Back to Journals » Cancer Management and Research » Volume 11
Long non-coding RNA TUG1-mediated down-regulation of KLF4 contributes to metastasis and the epithelial-to-mesenchymal transition of colorectal cancer by miR-153-1
Authors Shao H, Dong D, Shao F
Received 12 March 2019
Accepted for publication 19 July 2019
Published 24 September 2019 Volume 2019:11 Pages 8699—8710
DOI https://doi.org/10.2147/CMAR.S208508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Hongjin Shao,1 Dianbo Dong,1 Feng Shao2
1Department of Proctology, Liaocheng People’s Hospital, Liaocheng, Shandong 252000, People’s Republic of China; 2Department of Gastrointestinal Surgery, Liaocheng People’s Hospital, Liaocheng, Shandong 252000, People’s Republic of China
Correspondence: Feng Shao
Department of Gastrointestinal Surgery, Liaocheng People’s Hospital, Liaocheng, Shandong 252000, People’s Republic of China
Tel +86 1 396 956 7952
Email [email protected]
Introduction: Taurine up-regulated 1 (TUG1) was reported to be over-expressed and involved in various human malignancies. However, its expression status and mechanistic importance in colorectal cancer (CRC) were yet to be defined.
Methods: Relative expressions of TUG1, miR-153-1 and Kruppel-like factor 4 (KLF4) were analyzed by real-time PCR. The potential influences of TUG1-proficiency and miR-153-1-deficiency on cell proliferation, migration and viability were determined by colony formation, wound healing and CCK-8 assays, respectively. Cell invasion was evaluated by transwell chamber assay. The regulatory effect of KLF4 on miR-153-1 was interrogated by luciferase reporter assay. Direct association between KLF4 and miR-153-1 promoter was measured by chromatin immunoprecipitation (ChIP) assay. Subcellular localization of TUG1 was determined by fractionization PCR. Enrichment of EZH2 on KLF4 promoter was analyzed by ChIP-PCR. The pro-tumoral activity of TUG1 was determined using xenograft tumor model.
Results: We demonstrated the over-expression of TUG1 and down-regulation of miR-153-1 in CRC. Knockdown of TUG1 or ectopic over-expression of miR-153-1 in SW480 significantly suppressed cell proliferation, migration and viability. TUG1 negatively modulated miR-153-1 expression, and simultaneous expression of TUG1 completely abolished the anti-tumor effect of miR-153-1. We further identified KLF4 as a transcription factor of miR-153-1, which was negatively regulated by TUG1 along with EZH2.
Conclusion: Our study unravels the critical involvement of TUG1/KLF4/miR-153-1 axis in CRC.
Keywords: TUG1, miR-153-1, colorectal cancer, KLF4, EZH2
Introduction
Colorectal cancer (CRC) is the third most common human malignancy worldwide, and the morbidity is continuously increasing in developed countries.1 In 2018, it is estimated that 140,000 new cases were diagnosed and 50,000 cancer-related deaths were claimed in the United States.2 Old age and unhealthy lifestyle significantly elevate the probability of CRC. Other recognized risk factors linked to the incidence of CRC include diet, obesity, smoking and lack of physical exercises.3 Another crucial risk factor is inflammatory bowel diseases including Crohn’s disease and ulcerative colitis.4 In addition, several hereditary genetic disorders have been identified as causal aberrance for the occurrence of CRC, such as Lynch syndrome, Gardner syndrome and familial adenomatous polyposis.5 The primary bowel cancer is usually diagnosed through biopsy during sigmoidoscopy or colonoscopy, and regularly followed by medical imaging examination to determine the metastatic status of disease.6 Preventative screening is effective and recommended to population over 50 years old. Currently, clinical options for this disease include surgical resection, radiotherapy, chemotherapy and targeted therapies.7 The prognostic outcomes of CRC heavily depend on cancer stage and individual health conditions, and 5-year survival rate in the US is around 65%. Still, insightful and comprehensive understanding of this disease at molecular level is urgently necessary.
Long non-coding RNA (lncRNA) is defined as oligonucleotide longer than 200 bases without protein-coding potential. The kaleidoscopic physiological roles of lncRNAs have been implicated in gene transcription regulation, post-transcriptional regulation and epigenetic programming.8 A variety of lncRNAs has been characterized to be playing critical roles in human cancers. Taurine up-regulated 1 (TUG1) recently attracted intensive attentions from the research community and its importance in cancer biology has been increasingly uncovered. Wang et al showed in osteosarcoma cells that TUG1 promoted cell migration and invasion by acting as a competing endogenous RNA of miR-335-5p.9 Li et al proposed that TUG1 predicted unfavorable prognosis and promoted cell proliferation and inhibited cell apoptosis in epithelial ovarian cancer.10 Li et al provided evidence that TUG1 promoted proliferation and inhibited apoptosis of osteosarcoma cells by sponging miR-132-3p and up-regulating SOX4 expression.11 In CRC, Li et al characterized TUG1-mediated methotrexate resistance via the miR-186/CPEB2 axis.12 Zhai13 and Wang14 et al demonstrated, respectively, that over-expression of TUG1 promoted colon cancer progression and metastasis via epithelial-to-mesenchymal transition (EMT) pathway. In line with this notion, here we set out to characterize the expression status and mechanistic involvement of TUG1 in CRC.
The tumor suppressor role of miR-153-1 has been recognized in some human cancers including melanoma, glioma, lung cancer and breast cancer. Zeng et al reported miR-153-1 suppressed cell proliferation and invasion by targeting SNAl1 in melanoma.15 Cui et al demonstrated miR-153-1 targeted mTORC2 component Rictor to inhibit glioma cells.16 Yuan et al proposed that miR-153-1 exerted anti-tumor activity via suppression of AKT signaling in lung cancer.17 The intimate regulatory mode-of-action between lncRNA and miRs has been increasingly recognized to be fundamentally involved in tumorigenesis and progression in a variety of human malignancies. Notably, Wang et al reported that knockdown of TUG1 inhibited the proliferation and cellular invasion of osteosarcoma cells by sponging miR-153-1,18 which prompted us to elucidate the potential interaction between these two non-coding RNAs in CRC.
Materials and methods
Patient samples
CRC tumor tissues and corresponding adjacent benign tissues were collected from 40 patients at Liaocheng People’s Hospital, Shandong Province between January 2012 and February 2017. All samples were confirmed by three independent pathologists and flash-cryopreserved in liquid nitrogen. Written informed consents were obtained from all enrolled patients. The study was conducted in accordance with the Declaration of Helsinki and the protocol was approved by the Liaocheng People’s Hospital, Shandong Province Ethic Review Committee.
Cell culture
Three CRC cell lines (HCT116, HT29 and SW480) and normal human colon mucosal epithelial cell line NCM460 were obtained from and authenticated by Cell Bank (Shanghai, China). Mycoplasma contamination was examined by PCR method regularly. Cells were maintained in Roswell Park Memorial Institute (RPMI) modified medium supplemented with 10% FBS (Gibco, Grand Island, NY, USA) and 100 U/mL penicillin plus 100 mg/mL streptomycin (Gibco) in humidified incubator at 37°C with 5% CO2.
Real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, MA, USA, USA), and real-time PCR was conducted with SYBR Green Reaction Mix (Qiagen, Valencia, CA, USA) for TUG1 quantitation, using GAPDH as internal reference. RNA was immediately converted into cDNA using SuperScript II reverse transcriptase (Invitrogen). The primer sequences were provided below:
TUG1 forward: 5′-CTGAAGAAAGGCAACATC-3′
TUG1 reverse: 5′-GTAGGCTACTACAGGATTTG-3′;
GAPDH forward: 5′-AGCCACATCGCTCAGACAC-3′
GAPDH reverse: 5′-GCCCAATACGACCAAATCC-3′.
The relative expression was determined using 2−ΔΔCt method. For TUG1 subcellular localization, the nuclear and cytosolic fractions from both SW480 and HCT116 cells were separated with PARIS Kit (Life Technologies, Pleasanton, CA, USA).
Cell transfection
miR-153-1, scramble control, si-TUG1 and negative control (si-NC) were synthesized by Ribobio (Guangzhou, China). The pcDNA-TUG1 expressing plasmid was constructed by PCR amplifying TUG1 and ligated into pcDNA3.1 (Invitrogen). Cell transfection was carried out using Lipofectamine 2000 (Invitrogen) as instructed by the manufacturer’s manual.
Colony formation assay
SW480 cells were first transfected with si-TUG1, miR-153-1 mimic, or scramble, and then seeded into 6-well plate in triplicates (200 cells/well) and allowed for consecutive culture. Ten days later, the culture medium was completely aspirated and cells were subjected to fixation with 4% PFA followed by crystal violet (0.05%) staining. Representative images were captured and colony numbers were counted under a light microscope (Leica, Heerbrugg, Switzerland).
Transwell assay
Transwell chambers (BD Biosciences, Franklin Lakes, NJ, USA) were applied to measure cell invasive capacity. The indicated cells (1×105) resuspended in serum-free medium were plated into the upper compartment pre-coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). The lower chamber was supplemented with 700 μL complete medium with 10% FBS. Invaded cells were visualized by crystal violet staining after 24 hrs. The images were photographed using a microscope (Olympus, Tokyo, Japan), and cells were counted in three random fields.
Cell viability
SW480 cells were transiently transfected with either si-TUG1 or miR-153-1. After 24 hrs, the indicated cells were placed into 96-well plates at the density of 103 cells/100 μL and allowed for culture for another 24 hrs. Fresh medium was replaced and 10 μL of CCK-8 reagent (Dojindo, Kumamoto, Japan) was added and incubated for 3 hrs. OD 450 was then measured on a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Luciferase reporter assay
Either wild-type or putative Kruppel-like factor 4 (KLF4)-recognition site-mutated miR-153-1 promoter was fused to the psi-CHECK-2 plasmid (Promega, Fitchburg, WI, USA), and co-transfected with KLF4 expressing plasmids. The relative luciferase activity was measured using the Dual-Glo Luciferase Assay kit (Promega) in accordance with the provider’s instruction on SpectraMax M5 (Molecular Devices).
Chromatin immunoprecipitation (ChIP)
ChIP was conducted with Pierce Agarose ChIP Kit (ThermoFisher, Waltham, MA, USA) following the manufacturer’s manual. Briefly, the indicated cells cultured in 100 mm petri dishes were first fixed and cross-linked in 0.8% formaldehyde for 15 mins. The reaction was quenched by addition of glycine solution and cells were collected by scraping. Cell pellet was lysed and digested with micrococcal nuclease at 37°C for 30 mins, the supernatant was collected and diluted. The anti-KLF4 antibody (Abcam, Cambridge, UK) or IgG control was added for incubation at 4°C overnight and the immunocomplex was precipitated with Protein A/G Agarose beads (ThermoFisher). The protein/DNA complex was eluted and crosslink was reversed by proteinase K at 65°C for 2 hrs. The genomic DNA fragments were retrieved and analyzed by qRT-PCR.
Western blot
Protein was lysed in RIPA buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and protein concentration was determined by BCA kit (Beyotime, Nantong, China). Equal amount of protein was resolved by 12% SDS-PAGE gel and transferred onto PVDF membrane (Roche, Penzberg, Upper Bavaria, Germany). After blocking with 5% milk, specific primary antibodies (anti-EZH2, #4905, 1:1000; anti-KLF4, #4308, 1:1000; anti-GAPDH, #2118, 1:1000; anti-E-cadherin, #3195, 1:1000; anti-N-cadherin, #4061, 1:1000; Cell Signaling Technology, MA, USA) were incubated at 4°C overnight, followed by washing and further incubation with appropriate secondary antibodies (anti-rabbit, #7074, 1:5000, Cell Signaling Technology, MA, USA). GAPDH was used for loading control and blots were visualized with ECL kit (Millipore, Billerica, MA, USA).
Xenograft tumor model
BALB/c nude mice (4-week-old, 18–20 g) were purchased from Vital River (Beijing, China) and housed in the SPF environment supplied with food and drinking water ad libitum. The animal-related study followed the Guide for the Care and Use of Laboratory Animals in Liaocheng People’s Hospital, and was approved by the Animal Welfare Committee of Liaocheng People’s Hospital, Shandong Province. Either TUG1-intact or -depleted SW480 cells (5×106 cells/100 μL, 8–12 mice per group) were subcutaneously inoculated into both lower flanks, and xenograft tumor progression was regularly monitored. All mice were sacrificed at day 30 post-injection and xenograft tumors were surgically extracted.
Statistical analysis
SPSS 23.0 (SPSS Inc., Chicago, IL, USA) was employed for statistical analysis. The unpaired Student’s t-test was applied for inter-group comparison and one-way ANOVA analysis was performed for multiple group comparison. Correlations between TUG1 and miR-153-1 as well as between TUG1 and KLF4 in CRC samples were analyzed with Spearman’s algorithm. P<0.05 were considered as statistical significance.
Results
Overexpression of TUG1 and down-regulation of miR-153-1 in CRC
We first set out to analyze the relative abundance of TUG1 and miR-153-1 in CRC both in vivo and in vitro. To this end, we collected 40 pairs of normal tissues and tumor tissues from CRC patients. Quantitative PCR results demonstrated significant higher expression of TUG1 in tumors in comparison with their normal counterparts (Figure 1A, P<0.01). Similarly, the relative abundance of TUG1 was remarkably increased in the CRC cells including SW480, HCT116 and HT29, compared to normal colorectal cell NCM460. At the same time, the relative expression of miR-153-1 was determined simultaneously by real-time PCR. In contrast to the significant overexpression of TUG1, we observed markedly down-regulation of miR-153-1 both in CRC tissue samples and cell lines (Figure 1B). Our data suggested an inverse correlation between TUG1 and miR-153-1 in CRC both in vivo and in vitro, which might indicate the negative regulatory mechanism underlying this phenotype.
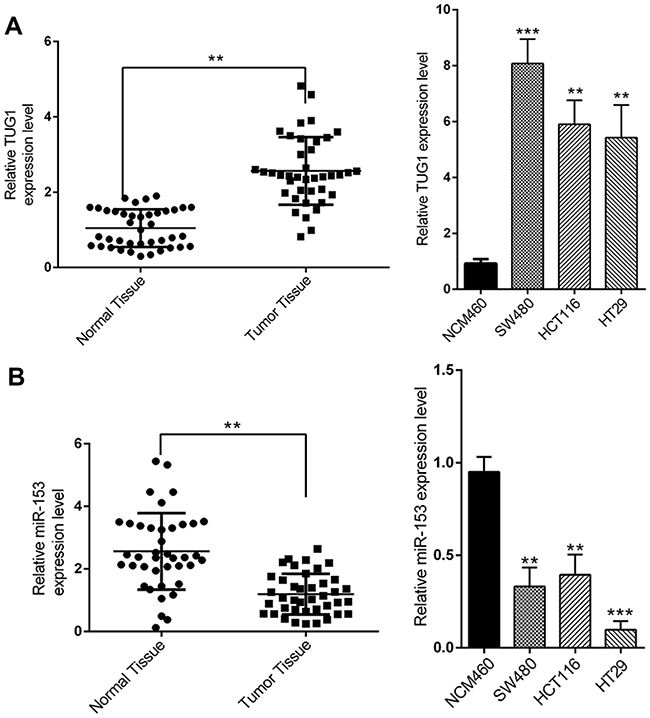 |
Figure 1 The expression levels of TUG1 and miR-153-1 in CRC tissues and cell lines. (A) TUG1 was detected in CRC tissues and adjacent normal mucosa tissues (left) and in different CRC cell lines (right) by qRT-PCR. (B) mi-R153 was detected in CRC tissues and adjacent normal mucosa tissues (left) and in different CRC cell lines (right) by qRT-PCR. Values were expressed as mean ± SEM. ***P<0.001, **P<0.01.Abbreviations: TUG1, taurine up-regulated 1; CRC, colorectal cancer. |
TUG1-deficiency and miR-153-1-proficiency inhibit proliferation and invasion of CRC cells
Next, we sought to elucidate the precise roles of TUG1 and miR-153-1 in the tumor biology of CRC. To this purpose, we specifically silenced TUG1 in SW480 cells, and success in establishing TUG1-depleted cell line was validated by real-time PCR (Figure 2A). Phenotypically, TUG1 silencing remarkably compromised the colony formation capacity in comparison with scramble control (Figure 2B). Cell migration as indicated by wound closure was significantly suppressed in the TUG1-silencing cells as well (Figure 2C). Likewise, cell viability was determined by CCK-8 assay, which showed an obvious reduction in TUG1-deficient SW480 cells as well (Figure 2D). On the other hand, we ectopically expressed miR-153-1 into SW480 cells, which was confirmed by real-time PCR (Figure 2E). Colony formation capacity was greatly compromised by miR-153-1 as indicated by the decreased colony numbers (Figure 2F). Wound healing was remarkably retarded by miR-153-1 overexpression in SW480 cells as well (Figure 2G). Consistently, cell viability was significantly suppressed by ectopic miR-153-1 (Figure 2H). Therefore, our data uncovered that both TUG1 knockdown and miR-153-1 overexpression-inhibited proliferation and migration of CRC cells.
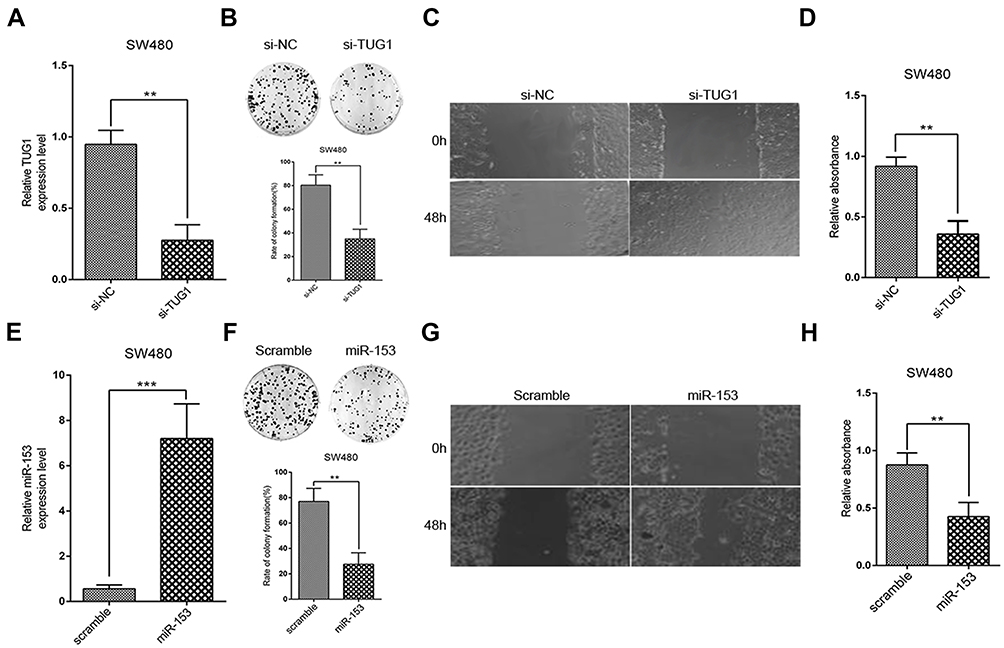 |
Figure 2 TUG1 knockdown and miR-153-1 overexpression inhibit proliferation and invasion of CRC cells. (A) SW480 and HCT116 (data not shown) were transfected with si-NC or si-TUG1 and transfection efficiency was determined with qRT-PCR. (B–C) Colony formation and Wound healing assays were performed to measure the effect of TUG1 on the proliferation and invasion of CRC cells. (D) CCK-8 assay was performed to determine the viability of CRC cells. (E) SW480 and HCT116 (data not shown) were transfected with scramble or miR-153-1 mimic and transfection efficiency was determined with qRT-PCR. (F–G) Colony formation and wound healing assays were performed to measure the effect of miR-153-1 on the proliferation and invasion of CRC cells. (D) CCK-8 assay was performed to determine the viability of CRC cells. Values were expressed as mean ± SEM of three independent experiments. Data were collected from at least three independent experiments. ***P<0.001, **P<0.01.Abbreviations: TUG1, taurine up-regulated 1; CRC, colorectal cancer. |
TUG1 overexpression reverses miR-153-1-mediated inhibition of proliferation and invasion in CRC cells
Next, we sought to clarify the potential regulatory relationship between TUG1 and miR-153-1 in CRC. The correlation between TUG1 and miR-153-1 expression was analyzed by Spearman’s method, and significantly inverse relation was observed (R=−0.674, P<0.001, Figure 3A). Specific knockdown of TUG1 led to remarkably increased endogenous miR-153-1 in SW480 cells, while the ectopic introduction of TUG1 greatly inhibited miR-153-1 expression (Figure 3B). Similar conclusion was consolidated in HCT116 cells as well (Figure 3C). Co-transfection with TUG1-expressing plasmids almost completely reversed the inhibitory effect on cell viability imposed by miR-153-1 overexpression (Figure 3D). Likewise, forced expression of TUG1 restored the colony formation capacity, which was greatly suppressed by miR-153-1 expression alone (Figure 3E). We further examined the cell invasive behavior in response to alterations in miR-153-1 and TUG1. In support of the tumor suppressor role of miR-153-1, our transwell assay results demonstrated that miR-153-1 overexpression could reduce the invaded cell number, which was partially restored by co-expression of TUG1 (Figure 3F). Taken together, our data suggested that TUG1 negatively regulated miR-153-1 expression in CRC cells, which consequently antagonized the anti-tumor activity of miR-153-1 in this context.
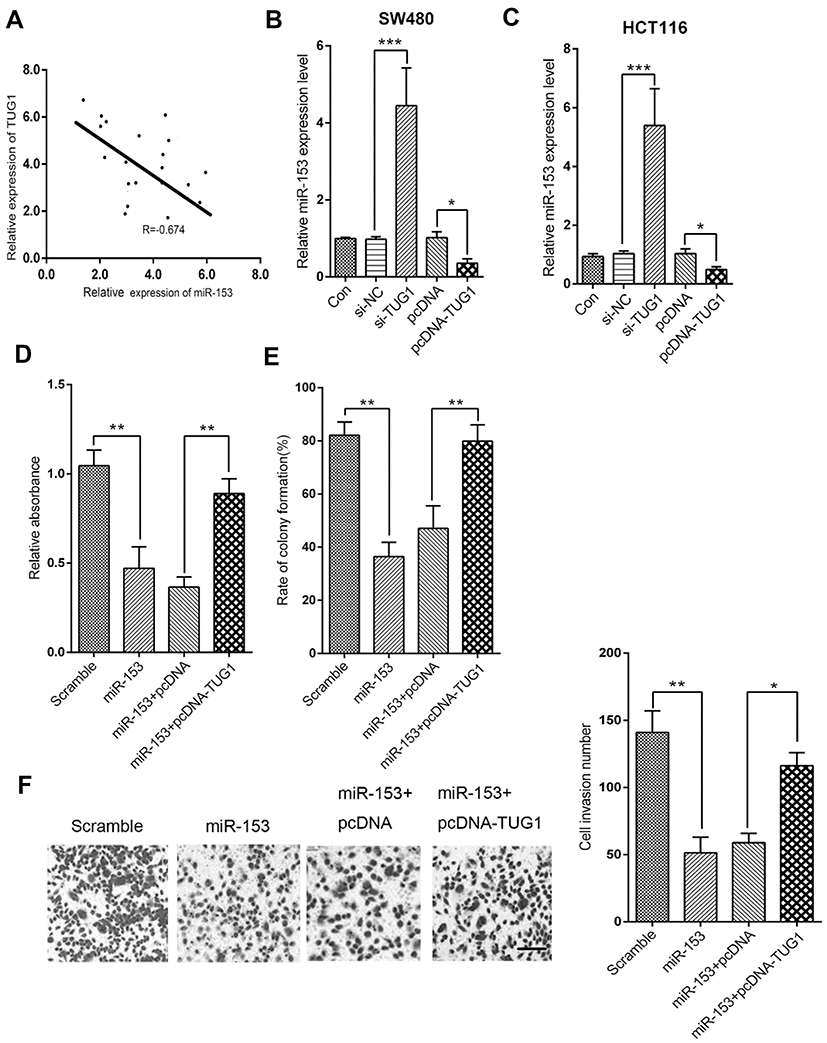 |
Figure 3 TUG1 overexpression reversed miR-153-1-mediated inhibition of proliferation and invasion in CRC cells. (A) The correlation between miR-153-1 and TUG1 was analyzed by Spearman’s correlation analysis (R=−0.674, P<0.001). (B–C) qRT-PCR analysis was performed to detect the expression of miR-153-1 in SW480 and HCT116 cells with si-TUG1 or pcDNA-TUG1. (D–F) SW480 cells were transfected with either miR-153-1 or in combination with pcDNA-TUG1. (D) The viability of SW480 cells was determined by CKK-8 assay. (E) Colony formation assay was performed to detect the colony numbers of SW480 cells. (F) The invasive ability of SW480 cells was determined by Transwell invasion assay. Scale bar =100 μm. Values were expressed as mean ± SEM. ***P<0.001, **P<0.01, *P<0.05.Abbreviations: TUG1, taurine up-regulated 1; CRC, colorectal cancer. |
Identification of KLF4 as a direct transcriptional factor for miR-153-1
Here we set out to elucidate the regulatory mechanism underlying the suppressed expression of miR-153-1 in CRC. With the aid of UCSC genome browser and JASPAR Bioinformatics Analyzer, we identified three putative KLF4 binding sites, which prompted us to hypothesize that KLF4 might be involved in miR-153-1 in this scenario. To experimentally validate this possibility, we first determined the relative expression of KLF4 in CRC cell lines. As shown in Figure 4A, we noticed that KLF4 expression was significantly down-regulated in CRC cell lines in comparison with normal control, and positive correlation between KLF4 and miR-153-1 was observed in the clinical samples as well (Figure 4B). Over-expression of KLF4 cells induced markedly increased endogenous miR-153-1 in SW480 (Figure 4C), and relative level of KLF4 was quantified in Figure 4D and E. We further employed luciferase reporter assay to interrogate the regulatory mode of KLF4 on miR-153-1 expression. Exogenous KLF4 stimulated remarkably increased luciferase activity driven by miR-153-1 in both SW480 (Figure 4F) and HCT116 cells (Figure 4G). The mutation introduced into the third putative binding site, rather than the other two, completely abolished the stimulatory effect of KLF4, which implied a direct recognition between KLF4 and this motif (Figure 4H and I). The physical association between KLF4 and miR-153-1 promoter was further confirmed by ChIP assay, wherein significant enrichment of miR-153-1 promoter was observed in the KLF4 immunoprecipitated complex (Figure 4J and K). Therefore, our data for the first time uncovered KLF4 as the direct transcription factor involved in the regulation of miR-153-1 expression.
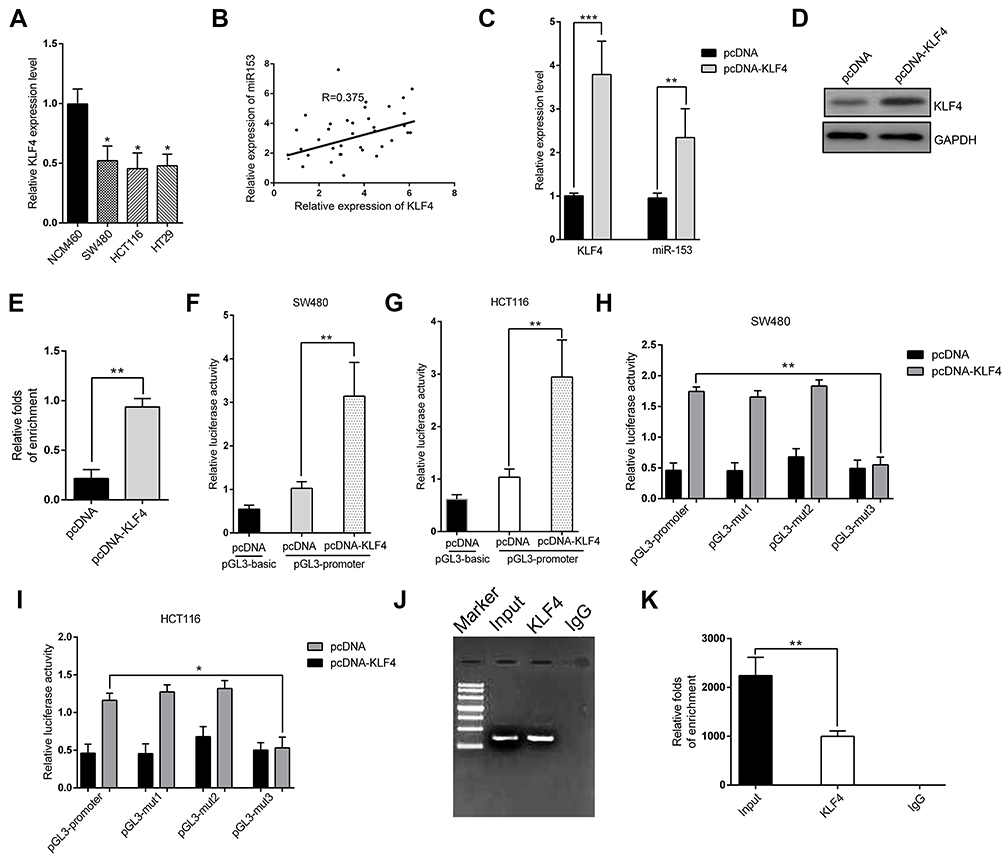 |
Figure 4 Identification of KLF4 as a direct transcriptional factor for miR-153-1. (A) KLF4 expression level was detected in different CRC cell lines by qRT-PCR. (B) The correlation between miR-153-1 and KLF4 was analyzed by Spearman’s correlation analysis (R=0.375, P=0.0102). (C) qRT-PCR analysis of the mRNA level of KLF4 and the effect of KLF4 plasmid on miR-153-1. (D) Western blot for KLF4 protein level after transfection with cloned vector. (E) Relative folds of enrichment from Western blot. (F–G) Relative luciferase activity of the miR-153-1 promoter in SW480 cells and HCT116 cells transfected with pcDNA-KLF4. Luciferase constructs containing the miR-153-1 promoter (pGL3-promoter) and the unmodified construct (pRL-TK) were co-transfected with pcDNA3.1 and pcDNA-KLF4 into SW480 cells and HCT116 cells. Firefly luciferase activity was normalized to Renilla luciferase activity. (H–I) Relative luciferase activity of the mutant miR-153-1 promoter constructs in SW480 and HCT116 cells transfected with pcDNA-KLF4. Firefly luciferase activity was normalized to Renilla luciferase activity. (J) Electrophoresis graph shows the result from ChIP qRT-PCR quantitative analysis. (K) Relative folds of enrichment from the electrophoresis image intensity. Values were expressed as mean ± SEM. ***P<0.001, **P<0.01, *P<0.05.Abbreviations: CRC, colorectal cancer; KLF, Kruppel-like factor 4. |
KLF4 inhibits CRC cell proliferation and migration
Our previous results disclosed the regulatory action of KLF4 on miR-153-1 transcription, which might fundamentally contribute to the proliferation and invasion of CRC. To clarify this issue, we transiently silenced KLF4 in SW480 cells while simultaneously overexpressed either scramble or miR-153-1 (Figure 5A), and ectopically expressed KLF4 in HCT116 cells while co-transfected with either negative control or miR-153-1-specific inhibitor (Figure 5B). Noteworthily, over-expression or down-regulation of miR-153-1 imposed no significant influences on KLF4 expression in both scenarios. KLF4-deficiency slightly, but significantly, increased cell invasion, which was abrogated by co-expression of miR-153-1 (Figure 5C). Ectopic over-expression of KLF4 inhibited invasion in HCT116, which was readily restored by miR-153-1 inhibitor (Figure 5D). Our data clearly unraveled the anti-tumor properties of KLF4 in CRC.
 |
Figure 5 KLF4 inhibit CRC cell proliferation and migration in vitro. (A) Transfection with siKLF4 promoted SW480 cell proliferation. Transfection with miR-153-1 mimic decreased cell proliferation in the cells transfected with si-KLF4. (B) KLF4 overexpression suppressed the proliferation of HCT116 cells transfected with miR-153-1 inhibitor. (C–D) Transwell motility assays of SW480 and HCT116 cells transfected with si-KLF4 and miR-153-1 mimic or KLF4 overexpression vector and miR-153-1 inhibitor. Values were expressed as mean ± SEM. ***P<0.001, **P<0.01, *P<0.05.Abbreviations: CRC, colorectal cancer; KLF, Kruppel-like factor 4. |
TUG1 negatively regulates the expression of KLF4 via interacting with EZH2
Next, we sought to clarify the relation between TUG1 and KLF4 with respect to miR-153-1 regulation. An inverse correlation was uncovered in our CRC samples as shown in Figure 6A. TUG1-silencing induced significant up-regulation of KLF4 transcription in both SW480 and HCT116 cells (Figure 6B). Consistent results were confirmed at protein level by Western blot (Figure 6C and D). We further silenced EZH2 in both SW480 and HCT116 by specific siRNA to investigate the suppressive effect on KLF4 expression. The knockdown efficiency was validated by Western blot as shown in the upper panel in Figure 6E. EZH2-deficiency remarkably compromised the enrichment of KLF4 promoter in the ChIP complex with anti-EZH2 antibody (lower panel in Figure 6E), and up-regulated KLF4 transcription (Figure 6F) and translation (Figure 6G and H) as well. The subcellular localization of TUG1 was examined by fractionization, which unambiguously displayed the predominant nucleus localization of TUG1, which supported its physiological role involved in transcriptional modulation (Figure 6I and J). Notably, TUG1 knockdown compromised the suppressive regulation of EZH2 on KLF4 as indicated by the reduced enrichment in both SW480 and HCT116 cells (Figure 6K and L). Therefore, our results suggested that TUG1 negatively regulated KLF4 expression via interacting with EZH2.
 |
Figure 6 TUG1 negatively regulates expression of KLF4 via interacting with EZH2. (A) The correlation between TUG1 and KLF4 was analyzed by Spearman’s correlation analysis (r=−0.549, P<0.001). (B) The levels of KLF4 mRNA were detected by qPCR when SW480 and HCT116 cells transfected with si-TUG1 and results are expressed relative to the corresponding values for control cells. (C–D) The levels of KLF4 protein levels were determined by Western Blotting when SW480 and HCT116 cells were transfected with si-TUG1. (E) The levels of EZH2 protein were detected by Western Blotting when SW480 and HCT116 cells transfected with si-EZH2 and results are expressed relative to the corresponding values for control cells. (F) The levels of KLF4mRNA were detected by qPCR when SW480 and HCT116 cells transfected with si-EZH2 and results are expressed relative to the corresponding values for control cells. (G–H) The levels of KLF4 protein were detected by Western Blotting when SW480 and HCT116 cells transfected with si-EZH2 and results are expressed relative to the corresponding values for control cells. (I–J) TUG1 expression levels in cell cytoplasm or nucleus of HCC cell lines SW480 and HCT116 were detected by qPCR. (K–L) ChIP-qPCR analysis of IgG, EZH2 occupancy in the KLF4 promoter region with or without TUG1 silencing in SW480 and HCT116 cells. Values were expressed as mean ± SEM. ***P<0.001, **P<0.01.Abbreviations: TUG1, taurine up-regulated 1; KLF, Kruppel-like factor 4. |
The TUG1-KLF4-miR-153-1 axis in CRC in vivo
Our previous data unraveled the TUG1-KLF4-miR-153-1 axis in vitro. Next, we sought to confirm our preliminary observations in vivo using xenograft tumor model. TUG1-deficient SW480 cells were subcutaneously inoculated into immunodeficient mice and tumor progression was monitored. In comparison with scramble control, TUG1 depletion significantly retarded xenograft tumor growth (Figure 7A). The regulatory axis was validated in the tumor samples at the endpoint of experiment, wherein remarkable up-regulation of both KLF4 and miR-153-1 was observed (Figure 7B). In addition, tumor metastasis-related EMT marks including E-Cadherin and N-Cadherin were examined in three pairs of xenograft tumors. Evident up-regulation of E-Cadherin and down-regulation of N-Cadherin were found in TUG1-deficient mice (Figure 7C), which indicated the inhibitory effect on EMT and metastatic processing in vivo. In summary, we demonstrated that TUG1/KLF4/miR-153-1 signaling axis contributed to the growth and EMT of CRC in vivo.
 |
Figure 7 TUG1/KLF4/miR-153-1 axis contributed to the proliferation and EMT in vivo. (A) Growth curve of tumor volumes and representative tumor photographs were generated and obtained after 24 days. n=8–12. (B) The levels of KLF4 and miR-153-1 in tumor tissues were detected by qRT-PCR. (C) Western blotting was utilized to detect the expression level of E-cadherin and N-cadherin. Values were expressed as mean ± SEM of three independent experiments. **P<0.01.Abbreviations: TUG1, taurine up-regulated 1; KLF, Kruppel-like factor 4; EMT, epithelial-to-mesenchymal transition. |
Discussion
Previous study reported that TUG1 regulated cell proliferation and invasion in osteosarcoma via miR-153-1, which prompted us to investigate this potential scenario in CRC. Through analyzing both clinical tissue samples and established cell lines, we clearly demonstrated aberrant up-regulation of TUG1 in CRC, which hinted an oncogenic role of TUG1 in this disease. Inversely, miR-153-1 was remarkably down-regulated in CRC, which suggested a potential negative regulation between miR-153-1 and TUG1 in CRC. Specific knockdown of TUG1 significantly compromised the colony formation and wound healing capacities in SW480 cells. Likewise, cell viability was greatly inhibited in the TUG1-deficient cells. In the same way, ectopic introduction of miR-153-1 suppressed cell proliferation, migration and viability in SW480 cells. Simultaneous over-expression of TUG1 completely reversed the inhibitory effects of miR-153-1 on proliferation and invasion in CRC cells. Noteworthily, our study for the first time predicted and identified KLF4 as a direct transcription factor of miR-153-1 in CRC. Forced expression of KLF4 induced marked up-regulation of endogenous miR-153-1, and stimulated luciferase activity driven by miR-153-1 promoter. We further identified the recognition site of KLF4 on miR-153-1 promoter, wherein mutation completely abolished inducing action of KLF4. Direct association between KLF4 and miR-153-1 promoter region was validated by ChIP-PCR as well. As the upstream signaling cue, we further demonstrated that over-expression of KLF4 greatly inhibited cell proliferation and invasion in HCT116 cells, while silencing KLF4 markedly stimulated cell viability and invasion in SW480 cells. The endogenous expression of KLF4 was negatively correlated with TUG1, depletion of which greatly promoted up-regulation of KLF4 at both transcript and protein levels. We also provided experimental evidence in support of the inhibitory regulation of TUG1 on KLF4 expression via interacting with EZH2, which was critically involved in the transcriptional suppression of KLF4. Notably, we consolidated our observations in vivo using xenograft tumor model, wherein TUG1-silencing significantly delayed tumor progression and blocked EMT process as indicated by increased E-Cadherin. Our study highlighted the importance of TUG1/KLF4/miR-153-1 signaling axis in the tumor biology of CRC and deepened our understanding of this disease at the molecular level.
KLF4 belongs to the Kruppel family of transcription factors, which encode zinc finger proteins required for normal development of the barrier function of skin.19 Accumulative evidence have uncovered the critical roles of KLF4 in a number of human malignancies. For instance, Nagata et al proposed that KLF4 and NANOG possessed prognostic biomarker value for triple-negative breast cancer.20 Zhao et al demonstrated that lncRNA SNHG5/miR-32 axis-regulated gastric cancer cell proliferation and migration by targeting KLF4.21 In bladder cancer cells, Minami et al uncovered that miR-145 negatively regulated Warburg effect by silencing KLF4 and PTBP1.22 Yan et al reported that KLF4-mediated suppression of CD44 signaling negatively impacted pancreatic cancer stemness and metastasis.23 Wei et al characterized that KLF4 was essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis.23 In colorectal carcinoma, Zhai et al showed miR-543 promoted cancer proliferation and metastasis by targeting KLF4.24 Ma et al unraveled that KLF4 inhibited CRC cell proliferation dependent on NDRG2 signaling.25 Xu et al provided evidence that over-expression of KLF4 promoted cell senescence through miR-203-survivin-p21 pathway.26 Consistent with the well-acknowledged anti-tumor properties of KLF4 in CRC, here we for the first time displayed that KLF4 was directly involved in miR-153-1 transcription, which eventually contributed to its tumor suppressor function. Noteworthily, while most investigations focused on the downstream regulatory network of dysregulated microRNAs, here we bioinformatically predicted and experimentally identified KLF4 as the transcription factor of miR-153-1.
Regarding the suppressive expression of KLF4 in CRC, we unraveled the TUG1 specifically inhibited KLF4 transcription via direct association with EZH2. The synergistic or serial regulatory scenario between KLF4 and TUG1 has been addressed by several investigations. For example, Chen et al disclosed that EZH2-mediated α-actin methylation required lncRNA TUG1 and promoted the cortex cytoskeleton formation in vascular smooth muscle cells.27 In osteosarcoma, Cao et al demonstrated TUG1 promoted tumorigenesis by up-regulating EZH2 expression via competing with miR-144-3p.28 Niu et al showed TUG1 was involved in cell growth and chemoresistance of small cell lung cancer by regulating LIMK2b via EZH2.29 Intriguingly, Xu et al reported that TUG1 conferred cisplatin resistance in esophageal squamous cell carcinoma by epigenetically suppressing PDCD4 expression via recruiting EZH2.30 Our study validated the same scenario in CRC, wherein TUG1 associated with EZH2 directly to be involved in the modulation of miR-153-1 expression.
In summary, our study highlights the importance of TUG1/KLF4/miR-153-1 signaling axis in the tumor biology of CRC, which might hold great promises for either diagnostic or therapeutic potentials.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Brody H. Colorectal cancer. Nature. 2015;521(7551):S1. doi:10.1038/521S1a
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
3. Song M, Garrett WS, Chan AT. Nutrients, foods, and colorectal cancer prevention. Gastroenterology. 2015;148(6):1244–1260.e1216. doi:10.1053/j.gastro.2014.12.035
4. Dulai PS, Sandborn WJ, Gupta S. Colorectal cancer and dysplasia in inflammatory bowel disease: a review of disease epidemiology, pathophysiology, and management. Cancer Prev Res (Phila). 2016;9(12):887–894. doi:10.1158/1940-6207.CAPR-16-0124
5. Stoffel EM, Yurgelun MB. Genetic predisposition to colorectal cancer: implications for treatment and prevention. Semin Oncol. 2016;43(5):536–542. doi:10.1053/j.seminoncol.2016.08.002
6. Spronk I, Korevaar JC, Burgers JS, Albreht T, Schellevis FG. Review of guidance on recurrence risk management for general practitioners in breast cancer, colorectal cancer and melanoma guidelines. Fam Pract. 2017;34(2):154–160. doi:10.1093/fampra/cmw140
7. Binefa G, Rodriguez-Moranta F, Teule A, Medina-Hayas M. Colorectal cancer: from prevention to personalized medicine. World J Gastroenterol. 2014;20(22):6786–6808. doi:10.3748/wjg.v20.i22.6786
8. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152(6):1298–1307. doi:10.1016/j.cell.2013.02.012
9. Wang Y, Yang T, Zhang Z, et al. Long non-coding RNA TUG1 promotes migration and invasion by acting as a ceRNA of miR-335-5p in osteosarcoma cells. Cancer Sci. 2017;108(5):859–867. doi:10.1111/cas.13201
10. Li TH, Zhang JJ, Liu SX, Chen Y. Long non-coding RNA taurine-upregulated gene 1 predicts unfavorable prognosis, promotes cells proliferation, and inhibits cells apoptosis in epithelial ovarian cancer. Medicine (Baltimore). 2018;97(19):e0575. doi:10.1097/MD.0000000000010575
11. Li G, Liu K, Du X. Long non-coding RNA TUG1 promotes proliferation and inhibits apoptosis of osteosarcoma cells by sponging miR-132-3p and upregulating SOX4 expression. Yonsei Med J. 2018;59(2):226–235. doi:10.3349/ymj.2018.59.2.226
12. Li C, Gao Y, Li Y, Ding D. TUG1 mediates methotrexate resistance in colorectal cancer via miR-186/CPEB2 axis. Biochem Biophys Res Commun. 2017;491(2):552–557. doi:10.1016/j.bbrc.2017.03.042
13. Zhai HY, Sui MH, Yu X, et al. Overexpression of long non-coding RNA TUG1 promotes colon cancer progression. Med Sci Monit. 2016;22:3281–3287. doi:10.12659/msm.897072
14. Wang L, Zhao Z, Feng W, et al. Long non-coding RNA TUG1 promotes colorectal cancer metastasis via EMT pathway. Oncotarget. 2016;7(32):51713–51719. doi:10.18632/oncotarget.10563
15. Zeng HF, Yan S, Wu SF. MicroRNA-153-3p suppress cell proliferation and invasion by targeting SNAI1 in melanoma. Biochem Biophys Res Commun. 2017;487(1):140–145. doi:10.1016/j.bbrc.2017.04.032
16. Cui Y, Zhao J, Yi L, Jiang Y. microRNA-153 targets mTORC2 component rictor to inhibit glioma cells. PLoS One. 2016;11(6):e0156915. doi:10.1371/journal.pone.0156915
17. Yuan Y, Du W, Wang Y, et al. Suppression of AKT expression by miR-153 produced anti-tumor activity in lung cancer. Int J Cancer. 2015;136(6):1333–1340. doi:10.1002/ijc.29103
18. Wang H, Yu Y, Fan S, Luo L. Knockdown of long noncoding RNA TUG1 inhibits the proliferation and cellular invasion of osteosarcoma cells by sponging miR-153. Oncol Res. 2018;26(5):665–673. doi:10.3727/096504017X14908298412505
19. Swamynathan S, Kenchegowda D, Piatigorsky J, Swamynathan S. Regulation of corneal epithelial barrier function by Kruppel-like transcription factor 4. Invest Ophthalmol Vis Sci. 2011;52(3):1762–1769. doi:10.1167/iovs.10-6134
20. Nagata T, Shimada Y, Sekine S, et al. KLF4 and NANOG are prognostic biomarkers for triple-negative breast cancer. Breast Cancer. 2017;24(2):326–335. doi:10.1007/s12282-016-0708-1
21. Zhao L, Han T, Li Y, et al. The lncRNA SNHG5/miR-32 axis regulates gastric cancer cell proliferation and migration by targeting KLF4. FASEB J. 2017;31(3):893–903. doi:10.1096/fj.201600994R
22. Minami K, Taniguchi K, Sugito N, et al. MiR-145 negatively regulates Warburg effect by silencing KLF4 and PTBP1 in bladder cancer cells. Oncotarget. 2017;8(20):33064–33077. doi:10.18632/oncotarget.16524
23. Wei D, Wang L, Yan Y, et al. KLF4 is essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis. Cancer Cell. 2016;29(3):324–338. doi:10.1016/j.ccell.2016.02.005
24. Zhai F, Cao C, Zhang L, Zhang J. miR-543 promotes colorectal cancer proliferation and metastasis by targeting KLF4. Oncotarget. 2017;8(35):59246–59256. doi:10.18632/oncotarget.19495
25. Ma Y, Wu L, Liu X, et al. KLF4 inhibits colorectal cancer cell proliferation dependent on NDRG2 signaling. Oncol Rep. 2017;38(2):975–984. doi:10.3892/or.2017.5736
26. Xu Q, Liu M, Zhang J, et al. Overexpression of KLF4 promotes cell senescence through microRNA-203-survivin-p21 pathway. Oncotarget. 2016;7(37):60290–60302. doi:10.18632/oncotarget.11200
27. Chen R, Kong P, Zhang F, et al. EZH2-mediated alpha-actin methylation needs lncRNA TUG1, and promotes the cortex cytoskeleton formation in VSMCs. Gene. 2017;616:52–57. doi:10.1016/j.gene.2017.03.028
28. Cao J, Han X, Qi X, Jin X, Li X. TUG1 promotes osteosarcoma tumorigenesis by upregulating EZH2 expression via miR-144-3p. Int J Oncol. 2017;51(4):1115–1123. doi:10.3892/ijo.2017.4110
29. Niu Y, Ma F, Huang W, et al. Long non-coding RNA TUG1 is involved in cell growth and chemoresistance of small cell lung cancer by regulating LIMK2b via EZH2. Mol Cancer. 2017;16(1):5. doi:10.1186/s12943-016-0575-6
30. Xu C, Guo Y, Liu H, Chen G, Yan Y, Liu T. TUG1 confers cisplatin resistance in esophageal squamous cell carcinoma by epigenetically suppressing PDCD4 expression via EZH2. Cell Biosci. 2018;8:61. doi:10.1186/s13578-018-0260-0
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.