Back to Journals » Infection and Drug Resistance » Volume 19
LILRB1 Modulates Neutrophil Migration, NETosis, and Inflammation in Drug-Resistant Pseudomonas aeruginosa-Associated Bronchiectasis
Authors Zheng S, Yang S, Huang Y, Hong K, Tang J, Wu X, Qing C, Jiang Y, Lu W, Bao C, Luo J, Kong J
Received 2 December 2025
Accepted for publication 19 February 2026
Published 23 February 2026 Volume 2026:19 581113
DOI https://doi.org/10.2147/IDR.S581113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hazrat Bilal
Shaochu Zheng,1,* Shihao Yang,1,* Yuting Huang,1 Kangkang Hong,2 Jinling Tang,1 Xiaopu Wu,1 Cao Qing,1 Yun Jiang,1 Wei Lu,1 Chongxi Bao,1 Jing Luo,1 Jinliang Kong1
1Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 2Department of Geriatric Medicine, The Fourth Affiliated Hospital of Guangxi Medical University, Liuzhou, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Luo; Email [email protected] Jinliang Kong, Email [email protected]
Background: Patients with drug-resistant Pseudomonas aeruginosa (P. aeruginosa)-associated bronchiectasis often exhibit persistent neutrophilic airway inflammation. The immunomodulatory receptor leukocyte immunoglobulin-like receptor B1 (LILRB1) is known to function as an inhibitory checkpoint in immune responses, yet its specific role in regulating neutrophil function in drug-resistant P. aeruginosa-associated bronchiectasis remains incompletely understood. This study aimed to characterize the function of LILRB1 in this clinical context.
Methods: Clinical samples were obtained from bronchiectasis patients with drug-resistant P. aeruginosa infection and control subjects. LILRB1 mRNA expression was quantified by reverse transcription-quantitative polymerase chain reaction (RT-qPCR), and soluble HLA-G (sHLA-G) levels were assessed by enzyme-linked immunosorbent assay (ELISA). Neutrophils isolated from peripheral blood were stimulated with a clinical isolate of drug-resistant P. aeruginosa. NETosis was evaluated using myeloperoxidase (MPO)/citrullinated histone H3 (CitH3) immunofluorescence staining and quantified via PicoGreen dsDNA assay. The modulatory effect of recombinant LILRB1 protein was examined in a Transwell co-culture system composed of neutrophils and P. aeruginosa-infected BEAS-2B bronchial epithelial cells, with neutrophil migration and inflammatory cytokine secretion assessed as outcome measures.
Results: LILRB1 expression was significantly lower in patients with drug-resistant P. aeruginosa infection than in controls (P = 0.0384) and was inversely associated with disease severity, as indicated by the Bronchiectasis Severity Index (P = 0.0089), and with peripheral neutrophil counts (r = − 0.35, P = 0.044). Levels of sHLA-G in bronchoalveolar lavage fluid were elevated and showed an inverse correlation with LILRB1 expression. Treatment with recombinant LILRB1 protein significantly attenuated P. aeruginosa-induced neutrophil migration in the co-culture model, reducing the number of migrated cells from 4.43± 0.26× 104 to 2.75± 0.19× 104. Furthermore, LILRB1 protein suppressed NETosis and significantly decreased the concentrations of IL1β, IL6, and IL8 in the co-culture supernatants.
Conclusion: Our findings indicate that LILRB1 acts as a significant regulator of neutrophilic inflammation in drug-resistant P. aeruginosa-associated bronchiectasis. By dampening neutrophil migration, NETosis, and pro-inflammatory cytokine release, LILRB1 may represent a potential target for mitigating inflammation in this patient population.
Keywords: bronchiectasis, LILRB1, neutrophilic inflammation, HLA-G, Pseudomonas aeruginosa, drug-resistant
Introduction
Bronchiectasis is a recurrent purulent infection caused by various etiologies, leading to repeated damage and/or obstruction of the medium and small airways, resulting in structural damage to the bronchial walls and causing abnormal and persistent bronchial dilation.1 In the past, bronchiectasis did not receive much attention, leading to minimal investment.2 However, in the past decade, the global incidence of bronchiectasis has shown a sharp upward trend, currently ranking third in chronic airway diseases, just after chronic obstructive pulmonary disease (COPD) and asthma.3,4 Although there has been an academic call to increase attention to bronchiectasis, there remains a significant gap in our comprehensive understanding of the disease and the implementation of evidence-based practices and personalized medicine. Compared to progress made in diseases like cystic fibrosis, research in bronchiectasis is in urgent need of further depth.5 Furthermore, bronchiectasis is a complex, heterogeneous inflammatory chronic respiratory disease, with a large proportion (approximately 50% to 70%) of patients failing to establish a clear etiology despite comprehensive examination, referred to as “idiopathic bronchiectasis.” Even among patients with the same etiology, there is significant clinical heterogeneity.6 Inflammation and infection are key driving factors in the pathophysiology of bronchiectasis, with persistent airway inflammation being a characteristic feature. Bronchiectasis has diverse etiologies, with severe or recurrent respiratory infections being the most common. Among these infections, Pseudomonas aeruginosa (P. aeruginosa) infection and colonization are particularly prevalent, and numerous studies have confirmed their association with worsening lung function and increased mortality.7 The propensity of P. aeruginosa to develop drug resistance poses a major therapeutic challenge in bronchiectasis. Choi et al’s integrated analysis of host inflammation and pulmonary microbiome data revealed correlations between inflammatory molecular subtypes and the risk of bronchiectasis exacerbation.8 Exploring the mechanisms of inflammation and immune responses in drug-resistant P. aeruginosa-associated bronchiectasis may provide directions for the development of new therapeutic strategies.
Immunotherapy has become a research hotspot, particularly showing remarkable effects in cancer treatment.9,10 Domblides et al reported a case of completely reversible capillary bronchitis and bronchiectasis induced by immunotherapy, despite its pathophysiological mechanisms previously being considered irreversible. This provides strong support for the study of immune checkpoints in bronchiectasis.11
Leukocyte immunoglobulin-like receptor B1 (LILRB1) plays a key role in various diseases, particularly in immune responses to infections and is considered a promising target in the field of immunotherapy.12,13 Aberrant expression and function of LILRB1 are associated with a variety of pathological conditions, including immune insufficiency such as infections and malignancies, suggesting that LILRB1 may participate in host immune defense against infections, including bacterial infections.14,15 Research has indicated that the LILRB1-HLA-G axis is a potential immune therapeutic target in chronic infectious diseases such as tuberculosis.16 However, the role of LILRB1 in bronchiectasis has not yet been investigated, and its association with P. aeruginosa infection remains unstudied.
Many clinical studies have indicated that neutrophil extracellular traps (NETs) formation is associated with increased severity of chronic airway inflammatory diseases, higher frequency of acute exacerbations, and disease progression. Bronchiectasis is inherently a neutrophilic inflammatory disease, and its treatment has always been challenging due to the delicate balance between reducing inflammation and increasing infection risk through immunosuppression.17
This study was designed to address the critical knowledge gap regarding the role of LILRB1 in drug-resistant P. aeruginosa-associated bronchiectasis. We first detected the expression of LILRB1 in the peripheral blood of patients with this disease and analyzed its clinical correlations with Bronchiectasis Severity Index (BSI),18 Bronchiectasis Aetiology Comorbidity Index (BACI),19 E-FACED (exacerbations, forced expiratory volume in one second, age, colonization, extension, dyspnoea) score,20 peripheral blood cell counts, and sHLA-G levels (the canonical ligand of LILRB1). Furthermore, we isolated primary neutrophils from patients to explore the regulatory effects of LILRB1 activation on neutrophil migration, NETosis, and pro-inflammatory cytokine secretion through in vitro cell stimulation and Transwell co-culture experiments. Ultimately, this study seeks to provide preliminary experimental evidence for clarifying whether LILRB1 may serve as a novel biomarker for disease severity and a potential candidate target in drug-resistant P. aeruginosa-associated bronchiectasis, laying a foundational basis for the further development of LILRB1 agonist-based therapeutic strategies in future research.
Materials and Methods
From February 2025 to September 2025, bronchoalveolar lavage fluid (BALF) samples were collected from drug-resistant P. aeruginosa-associated bronchiectasis patients and controls who underwent bronchoscopy at the First Affiliated Hospital of Guangxi Medical University. All samples were obtained before any treatment for the quantification of sHLA-G, and peripheral blood samples were collected simultaneously for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis. Meanwhile, clinical data of patients were collected, including age, gender, Body Mass Index (BMI), smoking history, frequency of acute exacerbations, and comorbidities. The BSI, E-FACED score, and BACI were calculated accordingly.
The inclusion criteria were as follows:
- Diagnosed with bronchiectasis.
- Age over 18 years.
- P. aeruginosa infection was confirmed by next-generation sequencing or sputum culture.
- Clinical drug susceptibility testing confirmed resistance to three or more classes of antibiotics with distinct mechanisms of action.
The exclusion criteria were: presence of immune system diseases; comorbidity with acquired immunodeficiency syndrome or other autoimmune diseases; comorbidity with diseases that may affect the study results, such as lung malignancy, active pulmonary tuberculosis, COPD, and bronchial asthma; comorbidity with severe dysfunction of major systems (heart, brain, kidney, etc); and inability to cooperate in completing the designated study procedures. Control subjects were also required to meet the above exclusion criteria except for being non-bronchiectasis patients.
PCR Validation of LILRB1 Expression
Total RNA was extracted from peripheral blood samples using RNAiso Plus (Takara Bio Inc., Dalian, China) following the manufacturer’s instructions. Reverse transcription was performed using the gDNA Eraser (Takara Bio Inc., Dalian, China). RT-qPCR was carried out using TB Green® Premix Ex Taq™ II (Takara Bio Inc., Dalian, China). The NCBI online tool Primer-BLAST was used for primer design.21 GAPDH was selected as the internal reference gene to normalize the expression level of LILRB1. The primers used were as follows: LILRB1: 5’ GAGACCCAGGAGTACCGTCT -3’ and 5’ CATCAGAGCCACACTGCAGA -3’; GAPDH: 5’ GAGAAGGCTGGGGCTCATTT -3’ and 5’ GTCAAAGGTGGAGGAGTGGG -3’. LILRB1 mRNA expression was presented as relative expression normalized to the internal control GAPDH and calculated using the 2−ΔΔCt method. Thermocycling conditions were as follows: 95 °C for 30s, followed by 40 cycles of 95 °C for 5s and 60 °C for 30s.
Detection of sHLA-G Concentration in Patients with Bronchiectasis
BALF samples were centrifuged at 300×g for 10 min at 4 °C to remove cell debris, and the supernatants were stored at −80 °C. The concentration of sHLA-G in the BALF of drug-resistant P. aeruginosa-associated bronchiectasis and non-bronchiectasis controls was detected using an enzyme-linked immunosorbent assay (ELISA) kit (JONLNBIO, China). All experimental procedures were performed strictly in accordance with the kit instructions. The absorbance was measured at 450 nm using a microplate reader, and sHLA-G concentrations were calculated via the standard curve and dilution factor. Subsequently, statistical analysis was conducted to explore the correlations between sHLA-G concentration, LILRB1 expression levels, and the BSI score.
Cultivation of P. aeruginosa Strain and Preparation of Bacterial Suspension
Drug-resistant P. aeruginosa was isolated from BALF or sputum of patients. The P. aeruginosa strain derived from a single colony was inoculated onto Luria-Bertani (LB) agar medium, followed by overnight incubation at 37 °C with shaking at 150 rpm for 16 hours. After incubation, the bacterial culture was centrifuged to collect the bacterial pellet, which was then resuspended in sterile LB broth. The bacterial concentration was determined using the McFarland turbidity method, and the suspension was adjusted to a final concentration of 1×108 colony-forming units (CFU)/mL for subsequent experiments.
Isolation of Neutrophils
Neutrophils were isolated from the peripheral blood of drug-resistant P. aeruginosa-associated bronchiectasis patients using a human neutrophil isolation kit (TBD, Tianjin, China), following the manufacturer’s instructions. Briefly, peripheral blood and layered onto separation medium ratio of 2:1, then centrifuged at 550×g for 30 min at 24 °C. The neutrophil layer was collected, and residual erythrocytes were lysed with lysis buffer for 5 min at room temperature. The cells were washed twice with phosphate-buffered saline (PBS), and the final pellet was resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum to obtain a neutrophil suspension.
Detection of Neutrophil Purity by Diff-Quick Staining Approximately
5 μL of the neutrophil suspension was dropped onto one-third of the end of a clean glass slide. A smooth spreader slide was held at a 45° angle to contact the droplet, allowing the suspension to spread along the spreader. The spreader was then pushed to the other end of the glass slide to prepare a thin blood smear. First, 100 μL of Reagent A was added to the smear and incubated for 20–30 seconds. The Reagent A was washed off with washing solution, and the slide was gently shaken to dry. Next, 100 μL of Reagent B was added for another 20–30 seconds of staining. After staining, the dye solution was rinsed off from one end of the slide with running water. The stained smear was allowed to dry completely before microscopic examination to determine neutrophil purity.
Cell Treatment
The isolated neutrophils were seeded into 6-well plates and divided into four groups based on different interventions: 1. Control group: Only culture medium was added. 2. P. aeruginosa group: Treated with P. aeruginosa alone. 3. LILRB1 + P. aeruginosa group: Pre-treated with recombinant LILRB1 protein (5 μg/mL) for 1 hour, followed by P. aeruginosa treatment. 4. Bovine serum albumin (BSA) + P. aeruginosa group: Pre-treated with BSA for 1 hour, followed by P. aeruginosa treatment. After the 1-hour pre-treatment with LILRB1 or BSA in the LILRB1 + P. aeruginosa and BSA + P. aeruginosa groups, 200 μL of prepared P. aeruginosa bacterial solution was added to the P. aeruginosa, LILRB1 + P. aeruginosa, and BSA + P. aeruginosa groups. All four groups were then incubated in a 37 °C, 5% CO2 incubator for 4 hours.
Extraction and Quantification of NETs
After the stimulation, the culture supernatant was collected for inflammatory cytokine detection. NETs were extracted following the method described in Reference.22 Briefly, after incubation, the supernatant was carefully removed, and the NETs layer adherent to the bottom of the wells was rinsed with pre-cooled PBS and transferred to centrifuge tubes. The samples were centrifuged at 450×g for 5 minutes at 4°C to obtain a cell-free supernatant enriched with soluble NETs-double-stranded DNA (dsDNA). For the collection of insoluble NETs-dsDNA, the aforementioned supernatant was further centrifuged at 18,000×g for 10 minutes at 4°C, the resulting NETs pellet was resuspended in pre-cooled sterile PBS. NETs-dsDNA was quantified using the Quant-iT PicoGreen dsDNA Quantitation Kit (Yeasen, Shanghai, China). Fluorescence intensity was detected using a multi-mode microplate reader with an excitation wavelength of 480 nm and an emission wavelength of 520 nm.
Immunofluorescence Staining of NETs
Glass coverslips were placed in 24-well plates. Neutrophil suspension (prepared as described previously) was added, and plates were incubated for 30 min to allow adhesion. Cells were subjected to the same four-group intervention protocol as described in Cell Treatment and incubated at 37 °C with 5% CO2 for 4 h. After incubation, both types of samples were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 10% goat serum. Primary antibodies against myeloperoxidase (MPO, 1:400 dilution, Abcam, Cambridge, UK) and citrullinated histone H3 (CitH3, 1:400 dilution, Abcam, Cambridge, UK) were added, followed by overnight incubation at 4°C. The next day, primary antibodies were recovered, and corresponding secondary antibodies were added. Cell nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI), and the glass slides were inverted and mounted on slides containing anti-fluorescence quencher. Observations were performed using an immunofluorescence microscope.
Assessment of Neutrophil Migration in a Co-Culture Model
Human bronchial epithelial cell line BEAS-2B (Procell, Wuhan, China) were seeded in the lower chamber of a 24-well plate and cultured until full confluence was achieved. The monolayer was then incubated for 2 hours with either P. aeruginosa strain, diluted in serum-free RPMI-1640 medium at a multiplicity of infection of 10:1, or with serum-free RPMI-1640 medium alone, to establish either a P. aeruginosa-infected or a normal bronchial epithelial system, respectively. Following this incubation, the medium in all groups was replaced with fresh serum-free RPMI-1640 medium to commence the neutrophil Transwell migration assay.
To precisely delineate the sources of inflammatory mediators and evaluate the role of LILRB1, the experiment included the following six groups: Group A: No neutrophils in the upper chamber; BEAS-2B cells in the lower chamber without prior P. aeruginosa intervention. Group B: No neutrophils in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. Group C: Neutrophils placed in the upper chamber; serum-free RPMI-1640 medium only in the lower chamber. Group D: Neutrophils placed in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. Group E: Neutrophils pre-treated with BSA (5 μg/mL) for 1 hour placed in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. Group F: Neutrophils pre-treated with a recombinant human LILRB1 agonist (5 μg/mL) for 1 hour placed in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa.
After a 4-hour incubation, the supernatant in the lower chamber was carefully collected. Neutrophils that migrated to the lower chamber were stained with trypan blue and counted.
Detection of Inflammatory Cytokines
The culture supernatants from each of the aforementioned groups were collected. Concentrations of IL1β, IL6, and IL8 were measured using ELISA kits (Cloud-Clone Corp., China). All experimental procedures were performed according to the kit manufacturer. The absorbance was measured at 450 nm, and cytokine concentrations were calculated via the standard curve.
Statistical Methods
All data calculations and statistical analyses were performed using R software and GraphPad Prism 9.5. A t-test was used between two groups. For multiple comparisons, one-way analysis of variance was applied. The monotonic relationship between the two variables was assessed using Spearman correlation analysis. Data were presented as the mean ± standard error of the mean (SEM), and a P-value < 0.05 was considered statistically significant.
Results
LILRB1 Expression
A total of 50 samples were collected, including 34 drug-resistant P. aeruginosa-associated bronchiectasis patients and 16 healthy controls. All of bronchiectasis patients met the study inclusion criterion of P. aeruginosa resistance to three or more antibiotic classes with distinct mechanisms of action via clinical drug susceptibility testing. The RT-qPCR results showed that the expression level of LILRB1 in the bronchiectasis group was significantly lower than in the control group (P = 0.0384), with statistical significance (Figure 1A). Additionally, the receiver operating characteristic curve for the LILRB1 expression levels between the bronchiectasis and healthy groups showed an area under the curve (AUC) of 0.658, with a 95% Confidence Interval (CI) of 0.485–0.825 (Figure 1B).
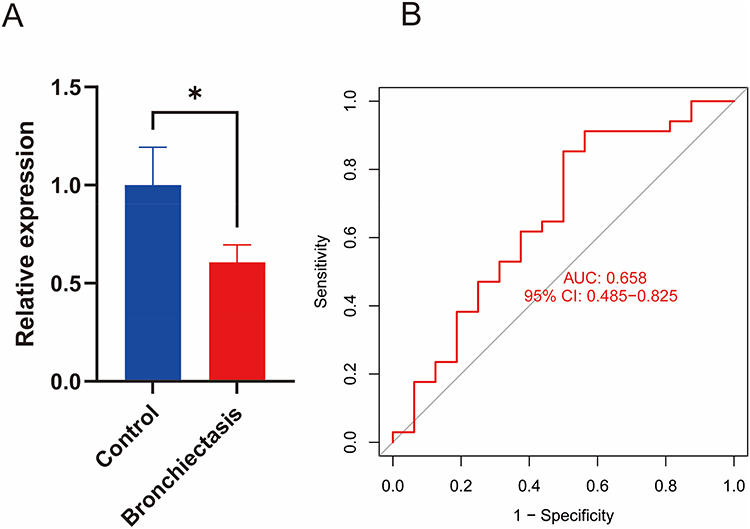 |
Figure 1 Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) detection of LILRB1 expression in peripheral blood. (A) RT-qPCR results of LILRB1 gene expression in peripheral blood of bronchiectasis and healthy control groups. (B) Area under the receiver operating characteristic curve (AUC) of LILRB1 expression in the bronchiectasis group vs the healthy control group. |
Clinical Correlation of LILRB1 Expression
Furthermore, we conducted a correlation analysis between the expression of LILRB1 and clinical factors. No statistically significant difference was observed in the groups categorized by age, gender, BMI, or smoking status (Figure 2A–D). The analysis revealed a significant difference in LILRB1 expression between the mild-moderate and severe BSI score groups (P = 0.0089), with an AUC of 0.761 and a 95% CI of 0.586–0.905 (Figure 3A and B). However, no statistically significant difference was observed in the groups categorized by BACI score or E-FACED score (Figure 3C and D).
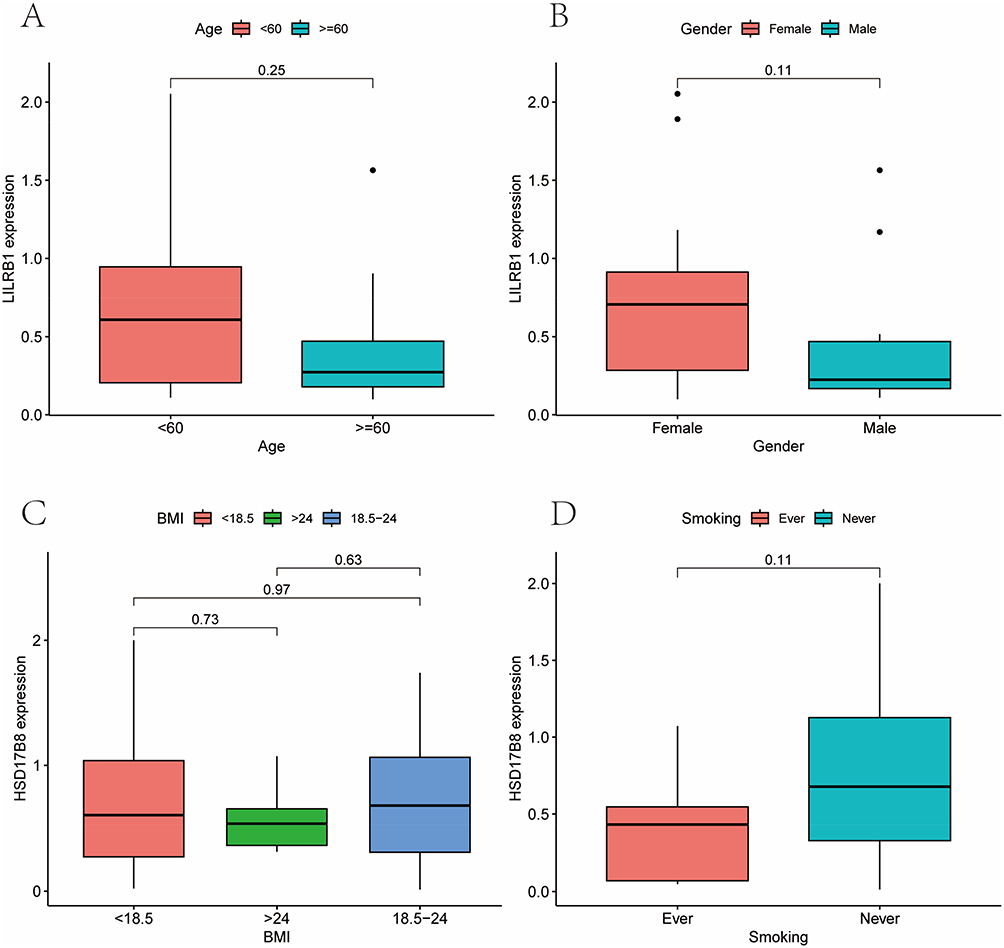 |
Figure 2 Clinical correlation analysis of LILRB1 in bronchiectasis group. The results showed that there were no statistically significant differences in LILRB1 expression with age (A), gender (B), Body Mass Index (BMI) (C), or smoking status (D). |
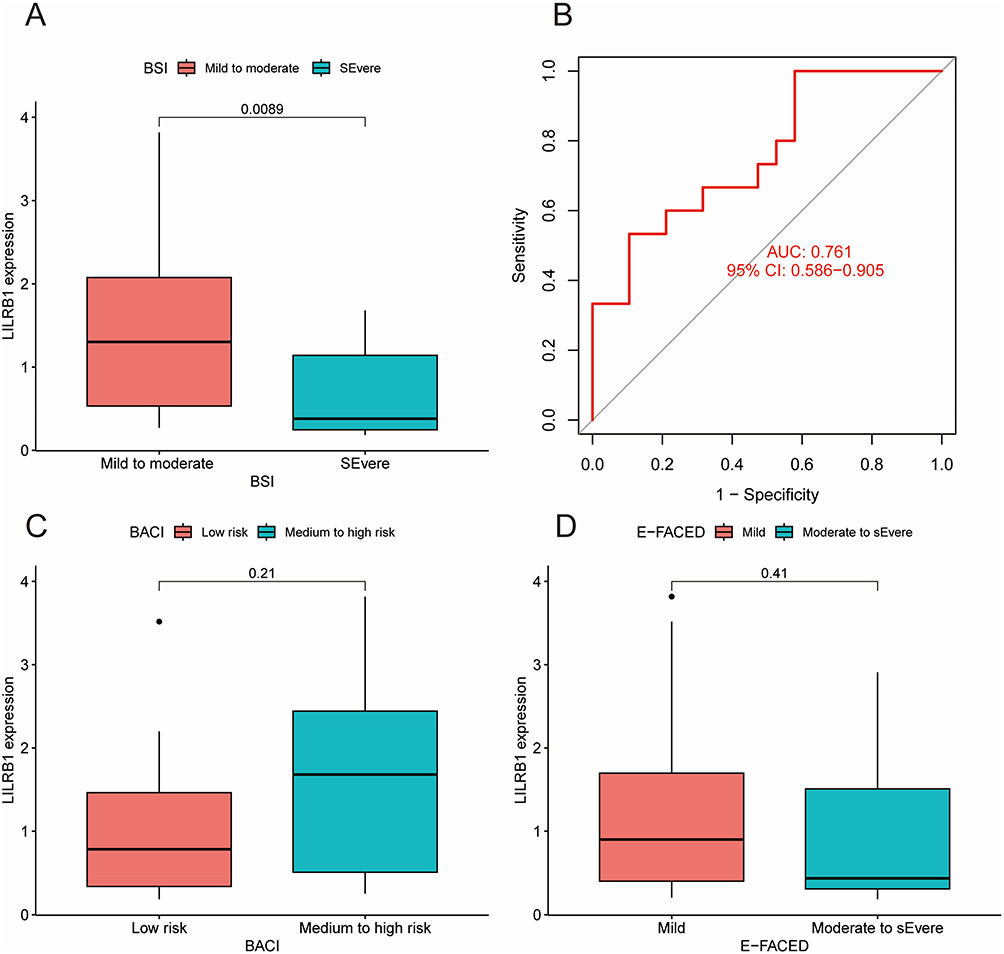 |
Figure 3 Association of LILRB1 with various scores in bronchiectasis group. (A) Correlation analysis of LILRB1 expression with Bronchiectasis Severity Index (BSI) in bronchiectasis patients. (B) AUC of LILRB1 expression and BSI in bronchiectasis patients. (C) Correlation analysis of LILRB1 expression with Bronchiectasis Aetiology Comorbidity Index (BACI) in bronchiectasis patients. (D) Correlation analysis of LILRB1 expression with E-FACED (exacerbations, forced expiratory volume in one second, age, colonization, extension, dyspnoea) score in bronchiectasis patients. |
Correlation of LILRB1 Expression with Peripheral Blood Cell Counts and Parameters
We also performed a correlation analysis between LILRB1 expression and various peripheral blood cell counts in drug-resistant P. aeruginosa-associated bronchiectasis patients. The results indicated a positive correlation between LILRB1 and the percentage of lymphocytes (r = 0.35, P = 0.045), and negative correlations with the absolute count of neutrophils (r = −0.35, P = 0.044), the percentage of neutrophils (r = −0.36, P = 0.036), and the white blood cell count (r = −0.36, P = 0.039) (Table 1 and Figure 4).
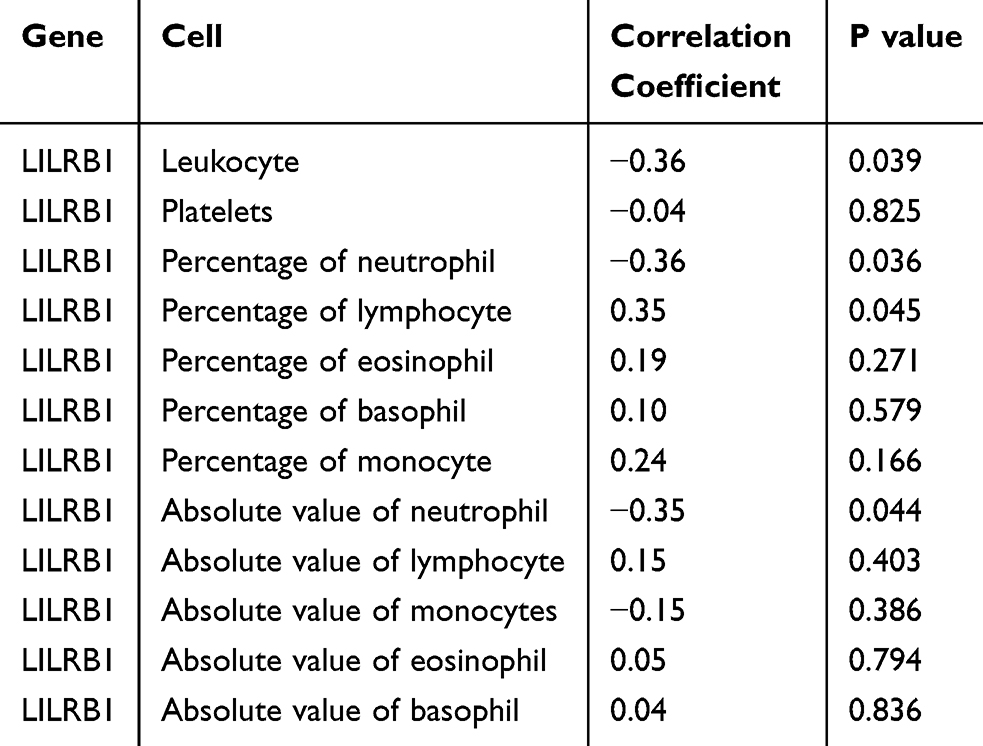 |
Table 1 Correlation of LILRB1 Expression with Peripheral Blood Cell Counts and Parameters |
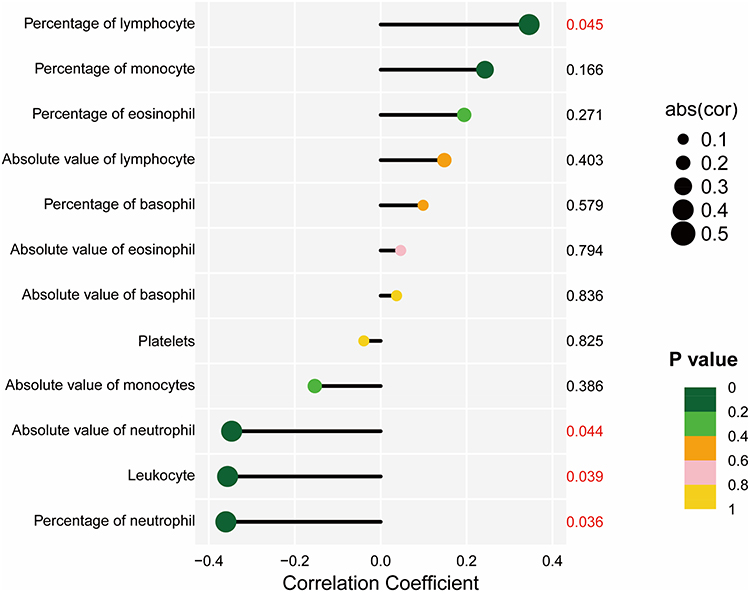 |
Figure 4 Correlation of LILRB1 expression with peripheral blood cell counts and parameters. |
Concentration of sHLA-G
Based on the aforementioned inclusion and exclusion criteria, we finally collected BALF samples from 50 drug-resistant P. aeruginosa-associated bronchiectasis patients and 30 control subjects for sHLA-G concentration quantification. In our results, the sHLA-G level in the bronchiectasis group was elevated compared with the control group (Figure 5A). sHLA-G concentration differed significantly among subgroups stratified by the BSI score (Figure 5B). Meanwhile, a negative correlation was found between sHLA-G concentration and LILRB1 expression levels (Figure 5C).
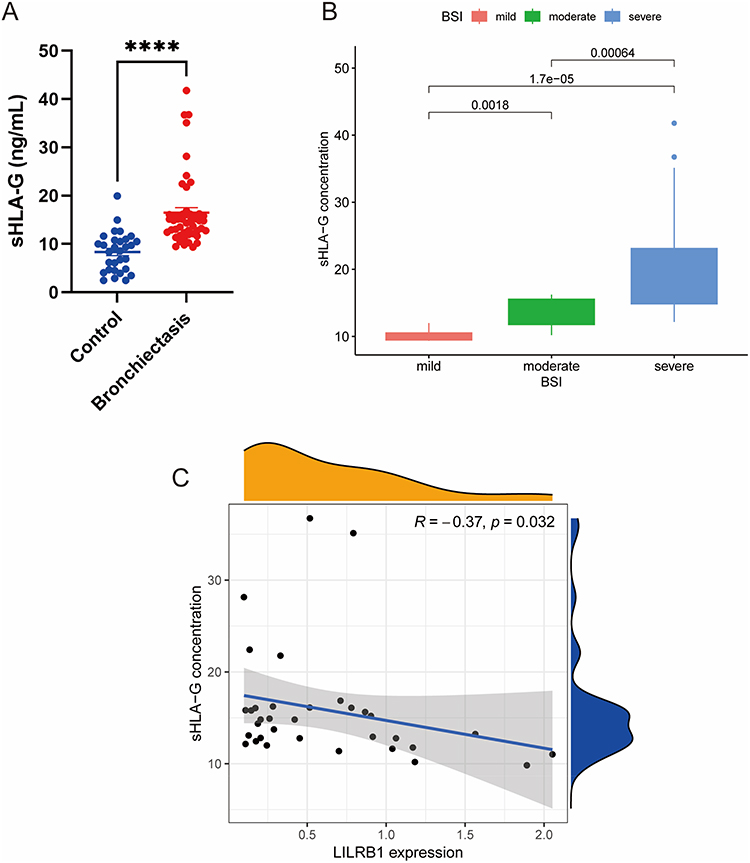 |
Figure 5 Analysis of soluble human leukocyte antigen-G (sHLA-G) concentration in bronchoalveolar lavage fluid (BALF) of patients with bronchiectasis. (A) Comparison of sHLA-G concentration between patients with bronchiectasis and controls. (B) Analysis of sHLA-G concentration of patients stratified by BSI score. (C) Correlation analysis between sHLA-G concentration and LILRB1 expression level. ****P < 0.0001. |
Isolation of Neutrophils
Neutrophils isolated by density gradient centrifugation were stained using the Diff-Quick method, then observed and imaged under a light microscope. Microscopic examination revealed the typical morphological characteristics of neutrophils: the cell nuclei were purple and lobulated. Purity analysis of the isolated neutrophils showed that their purity exceeded 90%, which met the experimental requirements (Figure 6).
 |
Figure 6 Detection of neutrophil purity by Diff-Quick staining approximately. |
Qualitative Observation and Quantification of NETs
In addition, immunofluorescence co-staining was used to observe NETs formation. The P. aeruginosa group and BSA + P. aeruginosa group exhibited enriched extracellular DNA filamentous connection structures, along with increased concentrations of MPO and CitH3 (P < 0.05). In contrast, in the LILRB1 + P. aeruginosa group, the extracellular DNA filamentous connection structures, as well as the concentrations of MPO and CitH3, were reduced compared with the P. aeruginosa and BSA + P. aeruginosa groups. Only a small amount of CitH3 and MPO was detected in the control group, with no formation of typical NETs structures (Figure 7A). Quantitatively, compared with the control group, the concentration of NETs-dsDNA was significantly increased in the P. aeruginosa group and BSA + P. aeruginosa group (P < 0.01), while it was decreased in the LILRB1 + P. aeruginosa group (Figure 7B).
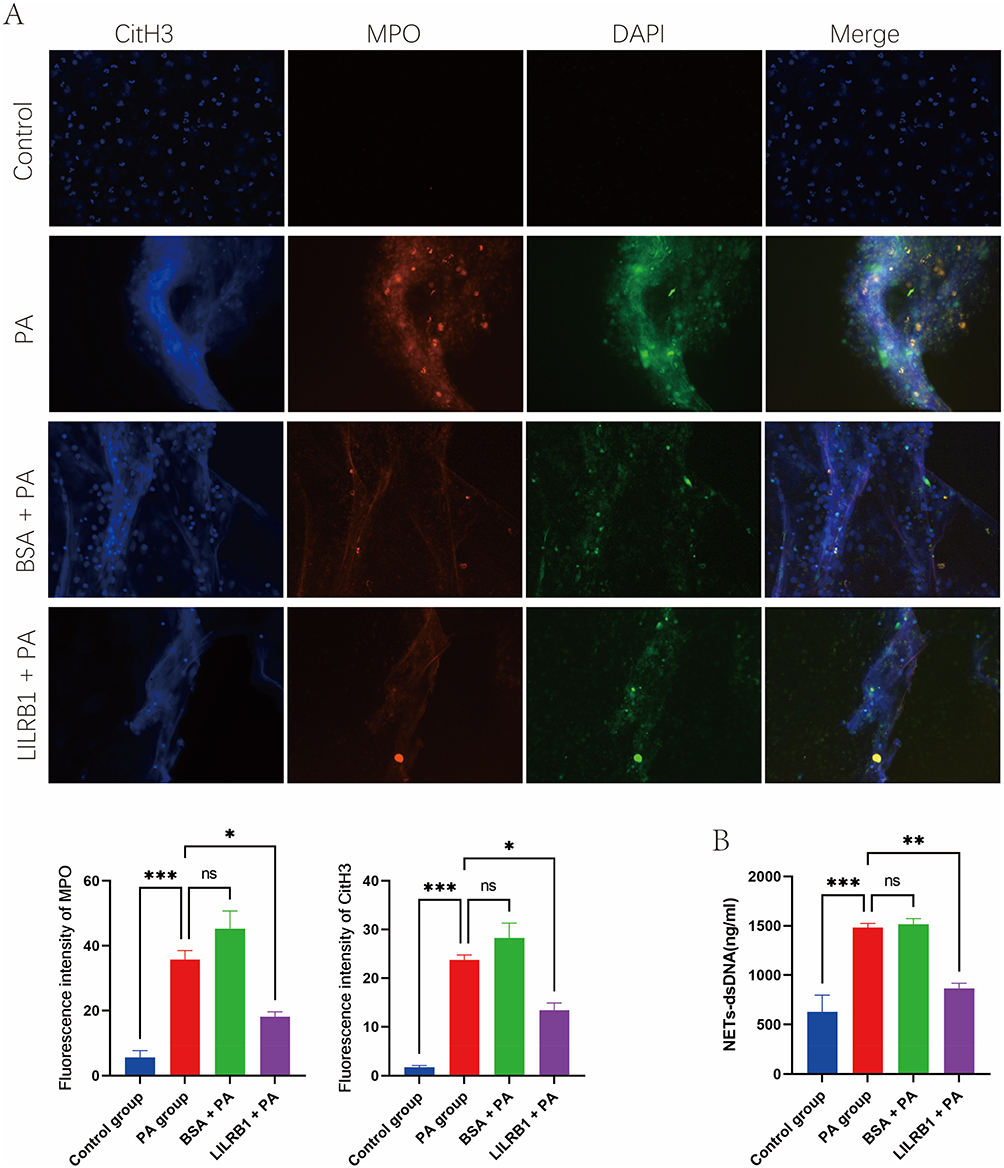 |
Figure 7 Qualitative and quantitative analysis of neutrophil extracellular traps (NETs) formation in neutrophils among different groups. (A) Immunofluorescence staining observation of NETs in neutrophils from different groups. (B) Comparison of NETs-dsDNA concentration in neutrophils among different. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: PA, Pseudomonas aeruginosa; BSA, bovine serum albumin; CitH3, citrullinated histone H3; MPO, myeloperoxidase; DAPI, 4’,6-diamidino-2-phenylindole; ns, not significant. |
Neutrophil Migration
As shown in Figure 8A, significant differences in neutrophil migration were observed among the experimental groups. Group C (basal migration control) exhibited a moderate level of migration (1.13 ± 0.15×104 cells), indicating the intrinsic migratory capacity of neutrophils. Group D (infected co-culture) showed a marked increase in migrated neutrophils compared to Group C (4.43 ± 0.26×104 cells, p < 0.001), demonstrating the potent chemotactic activity of P. aeruginosa-infected bronchial epithelial cells. Group E (BSA control) showed no significant difference from Group D (4.36 ± 0.18×104 cells, p > 0.05), effectively excluding non-specific effects of protein pretreatment on migration. Importantly, Group F (LILRB1 agonist-treated) displayed a substantial reduction in migrated neutrophils (2.75 ± 0.19×104 cells), which was significantly different from both Groups D and E (p < 0.001), indicating that LILRB1 activation effectively suppressed P. aeruginosa-induced neutrophil migration.
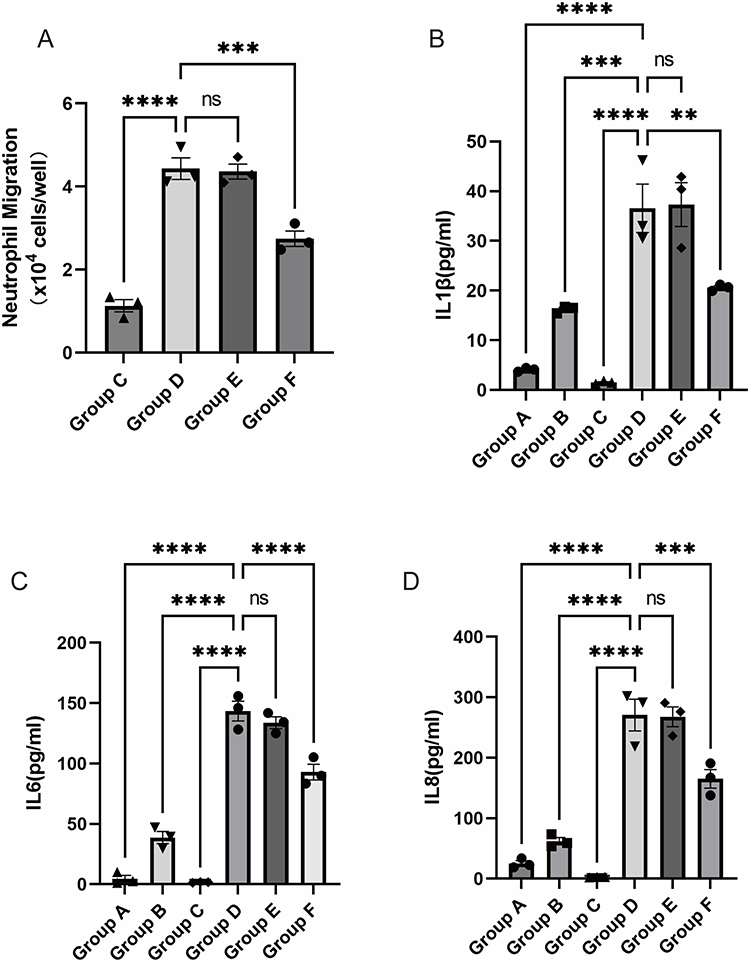 |
Figure 8 Comparison of neutrophil migration and inflammatory cytokine levels among different treatment groups. (A) Comparison of neutrophil migration. Comparison of IL1β (B), IL6 (C), and IL8 (D). Group A: No neutrophils in the upper chamber; BEAS-2B cells in the lower chamber without prior P. aeruginosa intervention. Group B: No neutrophils in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. Group C: Neutrophils placed in the upper chamber; serum-free RPMI-1640 medium only in the lower chamber. Group D: Neutrophils placed in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. Group E: Neutrophils pre-treated with BSA for 1 hour placed in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. Group F: Neutrophils pre-treated with a recombinant human LILRB1 agonist for 1 hour placed in the upper chamber; BEAS-2B cells in the lower chamber pre-treated with P. aeruginosa. **P < 0.01; ***P < 0.001; ****P < 0.0001. Abbreviation: ns, not significant. |
Inflammatory Cytokine
Quantitative analysis of three key inflammatory cytokines in the co-culture supernatants was performed. IL1β, a classical inflammasome activation marker, was dramatically elevated in Group D (36.58 ± 4.88 pg/mL), while Group B showed only a moderate level (16.23 ± 0.40 pg/mL). The IL1β level in Group F (20.61 ± 0.37 pg/mL) was significantly lower than in Groups D and E (37.31 ± 4.41 pg/mL, p < 0.001), suggesting that LILRB1 activation markedly reduced neutrophil-mediated IL1β production (Figure 8B). The IL6 level in Group D (143.4 ± 8.129 pg/mL) was significantly higher than in Group B (38.70 ± 5.011 pg/mL; p < 0.001). Group F showed a partial reduction in IL6 level (92.89 ± 6.479 pg/mL), which remained significantly higher than in Group B (p < 0.01), indicating that LILRB1 activation partially suppressed IL6 production (Figure 8C). IL8, a principal neutrophil chemotactic factor, reached its highest concentration in Group D (270.7 ± 26.19 pg/mL), significantly exceeding the level in Group B (epithelial infection alone, 62.22 ± 5.94 pg/mL; p < 0.001). The IL8 level in Group F (165.2 ± 15.32 pg/mL) was significantly lower than in Groups D and E (267.8 ± 16.41 pg/mL), demonstrating that LILRB1 activation specifically inhibited neutrophil-dependent IL8 production (Figure 8D).
Discussion
Despite the heterogeneity caused by various factors, bronchiectasis shares common features of airway remodeling and bronchial dilation. The initial stage of its pathogenesis is marked by various factors leading to bronchial obstruction or traction, damage to the mucociliary clearance function, and retention of airway secretions. This makes the respiratory tract more susceptible to pathogen infections and colonization, which in turn triggers chronic pulmonary inflammation. This leads to airway structural damage and wall remodeling, further impairing the clearance of airway secretions. This creates a vicious cycle that eventually results in permanent pathological bronchial dilation, a process referred to as the “vicious circle” of bronchiectasis.23 Notably, P. aeruginosa is one of the most commonly isolated bacteria in non-cystic fibrosis bronchiectasis patients, and numerous studies have confirmed its association with worsening lung function and increased mortality.7
LILRB1 is a cell surface receptor belonging to the immunoglobulin superfamily.23,24 As an immune-suppressive receptor, LILRB1 inhibits immune cell activation and immune responses by binding to MHC class I molecules.25–27 Consequently, inhibiting the MHC1/LILRB1 interaction can enhance immune responses, similar to the well-studied immune checkpoint PD-1/PD-L1, which has shown promising results in preclinical and clinical treatments for several cancer types.27
In our study, we observed a reduction in LILRB1 expression in the drug-resistant P. aeruginosa-associated bronchiectasis group. Furthermore, the level of sHLA-G—the canonical ligand of LILRB1—was significantly elevated in the BALF of drug-resistant P. aeruginosa-associated bronchiectasis patients and showed a negative correlation with LILRB1 expression. This finding suggests that the LILRB1–HLA-G axis may be involved in the immunopathological process of drug-resistant P. aeruginosa-associated bronchiectasis. Additionally, the reduction in LILRB1 expression was more pronounced in the group with severe BSI scores compared to the mild BSI group, thus, low LILRB1 expression in drug-resistant P. aeruginosa-associated bronchiectasis patients could suggest a worse prognosis. This may be due to the fact that low LILRB1 expression leads to a stronger immune response and higher levels of immune cell infiltration, which in turn promotes the development of chronic inflammation over time. This exacerbates airway structural damage, contributing to bronchial dilation and the progression of the disease. Many studies have shown that peripheral blood cell counts of certain immune cells can serve as alternative biomarkers for assessing the inflammatory status in COPD28–30 and asthma.31,32 In our study, we performed a correlation analysis between LILRB1 expression and various peripheral blood cell counts and markers. Our results indicated that LILRB1 expression was negatively correlated with white blood cell count, absolute neutrophil count, and neutrophil percentage, while it was positively correlated with lymphocyte percentage. As we know from previous sections, the pathophysiological hallmark of bronchiectasis involves chronic neutrophilic inflammation, with activated neutrophils playing a central role in the “vicious cycle” of lung damage.23 Neutrophils are innate immune phagocytes that play a crucial role in immune defense. They exert their effects mainly through three mechanisms: phagocytosis, degranulation, and the release of NETs. NETs are a complex, decondensed chromatin structure released by neutrophils, composed of antimicrobial histones, cytoplasmic proteins such as elastase, myeloperoxidase, and other granular proteins.23,33 Typically, when neutrophils are activated by pathogen stimuli, they release NETs to trap and neutralize bacteria, fungi, and viruses. However, excessive release of proteases, antimicrobial proteins, DNA, and histones can lead to tissue damage, impaired mucociliary clearance, impaired bacterial killing, and exacerbation of inflammation. This exacerbates disease progression and is associated with the risk of future deterioration of bronchiectasis, including severe exacerbations and declining lung function.34 On the other hand, lymphocytes have been more commonly reported in the literature as controllers of bronchial inflammation, playing a pathophysiological role in maintaining the balance between proteolytic and anti-proteolytic functions.35 Many studies have demonstrated that the neutrophil-to-lymphocyte ratio (NLR) in peripheral blood serves as a determinant of disease severity and prognosis in respiratory diseases. Martinez-García et al, in a study of 1369 patients, found that a higher NLR was associated with more severe bronchiectasis, worse quality of life, and more frequent pathogenic infections. Moreover, NLR showed better correlation with severity scores compared to other systemic inflammatory markers and was an independent predictor of event frequency and exacerbation severity.35 In our study, we found that LILRB1 expression was negatively correlated with neutrophils and NLR. We speculate that the decrease in LILRB1 expression may mediate an increase in neutrophils and NLR, which in turn leads to the onset and progression of bronchiectasis.
Based on these findings, to further investigate the relationship between LILRB1 and neutrophil-mediated inflammation, we conducted a series of in vitro functional experiments. The results demonstrated that treatment with a specific LILRB1 agonist significantly attenuated neutrophil migration toward the site of infection, reducing the number of migrated cells by approximately 38%. Concurrently, key markers of NETs, MPO and CitH3, showed a notable reduction, indicating that LILRB1 activation effectively suppresses the NETosis process. This discovery holds significant pathophysiological relevance, as excessive neutrophil activation and dysregulated NETosis have been established as central drivers of airway injury and remodeling in drug-resistant P. aeruginosa-associated bronchiectasis. Our experimental results provide evidence that LILRB1 may exert protective effects against airway inflammatory damage by negatively regulating neutrophil chemotaxis and NETs release.
Further mechanistic investigation revealed that LILRB1 activation plays a crucial role in modulating the inflammatory cytokine network, with particularly pronounced inhibitory effects on IL1β and IL8. The 44% decrease in IL1β levels, as a key product of inflammasome activation, suggests that LILRB1 may inhibit the maturation and release of IL1β by regulating the NLRP3 inflammasome pathway. Meanwhile, the 39% reduction in IL8 concentration, a major neutrophil chemoattractant, further confirms the central role of LILRB1 in suppressing neutrophil recruitment. These findings collectively demonstrate that LILRB1 modulates neutrophil-mediated inflammatory responses through multiple targets, providing new experimental evidence for understanding its protective mechanisms in drug-resistant P. aeruginosa-associated bronchiectasis.
Currently, preclinical studies and clinical trials on LILRB1 antagonists as a potential therapeutic target in cancer treatment are ongoing.36 Despite this progress in oncology, research on the role of LILRB1 in drug-resistant P. aeruginosa-associated bronchiectasis remains relatively sparse. Our findings provide evidence that LILRB1 expression is a previously unrecognized biomarker in the onset and progression of drug-resistant P. aeruginosa-associated bronchiectasis, and further studies are required to validate this potential role.
However, this study has certain limitations. First, the sample size was relatively limited, and the study was specifically designed to focus on the drug-resistant P. aeruginosa-associated bronchiectasis subtype—with strict inclusion criteria (clinical drug susceptibility testing confirmed resistance to three or more classes of antibiotics with distinct mechanisms of action). No other etiological subtypes of bronchiectasis were included, which may limit the generalizability of our findings to the broader bronchiectasis population. Second, the downstream molecular mechanisms underlying the LILRB1-mediated regulation of neutrophilic inflammation remain to be elucidated; further studies are needed to explore the specific signaling pathways involved in LILRB1-induced suppression of neutrophil migration, NETosis and inflammatory cytokine secretion. Additionally, this study only provided in vitro experimental evidence for the regulatory role of LILRB1, and the therapeutic efficacy of LILRB1 agonists needs to be further validated in in vivo animal models of drug-resistant P. aeruginosa-associated bronchiectasis. Future studies will expand the sample size to include multiple etiological subtypes of bronchiectasis and explore the aforementioned unresolved molecular and in vivo therapeutic questions.
Conclusions
In summary, this study found that LILRB1 expression was significantly decreased in the peripheral blood of patients with drug-resistant P. aeruginosa-associated bronchiectasis, and its low expression level was preliminarily correlated with disease severity scores, peripheral blood leukocyte counts, and sHLA-G levels, the canonical ligand of LILRB1. In vitro experimental results suggest that activation of LILRB1 may attenuate drug-resistant P. aeruginosa-induced neutrophil migration, NETs formation, and pro-inflammatory cytokine secretion. These preliminary observations indicate that LILRB1 may be involved in the regulation of neutrophilic inflammation in this disease and may serve as a candidate indicator for evaluating disease severity. This study provides basic experimental evidence and a preliminary foundation for future investigations into the role of LILRB1 in drug-resistant P. aeruginosa-associated bronchiectasis.
Abbreviations
COPD, chronic obstructive pulmonary disease; BSI, Bronchiectasis Severity Index; BACI, Bronchiectasis Aetiology Comorbidity Index; E-FACED, exacerbations, forced expiratory volume in one second, age, colonization, extension, dyspnoea; P. aeruginosa, Pseudomonas aeruginosa; LILRB1, leukocyte immunoglobulin-like receptor B1; sHLA-G, soluble human leukocyte antigen-G; BALF, bronchoalveolar lavage fluid; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay; BMI, body mass index; CFU, colony-forming unit; PBS, phosphate-buffered saline; BSA, bovine serum albumin; NETs, neutrophil extracellular traps; MPO, myeloperoxidase; CitH3, citrullinated histone H3; dsDNA, double-stranded DNA; DAPI, 4’,6-diamidino-2-phenylindole; NLR, neutrophil-to-lymphocyte ratio; MHC, major histocompatibility complex; LB, Luria-Bertani; SEM, standard error of the mean; AUC, area under the curve; CI, confidence interval.
Data Sharing Statement
Data are provided within the manuscript or available from the corresponding authors.
Ethics Approval and Consent to Participate
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (Approval Number: 2025-E0931). All participants provided informed consent before taking part in the study.
Consent for Publication
All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (grant number 82560016); High-Incidence Diseases Research of Guangxi Natural Science Foundation (grant number 2023GXNSFBA026146); the Health and Family Planning Commission of Guangxi Zhuang Autonomous Region, Self-funded Projects (grant number Z-A20240492, Z-A20240515); the Key Research Program of Guangxi Science and Technology Department (grant number AB21196010); Bethune Charitable Foundation (grant number BCF-QYWL-HX-2025-12); First-class Discipline Innovation-driven Talent Program of Guangxi Medical University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Koser U, Hill A. What’s new in the management of adult bronchiectasis? F1000Res. 2017;6:527. doi:10.12688/f1000research.10613.1
2. Wang N, Qu JM, Xu JF. Bronchiectasis management in China, what we can learn from European Respiratory Society Guidelines. Chinese Med J-Peking. 2018;131(16):1891–16. doi:10.4103/0366-6999.238134
3. Flume PA, Chalmers JD, Olivier KN. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet. 2018;392(10150):880–890. doi:10.1016/S0140-6736(18)31767-7
4. Zhang XX, Chen ZM, Zf H, Guan WJ. Advances in pharmacotherapy for bronchiectasis in adults. Expert Opin Pharmaco. 2023;24(9):1075–1089. doi:10.1080/14656566.2023.2210763
5. Chalmers JD, Chang AB, Chotirmall SH, Dhar R, McShane PJ. Bronchiectasis. Nat Rev Dis Primers. 2018;4(1):45. doi:10.1038/s41572-018-0042-3
6. Lin JL, Xu JF, Qu JM. Bronchiectasis in China. Ann Am Thorac Soc. 2016;13(5):609–616. doi:10.1513/AnnalsATS.201511-740PS
7. Reynolds D, Kollef M. The epidemiology and pathogenesis and treatment of pseudomonas aeruginosa infections: an update. Drugs. 2021;81(18):2117–2131. doi:10.1007/s40265-021-01635-6
8. Choi H, Ryu S, Keir HR, et al. Inflammatory molecular endotypes in bronchiectasis: a European Multicenter Cohort Study. Am J Resp Crit Care. 2023;208(11):1166–1176. doi:10.1164/rccm.202303-0499OC
9. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–287. doi:10.1038/nrc.2016.36
10. Micevic G, Bosenberg MW, Yan Q. The crossroads of cancer epigenetics and immune checkpoint therapy. Clin Cancer Res. 2023;29(7):1173–1182. doi:10.1158/1078-0432.CCR-22-0784
11. Domblides M, Geier M, Decroisette C, Descourt R. Durvalumab-induced lesions of bronchiolitis and fully reversible bronchiectasis in a patient with non-small cell lung cancer: a case report. Thorac Cancer. 2021;12(8):1240–1243. doi:10.1111/1759-7714.13862
12. Barkal AA, Weiskopf K, Kao KS, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19(1):76–84. doi:10.1038/s41590-017-0004-z
13. Zeller T, Münnich IA, Windisch R, et al. Perspectives of targeting LILRB1 in innate and adaptive immune checkpoint therapy of cancer. Front Immunol. 2023;14:1240275. doi:10.3389/fimmu.2023.1240275
14. Abdallah F, Coindre S, Gardet M, et al. Leukocyte immunoglobulin-like receptors in regulating the immune response in infectious diseases: a window of opportunity to pathogen persistence and a sound target in therapeutics. Front Immunol. 2021;12:717998. doi:10.3389/fimmu.2021.717998
15. Redondo-Garcia S, Barritt C, Papagregoriou C, et al. Human leukocyte immunoglobulin-like receptors in health and disease. Front Immunol. 2023;14:1282874. doi:10.3389/fimmu.2023.1282874
16. Wang J, Chai Q, Lei Z, et al. LILRB1-HLA-G axis defines a checkpoint driving natural killer cell exhaustion in tuberculosis. EMBO Mol Med. 2024;16(8):1755–1790. doi:10.1038/s44321-024-00106-1
17. Keir HR, Chalmers JD. Neutrophil extracellular traps in chronic lung disease: implications for pathogenesis and therapy. Eur Respir Rev. 2022;31(163):210241. doi:10.1183/16000617.0241-2021
18. Chalmers JD, Goeminne P, Aliberti S, et al. The bronchiectasis severity index. An international derivation and validation study. Am J Resp Crit Care. 2014;189(5):576–585. doi:10.1164/rccm.201309-1575OC
19. McDonnell MJ, Aliberti S, Goeminne PC, et al. Comorbidities and the risk of mortality in patients with bronchiectasis: an international multicentre cohort study. Lancet Resp Med. 2016;4(12):969–979. doi:10.1016/S2213-2600(16)30320-4
20. Martinez-Garcia MA, Athanazio RA, Girón R, et al. Predicting high risk of exacerbations in bronchiectasis: the E-FACED score. Int J Chronic Obstr. 2017;12:275–284. doi:10.2147/COPD.S121943
21. Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 2012;13(1):134. doi:10.1186/1471-2105-13-134
22. Najmeh S, Cools-Lartigue J, Giannias B, Spicer J, Ferri LE. Simplified human neutrophil extracellular traps (NETs) isolation and handling. J Vis Exp. 2015;(98). doi:10.3791/52687
23. Chen ZG, Li YY, Wang ZN, et al. Aberrant epithelial remodeling with impairment of cilia architecture in non-cystic fibrosis bronchiectasis. J Thorac Dis. 2018;10(3):1753–1764. doi:10.21037/jtd.2018.02.13
24. Banham AH, Colonna M, Cella M, et al. Identification of the CD85 antigen as ILT2, an inhibitory MHC class I receptor of the immunoglobulin superfamily. J Leukocyte Biol. 1999;65(6):841–845. doi:10.1002/jlb.65.6.841
25. Lamar DL, Weyand CM, Goronzy JJ. Promoter choice and translational repression determine cell type-specific cell surface density of the inhibitory receptor CD85j expressed on different hematopoietic lineages. Blood. 2010;115(16):3278–3286. doi:10.1182/blood-2009-09-243493
26. Zhao J, Zhong S, Niu X, Jiang J, Zhang R, Li Q. The MHC class I-LILRB1 signalling axis as a promising target in cancer therapy. Scand J Immunol. 2019;90(5):e12804. doi:10.1111/sji.12804
27. Hu Z, Zhang Q, He Z, Jia X, Zhang W, Cao X. MHC1/LILRB1 axis as an innate immune checkpoint for cancer therapy. Front Immunol. 2024;15:1421092. doi:10.3389/fimmu.2024.1421092
28. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. 2021;76(2):188–195. doi:10.1136/thoraxjnl-2020-215167
29. Zinellu A, Zinellu E, Mangoni AA, et al. Clinical significance of the neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio in acute exacerbations of COPD: present and future. Eur Respir Rev. 2022;31(166):220095. doi:10.1183/16000617.0095-2022
30. Lan CC, Su WL, Yang MC, Chen SY, Wu YK. Predictive role of neutrophil-percentage-to-albumin, neutrophil-to-lymphocyte and eosinophil-to-lymphocyte ratios for mortality in patients with COPD: evidence from NHANES 2011–2018. Respirology. 2023;28(12):1136–1146. doi:10.1111/resp.14589
31. Agarwal S, Jat KR. Peripheral blood counts in asthma control: is it relevant? Indian J Pediatr. 2023;90(6):535–536. doi:10.1007/s12098-023-04522-y
32. Ke J, Qiu F, Fan W, Wei S. Associations of complete blood cell count-derived inflammatory biomarkers with asthma and mortality in adults: a population-based study. Front Immunol. 2023;14:1205687. doi:10.3389/fimmu.2023.1205687
33. Keir HR, Shoemark A, Dicker AJ, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Resp Med. 2021;9(8):873–884. doi:10.1016/S2213-2600(20)30504-X
34. Chalmers JD, Moffitt KL, Suarez-Cuartin G, et al. Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am J Resp Crit Care. 2017;195(10):1384–1393. doi:10.1164/rccm.201605-1027OC
35. Martinez-García MÁ, Olveira C, Girón R, et al. Peripheral neutrophil-to-lymphocyte ratio in bronchiectasis: a marker of disease severity. Biomolecules. 2022;12(10):1399. doi:10.3390/biom12101399
36. Mandel I, Haves ZD, Goldshtein I, et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J Immunother Cancer. 2022;10(9):e004859. doi:10.1136/jitc-2022-004859
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Tackling Neutrophilic Inflammation in Bronchiectasis: From Macrolides to Cathepsin C Inhibitors
Gramegna A, Premuda C, Putti G, Piedepalumbo FV, Misuraca S, Ori M, Nigro M, Simonetta E, Aliberti S, Blasi F
Journal of Inflammation Research 2026, 19:558745
Published Date: 11 April 2026