including those for the detection of lncRNA–chromatin, lncRNA–protein, and lncRNA–RNA interactions, are summarized in Table 2.
 | Table 2 Experimental methods for the study of lncRNAs |
Mechanisms and functions of lncRNAs
LncRNAs are highly heterogeneous molecules and can act at multiple levels to affect gene expression. While a significant number of lncRNAs have been suggested to interact with chromatin-remodeling complexes, driving them to specific genomic loci, others have been implicated in the architectural conformation and activity of transcriptional enhancers.9,83 In other cases, lncRNAs have been shown to interfere with the transcriptional machinery, or even to maintain the structure of nuclear speckles.91,92 Furthermore, some lncRNAs have been shown to act post-transcriptionally as regulators of splicing, mRNA decay, protein translation, protein stability, or as molecular decoys for microRNAs.93 LncRNAs can be classified depending on the cellular compartment where they carry out their functions.
lncRNAs with nuclear functions
Trans-acting lncRNAs: interfacing with chromatin complexes and nuclear architecture
A significant number of nuclear lncRNAs has been shown to function in cooperation with chromatin modifiers, regulating distantly located genes at the epigenetic level. Some of these lncRNAs have been involved in transcriptional activation. For instance, the lncRNA Mistral (Mira) recruits the H3K4 methyltransferase MLL1 and activates the expression of the HOXA6-7 locus (Figure 4).94 However, the majority of the reported chromatin-interacting lncRNAs are found to bind to repressive chromatin modifiers, such as PRC2 or H3K9 methyltransferases.95–97 In several cases, the interaction of the lncRNAs with the chromatin complexes is required for the repression of specific gene loci. For example, the lncRNA HOX transcript antisense RNA (HOTAIR) is expressed from the HOXC gene cluster and represses the HOXD locus by interacting with the chromatin complexes PRC2 and LSD1.98,99 Also, Pint is a lncRNA that is able to promote PRC2-dependent repression of specific genes of the p53 pathway (Figure 4).100
 | Figure 4 Mechanisms of nuclear trans-acting lncRNAs. |
An emerging theme is the role that lncRNAs may play as structural elements, contributing to the nuclear architecture. Firre, a lncRNA located on the X chromosome, regulates transgenomic regions in cooperation with hnRNPU (Figure 4), a protein also previously shown to be associated with proper localization of Xist and the formation of highly structured chromatin territories.101–103 Also, the highly expressed nuclear lncRNAs, NEAT1 and MALAT1, have been related to nuclear architecture.104 Although their exact function remains unknown, NEAT1 has been previously shown to contribute to nuclear paraspeckle formation (Figure 4), while MALAT1 has been shown to interact with several splicing factors influencing alternative splicing of pre-mRNAs (Figure 4).105,106 In a recent study, it has been reported that both lncRNAs are co- and mutually regulated, and they are found be associated to 314 genomic sites, connected to histone modifications associated with active chromatin.104 Furthermore, a different study has reported that MALAT1 interacts with many nascent pre-mRNAs at active gene loci, suggesting the possibility that MALAT1 could recruit different RNA-processing proteins to these genomic sites.90 Finally, it has been suggested that NEAT1 and MALAT1 could act as the scaffolds that contribute to the assembly of DNA–RNA–protein interactions at specific transcribed locations. A question that remains open is whether these lncRNAs are able to directly interact with DNA or act as bridging molecules between proteins and DNA.
Cis-acting lncRNAs: genomic imprinting, dosage compensation, and enhancer functions
RNA molecules considered to act in cis are those involved in the regulation of neighbor gene expression either via direct binding and chromatin structure modification or by recruiting chromatin-associated proteins that modify the chromatin state of the vicinity region. Some of the classical examples of lncRNAs that act in cis are Airn and Kcnq1ot1, which are involved in genomic imprinting or dosage compensation.55,107,108 These two lncRNAs share some features: both are paternally expressed and repress the adjacent protein-coding gene. However, in the case of Airn, the lncRNA itself may not be required for gene silencing in cis. It has been shown that Airn can be replaced by an unrelated sequence, which still retains the same silencing activity on the neighbor gene. Transcription of the RNA, independent of its sequence, causes transcriptional interference and a lack of expression of the neighbor gene in the absence of repressive chromatin.109
In mammals, the process of dosage compensation, an epigenetic mechanism that has evolved to equalize X-linked gene expression between males and females, is regulated by the lncRNA Xist. Xist is expressed from one of the two female X chromosomes, and it induces chromatin changes on the X chromosome that is going to be inactivated; thus, the transcription of the majority of genes is silenced.110 Xist initiates X chromosome inactivation by spreading in cis across the future inactive X chromosome, recruiting PRC2, and forming a transcriptionally silent nuclear compartment enriched for repressive chromatin modifications including H3K27me3.111,112 As previously discussed, these functions of Xist (localization to chromatin and the silencing of gene expression) are mediated by distinct RNA domains: transcriptional silencing requires the A-repeat domain, which interacts with the PRC2 chromatin regulatory complex; whereas localization to chromatin requires several distinct domains and interactions with proteins associated with the nuclear matrix.55,113,114
Other nuclear cis-acting lncRNAs have been shown to carry activating functions as enhancer RNAs – ie, to promote the transcription of neighboring genes.9 Within this subset, two types of lncRNAs have been described: activating ncRNAs (ncRNA-a) and enhancer RNAs (eRNAs). ncRNA-a are included in the lincRNA category, and are expressed as independent transcription units with the typical chromatin signature of polymerase II transcripts (H3K4me3 at the promoter and H3K36me3 at the elongated region), predominantly spliced and polyadenylated.9,23,115,116 On the other hand, eRNAs are expressed from distal regulatory elements with typical enhancer chromatin marks (H3K4me1). Although responsive to stimulus-dependent activation, they contain a mixture of nonpolyadenylated and polyadenylated unspliced transcripts.27,117 While the differences between ncRNA-a and eRNAs at the biogenesis level are still unclear, at the functional level, both promote the activation of protein-coding genes localized in their vicinity.9 One study has shown that the ncRNA-a7 (also called LINC00651) promotes the formation of a chromatin loop between the long ncRNA locus and the regulated promoter, an interaction that requires the Mediator complex.118 Other studies have shown that lncRNAs could be involved in HOX gene-positive regulation, such as HOTTIP.23 HOTTIP is expressed from the 5′ end of the HOXA locus and activates the expression of HOXA genes by interacting with the WDR5/MLL complex through chromatin loop formation.23,94 The MLL complex catalyzes H3K4me3, the histone mark associated with active transcription.94
However, it still remains to be determined whether other RNAs with enhancer-like activity are required for the establishment of the long-range chromatin interaction, or if these interactions are independent of RNA expression.
lncRNAs with cytoplasmic functions
lncRNAs that function through RNA–RNA interactions
While several of the best-known lncRNAs exert their functions in the nucleus of the cell, other functional lncRNAs are localized in the cytoplasm and regulate gene expression at the post-transcriptional level.46,93 Some of the cytoplasmic lncRNAs have been reported to work through RNA–RNA interactions. Sequence complementarity-mediated interactions allow for the regulation of mRNA stability, transport, translation, and so on by lncRNAs. Cytoplasmic lncRNAs containing Alu short interspersed elements (SINE) have been named half-STAU1-binding site RNAs (1/2-sbsRNAs). Their mechanism of action is mediated by the repeated element, which forms imperfect complementary RNA duplexes with Alu elements of the 3′ untranslated regions of target mRNAs and recruit the double-stranded RNA binding protein Staufen1, which targets mRNA for degradation. Thus, mRNA degradation is mediated by lncRNAs, which promote the recruitment of proteins to mRNAs that mediate their decay.119 In contrast, other lncRNAs, such as lncRNA–UCHL1, enhance mRNA translation. LncRNA–UCHL1 expression is induced by mTOR and shuttled to the cytoplasm where, via an antisense complementary to the UCHL1 AUG initiation codon and combined inverted SINEB2 domains, it increases UCHL1 protein synthesis.120
The classic example of post-transcriptional gene regulation is that mediated by miRNAs.121 However, target mRNAs are not the only molecules containing miRNA-binding sites in their sequence. Also, ncRNAs, including lncRNAs, pseudogenes, and circular RNAs, compete for shared miRNAs, reducing their capability for acting on mRNAs.122,123 This functional category of ncRNAs has thus been named competing endogenous RNAs (ceRNAs).124 The extent of this type of regulation and the competing capability of the ceRNAs are still unknown, but some ceRNAs are preserved during evolution, denoting a regulatory role to control miRNA homeostasis and thus to enhance ceRNA crosstalk.125 For instance, lnc-MD1 is a lncRNA with ceRNA activity involved in muscle differentiation. It interacts with miR-133 and miR-135 to promote the upregulation of MAML1 and MEF2C, which are transcription factors targeted by these microRNAs that play a key role in muscle-specific gene expression.125
lncRNAs that function through protein–protein interactions
As previously discussed, both cis- and trans-acting lncRNAs, independent of their subcellular localization, can work in cooperation with proteins by recruiting, assembling, scaffolding, or regulating their stability. LncRNAs may either contribute to or inhibit the formation of macromolecular complexes by allowing or blocking protein–protein interactions.84–86 Additionally, some lncRNAs act as regulators of protein post-translational modifications.126 This is the case of lnc-DC, a lncRNA unique to human dendritic cells. Depletion of lnc-DC affects the differentiation process of monocytes to dendritic cells. Lnc-DC interacts with signal transducer and activator of transcription 3 (STAT3) in the cytoplasm, and allows for STAT3 phosphorylation at Tyr705 by hindering STAT3 binding to SHP1, a protein with tyrosine phosphatase activity. STAT3 is phosphorylated and then translocated to the nucleus where it acts as a transcription factor.127
In summary, lncRNAs present high functional versatility, which likely relies on their ability as long RNA molecules to acquire different structures and molecular interactions.
Conclusion and perspectives
The identification of thousands of lncRNAs involved in many aspects of gene expression has changed our perspective and understanding of basic processes of cell biology. These discoveries have also added a new level of complexity to the structural organization, function, and evolution of the genome. Interestingly, despite being functional genes, most lncRNAs present with poor conservation, even among phylogenetic closely related species. This suggests that the highly accelerated evolution of ncRNA regions could be related to the development of complex structures, such as the brain. The origin and evolution of lncRNAs will therefore be an active focus of research in this area.163
Despite the rapid increase in the catalog of functions reported for lncRNAs, one of the current challenges is to identify the sequence and structural elements that allow long RNA molecules to carry out their cellular functions. Technological advances are needed to perform reliable computational predictions and experimental validation of lncRNAs’ secondary structures and putative binding partners, including other nucleic acids and proteins.
One of the most recurrent themes regarding lncRNA functionality is their involvement in transcription regulation by interacting with chromatin complexes and changing the chromatin state of the target gene loci. However, one of the questions that remains unanswered is how lncRNAs recruit the chromatin-remodeling complexes and establish interactions with DNA. The application of methodologies that identify genome-wide DNA binding sites and protein partners of lncRNAs to a larger number of lncRNAs will predictably allow us to infer the principles that govern the function of lncRNAs at this level.
Another technical advancement that will undoubtedly foster our knowledge of lncRNA functions is the possibility of performing gene editing in a fast and easy manner with the TALEN or CRISPR/Cas9 methodologies. Not only can these tools facilitate the generation of animal models, but they could also be used in combination with RNAi or ASOs to understand whether a given lncRNA is a functional molecule, or if the functionality is contained in the act of transcription or in the DNA regulatory elements of its sequence. A systematic and rigorous experimental analysis is needed to avoid incorrect conclusions that could mislead future research.
Knowing that lncRNAs are involved in the pathogenesis of many syndromes and diseases allows for the possibility of developing new therapies and diagnostic methods centered on lncRNAs. Some of the features of lncRNAs as regulatory molecules, such as their ability to modulate specific facets of cellular pathways and their highly specific expression patterns, represent opportunities for the development of less toxic and tailored targeting therapies. Predictably, future advances in lncRNA research will clarify our current view of the noncoding genome, and it will provide scientists with new perspectives for biotechnological and clinical applications.
Acknowledgments
OMB and MH are supported by the European Research Council Starting Grant 281877 and the Spanish Ministry of Science Grants BFU2011-23485 and SRYC1100I008347XV0.
Disclosure
The authors report no conflicts of interest in this work.
References
Crick FH. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. | |
Eddy R. Non–coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2(12):919–929. | |
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | |
Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009;21(3):452–460. | |
Hartmann B, Valcárcel J. Decrypting the genome’s alternative messages. Curr Opin Cell Biol. 2009;21(3):377–386. | |
Carninci P, Kasukawa T, Katayama S, et al; RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group). The transcriptional landscape of the mammalian gene. Science. 2005;309(5740):1559–1563. | |
Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. | |
Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. | |
Ørom UA, Derrien T, Beringer M, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143(1):46–58. | |
Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNA reveals global properties and specific subclasses. Genes Dev. 2011;25(18):1915–1927. | |
Clark BS, Blackshaw S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet. 2014;5:164. | |
Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. | |
Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. | |
Kapranov P, Cheng J, Dike S, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316(5830):1484–1488. | |
Mattick JS. A new paradigm for developmental biology. J Exp Biol. 2007;210(Pt 9):1526–1547. | |
Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF. RNA regulation of epigenetic processes. Bioessays. 2009;31(1):51–59. | |
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. | |
Brockdorff N, Ashworth A, Kay GF, et al. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature. 1991;351(6324):329–331. | |
Brown CJ, Ballabio A, Rupert JL, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349(6304):38–44. | |
Brown CJ, Hendrich BD, Rupert JL, et al. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71(3):527–542. | |
Clark MB, Johnston RL, Inostroza-Ponta M, et al. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012;22(5):885–898. | |
Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145(2):178–181. | |
Wang KC, Yang YW, Liu B, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–124. | |
Kawai J, Shinagawa A, Shibata K, et al; RIKEN Genome Exploration Research Group Phase II Team and the FANTOM Consortium. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409(6821):685–690. | |
Kodzius R, Kojima M, Nishiyori H, et al. CAGE: cap analysis of gene expression. Nat Methods. 2006;3(3):211–222. | |
Sun J, Pan H, Lei C, et al. Genetic and genomic analyses of RNA polymerase II-pausing factor in regulation of mammalian transcription and cell growth. J Biol Chem. 2011;286(42):36248–36257. | |
De Santa F, Barozzi I, Mietton F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8(5):e1000384. | |
Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–181. | |
Rao B, Shibata Y, Strahl BD, Lieb JD. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol. 2005;25(21):9447–9459. | |
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. | |
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. | |
Guttman M, Garber M, Levin JZ, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28(5):503–510. | |
Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. | |
Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8(6):469–477. | |
Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22(9):1760–1774. | |
Kong L, Zhang Y, Ye ZQ, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Web Server issue):W345–W349. | |
Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4(11):e1000176. | |
Lin MF, Deoras AN, Rasmussen MD, Kellis M. Performance and scalability of discriminative metrics for comparative gene identification in 12 Drosophila genomes. PLoS Comput Biol. 2008;4(4):e1000067. | |
Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27(13):i275–i282. | |
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41(6):e74. | |
Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147(4):789–802. | |
Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154(1):240–251. | |
Bánfai B, Jia H, Khatun J, et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012;22(9):1646–1657. | |
Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17(5):556–565. | |
Bejerano G, Pheasant M, Makunin I, et al. Ultraconserved elements in the human genome. Science. 2004;304(5675):1321–1325. | |
Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. | |
Necsulea A, Soumillon M, Warnefors M, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505(7485):635–640. | |
Kutter C, Watt S, Stefflova K, et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet. 2012;8(7):e1002841. | |
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384. | |
Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012;13(11):R107. | |
de Souza FS, Franchini LF, Rubinstein M. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol Biol Evol. 2013;30(6):1239–1251. | |
Faulkner GJ, Kimura Y, Daub CO, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41(5):563–571. | |
Johnson R, Guigó R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA. 2014;20(7):959–976. | |
Elisaphenko EA, Kolesnikov NN, Shevchenko AI, et al. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One. 2008;3(6):e2521. | |
Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet. 2002;30(2):167–174. | |
da Rocha ST, Boeva V, Escamilla-Del-Arenal M, et al. Jarid2 is implicated in the initial Xist-induced targeting of PRC2 to the inactive X chromosome. Mol Cell. 2014;53(2):301–316. | |
Plath K, Fang J, Mlynarczyk-Evans SK, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300(5616):131–135. | |
Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146(1):119–133. | |
Hamada M, Sato K, Asai K. Prediction of RNA secondary structure by maximizing pseudo-expected accuracy. BMC Bioinformatics. 2010;11:586. | |
Hamada M, Sato K, Asai K. Improving the accuracy of predicting secondary structure for aligned RNA sequences. Nucleic Acids Res. 2011;39(2):393–402. | |
Rivas E, Lang R, Eddy SR. A range of complex probabilistic models for RNA secondary structure prediction that includes the nearest-neighbor model and more. RNA. 2012;18(2):193–212. | |
Novikova IV, Dharap A, Hennelly SP, Sanbonmatsu KY. 3S: shotgun secondary structure determination of long non-coding RNAs. Methods. 2013;63(2):170–177. | |
Pyle AM. Looking at LncRNAs with the ribozyme toolkit. Mol Cell. 2014;56(1):13–17. | |
Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A. 2009;106(1):97–102. | |
Mortimer SA, Trapnell C, Aviran S, Pachter L, Lucks JB. SHAPE-Seq: high-throughput RNA structure analysis. Curr Protoc Chem Biol. 2012;4(4):275–297. | |
Low JT, Weeks KM. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52(2):150–158. | |
Novikova IV, Hennelly SP, Sanbonmatsu KY. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 2012;40(11):5034–5051. | |
Novikova IV, Hennelly SP, Tung CS, Sanbonmatsu KY. Rise of the RNA machines: exploring the structure of long non-coding RNAs. J Mol Biol. 2013;425(19):3731–3746. | |
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147(7):1537–1550. | |
Sarma K, Levasseur P, Aristarkhov A, Lee JT. Locked nucleic acids (LNAs) reveal sequence requirements and kinetics of Xist RNA localization to the X chromosome. Proc Natl Acad Sci U S A. 2010;107(51):22196–22201. | |
Carmell MA, Zhang L, Conklin DS, Hannon GJ, Rosenquist TA. Germline transmission of RNAi in mice. Nat Struct Biol. 2003;10(2):91–92. | |
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518(7539):409–412. | |
Valenzuela DM, Murphy AJ, Frendewey D, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2003;21(6):652–659. | |
Sauvageau M, Goff LA, Lodato S, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife. 2013;2:e01749. | |
Miller JC, Holmes MC, Wang J, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25(7):778–785. | |
Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol. 2011;29(2):149–153. | |
Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–338. | |
Wu X, Sharp PA. Divergent transcription: a driving force for new gene origination? Cell. 2013;155(5):990–996. | |
Xiang JF, Yin QF, Chen T, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24(5):513–531. | |
Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5(10):877–879. | |
Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods. 2012;9(7):743–748. | |
Larsson C, Grundberg I, Söderberg O, Nilsson M. In situ detection and genotyping of individual mRNA molecules. Nat Methods. 2010;7(5):395–397. | |
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–346. | |
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15 Spec No 1:R17–R29. | |
Qureshi IA, Mattick JS, Mehler MF. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010;1338:20–35. | |
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21. | |
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44(4):667–678. | |
Simon MD, Wang CI, Kharchenko PV, et al. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. 2011;108(51):20497–20502. | |
Engreitz JM, Pandya-Jones A, McDonel P, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341(6147):1237973. | |
Engreitz JM, Sirokman K, McDonel P, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159(1):188–199. | |
Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. 2014;9(1):21–26. | |
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. | |
Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425(19):3723–3730. | |
Bertani S, Sauer S, Bolotin E, Sauer F. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol Cell. 2011;43(6):1040–1046. | |
Zhao J, Ohsumi TK, Kung JT, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40(6):939–953. | |
Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106(28):11667–11672. | |
Pandey RR, Mondal T, Mohammad F, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32(2):232–246. | |
Rinn JL, Kertesz M, Wang JK, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–1323. | |
Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329(5992):689–693. | |
Marín-Béjar O, Marchese FP, Athie A, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14(9):R104. | |
Göhring F, Schwab BL, Nicotera P, Leist M, Fackelmayer FO. The novel SAR-binding domain of scaffold attachment factor A (SAF-A) is a target in apoptotic nuclear breakdown. EMBO J. 1997;16(24):7361–7371. | |
Hasegawa Y, Brockdorff N, Kawano S, Tsutui K, Tsutui K, Nakagawa S. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev Cell. 2010;19(3):469–476. | |
Lobov IB, Tsutsui K, Mitchell AR, Podgornaya OI. Specificity of SAF-A and lamin B binding in vitro correlates with the satellite DNA bending state. J Cell Biochem. 2001;83(2):218–229. | |
West JA, Davis CP, Sunwoo H, et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell. 2014; 55(5):791–802. | |
Clemson CM, Hutchinson JN, Sara SA, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33(6):717–726. | |
Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–938. | |
Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415(6873):810–813. | |
Thakur N, Tiwari VK, Thomassin H, et al. An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. Mol Cell Biol. 2004;24(18):7855–7862. | |
Latos PA, Pauler FM, Koerner MV, et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2012;338(6113):1469–1472. | |
Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12(8):542–553. | |
Clemson CM, McNeil JA, Willard HF, Lawrence JB. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol. 1996;132(3):259–275. | |
Chaumeil J, Le Baccon P, Wutz A, Heard E. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 2006;20(16):2223–2237. | |
Beletskii A, Hong YK, Pehrson J, Egholm M, Strauss WM. PNA interference mapping demonstrates functional domains in the noncoding RNA Xist. Proc Natl Acad Sci U S A. 2001;98(16):9215–9220. | |
Senner CE, Nesterova TB, Norton S, et al. Disruption of a conserved region of Xist exon 1 impairs Xist RNA localisation and X-linked gene silencing during random and imprinted X chromosome inactivation. Development. 2011;138(8):1541–1550. | |
Gomez JA, Wapinski OL, Yang YW, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. 2013;152(4):743–754. | |
Onoguchi M, Hirabayashi Y, Koseki H, Gotoh Y. A noncoding RNA regulates the neurogenin1 gene locus during mouse neocortical development. Proc Natl Acad Sci U S A. 2012;109(42):16939–16944. | |
Kim TK, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–187. | |
Lai F, Orom UA, Cesaroni M, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494(7438):497–501. | |
Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470(7333):284–288. | |
Carrieri C, Cimatti L, Biagioli M, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491(7424):454–457. | |
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | |
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–1038. | |
Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. | |
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011; 146(3):353–358. | |
Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369. | |
Wang P, Xue Y, Han Y, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344(6181):310–313. | |
O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550. | |
Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3(107):ra8. | |
Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28(2):195–208. | |
Wang G, Li Z, Zhao Q, et al. LincRNA-p21 enhances the sensitivity of radiotherapy for human colorectal cancer by targeting the Wnt/β-catenin signaling pathway. Oncol Rep. 2014;31(4):1839–1845. | |
Hall JR, Messenger ZJ, Tam HW, Phillips SL, Recio L, Smart RC. Long noncoding RNA lincRNA-p21 is the major mediator of UVB-induced and p53-dependent apoptosis in keratinocytes. Cell Death Dis. 2015;6:e1700. | |
Wang J, Liu X, Wu H, et al. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38(16):5366–5383. | |
Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–8041. | |
Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007;26(6):851–858. | |
Luo JH, Ren B, Keryanov S, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006;44(4):1012–1024. | |
Hatchell EC, Colley SM, Beveridge DJ, et al. SLIRP, a small SRA binding protein, is a nuclear receptor corepressor. Mol Cell. 2006;22(5):657–668. | |
Yan Y, Penner CC, Skliris GP, et al. Steroid receptor RNA activator protein (SRAP) expression as a prognostic factor in ER+ human breast tumors. J Cancer Res Clin Oncol. 2013;139(10):1637–1647. | |
Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. | |
Kogo R, Shimamura T, Mimori K, et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71(20):6320–6326. | |
Prensner JR, Chen W, Iyer MK, et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014;74(6):1651–1660. | |
Yap KL, Li S, Muñoz-Cabello AM, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38(5):662–674. | |
Zhang EB, Yin DD, Sun M, et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014;5:e1243. | |
Quagliata L, Matter MS, Piscuoglio S, et al. Long noncoding RNA HOTTIP/HOXA13 expression is associated with disease progression and predicts outcome in hepatocellular carcinoma patients. Hepatology. 2014;59(3):911–923. | |
Chiesa N, De Crescenzo A, Mishra K, et al. The KCNQ1OT1 imprinting control region and non-coding RNA: new properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum Mol Genet. 2012;21(1):10–25. | |
Cabianca DS, Gabellini D. The cell biology of disease: FSHD: copy number variations on the theme of muscular dystrophy. J Cell Biol. 2010;191(6):1049–1060. | |
Mackay DJ, Coupe AM, Shield JP, Storr JN, Temple IK, Robinson DO. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet. 2002;110(2):139–144. | |
Lin N, Chang KY, Li Z, et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell. 2014;53(6):1005–1019. | |
Moseley ML, Zu T, Ikeda Y, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38(7):758–769. | |
Faghihi MA, Modarresi F, Khalil AM, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. | |
Parenti R, Paratore S, Torrisi A, Cavallaro S. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during beta-amyloid-induced apoptosis. Eur J Neurosci. 2007;26(9):2444–2457. | |
Massone S, Vassallo I, Fiorino G, et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol Dis. 2011;41(2):308–317. | |
Sai Y, Zou Z, Peng K, Dong Z. The Parkinson’s disease-related genes act in mitochondrial homeostasis. Neurosci Biobehav Rev. 2012;36(9):2034–2043. | |
Scheele C, Petrovic N, Faghihi MA, et al. The human PINK1 locus is regulated in vivo by a non-coding natural antisense RNA during modulation of mitochondrial function. BMC Genomics. 2007;8:74. | |
Barry G, Briggs JA, Vanichkina DP, et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. 2014;19(4):486–494. | |
Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003; 302(5648):1212–1215. | |
Hafner M, Landthaler M, Burger L, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141(1):129–141. | |
Lebedeva S, Jens M, Theil K, et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43(3):340–352. | |
König J, Zarnack K, Rot G, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17(7):909–915. | |
Huarte M, Guttman M, Feldser D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–419. | |
Marín-Béjar O, Huarte M. RNA pulldown protocol for in vitro detection and identification of RNA-associated proteins. Methods Mol Biol. 2015;1206:87–95. | |
Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149(6):1393–1406. | |
Quinn JJ, Ilik IA, Qu K, et al. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat Biotechnol. 2014;32(9):933–940. | |
Lindblad-Toh K, Garber M, Zuk O, et al; Broad Institute Sequencing Platform and Whole Genome Assembly Team; Baylor College of Medicine Human Genome Sequencing Center Sequencing Team; Genome Institute at Washington University. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478(7370):476–482. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.