Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
LINC00987 Ameliorates COPD by Regulating LPS-Induced Cell Apoptosis, Oxidative Stress, Inflammation and Autophagy Through Let-7b-5p/SIRT1 Axis
Authors Wang Y, Chen J, Chen W, Liu L, Dong M, Ji J, Hu D, Zhang N
Received 12 August 2020
Accepted for publication 6 November 2020
Published 4 December 2020 Volume 2020:15 Pages 3213—3225
DOI https://doi.org/10.2147/COPD.S276429
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Yuanyuan Wang,1 Jingjing Chen,1 Wei Chen,2 Ling Liu,2 Mei Dong,2 Juan Ji,2 Die Hu,3 Nianzhi Zhang2
1Graduate School, Anhui University of Chinese Medicine, Anhui, Hefei 230012, People’s Republic of China; 2Department of Respiratory Medicine, The First Affiliated Hospital of Anhui University of Chinese Medicine, Anhui, Hefei, 230031, People’s Republic of China; 3Department of Scientific Research, The First Affiliated Hospital of Anhui University of Chinese Medicine, Anhui, Hefei 230031, People’s Republic of China
Correspondence: Nianzhi Zhang Tel +86 551-62850057
Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is the third cause of disease-related death and brings a heavy burden to human health. Long non-coding RNA (lncRNA) was revealed to participate in COPD pathogenesis. This study aims to establish the effects and regulatory mechanism of lncRNA long intergenic non-coding 00987 (LINC00987) in lipopolysaccharide (LPS)-induced apoptosis, oxidative stress, inflammation and autophagy in BEAS-2B cells.
Methods: The expression levels of LINC00987 and let-7b-5p were detected by real-time quantitativepolymerase chain reaction (RT-qPCR). The expression of apoptosis-associated proteins, oxidative stress (ROS)-related proteins, autophagy-related proteins and sirtuin1 (SIRT1) protein was determined by Western blot. Cell viability was illustrated by cell counting kit-8 (CCK-8) assay. Cell apoptosis was investigated by caspase3 activity and apoptosis analysis assays. ROS, inflammation and autophagy were demonstrated by detecting reactive ROS level and superoxide dismutase (SOD) activity, enzyme-linked immunosorbent assay (ELISA) and Western blot analysis, respectively. The binding sites between let-7b-5p and LINC00987 or SIRT1 were predicted by lncBase or miRWalk online database, and identified by dual-luciferase reporter assay.
Results: LINC00987 expression was strikingly downregulated and let-7b-5p expression was obviously upregulated in COPD tissues and LPS-induced BEAS-2B cells compared with control groups. LINC00987 overexpression promoted BEAS-2B cells against LPS-mediated viability, apoptosis, oxidative stress, inflammation and autophagy, whereas these effects were attenuated by let-7b-5p mimic or SIRT1 knockdown. Furthermore, LINC00987 sponged let-7b-5p and let-7b-5p bound to SIRT1.
Conclusion: LINC00987 ameliorated COPD through modulating LPS-induced cell apoptosis, oxidative stress, inflammation and autophagy via sponging let-7b-5p to associate with SIRT1. This finding will provide a theoretical basis for the research of LncRNA-mediated treatment in COPD.
Keywords: COPD, LINC00987, let-7b-5p, SIRT1, LPS
Introduction
Chronic obstructive pulmonary disease (COPD), a pervasive chronic respiratory disease, is the third leading reason for disease-related death worldwide.1 COPD is characterized by heterogeneity, pneumonia, bronchial asthma and pneumonectasis.2 Although inhaled glucocorticoids and bronchodilators are the primary therapy, the incidence of COPD is still high.3,4 Therefore, extensive research into COPD pathogenesis is necessary to seek new therapeutic methods for COPD.
Long non-coding RNA (lncRNA) is more than 200 nucleotides (nts) in size with non-coding protein.5,6 Accumulating studies revealed that lncRNAs participate in the pathogenesis of diseases.7,8 LncRNA metastasis-associated lung adenocarcinoma transcript 1 (lncRNA MALAT1) was revealed to regulate acute respiratory distress syndrome by affecting the secretion of an inflammatory factor.9 LncRNA plasmacytoma variant translocation (PVT1) was employed to predict COPD susceptibility based on its link with tumor necrosis factor and interleukin (IL), IL-6, IL-8, and IL-17, and microRNA-146a (miR-146a).7 In addition, lnc-small nucleolar RNA host gene 1 (lnc-SNHG1) and lnc-pro-transition associated RNA (PTAR) were revealed to promote the progression of lung cancer by sponging miR-497 and miR-101, respectively.10,11 In this study, LINC00987 was found to be apparently downregulated in COPD tissues. However, there are few data on the mechanism of COPD regulated by LINC00987.
MiRNA, a non-coding RNA, is approximately 21 nts in length and negatively modulates the expression of binding molecules through associating with its 3ʹ-untranslated regions (3ʹUTR).12 Abnormal miRNA expression is involved in the progression of lung diseases. For example, miR-132 and miR-155 were explained to contribute to COPD pathophysiology;13,14 miR-145-5p was demonstrated to inhibit cigarette smoke (CS)-induced inflammatory response in COPD,15 suggesting miR-145-5p acted as a suppressor in COPD development. Additionally, Zheng et al. explained that let-7b-5p suppressed cell proliferative and migratory abilities in squamous cell carcinoma.16 Let-7b-5p was also reported to participate in endoplasmic reticulum (ER) stress response in acute pulmonary embolism.17 In this study, we found LINC00987 contained the putative binding sites of let-7b-5p, suggesting that LINC00987 might regulate COPD progression by sponging let-7b-5p.
Sirtuin1 (SIRT1) is a nuclear gene and has been widely reported in lifetime regulation .18,19 In COPD, He et al. demonstrated that melatonin could ameliorate COPD by repressing cell apoptosis and ER stress via promoting SIRT1 expression.20 Additionally, Wang et al. also showed that resveratrol mediated an inflammatory response by activating SIRT1 in COPD in a mice model.21 Overall, SIRT1 plays a primary part in the pathogenesis of COPD. Herein, SIRT1 was predicted to possess the targeting sequence of let-7b-5p.
In this research, the expression of let-7b-5p was firstly determined in COPD tissues and lipopolysaccharide (LPS)-induced BEAS-2B cells. The effects of LINC00987 on LPS-induced cell apoptosis, oxidative stress, inflammation and autophagy were subsequently revealed. Furthermore, the associated relationship between let-7b-5p and LINC00987 or SIRT1 was identified.
Materials and Methods
Tissues Acquirement and Ethics
Twenty-nine COPD tissues and 33 normal lung tissues were obtained from COPD patients from Anhui University of Chinese Medicine during surgery. Normal lung tissues were obtained 5 cm from the tissue margin, which were further confirmed by pathologists. Tissues were kept in liquid nitrogen and stored at −80°C in a refrigerator. The Ethics Committee of Anhui University of Chinese Medicine endorsed this study. Patients linked to this experiment accepted written informed consent.
Cell Culture and Lipopolysaccharide (LPS) Treatment
Procell (Wuhan, China) offered human healthy lung epithelial cell lines 16HBE and BEAS-2B. 16HBE and BEAS-2B cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Thermo Fisher) with 1% streptomycin/penicillin (Thermo Fisher) in an incubator at 37°C with 5% CO2.
Cell Transfection
The overexpression plasmid of LINC00987, let-7b-5p inhibitor (anti-let-7b-5p), let-7b-5p mimic (let-7b-5p), the small inferring RNAs against SIRT1 (si-SIRT1#1, si-SIRT1#2 and si-SIRT1#3) and control groups (vector, anti-NC, miR-NC and si-NC) were synthesized by GenePharma (Shanghai, China). Cell transfection was carried out with Lipofectamine 3000 (Thermo Fisher). Sequences used in this part were let-7b-5p inhibitor 5ʹ-AACCACACAACCUACUACCUCA-3ʹ, let-7b-5p mimic 5ʹ-UGAGGUAGUAGGUUGUGUGGUU-3ʹ, si-SIRT1#15ʹ-GCACAGATCCTCGAACAAT-3ʹ, si-SIRT1#25ʹ-GCTGATGAACCGCTTGCTA-3ʹ, si-SIRT1#3 5ʹ-CCATGGCGCTGAGGTATAT-3ʹ, anti-NC 5ʹ-CAGUACUUUUGUGUAGUACAAA-3ʹ, miR-NC 5ʹ-UUUGUACUACACAAAAGUACUG-3ʹ and si-NC 5ʹ-CCACGCGGAGTATGGTTAT-3ʹ.
Cell Counting Kit-8 (CCK-8) Assay
The cell viability of 16HBE and BEAS-2B cells was revealed by CCK-8 detection kit (Beyotime). In short, cells were grown in a 96-well plate (5×103 cells per well) for 16 h. Supernatant was discarded and washed using phosphate buffer solution (PBS; Thermo Fisher). 10 μL CCK-8 solution (Beyotime) was added at 24 h after diverse treatments. Cells continued to cultivate for 2 h. Medium was removed and cell viability was determined by measuring absorbance at 450 nm under microplate reader (Thermo Labsystems, Waltham, MA, USA).
Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
COPD tissues and cells were lysed with TRIzol reagent (TaKaRa, Dalian, China). RNA was extracted via RNAsimple extraction kit (Tiangen, Beijing, China). cDNA was amplified by prime Script™ RT kit (TaKaRa) or MiX-x™ miRNA synthesis kit (TaKaRa). For illustrating the expression of LINC00987, let-7b-5p and SIRT1, SYBR® Premix DimerEraser kit (TaKaRa) was performed. Data were assessed with the 2−∆∆Ct method. U6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were chosen as references. The sense and antisense of primers were LINC00987 5ʹ-ACGACGCACAATGCAAAGAC-3ʹ and 5ʹ-TGTTTTCTGCACTGACCCCA-3ʹ; let-7b-5p 5ʹ-CTCACGTGAGGTAGTAGGT-3ʹ; SIRT1 5ʹ-CGGAAACAATACCTCCACCTGA-3ʹ and 5ʹ-TCCACATGAAACAGACACCCC-3ʹ; U6 5ʹ-CTCGCTTCGGCAGCACA-3ʹ and 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ; GAPDH 5ʹ-CCATGGGGAAGGTGAAGGTC-3ʹ and 5ʹ-TGGAATTTGCCATGGGTGGA-3ʹ.
Caspase3 Activity Assay
Caspase3 activity of 16HBE and BEAS-2B cells was determined by caspase3 activity detection kit (Beyotime). In brief, cell supernatant and cells digested by trypsin (Thermo Fisher) were collected and centrifuged at 500 rpm for 5 min. Lysis buffer (Beyotime) was incubated with supernatant for 15 min. Supernatant was gathered by centrifugation at 17,000 rpm for 12 min. Results were illustrated by microplate reader (Thermo Labsystems).
Apoptosis Analysis Assay
Cell apoptosis was assessed via Annexin V-fluorescein isothiocyanate (Annexin V-FITC)/propidium iodide (PI) apoptosis detection kit (Yeasen Biotech, Shanghai, China).
Cells were digested using trypsin (Thermo Fisher) at 48 h after transfection and were then centrifuged at 200 rpm for 6 min and washed using PBS (Thermo Fisher). 100 μL binding buffer (Yeasen Biotech) was used to suspend cells. Following this, cells were incubated with 5 μL Annexin V-FITC and 10 μL PI (Yeasen Biotech) for 12 min. Results were disclosed by flow cytometry (BD Biosciences, San Diego, CA, USA).
Reactive Oxygen Species (ROS) Level Determination
Intracellular ROS production was determined by ROS detection kit (Beyotime). In brief, supernatant was discarded and 2, 7-dichlorodihydrofluorescein diacetate (DCFH-DA) (10 μM/L; Beyotime) diluted with DMEM was added into plate. Cells then continued to culture for 20 min in an incubator, following this cells were washed three times using DMEM without FBS (Thermo Fisher) to remove DCFH-DA in plate. ROS level was unveiled using flow cytometry (BD Biosciences).
Superoxide Dismutase (SOD) Activity Assay
SOD activity of 16HBE and BEAS-2B cells was detected with SOD activity kit (Abcam, Cambridge, UK). In brief, 16HBE and BEAS-2B cells were harvested and lysed on ice, followed by centrifugation at 12,000 rpm for 5 min. Supernatant was collected and kept on ice. The output of absorbance (OD=450 nm) was determined with microplate reader (Thermo Labsystems).
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA kit (Beyotime) was employed to determine the levels of IL-6 and IL-8. Briefly, supernatant was centrifuged at 300 rpm for 5 min. Following sample and standard substance were added into reaction well, respectively, and were incubated at room temperature for 2 h. After that, scrubbing solution was used to wash plate and biotinylated anti-IL-6 (Beyotime) or anti-IL-8 (Beyotime) was then placed into well for 1 h. Sample was incubated with streptavidin marked horseradish peroxidase (Beyotime) for 20 min. After stop solution was added, IL-6 and IL-8 levels were determined via measuring absorbance (450 nm) with microplate reader (Thermo Labsystems).
Western Blot
Tissues and cells were lysed with RIPA buffer (Beyotime). Lysates were boiled in water for 10 min, which was then mixed with loading buffer (Thermo Fisher). 5 μg sample was loaded by 12% sodium dodecyl sulfonate-polyacrylamide gel electrophoresis (SDS-PAGE) (Beyotime). Protein bands were transduced into nitrocellulose membranes (GE Healthcare, Westborough, MA, USA). Membranes were blocked in 5% non-fat milk (Solarbio, Beijing, China) for 4 h and were incubated with anti-pro-caspase3 (anti-pro-c3) (1:1000; Abcam), anti-cleaved-caspase3 (anti-cleaved-c3) (1:5000; Abcam), anti-SOD1 (1:1000; Affinity, Nanjing, China), anti-SOD2 (1:1000; Affinity), anti-SOD3 (1:1000; Affinity), anti-microtubule-associated protein 1 light chain 3I/II (anti-LC3-I/LC3-II) (1:1000; Affinity), anti-autophagy-related protein 5 (anti-ATG5) (1:1000; Affinity), anti-sequestosome 1 (anti-P62) (1:1000; Affinity), anti-SIRT1 (1:1500; Affinity) and anti-GAPDH (1:1000; Affinity), respectively. Membranes were washed with PBS (Thermo Fisher) and incubated with second antibody marked with horseradish peroxidase (1:5000; Affinity). Bands were visualized under enhanced chemiluminescence (KeyGen, Nanjing, China). GAPDH was selected as a control.
Dual-Luciferase Reporter Assay
The binding sites between let-7b-5p and LINC00987 or SIRT1 were predicted by lncBase or miRWalk online database. The wide-type sequences of LINC00987 or SIRT1 3ʹUTR were inserted into pmirGLO vector (Promega, Madison, WI, USA), and named as wild-type-LINC00987 and wild-type-3ʹUTR SIRT1. The binding sequences of let-7b-5p in LINC00987 and SIRT1 3ʹUTR were mutated and then were cloned into pmirGLO vector (Promega), and named as mutant-LINC00987 and mutant-3ʹUTR SIRT1. Cell transfection was performed with DharmaFECT 4 (Thermo Fisher). At 48 h after transfection, cells were collected, lysed and detected via dual luciferase reporter assay kit (Promega) with Ranilla Luciferase as a control. Mutant-LINC00987 and mutant-3ʹUTR SIRT1 were used as controls of wild-type-LINC00987 and wild-type-3ʹUTR SIRT1, respectively.
Statistical Analysis
SPSS 21.0 software (IBM, Somers, NY, USA) was performed to analyze data from 3 independent experiments. The linear relationship between LINC00987 and let-7b-5p was assessed by Spearman correlation analysis. The value of LINC00987 in COPD diagnosis was investigated by receiver-operating characteristic (ROC) curve. Data values were presented with means ± standard deviations (SD). Pairwise differences between groups were revealed by two-tailed Student’s t-tests. P value < 0.05 was considered statistically significant.
Results
LINC00987 Expression Was Downregulated and Let-7b-5p Expression Was Upregulated in COPD Tissues and LPS-Induced BEAS-2B Cells
In order to determine the feasibility of the LPS-induced COPD model, the effect of LPS treatment on cell viability was determined in 16HBE and BEAS-2B cells. Results showed that LPS exposure (1 and 2 μg/mL) could dramatically inhibit the viability of 16HBE and BEAS-2B cells (Supplementary Figure 1A). 1 μg/mL LPS was chosen for subsequent study based on significantly repressive impact on cell viability. Subsequently, cell viability was revealed after LPS treatment at various times. The data showed that 1 μg/mL LPS obviously suppressed cell viability at 2 h after LPS treatment in both 16HBE and BEAS-2B cells (Supplementary Figure 1B). These results illustrated that the LPS-induced COPD model could be chosen in this study. Additionally, in subsequent research related to LPS, cells were treated with 1 μg/mL LPS for 2 h before various treatments.
To reveal the role of LINC00987 and let-7b-5p in COPD, their expression levels were first detected. Results showed that LINC00987 expression was dramatically downregulated and let-7b-5p expression was significantly upregulated in COPD tissues (N=29) relative to normal lung tissues (N=33) (Figure 1A). In addition, LINC00987 and let-7b-5p expression were determined in LPS-induced BEAS-2B cells. Results illustrated that LINC00987 expression was also remarkably decreased and let-7b-5p expression was notably increased in LPS-induced BEAS-2B cells compared with control groups (Figure 1B). To reveal the value of LINC00987 in COPD diagnosis, ROC curve was graphed. Results disclosed that LINC00987 had the ability to distinguish COPD from healthy control with areas under the curve (AUC) of 0.7983 (Figure 1C). Subsequently, RT-qPCR data showed that let-7b-5p expression was significantly upregulated in COPD tissues (N=29) relative to normal lung tissues (N=33) (Figure 1D). The expression of LINC00987 was revealed to be negatively related to let-7b-5p expression (Figure 1E).
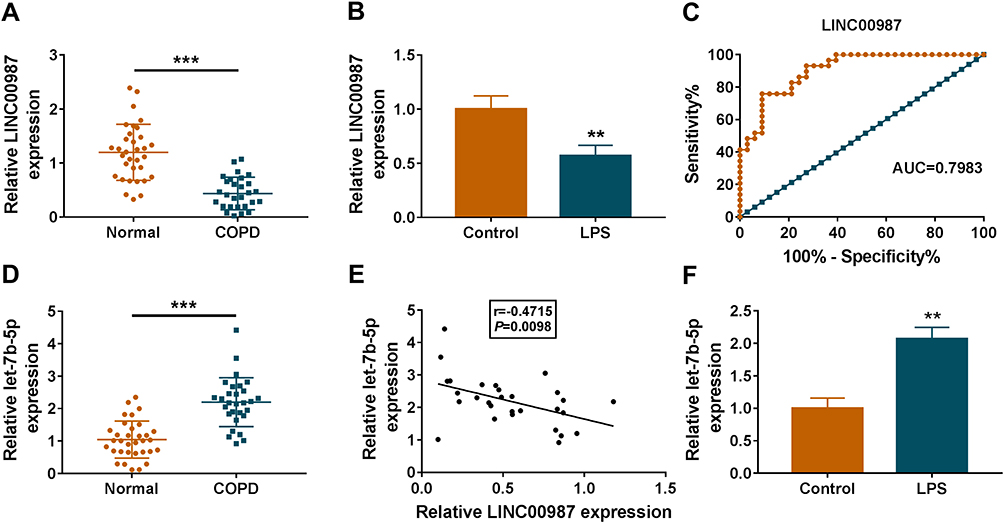 |
Figure 1 LINC00987 had low expression and let-7b-5p had high expression in LPS-induced BEAS-2B cells. (A and D) RT-qPCR was performed to detect LINC00987 and let-7b-5p expression in normal (N=33) and COPD tissues (N=29). (B and F) The expression levels of LINC00987 and let-7b-5p were determined by RT-qPCR in LPS-induced BEAS-2B cells. (C) ROC curve was used to investigate the value of LINC00987 in COPD diagnosis. (E) Spearman correlation analysis was employed to reveal the relationship between LINC00987 expression and let-7b-5p expression. **P<0.01 and ***P<0.001. |
Furthermore, let-7b-5p expression was notably increased in LPS-induced BEAS-2B cells compared with control groups (Figure 1F). These data showed that LINC00987 might play a vital part in COPD pathogenesis.
LINC00987 Overexpression Protected 16HBE and BEAS-2B Cells from LPS-Induced Cell Apoptosis, Oxidative Stress, Inflammation and Autophagy
The function of LINC00987 in COPD pathogenesis was further explored with an LPS-induced COPD model. The overexpression plasmid of LINC00987 was first built. RT-qPCR results showed that LINC00987 expression was strikingly upregulated after LINC00987 transfection (Figure 2A). In addition, the results revealed that LINC00987 expression was decreased after LPS treatment, whereas this effect was attenuated by LINC00987 overexpression (Figure 2B). The results also showed LPS treatment upregulated let-7b-5p expression, whereas enforced LINC00987 expression impaired this effect (Figure 2C). Subsequently, CCK-8 assay illustrated that cell viability was repressed in LPS-induced 16HBE and BEAS-2B cells, and LINC00987 overexpression restored this effect (Figure 2D). Caspase3 activity and apoptosis analysis assays demonstrated that caspase3 activity and cell apoptosis were promoted in LPS-induced 16HBE and BEAS-2B cells, respectively; however, these effects were abolished by LINC00987 overexpression (Figure 2E and F). Additionally, ROS level was upregulated and SOD activity was repressed in LPS-induced 16HBE and BEAS-2B cells; but these impacts were restored by LINC00987 overexpression (Figure 2G and H). Data also showed the ratio of cleaved-c3 and pro-c3 was upregulated and the protein levels of SOD1, SOD2 and SOD3 were downregulated in LPS-induced 16HBE and BEAS-2B cells, while LINC00987 overexpression restored these influences (Figure 2I). IL-6 and IL-8 levels were upregulated in LPS-induced 16HBE and BEAS-2B cells, and these influences were relieved by LINC00987 (Figure 2J and K). Furthermore, Western blot unveiled that IC3-II/IC3-I and ATG5 levels were upregulated and P62 protein expression was repressed in LPS-induced 16HBE and BEAS-2B cells; however, these impacts were restrained after LINC00987 overexpression (Figure 2L). These data indicated that LINC00987 alleviated cell apoptosis, oxidative stress, inflammation and autophagy induced by LPS in 16HBE and BEAS-2B cells.
 |
Figure 2 LINC00987 inhibited cell apoptosis, oxidative stress, inflammation and autophagy in LPS-mediated 16HBE and BEAS-2B cells. (A) The transfection efficiency of LINC00987 was detected by RT-qPCR in LPS-induced 16HBE and BEAS-2B cells. (B and C) The effects of LPS treatment and LINC00987 overexpression on LINC00987 and let-7b-5p expression were demonstrated by RT-qPCR. (D) CCK-8 assay was performed to illustrate the impacts between LPS and LINC00987 on cell viability in 16HBE and BEAS-2B cells. (E and F) Caspase3 activity and apoptosis analysis assays were conducted to present the influences between LPS and LINC00987 overexpression on caspase3 activity and cell apoptosis, respectively, in 16HBE and BEAS-2B cells. (G and H) ROS detection kit and SOD activity assays were carried out to uncover the impacts between LPS and enforced LINC00987 expression on oxidative stress in 16HBE and BEAS-2B cells. (I) Western blot assay was employed to determine the impacts of LINC00987 on the protein levels of pro-c3, cleaved-c3, SOD1, SOD2 and SOD3 in LPS-induced 16HBE and BEAS-2B cells. (J and K) ELISA kit assays were employed to exhibit the effects between LPS exposure and LINC00987 on the production of IL-6 and IL-8 in 16HBE and BEAS-2B cells. (L) The effects of LPS treatment and LINC00987 overexpression on the level of IC3-II/IC3-I and the protein expression of ATG5 and P62 were demonstrated by Western blot in 16HBE and BEAS-2B cells. *P<0.05, **P<0.01 and ***P<0.001. |
Let-7b-5p Inhibitor Restrained LPS-Induced Cell Apoptosis, Oxidative Stress, Inflammation and Autophagy in BEAS-2B Cells
The effects of let-7b-5p deletion on COPD pathogenesis were investigated in this part. RT-qPCR first verified the transfection efficiency of anti-let-7b-5p, and the result showed that let-7b-5p expression was dramatically downregulated by anti-let-7b-5p (Figure 3A). RT-qPCR data also revealed that let-7b-5p inhibitor attenuated the LPS-mediated effect on let-7b-5p expression, but not LINC00987 expression (Figure 3B and C). Subsequently, LPS treatment suppressed cell viability, whereas anti-let-7b-5p attenuated this impact in BEAS-2B cells (Figure 3D). Caspase3 activity and cell apoptosis were also promoted after LPS exposure, which were restored after let-7b-5p inhibitor transfection in BEAS-2B cells (Figure 3E and F). Additionally, LPS treatment upregulated ROS level and suppressed SOD activity in BEAS-2B cells; but these influences were abolished by anti-let-7b-5p (Figure 3G and H). Western blot analysis also showed that the value of cleaved-c3/pro-c3 was increased and the protein levels of SOD1, SDO2 and SOD3 were downregulated in LPS-induced BEAS-2B cells; however, these impacts were hindered after let-7b-5p repression (Figure 3I). IL-6 and IL-8 levels were also upregulated by LPS in BEAS-2B cells; however, these promoting effects were relieved by let-7b-5p repression (Figure 3J and K). Furthermore, Western blot revealed that IC3-II/IC3-I and ATG5 levels were upregulated and P62 protein expression was suppressed after LPS exposure in BEAS-2B cells, and these impacts were reversed by let-7b-5p absence (Figure 3L). Therefore, these results unveiled that let-7b-5p deletion ameliorated LPS-induced disorders in BEAS-2B cells.
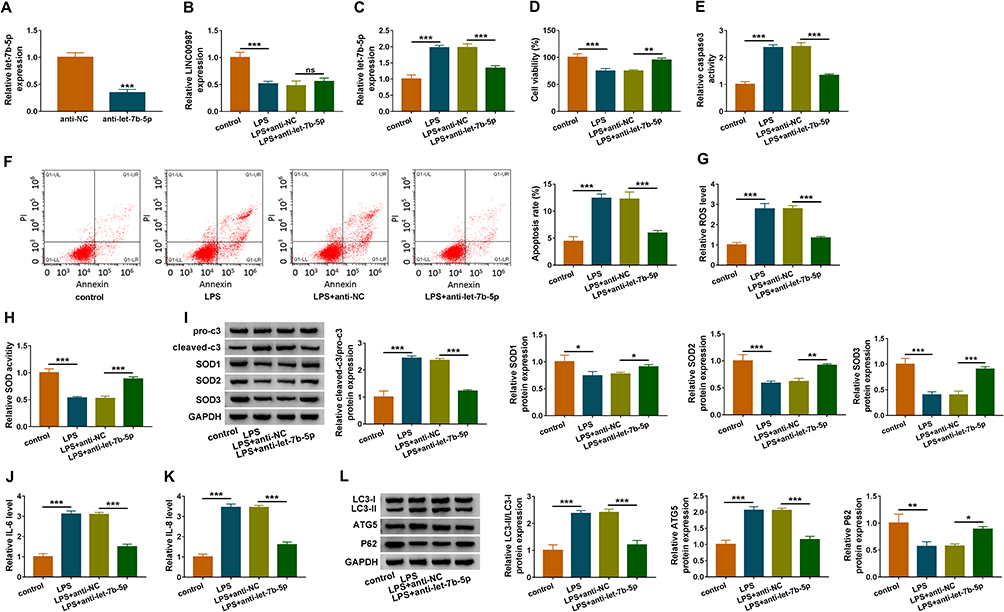 |
Figure 3 Let-7b-5p inhibitor attenuated LPS-induced apoptosis, oxidative stress, inflammation and autophagy in BEAS-2B cells. (A) RT-qPCR was performed to detect the transfection efficiency of anti-let-7b-5p in LPS-induced BEAS-2B cells. (B and C) The impact of anti-let-7b-5p on LINC00987 and let-7b-5p expression after LPS treatment was determined by RT-qPCR in BEAS-2B cells. (D) CCK-8 assay was carried out to illustrate the impacts between LPS and let-7b-5p absence on cell viability in BEAS-2B cells. (E and F) Caspase3 activity and apoptosis analysis assays were employed to reveal the influences between LPS exposure and let-7b-5p deletion on caspase3 activity and cell apoptosis, respectively, in BEAS-2B cells. (G and H) ROS detection kit and SOD activity assays were used to reveal the impacts between LPS treatment and let-7b-5p repression on oxidative stress in BEAS-2B cells. (I) Western blot assay was employed to determine the influences of let-7b-5p inhibitor on the protein levels of pro-c3, cleaved-c3, SOD1, SOD2 and SOD3 in LPS-induced BEAS-2B cells. (J and K) ELISA kits were purchased to disclose the influences of anti-let-7b-5p on the levels of IL-6 and IL-8 under LPS treatment in BEAS-2B cells. (L) Western blot was performed to detect the impacts between LPS and let-7b-5p inhibitor on the levels of IC3-II/IC3-I, ATG5 and P62 in BEAS-2B cells. *P<0.05, **P<0.01 and ***P<0.001. Abbreviation: Ns, no statistical significance. |
LINC00987 Repressed LPS-Induced Cell Apoptosis, Oxidative Stress, Inflammation and Autophagy by Sponging Let-7b-5p
Given the effects of LINC00987 overexpression and let-7b-5p inhibitor on COPD pathogenesis, whether LINC00987 regulated COPD pathogenesis by associating with let-7b-5p was explored. LncBase online database showed that LINC00987 contained the binding sites of let-7b-5p (Figure 4A). At the same time, several miRNAs possessing the binding sites of LINC00987 were exhibited in Figure 5A. RT-qPCR data illustrated that let-7b-5p mimic was successfully transfected into BEAS-2B cells based on its expression being dramatically upregulated after let-7b-5p mimic transfection (Figure 4B). And, dual-luciferase reporter assay showed that luciferase activity was obviously repressed after wild type-LINC00987 and let-7b-5p co-transfection, whereas there was no apparent change in mutant-LINC00987+let-7b-5p group (Figure 4C). Subsequently, the results demonstrated that LINC00987 repressed let-7b-5p expression, whereas let-7b-5p restrained this effect (Figure 4D). CCK-8 assay revealed that LINC00987 promoted cell viability in LPS-induced BEAS-2B cells, whereas this effect was restored by let-7b-5p (Figure 4E). LINC00987 inhibited caspase3 activity and downregulated cell apoptosis rate in LPS-induced BEAS-2B cells, and these effects were attenuated by let-7b-5p (Figure 4F and G). ROS detection kit and SOD activity assays disclosed that LINC00987 downregulated ROS level and promoted SOD activity in LPS-induced BEAS-2B cells; however, let-7b-5p abolished these impacts (Figure 4H and I). Meanwhile, Western blot analysis showed that LINC00987 obviously decreased the value of pro-caspase3/cleaved-caspase3 and upregulated the expression of SOD2 and SOD3; however, these effects were restrained after let-7b-5p transfection (Figure 4J). Of note, the expression level of SOD1 was obviously upregulated after LINC00987 transfection, but this effect was not affected by let-7b-5p (Figure 4J). These data illustrated that SOD2 and SOD3 were main participants in the COPD process mediated by LINC00987/let-7b-5p axis. In addition, IL-6 and IL-8 levels were downregulated after LINC00987 transfection, and let-7b-5p mimic restrained these impacts in LPS-mediated BEAS-2B cells (Figure 4K and L). Western blot unveiled that LC3-II/IC3-I and ATG5 levels were repressed and P62 protein expression was promoted by LINC00987 overexpression, but let-7b-5p relieved these influences (Figure 4M). Altogether the data showed that LINC00987 protected BEAS-2B cells from LPS-induced apoptosis, oxidative stress, inflammation and autophagy by sponging let-7b-5p.
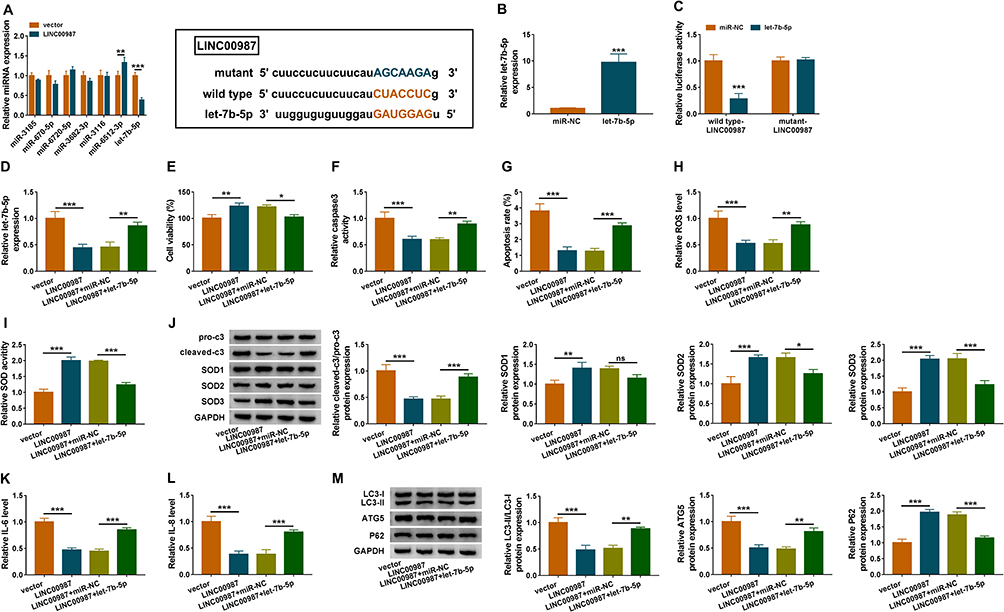 |
Figure 4 LINC00987 overexpression inhibited cell apoptosis, oxidative stress, inflammation and autophagy by binding to let-7b-5p in LPS-mediated BEAS-2B cells. (A) The expression levels of miRNAs possessing the binding sites of LINC00987 were determined by RT-qPCR in BEAS-2B cells transfected with LINC00987 or vector, and the binding sites between LINC00987 and let-7b-5p were predicted by lncBase online database. (B) The transfection efficiency of let-7b-5p mimic was determined by RT-qPCR. (C) Dual-luciferase reporter assay was performed to detect luciferase activity in LPS-induced BEAS-2B cells. (D) The effects between LINC00987 and let-7b-5p on let-7b-5p expression were revealed by RT-qPCR. (E) CCK-8 assay was employed to illustrate the impacts between LINC00987 and let-7b-5p on cell viability in LPS-induced BEAS-2B cells. (F and G) Caspase3 activity and apoptosis analysis assays were carried out to demonstrate the impacts between LINC00987 and let-7b-5p mimic on the apoptosis of LPS-induced BEAS-2B cells. (H and I) ROS detection kit and SOD activity assays were conducted to present the influences between LINC00987 overexpression and let-7b-5p mimic on oxidative stress in LPS-induced BEAS-2B cells. (J) The influences between LINC00987 and let-7b-5p on the expression of apoptosis-related proteins (pro-caspase3 and cleaved-caspase3) and ROS-associated proteins (SOD1, SOD2 and SOD3) were revealed by Western blot. (K and L) ELISA assays were used to disclose the impacts between LINC00987 overexpression and let-7b-5p mimic on inflammation response in LPS-induced BEAS-2B cells. (M) Western blot was performed to detect the influences between LINC00987 and let-7b-5p on the levels of LC3-II/IC3-I, ATG5 and P62 in LPS-mediated BEAS-2B cells. *P<0.05, **P<0.01 and ***P<0.001. |
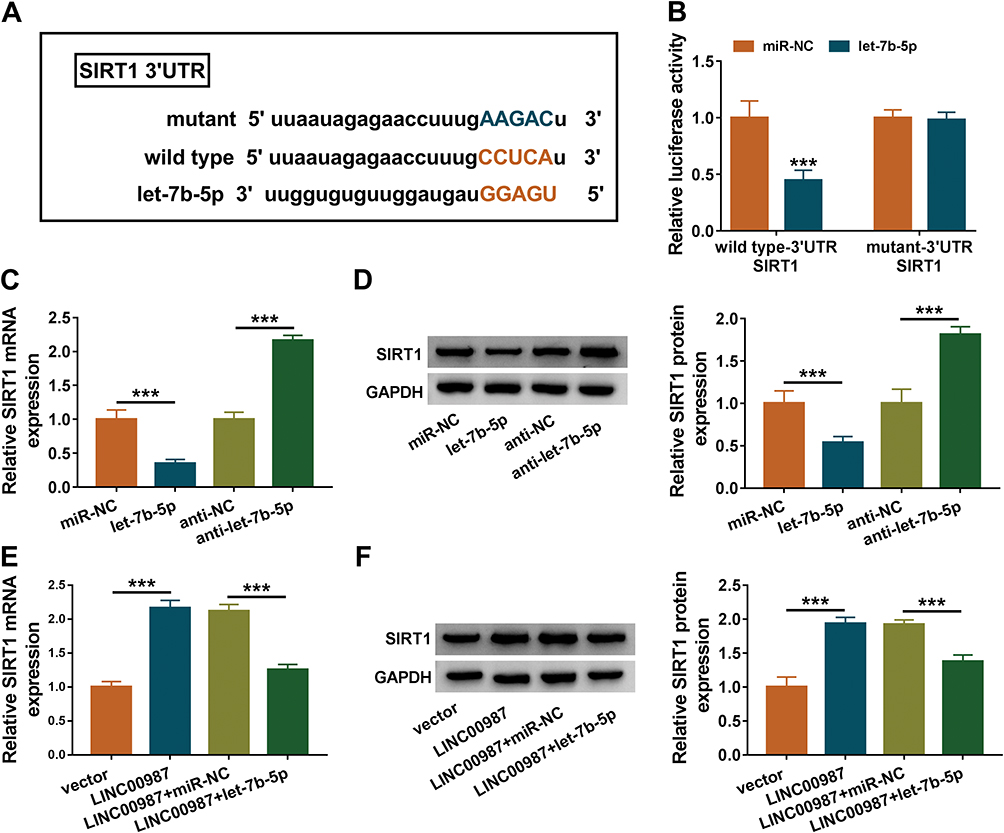 |
Figure 5 LINC00987 upregulated SIRT1 expression via binding to let-7b-5p. (A) MiRWalk online database was used to predict the binding sites between let-7b-5p and SIRT1 3ʹUTR. (B) Luciferase activities were detected by dual-luciferase reporter assay. (C) The effects of let-7b-5p mimic and inhibitor on SIRT1 mRNA level were revealed by RT-qPCR. (D) The effects of let-7b-5p mimic and inhibitor on SIRT1 protein level were investigated by Western blot. (E and F) The effects between LINC00987 and let-7b-5p on SIRT1 expression at mRNA and protein levels were illustrated via RT-qPCR and Western blot, respectively. ***P<0.001. |
LINC00987 Sponged Let-7b-5p to Bind to SIRT1
The underlying interaction between LINC00987 and let-7b-5p on COPD pathogenesis were revealed in this part. MiRWalk online database presented that SIRT1 3ʹUTR contained the targeting sites of let-7b-5p (Figure 5A). Dual-luciferase reporter assay displayed that the luciferase activity of wild type-3ʹUTR SIRT1+let-7b-5p group was remarkably repressed, whereas there was no obvious change in mutant-3ʹUTR SIRT1+let-7b-5p group (Figure 5B). Subsequently, the results showed that let-7b-5p mimic dramatically repressed SIRT1 expression and let-7b-5p inhibitor obviously upregulated SIRT1 expression at both mRNA and protein levels in LPS-mediated BEAS-2B cells (Figure 5C and D). Data also revealed that LINC00987 overexpression increased the mRNA and protein expression levels of SIRT1, and these impacts were attenuated by let-7b-5p mimic in LPS-induced BEAS-2B cells (Figure 5E and F). These results suggested that LINC00987 regulated SIRT1 expression by associating with let-7b-5p.
SIRT1 Knockdown Hindered the Inhibition Effects of LINC00987 on LPS-Induced Apoptosis, Oxidative Stress, Inflammation and Autophagy in BEAS-2B Cells
To reveal whether LINC00987 mediated COPD pathogenesis by modulating SIRT1, the effects between LINC00987 and SIRT1 knockdown on LPS-induced apoptosis, oxidative stress, inflammation and autophagy were disclosed. The transfection efficiency of interfering plasmids of SIRT1 was determined first. The results showed that si-SIRT1#1, si-SIRT1#2 and si-SIRT1#3 dramatically repressed SIRT1 expression at mRNA and protein levels in LPS-induced BEAS-2B cells (Figure 6A and B), and si-SIRT1#2 was chosen for subsequent study owing to it having the most successful interfering efficiency. Data also disclosed that the mRNA and protein expression of SIRT1 was strikingly upregulated by LINC00987 after LPS exposure, and SIRT1 knockdown restored these effects (Figure 6C and D). Subsequently, it was found that LINC00987 overexpression promoted cell viability under LPS treatment, whereas this influence was abolished by SIRT1 silencing (Figure 6E). LINC00987 also repressed caspase3 activity and cell apoptosis, and these impacts were blocked after SIRT1 absence in LPS-induced BEAS-2B cells (Figure 6F and G). ROS level was downregulated and SOD activity was promoted by LINC00987 after LPS treatment, whereas these influences were relieved by SIRT1 absence (Figure 6H and I). Meanwhile, the results showed that LINC00987 greatly reduced the ratio between cleaved-c3 and pro-c3 and increased the protein levels of SOD2 and SOD3, while SIRT1 silencing restored these influences (Figure 6J). As expected, LINC00987-mediated upregulation in SOD1 expression was still not changed by si-SIRT1#2 (Figure 6J). In addition, ELISA kit assays revealed that LINC00987 repressed the production of IL-6 and IL-8 in LPS-induced BEAS-2B cells; however, SIRT1 repression impaired these effects (Figure 6K and L). Western blot analysis also unveiled that LINC00987 repressed LC3-II/IC3-I and ATG5 levels, and facilitated P62 protein expression in LPS-induced BEAS-2B cells; but these impacts were restrained by SIRT1 silencing (Figure 6M). These results illustrated that LINC00987 protected BEAS-2B cells against LPS-induced apoptosis, oxidative stress, inflammation and autophagy by regulating SIRT1.
 |
Figure 6 LINC00987 ameliorated LPS-induced disorders by regulating SITR1. (A and B) The knockdown efficiency of si-SIRT1#1, si-SIRT1#2 and si-SIRT1#3 was determined by detecting the mRNA and protein levels of SIRT1 via RT-qPCR and Western blot, respectively. (C) The effects of LINC00987 and SIRT1 knockdown on the mRNA level of SIRT1 were illustrated by RT-qPCR. (D) The effects of LINC00987 and SIRT1 knockdown on the protein expression of SIRT1 were illustrated by Western blot. (E) CCK-8 assay was performed to illustrate the effects between LINC00987 and SIRT1 silencing on cell viability in LPS-induced BEAS-2B cells. (F and G) Caspase3 activity and apoptosis analysis assays were employed to demonstrate the impacts between LINC00987 and SIRT1 deletion on cell apoptosis in LPS-induced BEAS-2B cells. (H and I) ROS detection kit and SOD activity assays were carried out to display the influences between LINC00987 overexpression and SIRT1 absence on oxidative stress in LPS-induced BEAS-2B cells. (J) The impacts between ectopic LINC00987 expression and SIRT1 downregulation on the protein expression of pro-c3, cleaved-c3, SOD1, SOD2 and SOD3 were presented by Western blot assay. (K and L) ELISA kit assays were conducted to investigate the impacts between LINC00987 overexpression and SIRT1 deletion on IL-6 and IL-8 production in LPS-induced BEAS-2B cells. (M) The effects between LINC00987 overexpression and SIRT1 knockdown on the levels of LC3-II/IC3-I, ATG5 and P62 were presented by Western blot in LPS-induced BEAS-2B cells. *P<0.05, **P<0.01 and ***P<0.001. |
Discussion
COPD seriously endangers human health owing to an incomplete therapy schedule and the presence of comorbidities.22 LncRNA has been enrolled in regulating COPD pathogenesis.23 The COPD model was commonly built on treating CS, LPS or intranasal elastase.24 However, studies found that LPS could enlarge alveolar spaces and reduce emphysema after treatment with elastase when employed solely or concomitant with CS,25,26 suggesting a LPS-induced COPD model could be used instead of a CS/elastase-induced COPD model in studying COPD progression. In this study, the effects of LncRNA LINC00987 on COPD pathogenesis were studied using a LPS-mediated COPD model and the underlying mechanism was further revealed. Our findings showed that LINC00987 protected against LPS-induced apoptosis, oxidative stress, inflammation and autophagy through regulating let-7b-5p/SIRT1 axis in BEAS-2B cells.
LncRNAs play a key role in the pathogenesis of COPD. For example, lncRNA taurine up-regulated 1 (TUG1) suppressed cell proliferation in COPD.27 LncRNA small nucleolar RNA host gene 5 regulated the apoptosis and inflammation of COPD cells.28 In this study, the expression of LINC00987 was downregulated in COPD tissues and LPS-induced BEAS-2B cells. In order to reveal the effects of LINC00987 on COPD pathogenesis, LINC00987 was transfected into LPS-induced 16HBE and BEAS-2B cells with control groups. Results showed that LINC00987 protected 16HBE and BEAS-2B cells against LPS-induced apoptosis, oxidative stress, inflammation and autophagy. To disclose the underlying regulatory mechanism, the associated miRNA with LINC00987 was predicted with the lncBase online database. Results showed that LINC00987 acted as a sponge of let-7b-5p, and LINC00987 overexpression downregulated let-7b-5p expression, but let-7b-5p inhibitor could not regulate LINC00987 expression, implicating LINC00987 might regulate COPD processes by sponging let-7b-5p.
In order to illustrate this, loss-of-function experiments were performed. We found that the absence of let-7b-5p relieved the inhibition effects of LPS stimulation on cell viability, and the promotion effects of LPS pre-treatment on cell apoptosis and inflammation, which meant that let-7b-5p inhibited cell viability and induced cell apoptosis and inflammation. Xu et al. revealed that enforced let-7b-5p expression contributed to cell apoptosis in multiple myeloma.29 Li et al. indicated that let-7b-5p restored the promotion effect of cyanidin-3-O-glucoside on cell viability, and the inhibition effects of that on inflammation response after LPS treatment in corneal epithelial cells,30 suggesting let-7b-5p could repress cell viability and induce cell inflammation. Our findings were consistent with the above data. In addition, let-7b-5p expression was upregulated in COPD tissues and LPS-induced BEAS-2B cells. Also, let-7b-5p induced oxidative stress and autophagy in BEAS-2B cells. Meanwhile, reverse experiments demonstrated that let-7b-5p mimic could restrain the impacts of LINC00987 overexpression on LPS-induced BEAS-2B cells, suggesting LINC00987 could modulate COPD process by binding to let-7b-5p. Furthermore, let-7b-5p was found to target SIRT1 in BEAS-2B cells.
Chun et al. indicated that SIRT1 could protect against COPD and repress autophagy and inflammation in lungs.31 Gu et al. reported that SIRT1 ameliorated COPD via repressing cell apoptosis in rats.32 Additionally, it was shown that the enforced expression of SIRT1 repressed cigarette smoke-induced oxidative stress in a COPD model.33 Similarly, we found that SIRT1 knockdown blocked the effects of LINC00987 on LPS-induced cell apoptosis, oxidative stress, inflammation and autophagy, which suggests that SIRT1 was capable of ameliorating COPD. Furthermore, we found let-7b-5p mimic impaired the promotion impact of LINC00987 on SIRT1 expression. These results meant LINC00987 could regulate SIRT1 expression by sponging let-7b-5p. Of note, we found LINC00987 could upregulate the expression of SOD2 and SOD3, not SOD1 expression, by controlling let-7b-5p or SIRT1, indicating that SOD2 and SOD3 were main regulators in LPS-induced ROS by LINC00987/let-7b-5p/SIRT1 axis. The shortcoming of this experiment was a lack of data on the effects of LINC00987/let-7b-5p/SIRT1 axis on COPD pathogenesis in vivo. We will explore this in a subsequent study.
Collectively, LINC00987 expression was remarkably downregulated and let-7b-5p expression was notably upregulated in COPD tissues and LPS-induced BEAS-2B cells. LINC00987 overexpression or let-7b-5p inhibitor hindered the inhibition effect of LPS treatment on cell viability and promotion effects in cell apoptosis, oxidative stress, inflammation and autophagy. Furthermore, LINC00987 functioned as a sponge of let-7b-5p and let-7b-5p was associated with SIRT1. The illustration of LINC00987 in regulating LPS-induced COPD is presented in Supplementary Figure 2. We found a new mechanism of LINC00987 in regulating COPD pathogenesis such that LINC00987 can hinder LPS-induced cell apoptosis, oxidative stress, inflammation and autophagy by upregulating SIRT1 expression through sponging let-7b-5p, which suggests LINC00987 may be a potential target for COPD therapy and may be used to make oligonucleotide drugs to treat COPD to further improve clinical patients.
Disclosure
The authors declare that they have no financial or non-financial conflicts of interest.
References
1. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2018;18(7):454–466. doi:10.1038/s41577-018-0006-6
2. Osei ET, Florez-Sampedro L, Timens W, Postma DS, Heijink IH, Brandsma CA. Unravelling the complexity of COPD by microRNAs: it’s a small world after all. Eur Respir J. 2015;46(3):807–818. doi:10.1183/13993003.02139-2014
3. Roffel MP, Bracke KR, Heijink IH, Maes T. miR-223: a key regulator in the innate immune response in asthma and COPD. Front Med (Lausanne). 2020;7:196. doi:10.3389/fmed.2020.00196
4. Jemal A, Ward E, Hao Y, Thun M. Trends in the leading causes of death in the United States, 1970-2002. JAMA. 2005;294(10):1255–1259. doi:10.1001/jama.294.10.1255
5. Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. doi:10.1038/nature11233
6. Liu X, Xiao ZD, Han L, et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18(4):431–442. doi:10.1038/ncb3328
7. Wang Y, Lyu X, Wu X, Yu L, Hu K. Long non-coding RNA PVT1, a novel biomarker for chronic obstructive pulmonary disease progression surveillance and acute exacerbation prediction potentially through interaction with microRNA-146a. J Clin Lab Anal. 2020;e23346. doi:10.1002/jcla.23346
8. Melissari MT, Grote P. Roles for long non-coding RNAs in physiology and disease. Pflugers Arch. 2016;468(6):945–958. doi:10.1007/s00424-016-1804-y
9. Huang X, Zhao M. High expression of long non-coding RNA MALAT1 correlates with raised acute respiratory distress syndrome risk, disease severity, and increased mortality in sepstic patients. Int J Clin Exp Pathol. 2019;12(5):1877–1887.
10. Yu W, Sun Z, Yang L, et al. lncRNA PTAR promotes NSCLC cell proliferation, migration and invasion by sponging microRNA‑101. Mol Med Rep. 2019;20(5):4168–4174. doi:10.3892/mmr.2019.10646
11. Li Z, Lu Q, Zhu D, Han Y, Zhou X, Ren T. Lnc-SNHG1 may promote the progression of non-small cell lung cancer by acting as a sponge of miR-497. Biochem Biophys Res Commun. 2018;506(3):632–640. doi:10.1016/j.bbrc.2018.10.086
12. Shukla GC, Singh J, Barik S. MicroRNAs: processing, maturation, target recognition and regulatory functions. Mol Cell Pharmacol. 2011;3(3):83–92.
13. Diao X, Zhou J, Wang S, Ma X. Upregulation of miR-132 contributes to the pathophysiology of COPD via targeting SOCS5. Exp Mol Pathol. 2018;105(3):285–292. doi:10.1016/j.yexmp.2018.10.002
14. De Smet EG, Van Eeckhoutte HP, Avila Cobos F, et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. 2020;13(3):423–436. doi:10.1038/s41385-019-0241-6
15. Dang X, Yang L, Guo J, et al. miR-145-5p is associated with smoke-related chronic obstructive pulmonary disease via targeting KLF5. Chem Biol Interact. 2019;300:82–90. doi:10.1016/j.cbi.2019.01.011
16. Zheng S, Liu Q, Ma R, et al. Let-7b-5p inhibits proliferation and motility in squamous cell carcinoma cells through negative modulation of KIAA1377. Cell Biol Int. 2019;43(6):634–641. doi:10.1002/cbin.11136
17. Liu T-W, Liu F, Kang J. Let-7b-5p is involved in the response of endoplasmic reticulum stress in acute pulmonary embolism through upregulating the expression of stress-associated endoplasmic reticulum protein 1. IUBMB Life. 2020;72(8):1725–1736. doi:10.1002/iub.2306
18. Pucci B, Villanova L, Sansone L, et al. Sirtuins: the molecular basis of beneficial effects of physical activity. Intern Emerg Med. 2013;8(Suppl 1):S23–25. doi:10.1007/s11739-013-0920-3
19. Taka C, Hayashi R, Shimokawa K, et al. SIRT1 and FOXO1 mRNA expression in PBMC correlates to physical activity in COPD patients. Int J Chron Obstruct Pulmon Dis. 2017;12:3237–3244. doi:10.2147/COPD.S144969
20. He B, Zhang W, Qiao J, Peng Z, Chai X. Melatonin protects against COPD by attenuating apoptosis and endoplasmic reticulum stress via upregulating SIRT1 expression in rats. Can J Physiol Pharmacol. 2019;97(5):386–391. doi:10.1139/cjpp-2018-0529
21. Wang XL, Li T, Li JH, Miao SY, Xiao XZ. The effects of resveratrol on inflammation and oxidative stress in a rat model of chronic obstructive pulmonary disease. Molecules. 2017;22:9. doi:10.3390/molecules22091529
22. Smith MC, Wrobel JP. Epidemiology and clinical impact of major comorbidities in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:871–888. doi:10.2147/copd.S49621
23. Ming X, Duan W, Yi W. Long non-coding RNA NEAT1 predicts elevated chronic obstructive pulmonary disease (COPD) susceptibility and acute exacerbation risk, and correlates with higher disease severity, inflammation, and lower miR-193a in COPD patients. Int J Clin Exp Pathol. 2019;12(8):2837–2848.
24. Ghorani V, Boskabady MH, Khazdair MR, Kianmeher M. Experimental animal models for COPD: a methodological review. Tob Induc Dis. 2017;15:25. doi:10.1186/s12971-017-0130-2
25. Ishii T, Hosoki K, Nikura Y, Yamashita N, Nagase T, Yamashita N. IFN regulatory Factor 3 potentiates emphysematous aggravation by lipopolysaccharide. J Immunol. 2017;198(9):3637–3649. doi:10.4049/jimmunol.1601069
26. Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L1–15. doi:10.1152/ajplung.90200.2008
27. Tang W, Shen Z, Guo J, Sun S. Screening of long non-coding RNA and TUG1 inhibits proliferation with TGF-β induction in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2951–2964. doi:10.2147/copd.S109570
28. Shen Q, Zheng J, Wang X, Hu W, Jiang Y, Jiang Y. LncRNA SNHG5 regulates cell apoptosis and inflammation by miR-132/PTEN axis in COPD. Biomed Pharmacother. 2020;126:110016. doi:10.1016/j.biopha.2020.110016
29. Xu H, Liu C, Zhang Y, et al. Let-7b-5p regulates proliferation and apoptosis in multiple myeloma by targeting IGF1R. Acta Biochim Biophys Sin (Shanghai). 2014;46(11):965–972. doi:10.1093/abbs/gmu089
30. Li X, Sun M, Long Y. Cyanidin-3-O-glucoside attenuates lipopolysaccharide-induced inflammation in human corneal epithelial cells by inducing Let-7b-5p-mediated HMGA2/PI3K/Akt pathway. Inflammation. 2020;43(3):1088–1096. doi:10.1007/s10753-020-01194-0
31. Chun P. Role of sirtuins in chronic obstructive pulmonary disease. Arch Pharm Res. 2015;38(1):1–10. doi:10.1007/s12272-014-0494-2
32. Gu C, Li Y, Xu WL, et al. Sirtuin 1 activator SRT1720 protects against lung injury via reduction of type ii alveolar epithelial cells apoptosis in emphysema. Copd. 2015;12(4):444–452. doi:10.3109/15412555.2014.974740
33. Yao H, Sundar IK, Ahmad T, et al. SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):L816–828. doi:10.1152/ajplung.00323.2013
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.