Back to Journals » Journal of Inflammation Research » Volume 16
Leptin Induces MMP-1 Expression Through the RhoA/ERK1/2/NF-κB Axis in Human Intervertebral Disc Cartilage Endplate-Derived Stem Cells
Authors Hua KF ![]() , Li LH, Yu HC, Wong WT, Hsu HT
, Li LH, Yu HC, Wong WT, Hsu HT
Received 17 July 2023
Accepted for publication 31 October 2023
Published 15 November 2023 Volume 2023:16 Pages 5235—5248
DOI https://doi.org/10.2147/JIR.S431026
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Kuo-Feng Hua,1,2,* Lan-Hui Li,3,* Hsin-Chiao Yu,4 Wei-Ting Wong,1 Hsien-Ta Hsu4,5
1Department of Biotechnology and Animal Science, National Ilan University, Yilan, 26047, Taiwan; 2Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, 404333, Taiwan; 3Department of Laboratory Medicine, Linsen, Chinese Medicine and Kunming Branch, Taipei City Hospital, Taipei, 108, Taiwan; 4Division of Neurosurgery, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City, 231, Taiwan; 5School of Medicine, Buddhist Tzu Chi University, Hualien, 970, Taiwan
*These authors contributed equally to this work
Correspondence: Hsien-Ta Hsu, Division of Neurosurgery, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, No. 289, Jianguo Road, Xindian Dist, New Taipei City, 231016, Taiwan, Tel +866-2-6628-9779, Email [email protected]
Purpose: Intervertebral disc (IVD) degeneration, associated with aging, may cause low back pain and disability, with obesity as a significant risk factor. In a prior study, we found a positive correlation between IVD degeneration and levels of matrix metalloproteinase-1 (MMP-1) and leptin. Yet, the interaction between MMP-1 and leptin in IVD degeneration is unclear. Our research seeks to explore leptin’s influence on MMP-1 expression and the underlying mechanisms in human intervertebral disc cartilage endplate-derived stem cells, specifically SV40 cells.
Methods: The mRNA and protein expression in leptin-stimulated SV40 cells were assessed using RT-real-time PCR and Western blotting or ELISA, respectively. We examined leptin-mediated RhoA activation through a GTP-bound RhoA pull-down assay. Furthermore, the phosphorylation levels of mitogen-activated protein kinases and AKT in leptin-stimulated SV40 cells were analyzed using Western blotting. The activation of NF-κB by leptin was investigated by assessing phosphorylation of IKKα/β, IκBα, and NF-κB p65, along with the nuclear translocation of NF-κB p65. To understand the underlying mechanism behind leptin-mediated MMP-1 expression, we employed specific inhibitors.
Results: Leptin triggered the mRNA and protein expression of MMP-1 in SV40 cells. In-depth mechanistic investigations uncovered that leptin heightened RhoA activity, promoted ERK1/2 phosphorylation, and increased NF-κB activity. However, leptin did not induce phosphorylation of JNK1/2, p38, or AKT. When we inhibited RhoA, ERK1/2, and NF-κB, it resulted in a decrease in MMP-1 expression. Conversely, inhibition of reactive oxygen species and NADPH oxidase did not yield the same outcome. Additionally, inhibiting RhoA or ERK1/2 led to a reduction in leptin-induced NF-κB activation. Moreover, inhibiting RhoA also decreased leptin-mediated ERK1/2 phosphorylation.
Conclusion: These results indicated that leptin induced MMP-1 expression in SV40 cells through the RhoA/ERK1/2/NF-κB axis. This study provided the pathogenic role of leptin and suggested the potential therapeutic target for IVD degeneration.
Keywords: intervertebral disc degeneration, leptin, matrix metalloproteinase, intervertebral disc cartilage endplate-derived stem cells, RhoA, ERK1/2
Graphical Abstract:

Introduction
Intervertebral disc (IVD) degeneration is believed to be an age-related process resulting from the depletion of proteoglycans in the central core of the vertebral disc. This depletion ultimately reduces the disc’s ability to withstand compressive forces.1 This condition is commonly observed in the cervical and lumbar spine, especially among the elderly population. Patients suffering from IVD degeneration often experience low back pain, which can lead to disability.2 Although the pathophysiology of IVD degeneration is intricate and not yet fully understood, several risk factors have been identified as significant contributors to the process. These factors include aging,3 genetics,4 smoking,5 and physically demanding occupational activities.6 Furthermore, mounting evidence suggests that IVD degeneration is associated with profound disruptions in metabolic balance, such as obesity and diabetes.7,8
Obesity stands as a global health crisis primarily stemming from excessive food consumption and a sedentary lifestyle, impacting over 600 million individuals across the globe.9 Reports have indicated a substantial and direct correlation between obesity and the severity of IVD degeneration.10–12 Moreover, obesity is a recognized pivotal risk factor that triggers the onset of type 2 diabetes mellitus through the induction of insulin resistance.13 IVD degeneration is further exacerbated by diabetes due to the creation of pathogenic conditions. For instance, elevated blood glucose levels prompt the apoptosis of cartilaginous end-plate cells by elevating the production of reactive oxygen species (ROS).14 Additionally, advanced glycation end-products stimulate matrix degradation by upregulating the expression of metalloproteinases in nucleus pulposus cells.15 Adipose tissue comprises adipocytes, fibroblasts, endothelial cells, and various types of immune cells, functioning not only as an energy reservoir but also as an endocrine organ. In the context of obesity and diabetes, this adipose tissue becomes an overproducer of adipokines and pro-inflammatory cytokines by adipocytes and immune cells, respectively.16 Adipokines constitute a class of cytokine-like hormones, including leptin, adiponectin, resistin, interleukin (IL)-6, and tissue necrosis factor, while pro-inflammatory cytokines such as tumour necrosis factor α (TNF-α) and IL-1β contribute to the chronic low-grade inflammation observed in obese and diabetic individuals.17,18 While it is hypothesised that the pathogenic process of disc degeneration is significantly influenced by chronic low-grade inflammation driven by pro-inflammatory cytokines and adipokines released from adipose tissue and immune cells of obese and diabetic individuals, the precise mechanisms linking these factors remain elusive.18–20
Matrix metalloproteinases (MMPs) form a family of extracellular proteinases responsible for breaking down collagens and aggrecan, which are implicated in the development of various diseases.21 It has been established that the expression levels of MMPs in degenerative IVD tissue significantly exceed those in normal IVD tissue.22,23 Leptin, a 16 kDa non-glycosylated adipokine derived from adipose tissue and encoded by the obese (ob) gene, plays a crucial role in maintaining energy balance and also holds significance in human physiology and pathophysiology.24,25 In a previous study, we demonstrated a positive correlation between the histological degeneration score of IVD degeneration and immunohistochemical scores for leptin and MMP-1.26 Leptin has been shown to enhance MMPs expression in bovine intervertebral disc cells and human osteoarthritic cartilage.27,28 However, the impact of leptin on the intracellular signaling of human intervertebral disc cartilage endplate-derived stem cells, which governs MMP-1 expression, remains unclear. In this study, we elucidate that leptin triggers MMP-1 expression through the RhoA/ERK1/2/NF-κB pathway in human intervertebral disc cartilage endplate-derived stem cells.
Materials and Methods
Chemicals and Reagents
Recombinant human leptin protein (398-LP) was obtained from R&D Systems (Minneapolis, MN). The ELISA kit for human MMP-1 and enhanced chemiluminescence (ECL) substrates were procured from Thermo Fisher Scientific (Waltham, MA). The Coomassie stain kit (iBlue) was purchased from GeneDireX (Taoyuan, Taiwan). We acquired the antibody against human MMP-1 from GeneTex (Irvine, CA). Antibodies against phospho-p38 (T180/Y182), phospho-JNK1/2/3 (T183/Y185), JNK1/2/3, phospho-AKT (T308), AKT, ERK1/2, and phospho-NF-κB p65 (Ser536) were sourced from Taiclone (Taipei, Taiwan). Antibodies against phospho-ERK1/2, phospho-IKKα/β, phospho-IκBα, p38, NF-κB p65, and GAPDH were obtained from Cell Signaling Technology (Danvers, MA). We acquired Nuclear Matrix Protein p84 from ABclonal (Boston, USA). Prigrow IV medium (TM004) and fetal bovine serum were purchased from Applied Biological Materials (BC, Canada). Rho Assay Reagent (Rhotekin RBD, agarose) and Amicon® Ultra-4 Centrifugal Filter Unit (UFC801024) were procured from Merck (Taipei, Taiwan). The Nuclear/Cytosol Fractionation Kit was purchased from BioVision (Milpitas, CA). N-acetyl-l-cysteine (NAC) and diphenyleneiodonium (DPI) were sourced from Sigma-Aldrich (St. Louis, MO).
Cell Culture
The human intervertebral disc cartilage endplate-derived stem cell line (SV40 cells) was procured from Applied Biological Materials (BC, Canada). SV40 cells were cultivated and subcultured every 2–3 days in Prigrow IV medium, supplemented with 10% heat-inactivated FBS, and maintained at 37 °C in a 5% CO2 incubator.
Induction of MMP-1 by Leptin
SV40 cells were exposed to 100 ng/mL leptin for 6–24 hours or a vehicle for 24 hours. Protein expression levels of MMP-1 in the supernatants or cell lysate were assessed using Western blotting and ELISA. Furthermore, SV40 cells were treated with 100 ng/mL leptin for 2–24 hours or a vehicle for 24 hours. The mRNA expression levels of MMP-1 were examined through reverse transcription and real-time polymerase chain reaction (RT-real-time PCR). To explore the impact of inhibitors on leptin-induced MMP-1 expression, SV40 cells were pre-incubated with the inhibitor or a vehicle for 0.5 hours and then stimulated with 100 ng/mL leptin for 2 hours (for mRNA expression) or 24 hours (for protein expression). Subsequently, both mRNA and protein expression levels of MMP-1 were analyzed using RT-real-time PCR and Western blotting, respectively.
Activation of RhoA by Leptin
SV40 cells were exposed to 100 ng/mL leptin for 15–60 minutes or a vehicle for 60 minutes. The levels of GTP-bound RhoA were determined using the Rho Assay Reagent, following the manufacturer’s instructions. In a nutshell, 0.5 mL of cell lysates was mixed with 20 µg of Rho Assay Reagent (Rhotekin RBD, agarose) and incubated at 4 °C for 45 minutes with gentle shaking. GTP-bound RhoA was isolated through centrifugation for 10 seconds at 14,000 x g at 4 °C. The supernatant was discarded, and 500 µL of wash buffer was added. This centrifugation and washing procedure was repeated three times. After removing the supernatant during the final wash, 40 µL of 2X Laemmli buffer and 2 µL of 1M dithiothreitol were introduced and heated in boiling water for 5 minutes. The mixture was centrifuged for 5 minutes at 14,000 x g at 4 °C, and 20 µL of the supernatant was loaded onto SDS-PAGE for Western blotting using the RhoA antibody.
Activation of Mitogen-Activated Protein Kinases (MAPKs) and NF-κB by Leptin
SV40 cells were treated with 100 ng/mL leptin for 15–60 minutes or a vehicle for 60 minutes. We analyzed the phosphorylation levels of ERK1/2, JNK1/2/3, p38, IKKα/β, IκBα, and NF-κB p65 using Western blotting. To assess NF-κB p65 nuclear translocation, SV40 cells were exposed to 100 ng/mL leptin for 2–6 hours or a vehicle for 6 hours. Nuclear and cytosolic fractionation was performed following the manufacturer’s instructions. To investigate the impact of inhibitors on leptin-induced phosphorylation of ERK1/2, IKKα/β, and IκBα, SV40 cells were pre-incubated with the inhibitor or a vehicle for 0.5 hours and then stimulated with 100 ng/mL leptin for 30 minutes. The phosphorylation levels of ERK1/2, IKKα/β, and IκBα were analyzed using Western blotting.
ELISA and Western Blotting
ELISA was conducted according to the manufacturer’s instructions as described previously. Western blotting was performed as described previously.29
RT and Quantitative Real-Time PCR Analysis for MMP-1
SV40 cells were exposed to 100 ng/mL leptin for 2–24 hours, or they were pre-incubated with an inhibitor for 0.5 hours before being stimulated with 100 ng/mL leptin for 2 hours. RNA extracted from SV40 cells was reverse-transcribed prior to quantitative PCR analysis. Human MMP-1 mRNA expression data were presented as relative expression levels normalized to those of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The following primers were employed: MMP-1, forward: 5’-ATGCTTTTCAACCAGGCCCA-3’; MMP-1, reverse: 5’-AGTCCAAGAGAATGGCCGAG-3’; GAPDH, forward: 5’-TTCCAGGAGCGAGATCCCT-3’; GAPDH, reverse: 5’-CACCCATGACGAACATGGG-3’.
Statistical Analysis
Statistical analysis involved two-tailed t-tests for two groups and ANOVA with Dunnett’s multiple comparison test for three or more groups. The error bars depict the standard deviation from three distinct experiments. The symbols *, **and *** correspond to p-values of < 0.05, < 0.01, and < 0.001, respectively.
Results
Leptin Induces MMP-1 Expression in SV40 Cells
To investigate whether leptin triggers MMP-1 expression, SV40 cells were exposed to leptin for 6–24 hours. We observed that intracellular MMP-1 levels increased between 6 and 12 hours, gradually returning to the baseline by the 24-hour mark (Figure 1A). Moreover, leptin induced the time-dependent release of MMP-1 into the supernatant (Figure 1B). The induction of MMP-1 by leptin was further validated through the detection of MMP-1 in the supernatant using ELISA (Figure 1C). Additionally, the mRNA levels of MMP-1 were elevated by leptin (Figure 1D). These findings demonstrate that leptin prompts MMP-1 expression at the transcriptional level and stimulates the extracellular release of MMP-1 in SV40 cells.
 |
Figure 1 Effect of leptin on MMP-1 expression. (A–C) SV40 cells were exposed to 100 ng/mL of leptin for 6–24 hours. Subsequently, the MMP-1 protein expression in both cell lysates (A) and supernatants (B) was assessed through Western blotting. Additionally, (C) the MMP-1 protein expression in supernatants was quantified using ELISA. (D) In a separate experiment, SV40 cells were incubated with 100 ng/mL leptin for 2–24 hours, and the MMP-1 mRNA expression was determined via RT-real-time PCR. Data from the ELISA and RT-real-time PCR are presented as means ± SD from three distinct experiments. Images obtained from Western blotting are representative of individual experiments. The significance of the findings is denoted by ***p < 0.001 when compared to control cells. |
Leptin Induces MMP-1 Expression Through RhoA-Dependent Pathways
Leptin triggers a reorganization of the cytoskeleton in chondrocytes and nucleus pulposus cells by engaging RhoA-dependent signaling pathways, as previously shown.30,31 These findings prompted us to investigate whether RhoA plays a role in MMP-1 expression in leptin-stimulated SV40 cells. To evaluate the impact of leptin on RhoA activity, we conducted a pull-down assay to measure the levels of GTP-bound RhoA (RhoA-GTP). Our results revealed a significant increase in RhoA-GTP levels in SV40 cells at 15 and 30 minutes after leptin activation, which declined at the 60-minute mark, while the total RhoA levels remained unchanged (Figure 2A). To further explore the involvement of RhoA activation in MMP-1 expression in leptin-activated SV40 cells, we examined the effects of Rhosin, a specific inhibitor of the RhoA subfamily of Rho GTPases, on MMP-1 expression. Our study demonstrated that Rhosin dose-dependently inhibited both MMP-1 mRNA (Figure 2B) and protein (Figure 2C) expression in leptin-activated SV40 cells. These results provide evidence that leptin induces MMP-1 expression in SV40 cells through RhoA-dependent pathways.
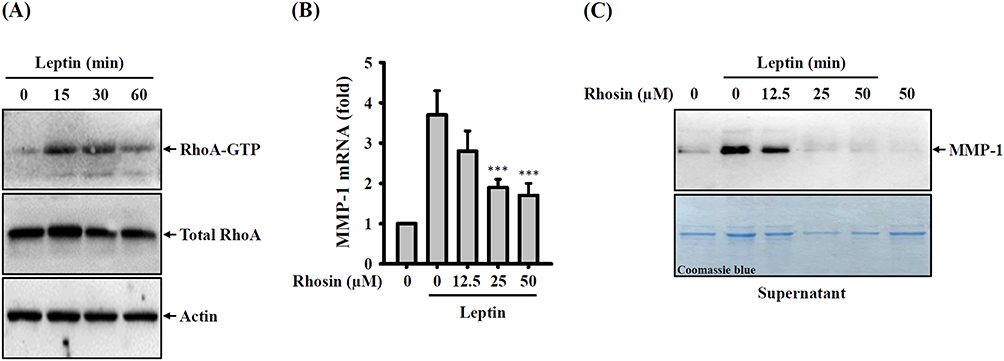 |
Figure 2 Effect of RhoA on leptin-induced MMP-1 expression. (A) SV40 cells were exposed to 100 ng/mL of leptin for 15–60 minutes, and the levels of GTP-bound RhoA were assessed through pull-down and Western blotting. (B) To investigate the effect of Rhosin, SV40 cells were first incubated with Rhosin for 0.5 hours, followed by incubation with 100 ng/mL of leptin for 2 hours. Subsequently, MMP-1 mRNA expression was determined using RT-real-time PCR. (C) For a similar investigation, SV40 cells were first incubated with Rhosin for 0.5 hours, followed by incubation with 100 ng/mL of leptin for 24 hours. The MMP-1 protein expression in supernatants was then analyzed through Western blotting. Data obtained from RT-real-time PCR is presented as the means ± SD from three separate experiments. The Western blotting images are representative of individual experiments. The significance of the findings is indicated by ***p < 0.001 when compared to leptin-activated cells. |
Leptin Induces MMP-1 Expression Through ERK1/2-Dependent Pathways
MAPKs play a pivotal role in the intricate signaling network of leptin, governing its multifaceted biological functions.32 However, their involvement in leptin-induced MMP-1 expression within SV40 cells has remained unexplored. Our investigation revealed that leptin substantially increased the phosphorylation levels of ERK1/2 at both the 15 and 30-minute marks after treatment, only to return to basal levels at the 60-minute point (Figure 3A). Conversely, leptin exhibited no ability to augment the phosphorylation levels of JNK1/2 (Figure 3B) or p38 (Figure 3C) in SV40 cells. To delve into whether leptin induces MMP-1 expression through ERK1/2-associated pathways, we tested the impact of PD98059, a selective MEK inhibitor that impedes the phosphorylation and activation of ERK1/2, on MMP-1 expression. Our findings demonstrated that PD98059 dose-dependently suppressed MMP-1 mRNA (Figure 3D) and protein (Figure 3E) expression in leptin-activated SV40 cells. These outcomes strongly suggest that leptin triggers MMP-1 expression through ERK1/2-dependent pathways in SV40 cells. Moreover, leptin is known to induce AKT activation in various cell types.33–35 However, in SV40 cells, leptin did not elicit AKT phosphorylation (Figure 3F).
 |
Figure 3 Effect of ERK1/2 on leptin-induced MMP-1 expression. (A-C) SV40 cells were treated with 100 ng/mL of leptin for 15–60 minutes. Subsequently, the phosphorylation levels of ERK1/2 (A), JNK1/2/3 (B), and p38 (C) in cell lysates were assessed through Western blotting. (D) To investigate the impact of PD98059, SV40 cells were pre-incubated with PD98059 for 0.5 hours, followed by treatment with 100 ng/mL of leptin for 2 hours. The subsequent analysis focused on MMP-1 mRNA expression, which was evaluated via RT-real-time PCR. (E) In a similar experimental setup, SV40 cells were pre-incubated with PD98059 for 0.5 hours, followed by exposure to 100 ng/mL of leptin for 24 hours. This time, the analysis involved MMP-1 protein expression in supernatants, assessed using Western blotting. (F) In a different series of experiments, SV40 cells were treated with 100 ng/mL of leptin for 15–60 minutes, and the phosphorylation levels of AKT in cell lysates were examined through Western blotting. Data derived from RT-real-time PCR is presented as the means ± SD from three separate experiments. The Western blotting images represent individual experiments. Findings indicating significance are marked as *p < 0.05 and ***p < 0.001 in comparison to leptin-activated cells. |
Leptin Induces MMP-1 Expression Through NF-κB-Dependent Pathways
We explored the involvement of NF-κB in MMP-1 expression within leptin-activated SV40 cells. Our findings revealed that leptin increased the phosphorylation levels of IKKα/β (Figure 4A), IκBα (Figure 4B), and NF-κB p65 (Figure 4C) in SV40 cells. Furthermore, leptin induced the translocation of NF-κB p65 into the cell nucleus (Figure 4D). To gain deeper insights into the role of NF-κB in leptin-mediated MMP-1 expression, we examined the impact of the NF-κB inhibitor PDTC on MMP-1 expression in SV40 cells. Our results indicated that PDTC significantly inhibited leptin-induced mRNA (Figure 4E) and protein (Figure 4F) expression of MMP-1. Furthermore, another NF-κB inhibitor, SN50, also confirmed the role of NF-κB in reducing MMP-1 protein expression (Figure 4G). These outcomes underscore that leptin induces MMP-1 expression through NF-κB-dependent pathways. Additionally, ROS are known to play crucial roles in leptin-mediated responses.36,37 However, we observed that the inhibition of ROS using NAC (Figure 4H) or the inhibition of the ROS production enzyme NADPH oxidase with DPI (Figure 4I) did not affect leptin-induced MMP-1 expression in SV40 cells.
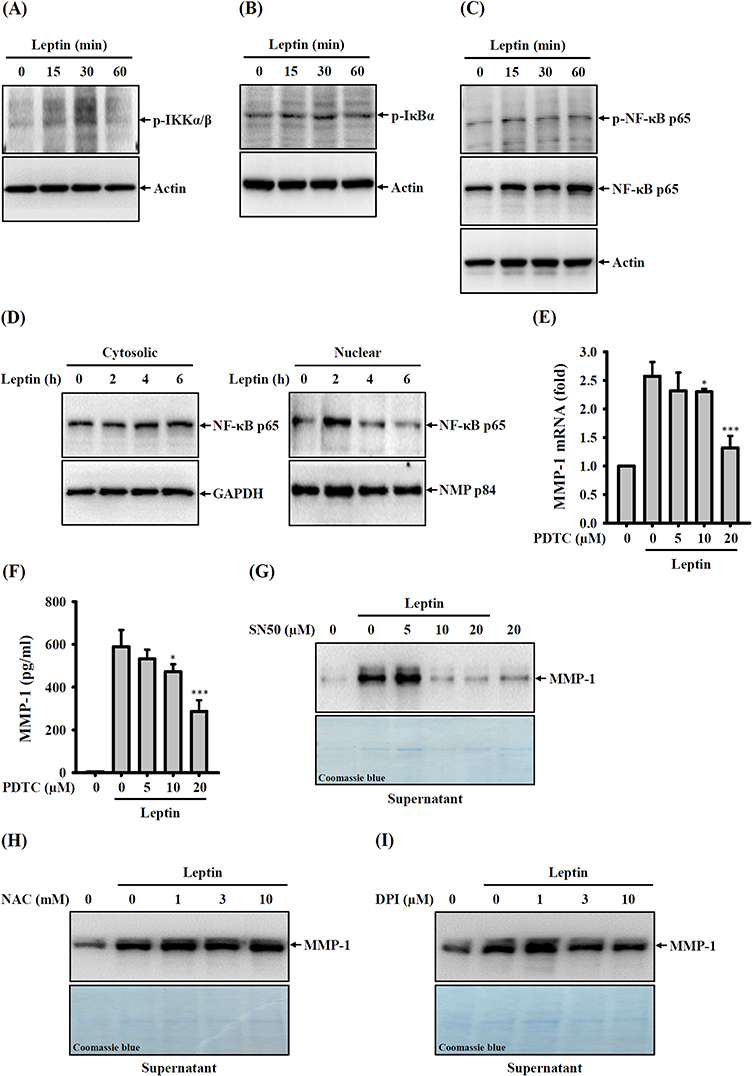 |
Figure 4 Effect of NF-κB on leptin-induced MMP-1 expression. (A–C) SV40 cells were exposed to 100 ng/mL of leptin for 15–60 minutes. Following this, phosphorylation levels of IKKα/β (A), IκBα (B), and NF-κB p65 (C) in cell lysates were examined via Western blotting. (D) For an extended treatment period, SV40 cells were incubated with 100 ng/mL of leptin for 2–6 hours, and the MMP-1 protein expression in both cytosolic and nuclear fractions was evaluated through Western blotting. (E) Investigating the impact of PDTC, SV40 cells were pre-treated with PDTC for 0.5 hours, followed by incubation with 100 ng/mL of leptin for 2 hours. The subsequent analysis focused on MMP-1 mRNA expression, assessed using RT-real-time PCR. (F) In a related experiment, SV40 cells were pre-treated with PDTC for 0.5 hours, followed by exposure to 100 ng/mL of leptin for 24 hours. The analysis centered on MMP-1 protein expression in supernatants and was assessed by ELISA. (G) Another pre-treatment experiment involved SV40 cells being incubated with SN50 for 0.5 hours, followed by incubation with 100 ng/mL of leptin for 24 hours. MMP-1 protein expression in supernatants was examined through Western blotting. (H) A separate pre-treatment study had SV40 cells being incubated with NAC for 0.5 hours, followed by exposure to 100 ng/mL of leptin for 24 hours. The analysis focused on MMP-1 protein expression in supernatants and was assessed by Western blotting. (I) In a different pre-treatment setup, SV40 cells were exposed to DPI for 0.5 hours, followed by incubation with 100 ng/mL of leptin for 24 hours. The subsequent analysis involved the assessment of MMP-1 protein expression in supernatants and was conducted through Western blotting. Data from ELISA and RT-real-time PCR are presented as the means ± SD from three separate experiments. The Western blotting images represent individual experiments. Findings denoting significance are marked as *p < 0.05 and ***p < 0.001 in comparison to leptin-activated cells. |
Leptin Activates NF-κB Through RhoA-ERK1/2 Axis
We established that RhoA operates upstream of ERK1/2, as the phosphorylation of ERK1/2 induced by leptin was significantly hindered by the RhoA inhibitor Rhosin (Figure 5A). Additionally, Rhosin inhibited the leptin-induced phosphorylation of IKKα/β (Figure 5B) and IκBα (Figure 5C), indicating that RhoA also functions upstream of NF-κB. To further unravel the interplay between ERK1/2 and NF-κB, we investigated the impact of the MEK inhibitor PD98059 on NF-κB activation. Our findings revealed that PD98059 inhibited the leptin-induced phosphorylation of IKKα/β (Figure 5D) and IκBα (Figure 5E). Collectively, these results point to leptin’s activation of NF-κB through the RhoA-ERK1/2 axis in SV40 cells.
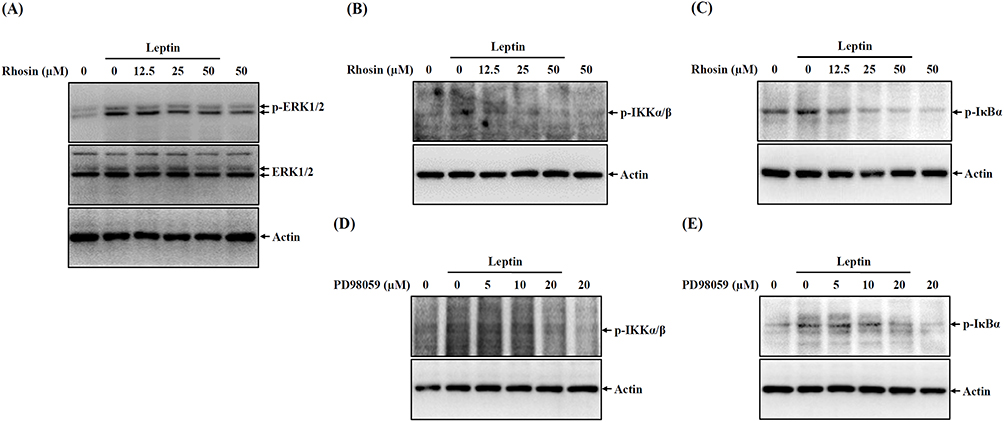 |
Figure 5 Leptin activates NF-κB through RhoA-ERK1/2 axis. (A–C) SV40 cells were pre-treated with Rhosin for 0.5 hours, followed by incubation with 100 ng/mL of leptin for 30 minutes. Subsequently, the phosphorylation levels of ERK1/2 (A), IKKα/β (B), and IκBα (C) in cell lysates were examined through Western blotting. (D and E) In a similar experimental setup, SV40 cells were pre-treated with Rhosin for 0.5 hours, followed by exposure to 100 ng/mL of leptin for 30 minutes. The subsequent analysis focused on the phosphorylation levels of IKKα/β (D) and IκBα (E) in cell lysates, which were assessed using Western blotting. The Western blotting images represent individual experiments. |
Discussion
Maintaining an appropriate extracellular matrix level is vital for the well-being of several physiological processes, particularly the health of the IVD. MMP-1, a pivotal enzyme responsible for extracellular matrix degradation in the IVD, plays a crucial role in this context. Studies have shown that MMP-1 levels in intervertebral disc tissues significantly increase in patients suffering from IVD degeneration, with higher expression levels observed in more severe cases.23,26 Furthermore, experimentally induced scoliotic deformity can induce MMP-1, resulting in its over-expression in disc cells of the annulus fibrosus, potentially hastening the degeneration of the intervertebral disc.38 Additionally, infection by Propionibacterium acnes can elevate MMP-1 expression, contributing to the loss of aggrecan and collagen II in nucleus pulposus cells, underscoring the role of P. acnes infection as a risk factor for IVD degeneration.39 In this study, we present a groundbreaking finding that MMP-1 can be induced by leptin in human intervertebral disc cartilage endplate-derived stem cells. It’s worth noting that MMP-1 induction can result from various factors in different tissues. For instance, in human chondrosarcoma cells and articular chondrocytes, IL-1β and TNF-α have been found to increase MMP-1 expression.40 In another scenario, mechanical stretch has been shown to induce MMP-1 expression in human keratoconus fibroblasts, highlighting its significance in corneal extracellular matrix remodeling.41 Furthermore, up-regulation of MMP-1 has been observed in Mycobacterium tuberculosis-infected macrophages, where MMP-1 appears to promote M. tuberculosis infection.42 Understanding the mechanisms through which MMP-1 is upregulated in disease conditions holds promise for gaining better insights and targeting therapeutics for IVD degeneration and MMP-1-associated disorders.
RhoA, a small GTPase protein, assumes pivotal roles in orchestrating leptin-mediated responses across a range of cell types. It’s been shown that leptin activates RhoA, instigating cytoskeleton remodeling in chondrocytes and nucleus pulposus cells, underscoring its critical involvement in conditions like obesity-associated lumbar disc degeneration and osteoarthritis.30,31, Furthermore, RhoA has been identified as a regulator of leptin-induced cardiomyocyte hypertrophy in cultured neonatal rat ventricular myocytes.43,44 Notably, p38 MAPK serves as an important downstream signaling pathway of RhoA in controlling leptin-induced cardiomyocyte hypertrophy;43,44 however, we have demonstrated that ERK1/2, not p38 MAPK, serves as the downstream signal of RhoA that is indispensable for leptin-induced MMP-1 expression in SV40 cells. ERK1/2 also participates in leptin-mediated osteoblastic differentiation of cartilage endplate cells.45 Moreover, serum leptin levels have shown a positive correlation not only with IVD degeneration syndrome but also with the increased presence of dysfunctional B cells in patients with systemic lupus erythematosus.26 This observation can be elucidated by the fact that leptin activates B cells to produce cytokines and induces B cell differentiation into plasma cells via the ERK1/2 pathway.46 These findings highlight ERK1/2 as a potential therapeutic target for conditions induced by leptin.
Excessive production of ROS can lead to oxidative damage. However, ROS also serve as critical elements in living organisms, functioning as signaling molecules and participating in defense against pathogens.47 In the context of IVD health, ROS can influence matrix metabolism, inflammatory processes, and autophagy in IVD cells, thereby promoting IVD degeneration.48 Additionally, leptin’s activation of the NLRP3 inflammasome, a key contributor to IVD degeneration, occurs through ROS production in osteoarthritic chondrocytes.37 On the flip side, in certain scenarios, such as hyperglycemia-induced apoptosis in pheochromocytoma cells, leptin reduces cell death by mitigating ROS production.49 It’s worth noting that in our study, we observed that the use of ROS scavengers like NAC and NADPH oxidase inhibitor DPI did not diminish MMP-1 expression in leptin-activated SV40 cells, suggesting that ROS play a less prominent role in this specific model. Furthermore, while leptin activates AKT, a regulator of various biological functions, in multiple cell types,33–35,50 we did not observe AKT activation in leptin-stimulated SV40 cells.
The inhibition of NF-κB had a significant impact on reducing the expression levels of MMP-3, MMP-9, and MMP-13 in IL-1β-stimulated nucleus pulposus cells.51 Moreover, when NF-κB was systemically inhibited in a mouse model of accelerated aging, it resulted in an increase in disc proteoglycan synthesis and a reduction in the loss of disc cellularity and matrix proteoglycan.52 Our own research has demonstrated that leptin activates NF-κB, and inhibiting NF-κB significantly reduces leptin-induced MMP-1 expression in SV40 cells. These findings underscore the critical role of NF-κB as a key pathogenic factor in IVD degeneration. Consequently, targeting NF-κB represents a potential therapeutic strategy for mitigating IVD degeneration.53
In this study, we present evidence demonstrating the crucial roles of RhoA, ERK1/2, and NF-κB in regulating MMP-1 expression in leptin-stimulated SV40 cells. Targeting molecules or pathways associated with RhoA, ERK1/2, and NF-κB holds the potential to modulate leptin-induced MMP-1 expression. Previous studies have shown various methods for the regulation of RhoA activity. For instance, RhoA activity can be negatively controlled by S-nitrosylation of cysteine residues in the GTP-binding domain of RhoA in smooth muscle cells,54 or through cAMP-mediated phosphorylation of serine residue 188 of RhoA in protein kinase A-activated human natural killer cells. This phosphorylation promotes a stable binding between RhoA-GTP and RhoGDIα, sequestering RhoA to the cytosol and eventually leading to RhoA inactivation.55 Activation of the G protein-coupled bile acid receptor TGR5 has been linked to the improvement of diabetic retinopathy, as it inhibits the RhoA signaling pathway by stabilizing F-actin.56 Moreover, the exogenous expression of phospholipase Cδ3 has been shown to down-regulate RhoA protein and inhibit RhoA signaling in cerebellar granule cells, thereby promoting neurite extension.57 Long noncoding RNAs (lncRNAs) are significant regulators of signal transduction and gene expression in cells. For example, lncRNA-01126 was significantly increased in periodontitis patients, and overexpression of lncRNA-01126 reduced the migration of human periodontal ligament cells by reducing the phosphorylation levels of ERK1/2.58 Additionally, lncRNA-NEF has been found to suppress epithelial-mesenchymal transition in colorectal cancer cells in vitro and inhibit metastasis in vivo by inhibiting ERK1/2.59 Rac1b depletion has been associated with increased ERK1/2 activation and an epithelial-mesenchymal transition phenotype in pancreatic ductal adenocarcinoma cells, suggesting that Rac1b negatively regulates ERK1/2 signaling.60 Furthermore, negative regulation of NF-κB signaling can be achieved at multiple levels, including transcriptional, post-transcriptional, and post-translational levels.61 For instance, the pseudo-kinase interleukin 1 receptor-associated kinase (IRAK)-3 (also known as IRAK-M) inhibits NF-κB activation by reducing the phosphorylation of IRAK-1 and inhibiting TNF receptor-associated factors 6 (TRAF6) at the transcriptional level.62 MicroRNAs, a specific class of RNA molecules, are involved in targeting RNA for degradation. MicroRNA-146, for example, can be induced by inflammatory stimulation and binds to the 3’ untranslated regions of both IRAK-1 and TRAF6 mRNA, preventing their translation and thereby inactivating NF-κB.63 The zinc finger protein A20 (also known as TNF-α-induced protein 3) is an important negative feedback regulator of NF-κB at the post-translational level by inhibiting the ubiquitination of TRAF6.64 These results suggest that the induction or activation of negative regulators of RhoA, ERK1/2, or NF-κB could serve as important therapeutic targets for mitigating IVD degeneration.
In addition to the pathways we examined in this study, numerous molecules and pathways have been reported to regulate MMP-1 expression in various cell types under different conditions. For instance, leptin has been found to promote MMP-1 expression in rat nucleus pulposus cells through the JAK2/STAT3 pathway.65 Glycogen synthase kinase-3α/β (GSK-3α/β) has been shown to positively regulate MMP-1 expression in Mycobacterium tuberculosis-infected human THP-1 macrophages.42 Additionally, protein kinase C-α (PKC-α) is involved in the induction of MMP-1 expression in response to phorbol ester 12-O-tetra-decanoylphorbol-13-acetate in MCF-7 breast cancer cells and heat shock-induced MMP-1 expression in human keratinocytes HaCaT cells.66,67 These findings suggest that JAK2/STAT3, GSK-3α/β, and PKC-α are potential targets that warrant further investigation to elucidate their roles in MMP-1 expression in leptin-activated SV40 cells.
While our analysis of MMP-1 expression included ELISA, Western blotting, and RT-PCR, it is important to consider the inclusion of other relevant evaluation methodologies in future studies to further substantiate the reliability of our results. In addition to assessing mRNA and protein expression, quantitative measurements of MMP-1 enzyme activity can be performed using collagen zymography and fluorimetric assays.68,69 Moreover, intracellular MMP-1 expression can be visualized by employing a fluorescence-conjugated MMP-1 antibody and observed using a fluorescent microscope.70 Given the crucial role of ERK1/2 in leptin-mediated MMP-1 induction, evaluating the impact of leptin on ERK1/2 can involve not only the assessment of phosphorylation levels but also in vitro kinase assays.71 In our present study, we scrutinized the effect of leptin on NF-κB signaling by examining the phosphorylation levels of IKKα/β, IκBα, and NF-κB p65, as well as the nuclear translocation of NF-κB p65 in SV40 cells. To enhance our understanding, it would be worthwhile to investigate the influence of leptin on NF-κB transcriptional activity using a reporter assay.72
A limitation of this study lies in the absence of genetic inhibition to validate our findings, despite demonstrating the significance of RhoA, ERK1/2, and NF-κB in leptin-induced MMP-1 expression in SV40 cells through the use of specific inhibitors. Our attempts to downregulate the expression of RhoA, ERK1/2, and NF-κB in SV40 cells via shRNA were unsuccessful, as these proteins’ levels were not significantly reduced in antibiotics-selected SV40 cells. This suggests that SV40 may have low transfection efficiency when using liposome-based transfection methods. Another limitation is the absence of an animal model to provide in vivo confirmation. To address this limitation, the impact of leptin on MMP-1 expression and IVD degeneration could be assessed in mice through weekly intraperitoneal injections of leptin over a 2-month period, with or without the administration of RhoA, ERK1/2, or NF-κB inhibitors. Subsequently, the L3-6 segments of the mice can be collected, and MMP-1 expression in the cartilage endplate can be analyzed via immunohistochemistry, while the severity of IVD degeneration can be evaluated.73 Furthermore, aged mice (23 months old) typically exhibit age-dependent IVD degeneration, marked by changes in phenotype and reduced viability of central nucleus pulposus cells.74 Investigating the impact of leptin on age-dependent IVD degeneration in mice could be achieved by reducing leptin in vivo using a leptin-neutralizing antibody administered intravenously.75
Conclusion
We conducted a comprehensive investigation into the function and regulatory mechanisms underlying leptin-mediated MMP-1 expression in human intervertebral disc cartilage endplate-derived stem cells. These findings have substantial potential for guiding the development of targeted therapeutic strategies for addressing intervertebral disc disease.
Abbreviations
IVD, intervertebral disc; MMP-1, matrix metalloproteinase-1; SV40 cells, human intervertebral disc cartilage endplate-derived stem cells; ROS, reactive oxygen species; PBS, phosphate buffered saline; NAC, N-acetyl-l-cysteine; DPI, diphenyleneiodonium; MAPKs, mitogen-activated protein kinases; IL, interleukin; TNF-α, tumour necrosis factor α; RT-real-time PCR, reverse transcription and real-time polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; lncRNAs, long noncoding RNAs; IRAK, interleukin 1 receptor-associated kinase; TRAF6, TNF receptor-associated factors 6; GSK-3α/β, glycogen synthase kinase-3α/β; PKC-α, protein kinase C-α.
Acknowledgments
This research was funded by Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, grant number TCRD-TPE-112-27 (1/3).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roughley PJ, Alini M, Antoniou J. The role of proteoglycans in aging, degeneration and repair of the intervertebral disc. Biochem Soc Trans. 2002;30(Pt 6):869–874. doi:10.1042/bst0300869
2. Ohnishi T, Iwasaki N, Sudo H. Causes of and molecular targets for the treatment of intervertebral disc degeneration: a review. Cells. 2022;11(3):394. doi:10.3390/cells11030394
3. de Schepper EI, Damen J, van Meurs JB, et al. The association between lumbar disc degeneration and low back pain: the influence of age, gender, and individual radiographic features. Spine. 2010;35(5):531–536. doi:10.1097/BRS.0b013e3181aa5b33
4. Kalb S, Martirosyan NL, Kalani MY, et al. Genetics of the degenerated intervertebral disc. World Neurosurg. 2012;77(3–4):491–501. doi:10.1016/j.wneu.2011.07.014
5. Nasto LA, Ngo K, Leme AS, et al. Investigating the role of DNA damage in tobacco smoking-induced spine degeneration. Spine J. 2014;14(3):416–423. doi:10.1016/j.spinee.2013.08.034
6. Williams FM, Sambrook PN. Neck and back pain and intervertebral disc degeneration: role of occupational factors. Best Pract Res Clin Rheumatol. 2011;25(1):69–79. doi:10.1016/j.berh.2011.01.007
7. Francisco V, Pino J, González-Gay MÁ, et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat Rev Rheumatol. 2022;18(1):47–60. doi:10.1038/s41584-021-00713-z
8. Cannata F, Vadalà G, Ambrosio L, et al. Intervertebral disc degeneration: a focus on obesity and type 2 diabetes. Diabetes Metab Res Rev. 2020;36(1):e3224. doi:10.1002/dmrr.3224
9. WHO. Obesity and Overweight Fact Sheet; 2018; Available from: http://www.who.int/news-room/factsheets/detail/obesity-and-overweight.
10. Zhou J, Mi J, Peng Y, et al. Causal associations of obesity with the intervertebral degeneration, low back pain, and sciatica: a two-sample Mendelian randomization study. Front Endocrinol. 2021;12:740200. doi:10.3389/fendo.2021.740200
11. Xu X, Li X, Wu W. Association between overweight or obesity and lumbar disk diseases: a meta-analysis. J Spinal Disord Tech. 2015;28(10):370–376. doi:10.1097/BSD.0000000000000235
12. Samartzis D, Karppinen J, Chan D, et al. The association of lumbar intervertebral disc degeneration on magnetic resonance imaging with body mass index in overweight and obese adults: a population-based study. Arthritis Rheum. 2012;64(5):1488–1496. doi:10.1002/art.33462
13. Malone JI, Hansen BC. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr Diabetes. 2019;20(1):5–9. doi:10.1111/pedi.12787
14. Jiang Z, Lu W, Zeng Q, et al. High glucose-induced excessive reactive oxygen species promote apoptosis through mitochondrial damage in rat cartilage endplate cells. J Orthop Res. 2018;36(9):2476–2483. doi:10.1002/jor.24016
15. Tsai TT, Ho NY, Lin YT, et al. Advanced glycation end products in degenerative nucleus pulposus with diabetes. J Orthop Res. 2014;32(2):238–244. doi:10.1002/jor.22508
16. Kiran S, Kumar V, Kumar S, Price RL, Singh UP. Adipocyte, immune cells, and miRNA crosstalk: a novel regulator of metabolic dysfunction and obesity. Cells. 2021;10(5):1004. doi:10.3390/cells10051004
17. Rohm TV, Meier DT, Olefsky JM, et al. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55(1):31–55. doi:10.1016/j.immuni.2021.12.013
18. Ruiz-Fernández C, Francisco V, Pino J, et al. Molecular relationships among obesity, inflammation and intervertebral disc degeneration: are adipokines the common link? Int J Mol Sci. 2019;20(8):2030. doi:10.3390/ijms20082030
19. Navone SE, Marfia G, Giannoni A, et al. Inflammatory mediators and signalling pathways controlling intervertebral disc degeneration. Histol Histopathol. 2017;32(6):523–542. doi:10.14670/HH-11-846
20. Molinos M, Almeida CR, Caldeira J, et al. Inflammation in intervertebral disc degeneration and regeneration. J R Soc Interface. 2015;12(104):20141191. doi:10.1098/rsif.2014.1191
21. Wang WJ, Yu XH, Wang C, et al. MMPs and ADAMTSs in intervertebral disc degeneration. Clin Chim Acta. 2015;448:238–246. doi:10.1016/j.cca.2015.06.023
22. Deng B, Ren JZ, Meng XQ, et al. Expression profiles of MMP-1 and TIMP-1 in lumbar intervertebral disc degeneration. Genet Mol Res. 2015;14(4):19080–19086. doi:10.4238/2015.December.29.16
23. Mantzoros CS, Magkos F, Brinkoetter M, et al. Leptin in human physiology and pathophysiology. Am J Physiol Endocrinol Metab. 2011;301(4):E567–84. doi:10.1152/ajpendo.00315.2011
24. Francisco V, Pino J, Campos-Cabaleiro V, et al. Obesity, fat mass and immune system: role for leptin. Front Physiol. 2018;9:640. doi:10.3389/fphys.2018.00640
25. Hsu HT, Yue CT, Teng MS, et al. Immuohistochemical score of matrix metalloproteinase-1 may indicate the severity of symptomatic cervical and lumbar disc degeneration. Spine J. 2020;20(1):124–137. doi:10.1016/j.spinee.2019.08.004
26. Segar AH, Fairbank JCT, Urban J. Leptin and the intervertebral disc: a biochemical link exists between obesity, intervertebral disc degeneration and low back pain—an in vitro study in a bovine model. Eur Spine J. 2019;28(2):214–223. doi:10.1007/s00586-018-5778-7
27. Koskinen A, Vuolteenaho K, Nieminen R, et al. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin Exp Rheumatol. 2011;29(1):57–64.
28. Liao P-C, Chao LK, Chou J-C, et al. Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1β secretion. Inflamm Res. 2013;62(1):89–96. doi:10.1007/s00011-012-0555-2
29. Liang J, Feng J, Wu WKK, et al. Leptin-mediated cytoskeletal remodeling in chondrocytes occurs via the RhoA/ROCK pathway. J Orthop Res. 2011;29(3):369–374. doi:10.1002/jor.21257
30. Li Z, Liang J, Wu WK, et al. Leptin activates RhoA/ROCK pathway to induce cytoskeleton remodeling in nucleus pulposus cells. Int J Mol Sci. 2014;15(1):1176–1188. doi:10.3390/ijms15011176
31. Park HK, Ahima RS. Leptin signaling. F1000Prime Rep. 2014;6:73. doi:10.12703/P6-73
32. Zeng Q, Luo X, Tang Y, et al. Leptin regulated ILC2 cell through the PI3K/AKT pathway in allergic rhinitis. Mediators Inflamm. 2020;2020:4176082. doi:10.1155/2020/4176082
33. Beales ILP, Ogunwobi OO. Leptin activates Akt in oesophageal cancer cells via multiple atorvastatin-sensitive small GTPases. Mol Cell Biochem. 2021;476(6):2307–2316. doi:10.1007/s11010-021-04067-8
34. Wang J, Zhou F, Li F, et al. Autocrined leptin promotes proliferation of non-small cell lung cancer (NSCLC) via PI3K/AKT and p53 pathways. Ann Transl Med. 2021;9(7):568. doi:10.21037/atm-20-7482
35. Mourmoura E, Papathanasiou I, Trachana V, et al. Leptin-depended NLRP3 inflammasome activation in osteoarthritic chondrocytes is mediated by ROS. Mech Ageing Dev. 2022;208:111730. doi:10.1016/j.mad.2022.111730
36. Grivas TB, Vasiliadis ES, Kaspiris A, et al. Expression of matrix metalloproteinase-1 (MMP-1) in Wistar rat’s intervertebral disc after experimentally induced scoliotic deformity. Scoliosis. 2011;6(1):9. doi:10.1186/1748-7161-6-9
37. Zheng Y, Lin Y, Chen Z, et al. Propionibacterium acnes induces intervertebral discs degeneration by increasing MMP-1 and inhibiting TIMP-1 expression via the NF-κB pathway. Int J Clin Exp Pathol. 2018;11(7):3445–3453.
38. Pei Y, Harvey A, Yu XP, Chandrasekhar S, Thirunavukkarasu K. Differential regulation of cytokine-induced MMP-1 and MMP-13 expression by p38 kinase inhibitors in human chondrosarcoma cells: potential role of Runx2 in mediating p38 effects. Osteoarthritis Cartilage. 2006;14(8):749–758. doi:10.1016/j.joca.2006.01.017
39. Du G-L, Chen W-Y, Li X-N, et al. Induction of MMP-1 and −3 by cyclical mechanical stretch is mediated by IL-6 in cultured fibroblasts of keratoconus. Mol Med Rep. 2017;15(6):3885–3892. doi:10.3892/mmr.2017.6433
40. Zhou X, Lie L, Liang Y, et al. GSK-3α/β activity negatively regulates MMP-1/9 expression to suppress Mycobacterium tuberculosis infection. Front Immunol. 2022;12(752466). doi:10.3389/fimmu.2021.752466.
41. Zeidan A, Javadov S, Chakrabarti S, et al. Leptin-induced cardiomyocyte hypertrophy involves selective caveolae and RhoA/ROCK-dependent p38 MAPK translocation to nuclei. Cardiovasc Res. 2008;77(1):64–72. doi:10.1093/cvr/cvm020
42. Rajapurohitam V, Izaddoustdar F, Martinez-Abundis E, et al. Leptin-induced cardiomyocyte hypertrophy reveals both calcium-dependent and calcium-independent/RhoA-dependent calcineurin activation and NFAT nuclear translocation. Cell Signal. 2012;24(12):2283–2290. doi:10.1016/j.cellsig.2012.07.025
43. Han YC, Ma B, Guo S, et al. Leptin regulates disc cartilage endplate degeneration and ossification through activation of the MAPK-ERK signalling pathway in vivo and in vitro. J Cell Mol Med. 2018;22(4):2098–2109. doi:10.1111/jcmm.13398
44. Chen H, Qi J, Liu T, et al. Leptin accelerates B cell dysfunctions via activating JAK/STAT3/5 and ERK1/2 pathways in patients with systemic lupus erythematosus. Clin Exp Rheumatol. 2022;40(11):2125–2132.
45. Halliwell B. Reactive oxygen species (ROS), oxygen radicals and antioxidants: where are we now, where is the field going and where should we go? Biochem Biophys Res Commun. 2022;633:17–19. doi:10.1016/j.bbrc.2022.08.098
46. Cao G, Yang S, Cao J, et al. The role of oxidative stress in intervertebral disc degeneration. Oxid Med Cell Longev. 2022;2022:2166817. doi:10.1155/2022/2166817
47. Kaeidi A, Hajializadeh Z, Jahandari F, et al. Leptin attenuates oxidative stress and neuronal apoptosis in hyperglycemic condition. Fundam Clin Pharmacol. 2019;33(1):75–83. doi:10.1111/fcp.12411
48. Fu R, Han F, Liu L, et al. The effects of leptin on the proliferation and differentiation of primary chondrocytes in vitro and cartilage regeneration in vivo. ACS Biomater Sci Eng. 2019;5(4):1907–1919. doi:10.1021/acsbiomaterials.8b01168
49. Zhongyi S, Sai Z, Chao L, et al. Effects of nuclear factor kappa B signaling pathway in human intervertebral disc degeneration. Spine. 2015;40(4):224–232. doi:10.1097/BRS.0000000000000733
50. Nasto LA, Seo H-Y, Robinson AR, et al. ISSLS prize winner: inhibition of NF-κB activity ameliorates age-associated disc degeneration in a mouse model of accelerated aging. Spine. 2012;37(21):1819–1825. doi:10.1097/BRS.0b013e31824ee8f7
51. Zhang GZ, Liu MQ, Chen HW, et al. NF-κB signalling pathways in nucleus pulposus cell function and intervertebral disc degeneration. Cell Prolif. 2021;54(7):e13057. doi:10.1111/cpr.13057
52. Lin L, Xu C, Carraway MS, Piantadosi CA, Whorton AR, Li S. RhoA inactivation by S-nitrosylation regulates vascular smooth muscle contractive signaling. Nitric Oxide. 2018;74:56–64. doi:10.1016/j.niox.2018.01.007
53. Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15(3):510–519. doi:10.1002/j.1460-2075.1996.tb00383.x
54. Zhu L, Wang W, Xie TH, et al. TGR5 receptor activation attenuates diabetic retinopathy through suppression of RhoA/ROCK signaling. FASEB J. 2020;34(3):4189–4203. doi:10.1096/fj.201902496RR
55. Kouchi Z, Igarashi T, Shibayama N, et al. Phospholipase Cdelta3 regulates RhoA/Rho kinase signaling and neurite outgrowth. J Biol Chem. 2011;286(10):8459–8471. doi:10.1074/jbc.M110.171223
56. Zhu Y, Ai R, Ding Z, et al. LncRNA-01126 inhibits the migration of human periodontal ligament cells through MEK/ERK signaling pathway. J Periodontal Res. 2020;55(5):631–641. doi:10.1111/jre.12749
57. Shi CJ, Xue ZH, Zeng WQ, et al. LncRNA-NEF suppressed oxaliplatin resistance and epithelial-mesenchymal transition in colorectal cancer through epigenetically inactivating MEK/ERK signaling. Cancer Gene Ther. 2023;30(6):855–865. doi:10.1038/s41417-023-00595-1
58. Witte D, Otterbein H, Förster M, et al. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci Rep. 2017;7(1):17313. doi:10.1038/s41598-017-15170-6
59. Rothschild DE, McDaniel DK, Ringel-Scaia VM, Allen IC. Modulating inflammation through the negative regulation of NF-κB signaling. J Leukoc Biol. 2018. ;103(6):1131–50.
60. Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110(2):191–202. doi:10.1016/S0092-8674(02)00827-9
61. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27(42):5643–5647. doi:10.1038/onc.2008.171
62. Pujari R, Hunte R, Khan WN, Shembade N. A20-mediated negative regulation of canonical NF-κB signaling pathway. Immunol Res. 2013;57(1–3):166–171. doi:10.1007/s12026-013-8463-2
63. Miao D, Zhang L. Leptin modulates the expression of catabolic genes in rat nucleus pulposus cells through the mitogen-activated protein kinase and Janus kinase 2/signal transducer and activator of transcription 3 pathways. Mol Med Rep. 2015;12(2):1761–1768. doi:10.3892/mmr.2015.3646
64. Kim S, Han J, Lee SK, et al. Berberine suppresses the TPA-induced MMP-1 and MMP-9 expressions through the inhibition of PKC-α in breast cancer cells. J Surg Res. 2012;176(1):e21–29. doi:10.1016/j.jss.2011.11.1041
65. Lee YM, Li WH, Kim YK, Kim KH, Chung JH. Heat-induced MMP-1 expression is mediated by TRPV1 through PKCalpha signaling in HaCaT cells. Exp Dermatol. 2008;17(10):864–870. doi:10.1111/j.1600-0625.2008.00738.x
66. Lee JH, Chellasamy G, Yun K, Nam MJ. EGF-expressed human mesenchymal stem cells inhibit collagenase1 expression in keratinocytes. Cell Signal. 2023;110:110827. doi:10.1016/j.cellsig.2023.110827
67. Naveed SU, Clements D, Jackson DJ, et al. Matrix metalloproteinase-1 activation contributes to airway smooth muscle growth and asthma severity. Am J Respir Crit Care Med. 2017;195(8):1000–1009. doi:10.1164/rccm.201604-0822OC
68. Herrera I, Cisneros J, Maldonado M, et al. Matrix metalloproteinase (MMP)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J Biol Chem. 2013;288(36):25964–25975. doi:10.1074/jbc.M113.459784
69. Yang FL, Yang YL, Liao PC, et al. Structure and immunological characterization of the capsular polysaccharide of a pyrogenic liver abscess caused by Klebsiella pneumoniae: activation of macrophages through Toll-like receptor 4. J Biol Chem. 2011;286(24):21041–21051. doi:10.1074/jbc.M111.222091
70. Lin WY, Li LH, Hsiao YY, et al. Repositioning of the angiotensin II receptor antagonist candesartan as an anti-inflammatory agent with NLRP3 inflammasome inhibitory activity. Front Immunol. 2022;13:870627. doi:10.3389/fimmu.2022.870627
71. Wang W, Jing X, Du T, et al. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic Biol Med. 2022;190:234–246. doi:10.1016/j.freeradbiomed.2022.08.018
72. Novais EJ, Tran VA, Johnston SN, et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun. 2021;12(1):5213. doi:10.1038/s41467-021-25453-2
73. Petrescu AD, Grant S, Williams E, et al. Leptin enhances hepatic fibrosis and inflammation in a mouse model of cholestasis. Am J Pathol. 2022;192(3):484–502. doi:10.1016/j.ajpath.2021.11.008
74. Zitka O, Kukacka J, Krizkova S, et al. Matrix metalloproteinases. Curr Med Chem. 2010;17(31):3751–3768. doi:10.2174/092986710793213724
75. Mahbouli S, Der Vartanian A, Ortega S, et al. Leptin induces ROS via NOX5 in healthy and neoplastic mammary epithelial cells. Oncol Rep. 2017;38(5):3254–3264. doi:10.3892/or.2017.6009
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.