Back to Journals » The Application of Clinical Genetics » Volume 12
Ketogenic diet attenuates cerebellar atrophy progression in a subject with a biallelic variant at the ATAD3A locus
Authors Al Madhoun A ![]() , Alnaser F, Melhem M
, Alnaser F, Melhem M ![]() , Nizam R, Al-Dabbous T, Al-Mulla F
, Nizam R, Al-Dabbous T, Al-Mulla F ![]()
Received 11 November 2018
Accepted for publication 28 February 2019
Published 5 June 2019 Volume 2019:12 Pages 79—86
DOI https://doi.org/10.2147/TACG.S194204
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Ashraf Al Madhoun,1,2 Fahad Alnaser,3 Motasem Melhem,1 Rasheeba Nizam,1 Tala Al-Dabbous,4 Fahd Al-Mulla1
1Genetics and Bioinformatics Department, Dasman Diabetes Institute, Kuwait City, Kuwait; 2Animal and Imaging Core Facility Department, Dasman Diabetes Institute, Dasman, Kuwait City, 15462, Kuwait; 3Radiology Department, Ibn Sina Hospital, Ministry of Health, Kuwait City, Kuwait; 4Bayt Abdullah Children’s Hospice, NBK Children’s Cancer Hospital, Al- Adan ICU, Kuwait City, Kuwait
Abstract: The ATPase AAA-domain protein 3 (ATAD3) is a ubiquitously expressed mitochondrial protein involved in mitochondrial dynamics, DNA-nucleoid structural organization, cholesterol transport and steroidogenesis. Mutations within the ancestral ATAD3A gene are strongly associated with neurological abnormalities due to alterations in the mitochondrial function and homeostasis. Here, we report the case of a subject diagnosed with developmental delay associated with ataxia and progressive atrophy of both cerebellar hemispheres and cerebellar vermis, despite exhibiting a normal biochemical profile. By whole exome sequencing, we identified two biallelic single nucleotide variants within the coding region of ATAD3A in the affected subject. Both variants were previously reported as monoallelic variants with uncertain clinical significance. Importantly, the variant ATAD3A c.251T>C leads to an amino acid change of a highly conserved residue across species and in silico analysis revealed structural alteration in the ATAD3A protein. Ketogenic diet was administered to the subject as a novel therapeutic approach. Notably, the treatment correlated with a reduction in cerebellum atrophy progression and the gradual enhancement of the subject’s physical skills, vitality and personal interactions. Thus, we report the first subject with a homozygous status for the ATAD3A c.251T>C (p.Thr84Met) variant. We propose that this mutation led to an alteration of the mitochondrial function, causing the neurological symptoms observed in the subject. The symptoms were partially alleviated following ketogenic diet, improving the subject’s quality of life.
Keywords: cerebellar atrophy, ATAD3A, SNV, ATAD3A c.251T>C (p.Thr84Met) rs546711654, ketogenic diet, whole exome sequencing
A Letter to the Editor has been received and published for this article.
A Response to Letter has been published for this article.
Introduction
A number of neurological disorders arise from mitochondrial dysfunctions due to the neurons’ high demand for oxygen and adenosine triphosphate (ATP) and their increased susceptibility to oxidative stress.1 The ATPase AAA + domain containing protein 3A (ATAD3A) is a mitochondrial protein, which has been associated with mitochondrial DNA (mtDNA) nucleoid organization and replication,2 mitochondrial translation3 and cholesterol metabolism.4–6 ATAD3A is part of the ATAD3 gene family that comprises three paralog genes in humans (ATAD3A, ATAD3B and ATAD3C), spanning a 85 kb tandem stretch on chromosome 1p36.33.7 In mice, ATAD3A knockout is lethal and causes early developmental retardation,8 whereas mutations in the gene cause mitochondrial DNA fragmentation and abnormalities.2 In humans, de novo mutations in ATAD3A were found to be associated with a neurologic syndrome characterized by global developmental delay, hypotonia, optic atrophy, axonal neuropathy and hypertrophic cardiomyopathy9 and associated with hereditary spastic paraplegia.10 In addition, deletions in the ATAD3 gene cluster were reported to cause cerebellar dysfunction due to altered mtDNA and cholesterol metabolism.4
Case report
We report the case of a subject who was diagnosed with progressive cerebellar atrophy. The affected subject was a 4.5-year-old female who presented with developmental delay and failure to thrive since the age of 5 months old (Figure S1). Morphologically, the subject had triangular face, micrognathia and low-set ears. The subject was interactive with unsteady gait, intention tremor, head titubation, progressive hand pill-rolling and ocular motor apraxia. She did not display any signs of cognitive delay or mental retardation. Biochemical, hematological and hormonal profiles were normal. Electromyogram, electrocardiogram and echocardiogram examinations were also normal (data not shown).
Magnetic resonance imaging (MRI), performed at age 18 months, indicated normal brain parenchyma, without focal lesions or heterotopias. Ventricles and cerebrospinal fluid appeared normal and there were no midline shifts. Sinuses and orbits appeared unremarkable. Detailed inspection of the MRI revealed a mild progressive cerebellum atrophy, as indicated by patchy high signal intensity on axial T2–weighted imaging (T2WI [Figure 1A] and Supplementary materials), and fluid-attenuated inversion recovery (FLAIR) sequences within the cerebellar cortex and vermis (Figure S2A) but not the cerebellar white matter. Hyperintensities were also observed in coronal–T2WI and sagittal–T1 FLAIR sequence images (Figure 1B and C, respectively). Brain diffusion-weighted imaging (DWI) sequences showed no diffusion, suggesting absence of brain malignancy or infarction (Figure S2B). Twenty-eight months later (at age 3 years and 10 months), MRI of the axial and coronal–T2WI sequences displayed progressive atrophy of the cerebellar cortex and vermis (Figure 1D and E) with 7.5% reduction in cerebellar hemispheres size, fracture enlargements and 20% reduction in the relative sagittal length of cerebellum (Figure 1F). No diffusion restrictions were observed in the brain DWI sequences (Figure S2B).
 | Figure 1 Magnetic resonance imaging (MRI) scans (A-F) of the subject’s brain. Progressive cerebellar and vermis atrophy were observed within a period of 28 months. |
The subject was born from consanguineous Palestinian parents and had two healthy elder siblings. The pregnancy was full-term with normal vaginal delivery and no remarkable antenatal history. Prior to the subject’s birth, the mother had experienced three first-trimester miscarriages without registered detailed examination (Figure 2A). The remarkable phenotypic profile and pedigree of the subject prompted us to perform trio whole exome sequencing (WES) on genomic DNA of the family. WES identified a total of 23,935 exonic variants at high quality. Of these, 13,003 (54.3%) were preserved in the family, 4,413 (18.4%) and 4,660 (19.5%) were identical in the subject and one of her parents (mother and father, respectively), and 1,859 (7.8%) were unique to the subject. Using a frequency filter of minor allele frequency <0.1% in public databases, we identified one homozygous single nucleotide variant (SNV) in the ATAD3A locus of the subject (c.251C>T; p.Thr84Met [rs546711654 g.chr1:1451437C>T [hg19] (2009)]) (Figure 2B). To explore whether this variant was a result of autozygosity, we analyzed homozygous stretches within chromosome 1. Regions of homozygosity did not span the ATAD3A gene, and there were no large regions of homozygosity detected involving the ATAD3 cluster (data not shown and Supplementary materials). For that reason, we examined other prospective variants within chromosome 1 and identified an additional homozygous SNV in the ATAD3A locus (c.206-12C>T [rs9439458; g.ch1:1451380C>T [hg19] (2009)]). No homozygous SNVs were found in the other ATAD3 paralogue genes (ATAD3B or ATAD3C) (data not shown). Both SNVs were confirmed by Sanger sequencing using genomic DNA isolated from all family members and primers specific to ATAD3A, which did not amplify any of the paralog genes (Figure 2C, Figure S3). Except for the affected subject, all family members were monoallelic (heterozygous) for both SNVs, and did not present with any neurological disorder. Notably, both SNVs identified in the subject have been previously reported in the Single Nucleotide Polymorphism (dbSNP) database with uncertain clinical significance. The ATAD3A c.206-12C>T variant, located in intron 1, is not associated with any prospective transcript, transcription factor binding motif nor non-coding RNA products. However, the c.251C>T variant, located in exon 2, led to a missense amino acid change in the ATAD3A protein (p.Thr84Met).
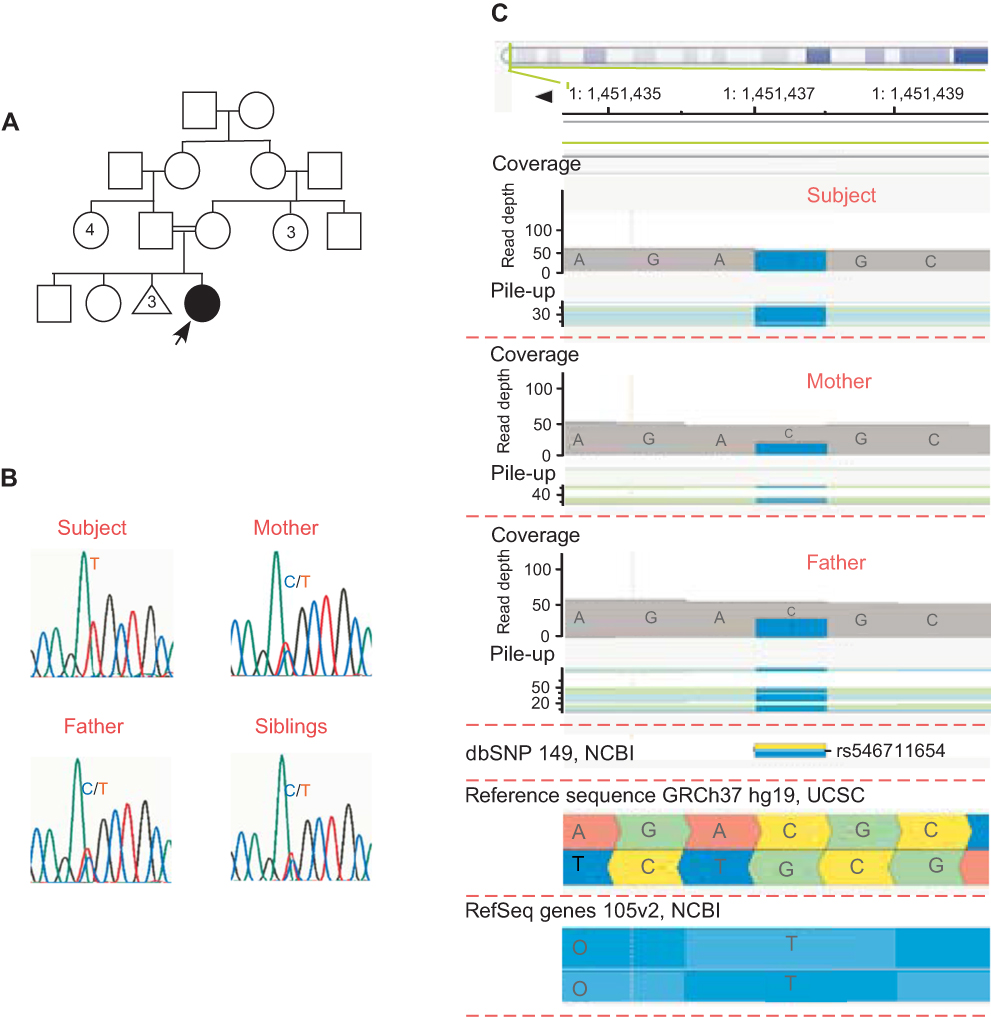 | Figure 2 Family Pedigree and ATAD3A c.251C>T (p.Thr84Met) single nucleotide variant (SNV). (A) Pedigree of the studied family, indicating the affected female subject born from consanguineous non-affected parents and with two non-affected elder siblings. Prior to the subject’s birth, the mother had experienced three miscarriages. (B) The ATAD3A c.251C>T variation leads to an amino acid substitution at position 84 (p.Thr84Met). (C) Sanger validation of the ATAD3A variant in the family. The affected subject carries a homozygous variation on ATAD3A (rs546711654 c.251C>T; p.Thr84Met), while all other family members are carriers. |
Alignment of ATAD3A proteins from different species revealed conservation of Thr84 and most of its adjacent amino acids (Figure 3A). Bioinformatic analysis predicted the phosphorylation of ATAD3A at Thr84 by several kinases, including Pyruvate dehydrogenase kinase (PDK), Protein Kinase C (PKC), Casein kinase 2 (CK2) and cyclin-dependent kinases (CDKs) (score rates >0.70, data not shown). Additionally, in silico protein structure modeling revealed that this amino acid substitution affected the overall structure of the protein, as these residues differ in size, polarity and charge (Figure 3B).
 | Figure 3 ATAD3A protein structure. (A) Localization of the ATAD3 gene cluster in chromosome 1p36.33. Schematic illustration of the ATAD3A protein showing the coiled-coil domain, the transmembrane domain and the AAA domain, which includes the Walker A&B ATP-binding and ATPase domains. The alignment of ATAD3A proteins from different species shows that Thr84 is highly conserved. (B) 3D protein structure modeling showing conformational changes in ATAD3A protein in the Thr84Met mutant relative to wild type. Arrows indicate the Thr (T) and Met (M) residues within the wild type and mutant proteins. |
Recently, a de novo recurrent mutation of the ATAD3A gene (c.1582C>T; p.Arg528Trp) was identified in individuals presenting with global developmental delay, hypotonia, spasticity, optic atrophy, axonal neuropathy and hypertrophic cardiomyopathy.9 The symptoms displayed by our patient were similar to those reported by Harel and colleagues; however, our subject did not present with any signs of cardiomyopathy or optic atrophy, though a minor reduction in retinal vessels was observed (data not shown). Harel et al. also reported other biallellic mutations and deletions in the ATAD3A gene and their associated phenotypes in two additional families. In another study, Cooper et al. describe the phenotype of two subjects from one family with a biallelic mutation at the ATAD3A locus (c.1064G>A; p.Gly355Asp).10 We have compared the phenotype of our subject with these recently published reports (Table 1). Overall, the symptoms and physical characteristics presented by our subject are similar to those presented by subjects with ATAD3A mutations.
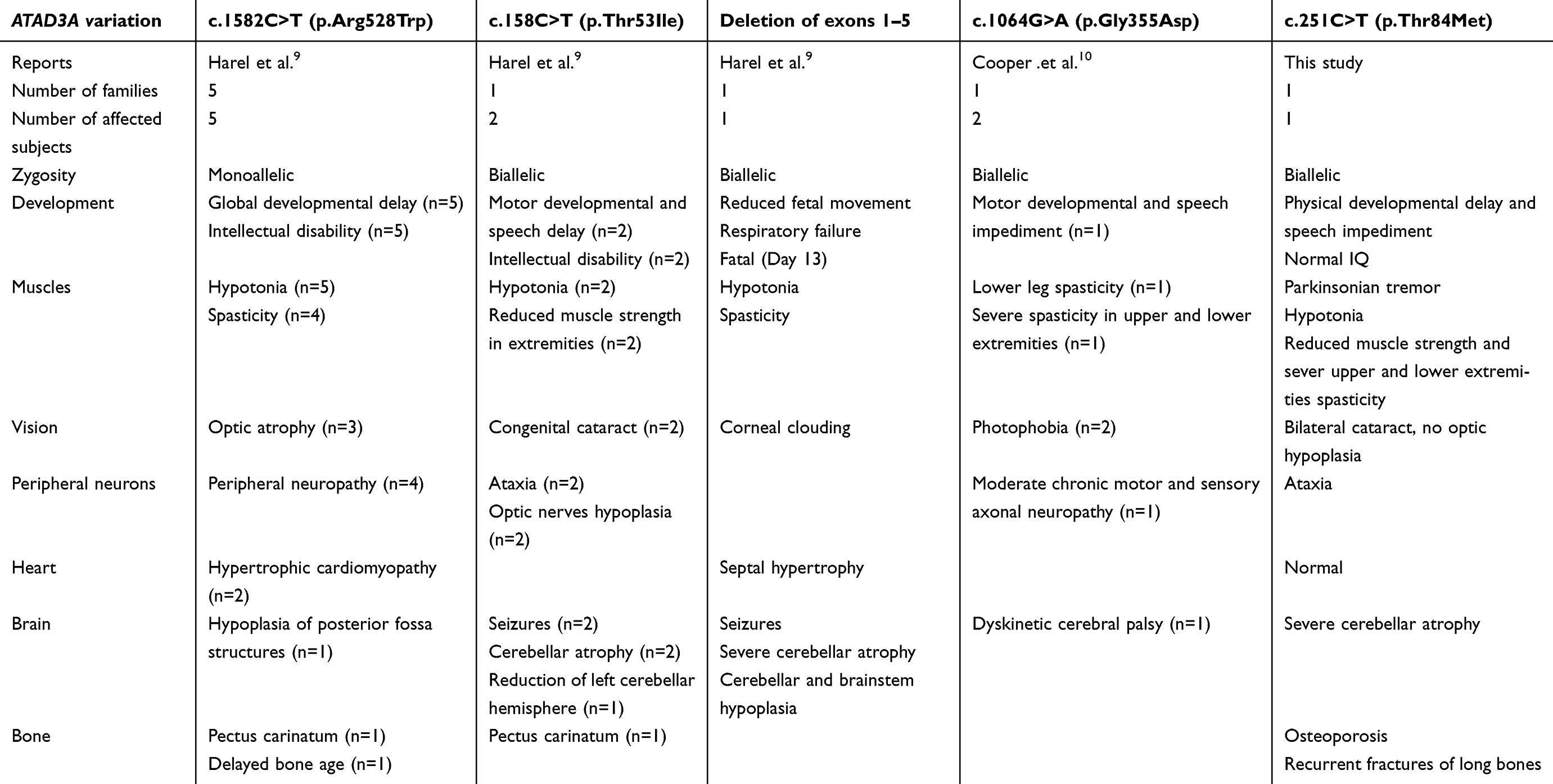 | Table 1 Comparison of phenotypes associated with mutations in the ATAD3 gene family |
ATAD3A regulates the mitochondrial dynamics at the interface of the inner and outer membranes and has been associated with nucleoid organization and mtDNA stability.3,4 ATAD3A variants or locus deletions have been found to be linked with mtDNA fragmentation and changes in cholesterol metabolism.4–6,10,11 The exonic SNV identified in the present report (c.251C>T; p.Thr84Met), which varies from that noted by Harel et al., has not been reported in a homozygous state in any public database, and its heterozygous reported frequency is rare (1/30,780).12 Furthermore, the missense mutation leading to a change in residue 84 within a highly conserved region significantly altered the structure of the ATAD3A protein. The conserved Thr84 is located in the N-terminus of the ATAD3A protein, adjacent to the coiled-coil domain, which is essential for mitochondrial protein interactions.13 Thus, the biallelic mutation identified in our subject may potentially lead to a functional alteration of ATAD3A that may render the protein non-functional, ultimately resulting in mitochondrial malfunction. Ketogenic diet (KD), characterized by high-fat and low-carbohydrate food intake, is well established in the treatment of pharmaco-resistant epilepsy,14 and its use for other neurological conditions is emerging.15 Particularly, evidence suggests that KD diets may benefit patients with neurological disorders resulting from mitochondriopathies by improving mitochondrial function and stimulating mitochondriogenesis.15 Indeed, KD has been shown to improve the brain energy metabolism and upregulate mitochondrial biogenesis,16 reduce the production of reactive oxygen species17 and increase ATP production.18 Given its potential therapeutic benefit in patients with mitochondrial diseases, we introduced the subject to KD.
During KD therapy, the subject’s behavior and physical performance were evaluated, using therapist-designed assessment forms (Figure 4 and Supplementary materials). Following KD initiation, there was a statistically significant improvement in the subject’s vitality and interaction with others relative to baseline observations. The subject also moaned significantly less during this period. Compared with baseline observations, there was a statistically significant improvement in the subject’s ability to walk and carry objects during KD treatment. The subject also displayed a visible, yet non-significant, reduction in hand tremor during KD treatment. Noticeably, when the subject felt ill due to an unrelated reason and stopped the treatment, her behavior and physical abilities significantly worsened (by 1.2- to 2.2-fold, relative to the KD treatment period). When the subject recovered and KD was re-introduced, the subject regained her vitality, interaction with peers, and showed reduced moaning (improving 2.1-, 1.5- and 1.35- fold, respectively, relative to the non-treatment period). Her physical abilities also recovered after KD re-introduction, with the subject displaying a significantly improved ability to walk and to carry objects, by 1.57- and 1.42- fold, respectively, compared with the non-treatment period.
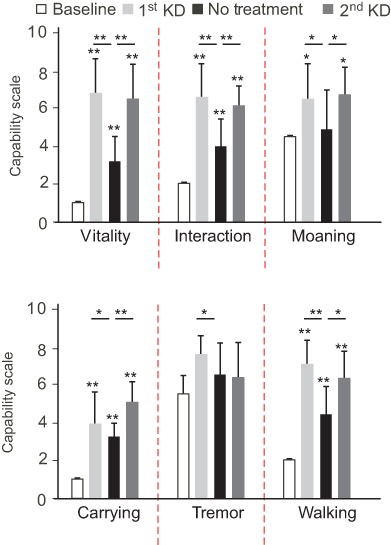 | Figure 4 Progression of the subject’s physical skills and behavior before and after ketogenic diet (KD) over the course of 127 days. Assessment was made on a discrete scale of 0 to 10, where 0 defined visible worsening and 10 visible improvement. Data are displayed as mean ± standard deviation. *P < 0.05; **P < 1X10−6 as calculated by a one-tailed Student’s t-test assuming unequal variance. |
We then investigated whether the general improvement observed in our subject following KD treatment was associated with changes in the cerebellum. MRI sections of both the axial– and coronal–T2WI taken 12 months post-KD initiation (age 5 years and 5 months, Figure 5A and B) showed no progression of the cerebellar and vermis atrophy, compared with the previous MRI (June 2016, Figure 1D and E). There were also no significant changes in the distribution of the patchy high signal intensity involving these tissues (Figure 5A and B vs Figure 1D and E). Furthermore, compared with the MRI taken in 2016, the sizes of the right and left cerebellar hemispheres were maintained (Figure 5B vs Figure 1E), and there was a slight increase in the relative sagittal length of cerebellum (Figure 5C vs Figure 1F). Brain DWI sequences showed no diffusion restrictions (Figure S2B). In contrast with the previous MRI data, which showed a significant deterioration of the cerebellum between 2014 and 2016, the MRI taken following KD initiation showed a slower progression of the cerebellum atrophy.
 | Figure 5 Magnetic resonance imaging (MRI) scans of the subject’s brain after 12 months of ketogenic diet. |
In conclusion, we describe for the first time a case of a subject with biallelic variation, rs546711654, at the ATAD3A locus, associated with progressive cerebellar and vermis atrophy, which caused developmental and cognitive delay. While the ATAD3A c.251C>T (p.Thr84Met) SNV had been previously reported, no clinical phenotypes had been associated with it, nor has it been reported in a homozygous state in any database. We propose that this SNV leads to the structural and functional alteration of the ATAD3A protein, compromising normal mitochondrial dynamics and function. Functional studies to further investigate the effects of this mutation are currently underway in our research group. Remarkably, improved motor and mental capacities were seen after KD treatment, suggesting that KD may provide an effective short-term therapy to improve quality of life in patients with mitochondrial disorders.
Abbreviations list
ATAD3A, ATPase AAA-domain containing 3 gene A; dbSNP, The Single Nucleotide Polymorphism database; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery sequences; KD, ketogenic diet; MRI, magnetic resonance imaging; T2WI, T2-weighted imaging; Thr84Met, threonine 84 methionine.
Ethics and consent
The study was pre-approved by the Ethical Review Committee of Dasman Diabetes Institute (protocol No: RA-2011-007) in accordance with the World Medical Association Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Informed written consent and minor assent (mandatory for participants below 21 years old) were obtained from the family and the affected subject, respectively, with permission to use the case details and accompanying images for publication purposes.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Facecchia K, Fochesato LA, Ray SD, Stohs SJ, Pandey S. Oxidative toxicity in neurodegenerative diseases: role of mitochondrial dysfunction and therapeutic strategies. J Toxicol. 2011;2011:683728. doi:10.1155/2011/683728
2. He J, Mao CC, Reyes A, et al. The AAA + protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J Cell Biol. 2007;176(2):141–146. doi:10.1083/jcb.200609158
3. He J, Cooper HM, Reyes A, et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012;40(13):6109–6121. doi:10.1093/nar/gks266
4. Desai R, Frazier AE, Durigon R, et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain. 2017;140(6):1595–1610. doi:10.1093/brain/awx094
5. Issop L, Fan J, Lee S, et al. Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology. 2015;156(1):334–345. doi:10.1210/en.2014-1503
6. Rone MB, Midzak AS, Issop L, et al. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol. 2012;26(11):1868–1882. doi:10.1210/me.2012-1159
7. Li S, Rousseau D. ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J Bioenerg Biomembr. 2012;44(1):189–197. doi:10.1007/s10863-012-9424-5
8. Goller T, Seibold UK, Kremmer E, Voos W, Kolanus W. Atad3 function is essential for early post-implantation development in the mouse. PLoS One. 2013;8(1):e54799. doi:10.1371/journal.pone.0054799
9. Harel T, Yoon WH, Garone C, et al. Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am J Hum Genet. 2016;99(4):831–845. doi:10.1016/j.ajhg.2016.08.007
10. Cooper HM, Yang Y, Ylikallio E, et al. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum Mol Genet. 2017;26(8):1432–1443. doi:10.1093/hmg/ddx042
11. Gilquin B, Cannon BR, Hubstenberger A, et al. The calcium-dependent interaction between S100B and the mitochondrial AAA ATPase ATAD3A and the role of this complex in the cytoplasmic processing of ATAD3A. Mol Cell Biol. 2010;30(11):2724–2736. doi:10.1128/MCB.01468-09
12. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi:10.1038/nature19057
13. Gilquin B, Taillebourg E, Cherradi N, et al. The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol Cell Biol. 2010;30(8):1984–1996. doi:10.1128/MCB.00007-10
14. Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev. 2012;3:CD001903.
15. Paoli A, Bianco A, Damiani E, Bosco G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int. 2014;2014:474296. doi:10.1155/2014/474296
16. Bough KJ, Wetherington J, Hassel B, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60(2):223–235. doi:10.1002/ana.20899
17. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145(1):256–264. doi:10.1016/j.neuroscience.2006.11.065
18. Masino SA, Geiger JD. Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci. 2008;31(6):273–278. doi:10.1016/j.tins.2008.02.009
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.