Back to Journals » International Journal of Nanomedicine » Volume 19
Lipid-Based Self-Microemulsion of Niclosamide Achieved Enhanced Oral Delivery and Anti-Tumor Efficacy in Orthotopic Patient-Derived Xenograft of Hepatocellular Carcinoma in Mice
Authors Liu Y ![]() , Guerrero DQ, Lechuga-Ballesteros D, Tan M, Ahmad F, Aleiwi B, Ellsworth EL, Chen B, Chua MS
, Guerrero DQ, Lechuga-Ballesteros D, Tan M, Ahmad F, Aleiwi B, Ellsworth EL, Chen B, Chua MS ![]() , So S
, So S
Received 28 September 2023
Accepted for publication 20 February 2024
Published 14 March 2024 Volume 2024:19 Pages 2639—2653
DOI https://doi.org/10.2147/IJN.S442143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Farooq A. Shiekh
Yi Liu,1 David Quintanar Guerrero,2 David Lechuga-Ballesteros,3 Mingdian Tan,1 Faiz Ahmad,1 Bilal Aleiwi,4 Edmund Lee Ellsworth,4 Bin Chen,4 Mei-Sze Chua,1 Samuel So1
1Department of Surgery, School of Medicine, Stanford University, Stanford, CA, USA; 2Laboratorio de Investigación y Posgrado en Tecnologías Farmacéuticas, Facultad de Estudios Superiores Cuautitlán, Universidad Nacional Autónoma de México, Cuautitlán Izcalli, CP, 54745, Mexico; 3AstraZeneca Pharmaceuticals, Ltd. 4222 Emperor Boulevard, Durham, NC, USA; 4Department of Pharmacology and Toxicology, Michigan State University, East Lansing, MI, USA
Correspondence: Mei-Sze Chua, Email [email protected]
Introduction: We previously identified niclosamide as a promising repurposed drug candidate for hepatocellular carcinoma (HCC) treatment. However, it is poorly water soluble, limiting its tissue bioavailability and clinical application. To overcome these challenges, we developed an orally bioavailable self-microemulsifying drug delivery system encapsulating niclosamide (Nic-SMEDDS).
Methods: Nic-SMEDDS was synthesized and characterized for its physicochemical properties, in vivo pharmacokinetics and absorption mechanisms, and in vivo therapeutic efficacy in an orthotopic patient-derived xenograft (PDX)-HCC mouse model. Niclosamide ethanolamine salt (NEN), with superior water solubility, was used as a positive control.
Results: Nic-SMEDDS (5.6% drug load) displayed favorable physicochemical properties and drug release profiles in vitro. In vivo, Nic-SMEDDS displayed prolonged retention time and plasma release profile compared to niclosamide or NEN. Oral administration of Nic-SMEDDS to non-tumor bearing mice improved niclosamide bioavailability and Cmax by 4.1- and 1.8-fold, respectively, compared to oral niclosamide. Cycloheximide pre-treatment blocked niclosamide absorption from orally administered Nic-SMEDDS, suggesting that its absorption was facilitated through the chylomicron pathway. Nic-SMEDDS (100 mg/kg, bid) showed greater anti-tumor efficacy compared to NEN (200 mg/kg, qd); this correlated with higher levels (p < 0.01) of niclosamide, increased caspase-3, and decreased Ki-67 in the harvested PDX tissues when Nic-SMEDDS was given. Biochemical analysis at the treatment end-point indicated that Nic-SMEDDS elevated lipid levels in treated mice.
Conclusion: We successfully developed an orally bioavailable formulation of niclosamide, which significantly enhanced oral bioavailability and anti-tumor efficacy in an HCC PDX mouse model. Our data support its clinical translation for the treatment of solid tumors.
Keywords: niclosamide, self-microemulsifying drug delivery system, SMEDDS, oral bioavailability, drug repurposing, hepatocellular carcinoma
Introduction
We previously used a bioinformatics approach to identify drugs (that have been approved by the Food and Drug Administration (FDA)), that can reverse the gene expression profiles of HCC cells to that of normal hepatocytes. Our efforts revealed niclosamide as top repurposing drug candidate for HCC treatment.1 Niclosamide is an FDA-approved oral antiparasitic drug for treating tapeworm infection in humans.2 In this capacity, it acts primarily by uncoupling oxidative phosphorylation in the mitochondria to impede metabolism.3 Recent evidence indicates that niclosamide has a broad spectrum of action, including the inhibition of multiple signaling pathways that are critical in cancer cells.4 Although in vitro data suggest encouraging anti-tumor effects in HCC,1 colon cancer,5 and prostate cancer,6 subsequent animal studies and clinical trials in these cancer types have not been unsuccessful (NCT02687009; NCT03123978; NCT02807805; NCT02519582; NCT02532114). This is attributed to poor systemic exposure due to low water solubility and, consequently low bioavailability, preventing niclosamide from reaching the solid tumor sites. In order to achieve sufficiently high plasma concentrations required for anti-cancer effects, higher than approved doses of niclosamide are needed, which may lead to dose-limiting toxicities.6,7
Although niclosamide failed to achieve significant anti-HCC efficacy in mouse models, its more water-soluble ethanolamine salt, niclosamide ethanolamine (NEN), significantly reduced the growth of an orthotopic HCC patient-derived xenograft (PDX) in mice.1 This provided evidence that enhancing the water solubility of niclosamide is a practical approach to enhancing its anti-tumor efficacy in HCC. However, NEN is not FDA-approved for human use, prompting us to develop other means of enhancing water solubility and bioavailability of niclosamide. To overcome the translational challenges of poorly water-soluble drugs, such as niclosamide, recent efforts have focused on designing novel oral formulations and dosing strategies, for example, (1) modification with intestinal drug efflux inhibitory materials, such as pharmaceutical excipients (Pluronic and Tweens), and polymers (polyethylene glycols and derivatives);8,9 (2) formulating with nanocarriers that may prolong retention time in the gastrointestinal tract (GIT), and slow down GI transition and digestion;10 (3) by exploring more schedule-intensive treatment options,11,12 which aim to shorten dosing schedules to expose tumor cells to therapeutic agents more frequently, thereby reducing the likelihood of severe side effects while achieving maximum plasma levels.13,14
In this study, we report the development of an oral, lipid-based self-microemulsifying drug delivery system (SMEDDS) formulation of niclosamide (Nic-SMEDDS), as well as the characterization of its in vitro physicochemical and drug release properties, and its in vivo pharmacokinetic profiles and absorption mechanisms, organ biodistribution, and anti-tumor efficacy in HCC PDX mouse models (Schematics).
Methods and Materials
Niclosamide and Tween80® were purchased from Sigma-Aldrich (St. Louis, MO). Cycloheximide and D-luciferin potassium salt were from Thermo Fisher Scientific Inc. (Waltham, MA). PEG-8 CapRrylic/Capric glycerides (Labrasol® ALF), Plurol isostearique, and Labrafac lipophile WL 1349 were from Gattesfosse (Paramus, NJ).
Preparation of Niclosamide-Encapsulated Self-Microemulsifying Drug Delivery System
The solubility of niclosamide was first tested in various oils, surfactants, and cosurfactants using the shaking flask method.14 An excess of niclosamide was added to each solution and vortexed for 30s. The mixtures were shaken for 48h at 30°C, equilibrated for 24h, and centrifuged at 600xg for 10 min. The supernatants were collected and filtered through a 0.45µm pore-size membrane filter. The filtrates were dissolved in methanol, and niclosamide concentration was detected with ultraviolet (UV) spectrometry at 290 nm. Selection of the surfactant and cosurfactant was based on emulsion efficiency measurements as described previously.15
A ternary phase diagram of the selected oil, surfactant, and co-surfactant was constructed. The portion of oil and surfactant ranged from 30–70%, and the cosurfactant ranged from 0–30%.16 A total of 20 mixtures were prepared; the oil phase was blended with a surfactant/cosurfactant mixture, and the final mixture was then stirred in a shaking water bath at 50°C for 10 min. For each sample, 50 mg of the mixture was diluted with 50 mL water. Only transparent or slightly yellowish dispersions with particle size lower than 200 nm were considered in the emulsion region of the diagram.17 The particle size was measured using the Dynamic Light Scattering (DLS) instrument (Zetasizer Pro, Marvel Panalytical, UK).
Based on the ternary diagram method as described previously,18 a SMEDDS formulation based on 66.94% Labrasol ALF, 13.36% Plural oleique, and 14.09% Labrafac lipophile WL1349 was selected for niclosamide loading and further characterization. Niclosamide (300 mg) was dissolved in the surfactant/cosurfactant (Labrasol® ALF/Labrafac lipophile WL 1349) mixture and the dispersion was shaken at 50°C for 5 min. Then, the oil phase (Plurol isostearique) was added to the mixture and shaken for another 30 min at 50°C. The final, clear Nic-SMEDDS dispersion was obtained and sealed in a dark glass bottle at room temperature until further use.
Characterization of Physicochemical Properties of the Self-Microemulsifying Niclosamide Formulation
The Nic-SMEDDS dispersion was diluted with water to reach the appropriate particle concentration (approximately 0.05 mL of SMEDDS in 5 mL of water), and all characterizations were based upon this diluted dispersion. The mean particle size, polydispersity index, and zeta potential were then determined by the DLS method (Malvern Instruments NS ZEN 3600, Worcestershire, UK) at 90° fixed angle for 180 s, at 25°C. Measurements were performed in triplicates. The density of the dispersion was determined by a glass pycnometer (n = 3), and its pH was determined with a potentiometer (Philips Harris model E3039018G/K, Cheshire, UK) equipped with an electrode (model P43-120). All measurements were performed in triplicates. Its viscosity was determined using a Brookfield viscosimeter (LVT, Middleboro, MA). To determine the niclosamide loading ratio, the prepared Nic-SMEDDS was centrifuged at 30,000 rpm, 4°C for 3 h, and the sediment obtained was washed with water and dried for 24 h to 48 h at room temperature. Niclosamide-loading efficiency was determined by UV spectrophotometry at 290 nm after dissolving the dried sediment in methanol. Methanol was used as blank control. Niclosamide quantification was measured by comparing the absorbance of 290 nm of niclosamide sample to a calibration curve (R2= 0.0005). The niclosamide loading efficiency in SMEDDS was calculated by:
Drug loading efficiency (%) = (experimental drug payload/initial drug) x 100%.
The “experimental drug payload” was the measured drug in the dried sediment and the “initial drug” was the total amount of drug used for Nic-SMEDDS preparation.
The Analysis of Niclosamide Release from Its Self-Microemulsifying System Under in vitro Conditions
Niclosamide release profiles from Nic-SMEDDS formulations were evaluated by dialysis. Nic-SMEDDS (100 µL) was pre-diluted with deionized water (1:2 v/v%), equivalent to about 5.6 mg niclosamide, and added to the dialysis tube (D-TubeTM Dialyzer, MWCO 6–8 kDa, Novagen®, Merck KGaA, Billerica, MA), which was then placed in Dissolution Medium (0.2 M phosphate buffer at a pH of 7.4 and a 0.2M HCl/KCl buffer at a pH of 1.2). Dialysis was done in a shaker water bath equipped with a thermostat set at 37°C and 100 rotations per minute. Samples (100 µL) were collected from a 400 mL release media at pre-determined time points of 0, 0.5, 4, 8, and 24 h. An equal volume of Dissolution Medium was immediately added to each sample after collection. The collected samples were centrifuged at 3000×g for 5 min, and the released niclosamide concentration was measured by a validated liquid chromatography and tandem mass spectrometry (LC-MS/MS) method19 (Integrated Analytical Solutions, Berkeley, CA).
The Analysis of in vivo Pharmacokinetics of Niclosamide Released from Its Self-Microemulsifying System
All animal studies were performed strictly according to the guidelines and rules concerning laboratory animal care, and were approved by the Institutional Animal Care and Use Committee (IACUC) at Stanford University (Protocol number: APLAC-20167). Healthy, non-gender biased, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (8 ~10 weeks old, with body weight 30 ~ 35 g) were used for in vivo pharmacokinetic (PK) study. Mice were randomized into seven groups (n = 3 in each group), and the following single doses were administered by oral gavage to each group: saline (100 µL), niclosamide (40 mg/kg, equivalent to NEN), NEN (40 mg/kg), or multiple Nic-SMEDDS dosages (equivalent to 40 mg/kg, 60 mg/kg, 100 mg/kg or 140 mg/kg NEN). For administration of Nic-SMEDDS, the formulation was diluted with deionized water (1:2%v/v dilution) immediately prior to oral gavage. The dosing volume in each treatment group was calibrated to be no more than 10 mL/kg of the mouse body weight. Blood samples (50–80 µL) were collected from each mouse at 0.5, 1, 4, and 24 h post-administration in heparin-coated tubes (Tubes with K2E, Microtainer®, BD, NJ), and immediately centrifuged at 3000×g for 5 min to obtain plasma samples, which were transferred to a new tube and stored at −80°C for further analysis of niclosamide concentration using LC-MS/MS (Integrated Analytical Solutions, Berkeley, CA).
The Analysis of in vivo Biodistribution of Niclosamide Self-Microemulsifying System in the Gastrointestinal Tract
We used a 3D-fluorescence imaging computed tomography technique to monitor the passage of Nic-SMEDDS in vivo in non-tumor bearing mice, using the vivoTag680® dye (Perkin Elmer LLC, Waltham, MA).20 To prepare the dye stock solution, 10 mg of the dye was dissolved in 1 mL of dimethylsulfoxide (per the manufacturer’s protocol). For nine mice, we prepared the control dye mixture consisting of 405 µL of stock dye, 225 µL of buffer solution (50 mM NaHCO3), and 720 µL of water. A total of 1.35 mL PBS (pH 7.2) was added, vortexed, and centrifuged for 10 min at 2000×g. The resulting dye solution was washed three times according to the protocol, and the supernatant was discarded after each wash. In a similar manner, another mixture was prepared by adding 720 µL Nic-SMEDDS in place of water. The fluorescence-labeled Nic-SMEDDS solution or control dye solution (150 µL each) was then orally administered to two groups of non-tumor-bearing NSG mice (n = 6 per group). Mice were anesthetized with 5% isoflurane prior to imaging at pre-determined time points (0.5, 4-, and 24 h post-administration), using the LAGO X imaging system (Spectral Instruments Imaging, Tucson, AZ). VivoTag680® signal distribution was measured at excitation and emission wavelengths of 640 and 710 nm, respectively. An average radiance of the Nic-SMEDDS was measured during imaging to determine the concentration of fluorescence dye in the target tissues (based on the region of interest, ROI). Mice were sacrificed after image acquisition, and their GITs were removed for the measurement of fluorescent dye concentrations. Aura software (XQuarts 2.8.5) was used for image analysis.
Evaluation of the Influence of Chylomicron Flow on the Absorption of Niclosamide from Its Self-Microemulsifying System
Non-tumor-bearing NSG mice (n=15) were randomly divided into three groups of five each. Group 1 was orally gavaged with Nic-SMEDDS (equivalent to 60 mg/kg of NEN); Groups 2 and 3 were pre-treated (0.5 h or 2 h, respectively) with intraperitoneal (i.p.) cycloheximide (3 mg/kg, dissolved in saline at 15 µg/uL), before mice were orally gavaged with Nic-SMEDDS (equivalent to 60 mg/kg of NEN). Blood samples (n = 3) from each group were collected through the saphenous vein at 0.5, 4, 8, and 24 h after administration of Nic-SMEDDS, and centrifuged immediately at 2000×g for 5 min. Plasma was collected and immediately stored at −80°C before being analyzed by LC-MS/MS (Michigan State University, East Lansing, MI).
Evaluation of in vivo Efficacy of Niclosamide Self-Microemulsifying System in Orthotopic Patient-Derived Xenograft Animal Models
Patient-derived HCC tissues were obtained with written consent from HCC patients prior to tumor resection. The use of human subjects was approved by accordance with the Institutional Review Board at Stanford University, and complies with the Declaration of Helsinki. Orthotopic PDX was established as described previously.19 Briefly, luciferase-labeled patient tumor cells were suspended in complete culture medium DMEM, containing 50% Matrigel matrix (Corning®, NY), and injected subcutaneously to NSG mice to generate donor xenografts (6 ~ 8 weeks, non-gender biased, 20 ~ 25 g body weight). The growth of subcutaneous donor xenografts was monitored daily by visual inspection and harvested once they reached ~1.7 cm in diameter. These xenografts were then cut into 1~2 mm3 pieces before being surgically implanted into the left lobe of the liver of another batch of mice (NSG, male, 6~8 weeks old, 25 ~ 30 g body weight) to establish the orthotopic PDX model for subsequent treatment.
Five to seven days following orthotopic implantation, mice were randomly assigned into five treatment groups (n = 6 each) for oral gavage of: 1). saline control, 200 µL, once a day (qd); 2). NEN control, at a dose of 200 mg/kg, in 200 µL of 0.5% Methylcellulose, qd; 3). Nic-SMEDSS at the dose of 60 mg/kg, qd; 4). Nic-SMEDDS at the dose of 60 mg/kg, twice-a-day (bid), and 5). Nic-SMEDDS at the dose of 100 mg/kg, bid. All dosages were calibrated to be equivalent to the molecular weight of NEN. The treatments were administered over the course of four weeks. During the treatment period, the PDX growth was monitored weekly by bioluminescence imaging using LagoX in vivo imaging system (Spectral Instruments Imaging, Tucson, AZ). Luciferase imaging was acquired 10 min after i.p. administration of D-Luciferin (150 mg/kg), at 30s exposure time. The total bioluminescence (in ventral position) in photon units (photons/s/cm2) was measured to estimate the tumor burden. Mice were monitored for body weight three times a week until the end of the study. Tissues including livers and xenografts of the mice were harvested at the end of the treatment period for further histological and tumor volume evaluations. The ex vivo tumor size was measured using digital calipers, and the volume of the tumor was calculated using the formula π/6 × larger diameter × [smaller diameter]2.
Evaluation of Blood Biochemistry and Tissue Distribution of Niclosamide in Treated Mice
At the end of the treatment study described above, blood was collected, and tumor tissues and all major organs were harvested from each treated mouse (n = 6 per treatment group). The blood samples were centrifuged at 3000×g for 5 min, and plasma was isolated and transferred to fresh tubes and stored at −80°C for further biochemical analysis. The harvested tissues and organs were immediately frozen at −80°C. For each 100 mg of tissue, 0.1% sodium lauryl sulfate (SLS) solution (1 mL) was added and mixed thoroughly with the tissue sample. A 1 mL internal standard solution of niclosamide in acetonitrile (20 ng/mL) was added and thoroughly mixed with each tissue sample. The samples were vortexed for 30s, centrifuged at 18,000×g for 10 min at 4°C, and the supernatant was transferred to fresh tubes for further analysis of niclosamide concentrations using LC-MS/MS (Integrated Analytical Solutions, Berkeley, CA).
Histological Examination of Tumor Tissues After Treatment with Niclosamide Self-Microemulsifying System
For hematoxylin and eosin (H&E) staining, harvested organs were fixed using 10% Formalin (Formaldehyde, 50–00-0, Fisher Scientific, MA) at room temperature for 24 h, immersed in 70% ethanol prior to paraffin embedding, and sectioned at a thickness of 5 μm using a Leica cryo-microtome (RM2255, Leica, IL). Slides were stained with hematoxylin (Sigma-Aldrich, MO) for 2 min, washed in water, then immersed in 1% HCl acid/alcohol solution for 30s. The sections were then washed, subjected to bluing solution (Thermo Fisher Scientific, MA) for 1 min, rinsed in water, and treated with 10 drops of 95% ethanol. Eosin counterstaining was done by briefly immersing the slides into a solution of ethanol: eosin at 1:5 ratio (Thermo Fisher Scientific, MA). Slides were dehydrated by sequential immersion in 95% alcohol and xylene for 5 min each, then mounted using a xylene-based mounting medium (Permount, Sigma-Aldrich, MO). Images were taken using a Nanozoomer system (Hamamatsu, Japan).
Immunostaining was done the proliferation marker Ki-67, the apoptotic marker cleaved caspase-3, and P-glycoprotein (P-gp). Harvested tissues were formalin-fixed and paraffin-embedded, then sectioned at 3 ~5 µm, and stained according to the manufacturer’s instructions using streptavidin-biotin peroxidase complexes, using primary monoclonal antibody against Ki-67 (1:20 dilution) (MA5-14520, InvitrogenTM, Waltham, MA), polyclonal antibodies against caspase-3 (1:300 dilution) (700182, Invitrogen), and polyclonal antibodies against P-gp (1:500 dilution) (22336-1-AP, Proteintech®, IL) using 3,3′-diaminobenzidine (DAB) as the chromogen. A freely available software Fiji (Image J, version 2.9.0) was used for quantitative analysis of protein expression from IHC.21,22
Western Blot Analysis of P-Gp and Cleaved Caspase-3
For P-gp analysis, approximately 3×105 Huh7 or HepG2 cells were seeded into each well of six-well plates, and were treated with 1 µM of niclosamide for 48 h. Total cell lysates were then extracted from the cells. For cleaved Caspase-3, total cell lysates were extracted from frozen PDX tissues (10 mg per sample) from different treatment conditions. Protein (50 μg) from cells or PDX tissues were heat denatured with 5% β-mercaptoethanol (Thermo Fisher Scientific, MA) in 4 × NuPAGE LDS loading buffer (Invitrogen, CA), and resolved in a 4 −12% SDS-PAGE gradient gel (Invitrogen, Carlsbad, CA) at 80 V for 2 h. Resolved proteins were electrotransferred onto nitrocellulose membrane (pore size −0.2 μm, Schleicher & Schuell, NH), and blocked with 5% non-fat milk in tris-buffered saline containing 0.01% Tween-20 (TBST, pH 7.6) for 30 min and incubated at 4°C on a rocking platform overnight, with the primary antibodies against rabbit anti-P-gp antibody (catalog number 22,336-1-AP, dilution 1:1000; Proteintech, IL), or rabbit anti-cleaved caspase-3 antibody (catalog number 9661S, dilution 1:1000; Cell Signaling Technology, MA). The internal loading control used was GAPDH, detected using mouse anti-GAPDH (catalog number 60,004-1-Ig, dilution 1: 50,000; Proteintech, IL). After the primary antibody incubation, the membrane was washed three times with TBST buffer for 10 minutes each. Subsequently, the membrane was incubated with an IR Dye-conjugated goat anti-rabbit or anti-mouse IgG secondary antibody (IRDye® 680RD, LI-COR Biosciences, NE) for 1 h at room temperature on a rocking platform. The membranes were washed three times with TBST buffer, 10 min per wash, and then imaged using Odyssey DLx imager (LI-COR Biosciences, NE).
Statistical Methods and Software
Data were evaluated using one-way ANOVA with Tukey’s multiple comparisons test unless otherwise stated, and calculated by the Prism software (Version 9.0, GraphPad Software, Inc. CA).
Results
Physicochemical Properties and in vitro Drug Release Profile of Niclosamide Self-Microemulsifying System
Our optimized Nic-SMEDDS formulation contained niclosamide at a concentration of 5.61% wt/wt (Table 1). This formulation has a density of 1.05 g/cm3, a viscosity of 133.3 cp, a pH of 3.96, and a mean particle size of ~150 nm (Figure S1a), carrying a negative surface charge of −6.8mV (Figure S1b). In vitro release profiles of Nic-SMEDDS formulation in different release media (PBS at pH 7.4, or 0.2 M HCl/KCl buffer at pH 1.2) indicated gradual release (reaching >75% of drug load after 24 h) that was independent of pH of the release media (concentration released depicted in Figure S2a, percentage released depicted in Figure S2b).
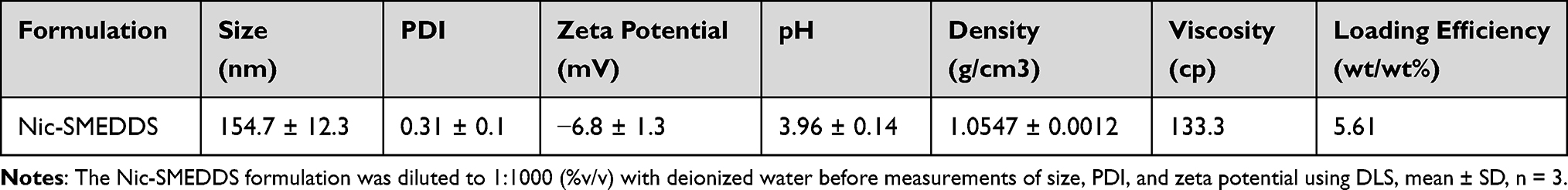 |
Table 1 Characterization of Nic-SMEDDS |
Oral Administration of Niclosamide Self-Microemulsifying System Enhanced the Bioavailability and Absorption of Niclosamide in Non-Tumor Bearing Mice
When Nic-SMEDDS was orally administered to non-tumor-bearing NSG mice, the resulting plasma levels of niclosamide was higher (both the averaged value and the values at individual time points) than that achieved by oral administration of dose equivalents of niclosamide or NEN (Figure 1a and b). There was a ~ 4.1-fold and ~1.8-fold increase in the area under the plasma concentration-time curve (AUC) and the Cmax value, respectively, resulting from oral administration of Nic-SMEDDS compared to niclosamide (Table 2). When compared to NEN which had greater AUC (and hence bioavailability) than niclosamide, Nic-SMEDDS achieved 2.5-fold greater AUC than NEN (Table 2), suggesting that Nic-SMEDDS further enhanced the bioavailability of niclosamide to beyond that achieved by NEN.
 |
Table 2 Comparison of in vivo Pharmacokinetic Parameters of Niclosamide, NEN, and Nic-SMEDDS Formulation Following Oral Administration in Non-Tumor-Bearing Mice |
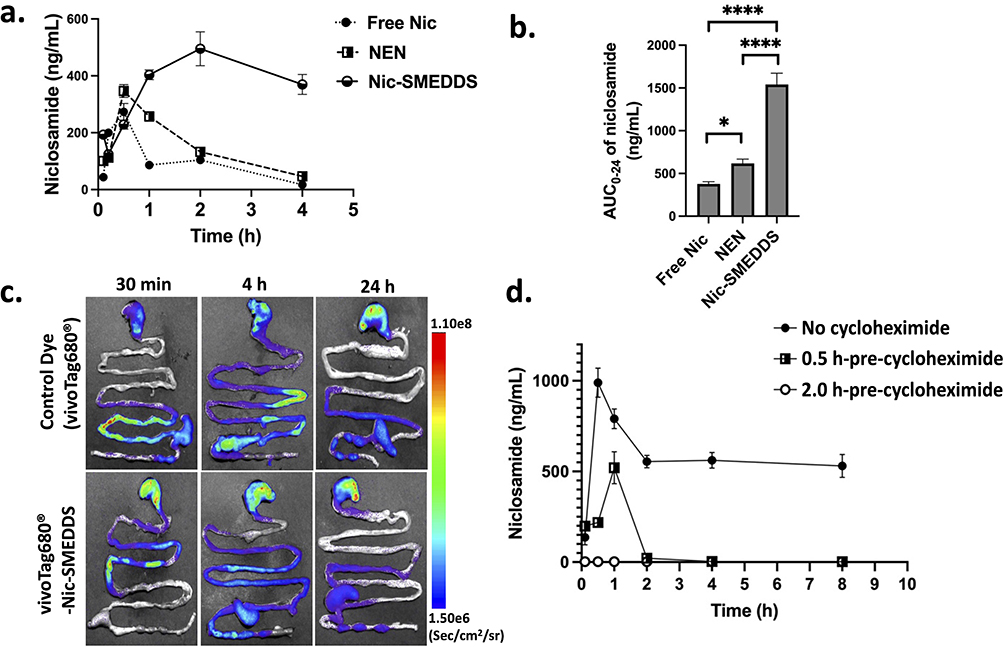 |
Figure 1 Pharmacokinetics and absorption properties of Nic-SMEDDS. (a) Plasma niclosamide concentration over time after administration of free niclosamide (Free Nic), Nen, or Nic-SMEDDS. (b) The area under the curve (AUC) analysis shows significantly enhanced bioavailability of niclosamide when administered as Nic-SMEDDS. (c) Ex vivo fluorescence imaging of GIT from mice at multiple time points following oral administration of Nic-SMEDDS. Nic-SMEDDS was labeled with vivoTag680® dye (Ex/Em Channel: 640/710), showing biodistribution of Nic-SMEDDS in the GIT. (d) Niclosamide absorption from Nic-SMEDDS was inhibited by pre-administration of cycloheximide. Plasma niclosamide concentration-time characteristics from mice administered with Nic-SMEDDS (60 mg/kg equivalent of Nen) in the absence of cycloheximide, or with 0.5 h or 2 h pre-administration of cycloheximide. n = 3 per group; data were plotted as mean ± SEM). One-way ANOVA with Tukey’s multiple comparisons test was performed for analysis; * represents p < 0.05, and ****represents p < 0.0001. |
A prolonged GIT emptying rate was observed in vivo when vivoTag680®-labeled Nic-SMEDDS (compared to control dye) was orally administered to mice, especially at the 30 min and 4 h time points at the jejunum (the main absorption site of vivoTag680®) (Figure 1c). This prolonged retention time of vivoTag680®-Nic-SMEDDS within the GIT suggests that more niclosamide may be available for systemic absorption,23,24 thereby leading to the enhanced plasma niclosamide levels in mice treated with Nic-SMEDDS compared to those treated with niclosamide (Figure 1a). Whole mice imaging showed that vivoTag680®-Nic-SMEDDS produced stronger fluorescence signal compared to vivoTag680® controls (Figure S3a, b), with accumulation of vivoTag680®-Nic-SMEDDS in the liver when assessed by ex vivo fluorescence imaging (Figure S3c, d).
Cycloheximide Blocked the Absorption of Niclosamide by Inhibiting Lymphatic Transport of Niclosamide Self-Microemulsifying System
To investigate the mechanism of Nic-SMEDDS transportation in the intestines, we pre-administered cycloheximide, a protein synthesis inhibitor that is known to inhibit chylomicron flow through the lymphatic system.25,26 In the absence of cycloheximide, plasma levels of niclosamide increased sharply within the first 30 min post oral gavage of Nic-SMEDDS (equivalent to 60 mg/kg of NEN) (Figure 1d). When cycloheximide was given 0.5 h prior to oral gavage of Nic-SMEDDS, we observed a delayed peak of niclosamide at 1 h post-administration, which decreased to near zero levels after 2 h post-administration. When cycloheximide was given 2 h prior to Nic-SMEDDS, the absorption of niclosamide was completely blocked. Our data suggest that cycloheximide inhibited the absorption of niclosamide, implying that Nic-SMEDDS transportation was facilitated by chylomicrons through the lymphatic system.
Niclosamide Self-Microemulsifying System Induced Greater in vivo Growth Inhibition of Patient-Derived Xenografts Than Niclosamide Ethanolamine Salt
To determine the optimal dose(s) of Nic-SMEDDS to be used for subsequent anti-tumor efficacy study in mice, we first evaluated the PK and bioavailability parameters of four different doses of Nic-SMEDDS – 40, 60, 100, or 140 mg/kg (all at molecular weight equivalent to NEN, given as a single oral dose), based upon previous studies on NEN.1,27 Analysis of the niclosamide AUC (0–24) achieved by these different doses of Nic-SMEDDS demonstrated that the three higher doses achieved comparatively high AUCs. The highest level was achieved by the 100 mg/kg dose, while the 60 and 140 mg/kg doses achieved similar (Figure S4). In order to minimize toxicities, we selected the lower doses, 60 mg/kg and 100 mg/kg, for subsequent in vivo efficacy study of Nic-SMEDDS in mice bearing orthotopic HCC PDX.
The study regimen is represented in Figure 2a. Besides the 60 mg/kg, qd dose, we chose a bid regimen for the 60 mg/kg and 100 mg/kg doses to maximize exposure of the tumor to niclosamide, since our PK study revealed a short plasma half-life of ~1.5 h (Figure S4). Oral NEN at 200 mg/kg, qd, was used as the positive treatment control.1 Based on bioluminescence intensity, we observed that the PDX increased in size in the saline-treated control group, starting from Day 6 after tumor implantation. The treatment groups (different dosages of Nic-SMEDDS, or NEN), showed different extents of tumor suppression up to 28 days post-tumor implantation (Figure 2b). Among the treatment groups, Nic-SMEDDS at 60 mg/kg, bid, and at 100 mg/kg, bid showed greater tumor inhibition than the 60 mg/kg, qd group, with significant inhibition of tumor volume observed with the 100 mg/kg, bid dose (Figure 2c). While NEN also achieved significant tumor inhibition, the effect seen with Nic-SMEDDS at 100 mg/kg, bid, was greater than that with NEN at 200 mg/kg, qd (Figure 2c).
 |
Figure 2 Evaluation of therapeutic efficacy of Nic-SMEDDS in orthotopic PDX-HCC mouse model. (a) Schematic outline of the animal study design including dosing and bioluminescent imaging schedules. (b) Whole animal bioluminescence images were acquired at various time points throughout the treatment period to assess tumor burden. (c) Ex vivo tumor volume (mm3) was calculated and compared between each treatment group on Day 28. (d) Niclosamide levels in plasma, and in the (e) liver, and (f) tumor tissue following 28 days of treatment (n = 3). (g) Histologic H&E-stained sections of HCC tissues of mice from each treatment group. Data were plotted as median ± SEM, and One-way ANOVA with Tukey’s multiple comparisons test was performed for analysis; *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001; “ns” represents not significant. |
On the last day of the treatment period, blood samples were collected from mice, and the liver and PDX tissues were harvested for measurement of niclosamide levels in these samples. We observed higher levels of niclosamide achieved by oral delivery of Nic-SMEDDS in all sample types, especially at the 60 mg/kg, bid, and 100 mg/kg, bid doses (Figure 2d–2f). Consistent with the greatest growth inhibitory activity seen with 100 mg/kg, bid dose, the niclosamide levels were highest in PDX tissues treated at this dose (Figure 2f). H&E staining of harvested PDX tissues indicated that, in the saline-treated group, tumor cells were tightly packed, and had a higher ratio of nuclear/cytoplasm than those in the Nic-SMEDDS- or NEN-treated groups. In mice treated with Nic-SMEDDS or NEN, the tumor cells displayed a dispersed arrangement, fragmentation, shrinkage, and chromatin destruction, indicative of multifocal necrosis and apoptosis (Figure 2g).
Body weight changes over the treatment period revealed a 19% drop in average body weight in the saline-treated group, primarily due to the increasing tumor burden and accompanying cachexia. In contrast, the Nic-SMEDDS or NEN-treated groups exhibited a decrease in body weight not exceeding 10%, with no statistical difference among the treatment groups (Figure S5). Additionally, H&E staining of excised major organs showed no discernable tissue damage caused by any of the Nic-SMEDDS dosing regimens (Figure S6), indicating their safety and tolerability.
Niclosamide Self-Microemulsifying System Induced Caspase-3 and Inhibited Ki-67 Expression in Treated Xenograft Tissues
Staining of proliferative marker Ki-6728 showed high intensity of positively stained (dark brown) HCC cells in the saline-treated group, indicating active cell proliferation. Treatment with various doses of Nic-SMEDDS and NEN reduced the expression of Ki-67, with significant reductions seen in the Nic-SMEDDS 60 mg/kg, bid, and 100 mg/kg, bid groups, and in the NEN treatment group. The most efficacious dose of Nic-SMEDDS, 100 mg/kg, bid, induced the greatest Ki-67 reduction, suggesting greatest inhibition of tumor cell proliferation (Figure 3a and b). Correlating with Ki-67 staining, treatment with all doses of Nic-SMEDDS and NEN increased the levels of activated Caspase-3 protein29 in PDX tissues, indicating an increased number of activated apoptotic cells; the greatest effect was similarly seen with the most efficacious dose of Nic-SMEDDS at 100 mg/kg, bid (Figure 3c and d). We additionally detected cleaved Caspase-3 protein levels in the PDX tissues, harvested at the treatment endpoint. Consistent with increased Caspase-3 observed on IHC, we observed increased expression of cleaved Caspase-3 in all treatment doses of Nic-SMEDDS and in the NEN treated groups, with the greatest induction caused by the most efficacious dose of Nic-SMEDDS 100mg/kg, bid (Figure S9). Our data demonstrated that Nic-SMEDDS reduced HCC PDX cell proliferation while increasing apoptotic activity.
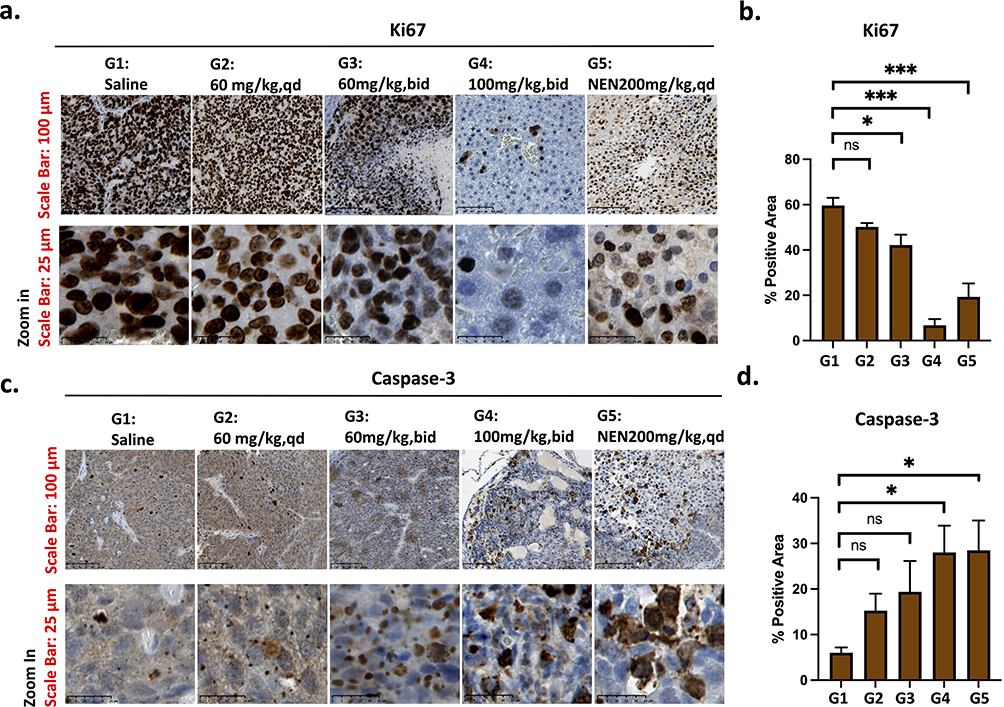 |
Figure 3 Immunohistochemical (IHC) staining and analysis of cell proliferation and apoptosis in PDX tissues from different treatment groups. (a) IHC staining for Ki-67 nuclei protein, a tumor cell proliferation and growth marker. (b) Quantitative analysis of Ki-67 staining using Image J method, (mean area detected: 120,212, threshold 0/100, n = 3, data were plotted as mean ± SEM, One-way ANOVA with Tukey’s multiple comparisons test was performed for analysis;*p < 0.05). (c) IHC staining for cleaved caspase-3 protein (an apoptosis biomarker). (d) Quantitative analysis of positively stained caspase-3 protein from (c) was performed using the Image J software. (The mean area detected was 120,117, threshold 0/100, n = 3, and the data were plotted as mean ± SEM. Statistical analysis using one-way ANOVA with Tukey’s multiple comparisons tests, *p < 0.05 represents significant, *** represents p < 0.001, and “ns” represents not significant. Magnification: 40x, Scale: 100 µm (upper row), and 25 µm (lower row). |
Niclosamide Self-Microemulsifying System Decreased Glucose Levels but Increased Lipid Levels in vivo
Non-significant changes in blood parameters such as white blood cell (WBC), red blood cell (RBC), hemoglobin (HbG), and platelet counts were observed after treatment with Nic-SMEDDS or NEN (Figure 4a–4d). Specifically, WBC counts were reduced in a dose-dependent manner in the Nic-SMEDDS treatment groups, as well as in the NEN-treated group; RBC and HbG counts were increased in all treatment groups. However, all parameters remained within acceptable clinical limits despite these treatment-induced changes. Analysis of biochemical parameters suggests that blood glucose levels were significantly reduced in both Nic-SMEDDS and NEN-treated groups compared to saline-treated mice (Figure 4e), with dose-dependent decreases observed within the Nic-SMEDDS treatment groups. AST and ALT levels were significantly reduced in niclosamide or NEN treatment groups (Figure 4f and g), indicating preserved liver function in response to the treatments; other parameters studied (such as albumin, BUN, and creatinine) were not significantly altered (Figure 4h–4j). Additionally, triglycerides and high-density lipoprotein (HDL) were found to be increased in all Nic-SMEDDS- treated groups in a dose-dependent manner, but not observed with NEN treatment (Figure 4k), suggesting that the lipid components of Nic-SMEDDS led to the observed increases in blood lipid levels.
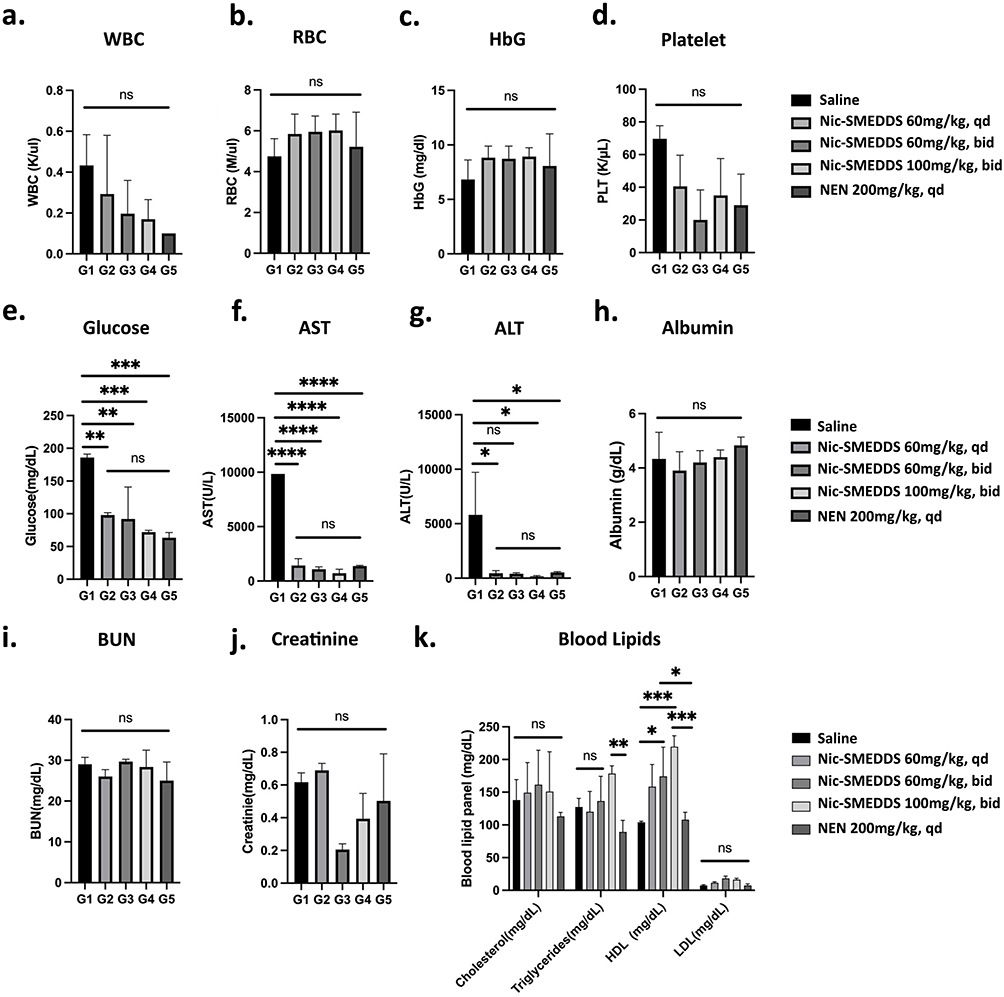 |
Figure 4 Biochemical analysis of blood at treatment endpoint. (a) White blood cell (WBC) counts, (b) Red blood cell (RBC) counts, (c) Hemoglobin (HbG) concentrations, (d) Platelet counts, (e) Glucose levels, (f) Aspartate aminotransferase (AST) levels, (g) Alanine transaminase (ALT), (h) Albumin concentrations, (i) Blood urea nitrogen levels, (j) Creatinine, and (k) Lipids levels were analyzed from mice in each treatment group at the end of the 28-day treatment period. Data were plotted as mean ± SEM (n = 3), in (a–j), one-way ANOVA with Tukey’s multiple comparisons test was performed for analysis, *represents p < 0.05. For (k), two-way ANOVA with Tukey’s multiple comparisons test was used for analysis. In the cholesterol subgroup, no significance was found in treatment groups; in the triglycerides subgroup, G1 vs G2 = ns, G1 vs G3: *p = 0.0328, G1 vs G4: ***p = 0.0002, G4 vs G5 **p = 0.0038 in the HDL subgroup, in LDL subgroup, no significance was observed between all treatment groups; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; “ns” represents not significant. |
Discussion
Niclosamide has attracted growing interest in its repurposed therapeutic applications for multiple diseases, including cancer, COVID-19, and Alzheimer’s disease.30–32 However, its low water solubility and resulting poor oral bioavailability have limited its efficacy in clinical applications. In this study, we developed an orally bioavailable lipid-based microemulsion system loaded with niclosamide, Nic-SMEDDS, and evaluated its absorption, bioavailability, and therapeutic efficacy in an orthotopic PDX-HCC mouse model.
Lipid-based SMEDDS formulations have been proven to be effective in enhancing the therapeutic profiles of poorly water-soluble drugs. Indeed, we observed enhanced plasma levels and bioavailability of niclosamide when it was administered as a SMEDDS formulation; these enhancements were superior to that observed with the more water-soluble NEN salt. Upon dispersion in water, SMEDDS formulations create greatly enhanced interfacial area which allows the easy partition of drugs from the oil phase into the aqueous phase. This may be one of several mechanisms by which an SMEDDS enhance the absorption of poorly water-soluble drugs.33 Secondarily, commonly used surfactants in the formulation of SMEDDS, such as cremophors and pluronics, are reported to inhibit drug efflux transporters such as P-gp, thereby improving the bioavailability of drugs which are substrates of the efflux pumps.33 Other possible mechanisms by which an SMEDDS increases drug bioavailability may be due to prolonged retention of the formulation within the GIT, and the promotion of lipoprotein turnover within enterocytes, consequently promoting lymphatic transportation of the drug.34,35
We observed that Nic-SMEDDS was retained for a prolonged time within the GIT, especially in the jejunum from 0.5 to 4 h after oral administration; this may in turn allow a greater time window for niclosamide to be absorbed, thus achieving enhanced plasma levels and bioavailability of niclosamide when administered as a SMEDDS. Additionally, our study used optimized levels of medium-chain lipids and high-potency surfactants (at levels of 60–80%) that are known to enhance intestinal lymphatic triglyceride secretion and drug transport.36–38 When we used cycloheximide as a chemical inhibitor of chylomicron flow,25,26 we observed complete blockade of niclosamide absorption when cycloheximide was administered to mice 2 h prior to Nic-SMEDDS administration. This suggests that the absorption of niclosamide when given as a SMEDDS occurred through the lymphatic transport system. It is possible that niclosamide micelles entering enterocytes as a result of SMEDDSylation may be processed similarly as chylomicrons.39–41 Chylomicrons are formed in the intestines and released into the lymphatic system, where they are further metabolized in the bloodstream to deliver fatty acids to the liver and other organs. Thus, the lipidized niclosamide may be recognized as a lipid particle and subsequently transported by lipoproteins, such as HDL. The lipoproteinated niclosamide micelles that are exocytosed from enterocytes may be primarily taken up by the mesenteric lymphatic system and subsequently enter the systemic circulation.42 This hypothesis may partially explain our observation that Nic-SMEDDS administration in mice increased HDL levels in a dose-independent manner.
When in vivo anti-tumor efficacy of Nic-SMEDDS was compared in orthotopic PDX mice at 60 mg/kg, bid, and at 100 mg/kg, bid, we observed anti-tumor efficacy in a dose-dependent manner, with significant tumor inhibition at the higher dose. Importantly, a 100 mg/kg bid dose of Nic-SMEDDS (at a dose equivalent to NEN) achieved greater tumor inhibition than 200 mg/kg of NEN, qd. Correspondingly, niclosamide levels were highest in the PDX tissues of mice treated with the most efficacious dose of Nic-SMEDDS at 100 mg/kg, bid. We infer that the higher niclosamide levels achieved in PDX tissues (as a result of delivery via Nic-SMEDDS) led to its observed superior anti-tumor efficacy. Biologically, the greatest decrease of Ki-67/cell proliferation and the greatest increase of Caspase-3/cell apoptosis were observed in the most efficacious dose of 100 mg/kg, bid of Nic-SMEDDS. The bid treatment of a lowered dose of niclosamide via the SMEDDS formulation (100 mg/kg) was superior to the equivalent once-daily dose of NEN (200 mg/kg). This may result from increased total exposure time from the bid regimen since niclosamide has a short plasma half-life. The bid regimen allows treatment at a reduced dose, which lowers the Cmax and reduces the incidence of potential side effects from higher doses.14
The mechanism of anti-tumor action of niclosamide has been widely reported, and largely attributed to the inhibition of signaling pathways that are critical in maintaining tumor growth, as well as the inhibition of mitochondrial function.30,43,44 We therefore did not perform an exhaustive study of its mechanism of action in our mouse model. However, when evaluating whether P-gp might be involved in Nic-SMEDDS absorption, we observed a significant decrease in immunopositivity of P-gp in PDX tissues from Nic-SMEDDS- or NEN-treated mice (Figure S7a, c). We did not observe any treatment-induced changes in P-gp expressions in the small intestines, a major site for drug/nutrients absorption (Figure S7a, b). To further examine our novel finding of P-gp reduction caused by niclosamide treatment, we detected the protein expression of P-gp in two HCC cell lines treated with niclosamide. While we observed a significant decrease in P-gp expression in niclosamide-treated Huh7 cells, this effect was not seen in HepG2 cells (Figure S8). It has been reported that P-gp is commonly expressed in HCC, and is inversely associated with chemotherapy response in inoperable HCC patients.45–47 The ability of niclosamide to reduce the expression of P-gp might therefore offer additional clinical benefits of enhancing chemotherapy response and therefore survival outcome. Our preliminary findings on P-gp modulation by niclosamide warrant further in-depth studies as a potentially novel and clinically relevant mechanism of action of niclosamide.
Despite the encouraging preclinical data, and promising mechanisms of anti-tumor action of niclosamide in HCC and other cancers (such as colon and prostate cancers), early clinical trials (NCT02687009; NCT03123978; NCT02807805; NCT02519582; NCT02532114) using free oral niclosamide have failed to achieve the expected efficacy. These failures are attributed to the inability of niclosamide to reach the solid tumor sites, as well as the dose-limiting toxicities.6,7 For example, a Phase I clinical trial evaluating the efficacy of niclosamide in prostate cancer determined that the dose could not be increased beyond 500 mg PO, three-times-daily (tid) due to toxicity concerns.6 However, in another trial of reformulated orally-bioavailable niclosamide/PDMX1001 in combination with abiraterone and prednisone in men with castration-resistant prostate cancer (CRPC), patients were treated with escalating doses of niclosamide/PDMX1001 and standard doses of abiraterone and prednisone, and no dose-limiting toxicities were observed, even at the dose of 1200 mg oral niclosamide/PDMX1001 three times daily (plus abiraterone 1000 mg PO once daily and prednisone 5 mg PO twice daily).48 This was despite high trough and peak niclosamide concentrations exceeding the therapeutic threshold of > 0.2 µM. The use of Nic-SMEDDS and other similar approaches to enhance tissue bioavailability of niclosamide (while minimizing toxicities with potentially higher achievable Cmax levels) will likely increase the success rates of future clinical trials. In our animal studies, we observed no overt signs of toxicities in mice treated with Nic-SMEDDS (100 mg/kg, bid) within the four-week treatment period. However, blood analysis revealed reduced glucose levels and increased triglyceride levels in these mice. Further optimization of dose and dosing regimen, as well as adjustment of SMEDDS composition may be necessary to achieve the balance of safety and efficacy in human cancer patients. We are encouraged by the current use of several FDA-approved SMEDDS formulations, containing drugs such as ritonavir, cyclosporin A, or saquinavir,49 that achieved improvements in bioavailability while minimizing side effects. Other SMEDDS-based systems for delivering non-water-soluble drugs, docosahexaenoic acid, and eicosapentaenoic acid have also been recently tested to be effective in clinical trials (NCT03592251, NCT03559361, NCT02661698) for improving the bioavailability,44 further validating our approach.
Conclusion
We successfully demonstrated that the formulation of niclosamide as a SMEDDS enhanced niclosamide bioavailability and its resulting anti-tumor efficacy. A significant reduction in the dose of niclosamide was able to achieve an efficacious tumor level of niclosamide. Individually, niclosamide and the SMEDDS formulation are each well-documented for their safety in humans;50 this will facilitate the clinical translation of our Nic-SMEDDS formulation. Given the broad mechanism of anti-tumor action reported for niclosamide, our successful development and validation of Nic-SMEDDS in HCC would enable its use in other forms of solid tumors, especially those that are dependent on signaling pathways that are regulated by niclosamide, and in which bioavailability to the tumor site would otherwise be an impediment to the use of niclosamide. A major focus of our future work is expected to include additional pre-clinical studies, and first-in-human clinical trials, for safety and toxicity evaluations of Nic-SMEDDS following FDA guidelines.
Acknowledgments
The authors would like to thank Stanford Center for Innovation in In vivo Imaging (SCi3) for providing the facilities and resources for executing this study; specifically, they thank Dr. Frezghi Habte, Dr. Laura J. Pisani, and Dr. Edwin Chan for their technical support with animal imaging and histological slides scanning. The authors would like to thank the Histology Service at the Department of Pathology, Dr. Pauline Chu, and Dr. Ferda Filiz for the preparation of histology slides and histological analysis of the stained slides.
Funding
This work was supported by CJ Huang Foundation, the TS Kwok Liver Foundation, and the Lui HM Foundation (Y.L., M.T., F.A., M.-S.C., and S.S.), and NIH NIGMS R01GM134307 (B.C., S.S., M.T., M.-S.C.).
Disclosure
The authors declare no potential conflicts of interest in this work.
References
1. Chen B, Wei W, Ma L., et al. Computational discovery of niclosamide ethanolamine, a repurposed drug candidate that reduces growth of hepatocellular carcinoma cells in vitro and in mice by inhibiting cell division cycle 37 signaling. Gastroenterology. 2017;152(8):2022–2036. doi:10.1053/j.gastro.2017.02.039
2. Corbel MJ, Das RG, Lei D, et al. WHO working group on revision of the manual of laboratory methods for testing dtp vaccines-report of two meetings held on 20-21 July 2006 and 28-30 march 2007, Geneva, Switzerland. Vaccine. 2008;26(16):1913–1921. doi:10.1016/j.vaccine.2008.02.013
3. Pearson RD, Hewlett EL. Niclosamide therapy for tapeworm infections. Ann Intern Med. 1985;102(4):550–551. doi:10.7326/0003-4819-102-4-550
4. Chen W, Mook RA, Premont RT, et al. Niclosamide: beyond an antihelminthic drug. Cell Signal. 2018;41:89–96. doi:10.1016/j.cellsig.2017.04.001
5. Shi L, Zheng H, Hu W, et al. Niclosamide inhibition of STAT3 synergizes with erlotinib in human colon cancer. Onco Targets Ther. 2017;10:1767–1776. doi:10.2147/OTT.S129449
6. Schweizer MT, Haugk K, McKiernan JS, et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS One. 2018;13(6):e0198389. doi:10.1371/journal.pone.0198389
7. Liu C, Lou W, Armstrong C, et al. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate. 2015;75(13):1341–1353. doi:10.1002/pros.23015
8. Werle M. Polymeric and low molecular mass efflux pump inhibitors for oral drug delivery. J Pharm Sci. 2008;97(1):60–70. doi:10.1002/jps.21090
9. Werle M. Natural and synthetic polymers as inhibitors of drug efflux pumps. Pharm Res. 2008;25(3):500–511. doi:10.1007/s11095-007-9347-8
10. Shen H, Zhong M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol. 2006;58(9):1183–1191. doi:10.1211/jpp.58.9.0004
11. Koolen SL, Oostendorp RL, Beijnen JH, et al. Population pharmacokinetics of intravenously and orally administered docetaxel with or without co-administration of ritonavir in patients with advanced cancer. Br J Clin Pharmacol. 2010;69(5):465–474. doi:10.1111/j.1365-2125.2010.03621.x
12. Andre T, Meyerhardt J, Iveson T, et al. Effect of duration of adjuvant chemotherapy for patients with stage III colon cancer (IDEA collaboration): final results from a prospective, pooled analysis of six randomised, Phase 3 trials. Lancet Oncol. 2020;21(12):1620–1629. doi:10.1016/S1470-2045(20)30527-1
13. Liston DR, Davis M. Clinically relevant concentrations of anticancer drugs: a guide for nonclinical studies. Clin Cancer Res. 2017;23(14):3489–3498. doi:10.1158/1078-0432.CCR-16-3083
14. Di Maio M, Perrone F, Chiodini P, et al. Individual patient data meta-analysis of docetaxel administered once every 3 weeks compared with once every week second-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2007;25(11):1377–1382. doi:10.1200/JCO.2006.09.8251
15. Elnaggar YS, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380(1–2):133–141. doi:10.1016/j.ijpharm.2009.07.015
16. Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1–2):166–172. doi:10.1016/j.ijpharm.2006.08.038
17. Zhang P, Liu Y, Feng N, et al. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355(1–2):269–276. doi:10.1016/j.ijpharm.2007.12.026
18. Llera-Rojas VG, Del Real L. A, Mendoza-Muñoz N, et al. Feasibility of obtaining in situ nanocapsules through modified self-microemulsifying drug delivery systems. A new manufacturing approach for oral route administration. Drug Dev Ind Pharm. 2017;43(6):925–931. doi:10.1080/03639045.2017.1285308
19. Wei W, Wu S, Wang X, et al. Novel celastrol derivatives inhibit the growth of hepatocellular carcinoma patient-derived xenografts. Oncotarget. 2014;5(14):5819–5831. doi:10.18632/oncotarget.2171
20. Liu Y, Sukumar UK, Jugniot N, et al. Inhaled gold nano-star carriers for targeted delivery of triple suicide gene therapy and therapeutic microRNAs to lung metastases: development and validation in a small animal model. Adv Ther. 2022;5(8):2200018.
21. Crowe AR, Yue W. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis: an integrated protocol. Biol Protoc. 2019;9(24). doi:10.21769/BioProtoc.3465
22. Law AMK, Yin JXM, Castillo L, et al. Andy’s Algorithms: new automated digital image analysis pipelines for Fiji. Sci Rep. 2017;7(1):15717. doi:10.1038/s41598-017-15885-6
23. Komesli Y, Burak Ozkaya A, Ugur Ergur B, et al. Design and development of a self-microemulsifying drug delivery system of olmesartan medoxomil for enhanced bioavailability. Drug Dev Ind Pharm. 2019;45(8):1292–1305. doi:10.1080/03639045.2019.1607868
24. Liu Y, Huang P, Hou X, et al. Hybrid curcumin-phospholipid complex-near-infrared dye oral drug delivery system to inhibit lung metastasis of breast cancer. Int J Nanomed. 2019;14:3311–3330. doi:10.2147/IJN.S200847
25. Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci. 2005;24(4):381–388. doi:10.1016/j.ejps.2004.12.006
26. Sabesin SM, Isselbacher KJ. Protein synthesis inhibition: mechanism for the production of impaired fat absorption. Science. 1965;147(3662):1149–1151. doi:10.1126/science.147.3662.1149
27. Tao H, Zhang Y, Zeng X, et al. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med. 2014;20(11):1263–1269. doi:10.1038/nm.3699
28. Menon SS, Guruvayoorappan C, Sakthivel KM, et al. Ki-67 protein as a tumour proliferation marker. Clin Chim Acta. 2019;491:39–45. doi:10.1016/j.cca.2019.01.011
29. Silva F, Padín-Iruegas M, Caponio V, et al. Caspase 3 and cleaved caspase 3 expression in tumorogenesis and its correlations with prognosis in head and neck cancer: a systematic review and meta-analysis. Int J Mol Sci. 2022;23(19):11937. doi:10.3390/ijms231911937
30. Li Y, Li P-K, Roberts MJ, et al. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett. 2014;349(1):8–14. doi:10.1016/j.canlet.2014.04.003
31. Singh S, Weiss A, Goodman J, et al. Niclosamide-A promising treatment for COVID-19. Br J Pharmacol. 2022;179(13):3250–3267. doi:10.1111/bph.15843
32. Taubes A, Nova P, Zalocusky KA, et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related alzheimer’s disease. Nat Aging. 2021;1(10):932–947. doi:10.1038/s43587-021-00122-7
33. Akula S, Gurram AK, Devireddy SR. Self-microemulsifying drug delivery systems: an attractive strategy for enhanced therapeutic profile. Int Sch Res Notices. 2014;2014:964051. doi:10.1155/2014/964051
34. O’Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15(5):405–415. doi:10.1016/S0928-0987(02)00051-9
35. Porter CJ, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 2001;50(1–2):61–80. doi:10.1016/S0169-409X(01)00151-X
36. Lupo N, Steinbring C, Friedl JD, et al. Impact of bile salts and a medium chain fatty acid on the physical properties of self-emulsifying drug delivery systems. Drug Dev Ind Pharm. 2021;47(1):22–35. doi:10.1080/03639045.2020.1851241
37. Marwah H, Garg T, Goyal AK, et al. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2016;23(2):564–578. doi:10.3109/10717544.2014.935532
38. Lind ML, Jacobsen J, Holm R, et al. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci. 2008;35(3):211–218. doi:10.1016/j.ejps.2008.07.003
39. Redgrave TG. Chylomicron metabolism. Biochem Soc Trans. 2004;32(Pt 1):79–82. doi:10.1042/bst0320079
40. Bisgaier CL, Glickman RM. Intestinal synthesis, secretion, and transport of lipoproteins. Annu Rev Physiol. 1983;45(1):625–636. doi:10.1146/annurev.ph.45.030183.003205
41. Ahmad J, Amin S, Rahman M, et al. Solid matrix based lipidic nanoparticles in oral cancer chemotherapy: applications and pharmacokinetics. Curr Drug Metab. 2015;16(8):633–644. doi:10.2174/1389200216666150812122128
42. Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. doi:10.1038/nrd2197
43. Park SJ, Shin J-H, Kang H, et al. Niclosamide induces mitochondria fragmentation and promotes both apoptotic and autophagic cell death. BMB Rep. 2011;44(8):517–522. doi:10.5483/BMBRep.2011.44.8.517
44. Peng SW, Ngo M-HT, Kuo Y-C, et al. Niclosamide revitalizes sorafenib through insulin-like growth factor 1 receptor (IGF-1R)/stemness and metabolic changes in hepatocellular carcinoma. Cancers. 2023;15(3):931. doi:10.3390/cancers15030931
45. Soini Y, Virkajarvi N, Raunio H, et al. Expression of P-glycoprotein in hepatocellular carcinoma: a potential marker of prognosis. J Clin Pathol. 1996;49(6):470–473. doi:10.1136/jcp.49.6.470
46. Takanishi K, Miyazaki M, Ohtsuka M, et al. Inverse relationship between P-glycoprotein expression and its proliferative activity in hepatocellular carcinoma. Oncology. 1997;54(3):231–237. doi:10.1159/000227694
47. Ng IO, Liu CL, Fan ST, et al. Expression of P-glycoprotein in hepatocellular carcinoma. A determinant of chemotherapy response. Am J Clin Pathol. 2000;113(3):355–363. doi:10.1309/AC1M-4TY4-U0TN-EN7T
48. Parikh M, Liu C, Wu C-Y, et al. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci Rep. 2021;11(1):6377. doi:10.1038/s41598-021-85969-x
49. Kohli K, Chopra S, Dhar D, et al. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today. 2010;15(21–22):958–965. doi:10.1016/j.drudis.2010.08.007
50. Gibaud S, Attivi D. Microemulsions for oral administration and their therapeutic applications. Expert Opin Drug Deliv. 2012;9(8):937–951. doi:10.1517/17425247.2012.694865
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.