Back to Journals » Journal of Inflammation Research » Volume 14
KCa3.1 Inhibition of Macrophages Suppresses Inflammatory Response Leading to Endothelial Damage in a Cell Model of Kawasaki Disease
Authors Zheng F, Tao Y ![]() , Liu J, Geng Z, Wang Y, Wang Y, Fu S, Wang W, Xie C, Zhang Y, Gong F
, Liu J, Geng Z, Wang Y, Wang Y, Fu S, Wang W, Xie C, Zhang Y, Gong F
Received 12 December 2020
Accepted for publication 10 February 2021
Published 5 March 2021 Volume 2021:14 Pages 719—735
DOI https://doi.org/10.2147/JIR.S297131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Fenglei Zheng,* Yijing Tao,* Jingjing Liu, Zhimin Geng, Ying Wang, Yujia Wang, Songling Fu, Wei Wang, Chunhong Xie, Yiying Zhang, Fangqi Gong
Department of Cardiology, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, 310052, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fangqi Gong
Department of Cardiology, Children’s Hospital, Zhejiang University School of Medicine; National Clinical Research Center for Child Health, No. 3333, Binsheng Road, Hangzhou, 310052, People’s Republic of China
Tel/Fax +86-571-86670008
Email [email protected]
Purpose: Macrophages-mediated inflammation is linked with endothelial damage of Kawasaki disease (KD). KCa3.1, a calcium-activated potassium channel, modulates inflammation of macrophages. However, little is known about the role of KCa3.1 in inflammation by macrophages involved in KD. Hence, this study is aimed to explore the potential role of KCa3.1 in regulating inflammatory response by macrophages and subsequent vascular injury in an in vitro model of KD.
Methods: RAW264.7 cells were stimulated with Lactobacillus casei cell wall extract (LCWE) with or without TRAM-34 or PDTC or AG490. Subsequently, mouse coronary artery endothelial cells (MCAECs) were incubated with RAW264.7 cells-conditioned medium to mimic local inflammatory lesions in KD. CCKi8 assay was used to evaluate cell viability. The mRNA levels of inflammatory mediators were detected by qRT-PCR. Expressions of KCa3.1, MCAECs injury-associated molecules, proteins involved in signal pathways of nuclear factor-κB (NF-κB), signal transducers and activators of transcription (STAT) 3 and p38 were evaluated by Western blot.
Results: Our study showed that LCWE increased KCa3.1 protein level in RAW264.7 macrophages and KCa3.1 inhibition by TRAM-34 notably suppressed the expression of pro-inflammatory molecules in LCWE-treated macrophages via blocking the activation of NF-κB and STAT3 pathways. Besides, the inflammation and damage of MCAECs were attenuated in the TRAM-34-treated group compared with the KD model group. This vascular protective role was dependent on the down-regulation of NF-κB and STAT3 signal pathways, which was confirmed by using inhibitors of NF-κB and STAT3.
Conclusion: This study demonstrates that KCa3.1 blockade of macrophages suppresses inflammatory reaction leading to mouse coronary artery endothelial cell injury in a cell model of KD by hampering the activation of NF-κB and STAT3 signaling pathway. These findings imply that KCa3.1 may be a potential therapeutic target for KD.
Keywords: Kawasaki disease, macrophages, inflammation, KCa3.1, vascular damage
Introduction
Kawasaki disease (KD), an acute febrile systemic vasculitis of children under 5 years old, is characterized by an overreaction of immune system and excessive release of inflammatory factors, which will lead to coronary arterial abnormalities.1 Up to 25% of untreated patients will develop coronary aneurysms, and 3–4% of patients who treated with intravenous immunoglobulin (IVIG) plus aspirin still suffer from coronary artery lesions.2,3 Moreover, 16.5% of KD patients are unresponsive to the initial IVIG therapy.4 Despite many additional therapies, such as IVIG re-administration,5 steroid or steroid pulse therapy,6,7 infliximab,8,9 cyclosporine A,10,11 and plasma exchange,12 are available, coronary artery lesions continue to develop in many KD patients. Therefore, there is an urgent need to develop an alternative therapeutic target for KD.
Emerging evidences suggest that KD is a macrophage-related vascular disease.13–17 Macrophages can be broadly classified into classically activated macrophage (M1) or alternately activated macrophage (M2) phenotypes.18 M1 macrophages accelerate atherosclerotic lesion development and complexity via its pro-inflammatory effects, whereas the M2 macrophages reduce the inflammatory reaction to promote tissue repair and healing.19 M1 phenotype is found to be more pronounced than M2 in coronary artery lesions of KD, which indicates that pro-inflammatory M1 macrophages may contribute to vascular damages in acute KD.17 In addition, the cytokine factors released by macrophages are responsible for coronary artery injury during acute stage of KD.20 In a mouse model of KD, it has been shown that the mice with defective macrophages fail to develop the coronary arteritis after a single injection of Lactobacillus casei cell wall extract (LCWE).21 Meanwhile, several studies have demonstrated that LCWE-induced inflammatory factors in macrophages are involved in vascular damage in the mouse model of KD.22–24 All these studies reveal that macrophages-mediated inflammation serves a crucial role in the pathogenesis of coronary arteritis in KD. Therefore, treatment aimed at repressing inflammatory mediators by macrophages provides a promising therapeutic option for KD.
KCa3.1, an intermediate conductance calcium-activated potassium channel, regulates the intracellular Ca2+ concentration via K+ efflux of maintaining a negative membrane potential.25 KCa3.1 activation helps to sustain calcium entry into the cell and is critical for the inflammatory response in macrophages.26–29 KCa3.1-mediated elevation of intracellular calcium is essential for the production of inflammatory cytokines and chemokines by macrophages.26 Most importantly, TRAM-34, a selective inhibitor of KCa3.1, dramatically reduces the expression of pro-inflammatory genes during macrophage polarization and inhibits macrophage differentiation toward the M1 phenotype.26 KCa3.1 is considered as an attractive molecular target in many macrophage-associated disorders, such as asthma, multiple sclerosis and stroke.28,30,31 As for cardiovascular diseases, previous evidences have demonstrated that pharmacological inhibition or gene silencing of KCa3.1 attenuated atherosclerotic lesion formation as well as inflammatory response in mouse models of atherosclerosis.32 However, the potential role and mechanism of KCa3.1 in KD is yet to be elucidated. We hypothesize that blockade of KCa3.1 could reduce the expression of pro-inflammatory mediators and then attenuate inflammation-mediated endothelial injury in an in vitro model of KD.
In this study, we used LCWE-treated RAW264.7 cells-conditioned medium (RAW-CM) to induce mouse coronary artery endothelial cells (MCAECs) injury as a cell model of KD to investigate the possible role and mechanisms of KCa3.1 in regulating inflammation leading to vascular damage in KD. Our results showed that KCa3.1 blockade attenuated the expression of pro-inflammatory mediators via interfering with nuclear factor-kappa B (NF-κB) and signal transducers and activators of transcription (STAT) 3 signaling in LCWE-stimulated macrophages. Moreover, KCa3.1 inhibitor TRAM-34-treated RAW-CM alleviated the inflammatory damage of the MCAECs compared with LCWE-treated RAW-CM via the inactivation of NF-κB and STAT3 signal pathways. All these results suggest that a promising beneficial effect of KCa3.1 inhibition for the treatment of KD.
Materials and Methods
Inhibitors and Antibodies
KCa3.1 inhibitor TRAM-34, NF-κB inhibitor PDTC and STAT3 inhibitor AG490 were purchased from MCE (Shanghai, China). All these inhibitors dissolved in DMSO (Sigma-Aldrich, St. Louis, MO, USA). Antibodies against KCa3.1 (60276-1-Ig) and Interleukin (IL)-1β (16806-1-AP) were purchased from Proteintech (Wuhan, China). Antibodies for inducible nitric oxide synthase (iNOS) (ab178945), matrix metalloproteinase (MMP)-9 (ab228402), vascular cell adhesion molecule (VCAM)-1 (ab134047), STAT3 (ab68153), phospho (p)-STAT3 (ab76315), horseradish peroxidase (HRP)-conjugated IgG (ab205718, ab205719) were purchased from Abcam (Cambridge, MA, USA). Antibodies against β-tubulin (15115S), NF-κB p65 (8242T), p-NF-κB p65 (3033T), p38 MAPK (8690T) and p-p38 MAPK (4511T) were purchased from CST (Danvers, MA, USA). Antibodies for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (BL006B) and β-actin (BL005B) were purchased from Biosharp (Hefei, China). Primary antibody for Histone H3 (EM30605) was purchased from HUABIO (Hangzhou, China)
Preparation of LCWE
LCWE was prepared as previously reported by Lehman et al21 In brief, L. casei (ATCC, Manassas, VA, USA) was cultured in Lactobacilli MRS broth (BD Systems, Franklin Lakes, NJ, USA). The bacteria were harvested by centrifugation during the log phase of growth and lysed with twice packed volumes of 4% SDS (Sigma-Aldrich, St. Louis, MO, USA) overnight. In order to remove any adherent material from the cell wall, sequential incubations with 250 mg/mL RNase, DNase I and trypsin (Sigma-Aldrich, St. Louis, MO, USA) were performed. The cell-wall fragments were then fragmented with JY92-IIN Ultrasonic Homogenizer (Scientz, Ningbo, China) in a dry ice/ethanol for 2 h. Following 1 h centrifugation at 20,000 rpm, the pellet was discarded and the supernatant was retained. The final concentration of rhamnose in the supernatant was determined using phenol-sulphuric colorimetric determination assay (GenMed Scientifics, Shanghai, China) according to the manufacturer’s instructions and expressed in mg/mL in phosphate-buffered saline (PBS).
Experimental Mice
A total of 5 wild-type 4-6-week-old male C57BL/6 mice were purchased from the Laboratory Animal Center of Zhejiang University and were housed under specific pathogen-free conditions (40–70% humidity, 20–26°C, 12-h light/dark cycle with full-valence granular mice feedstuff and sterile water). All animal experiments were performed under an animal protocol approved by the Animal Care Committee at Zhejiang University (approval number: ZJU2015-501-01).
Cell Culture and Treatment
Both mouse RAW264.7 cells (Cell Bank of the Chinese Academy of Science, Shanghai, China) and primary MCAECs (CHI Scientific Inc, Maynard, MA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), 1% penicillin-streptomycin (Gibco, Grand Island, NY, USA) at 37 °C in a humidified incubator with atmosphere of 5% CO2. RAW264.7 cells in 6-well plates or 75-cm2 flasks were stimulated by LCWE (1 μg/mL) 12 h with or without pretreatment with TRAM-34 (1, 5, 10 μM) or PDTC (100, 200, 500, 1000 μM) or AG490 (1, 5, 10 μM) for 30 min.
Co-Culture of RAW264.7 Cells and MCAECs
RAW264.7 cells in 6-well plates were pretreated with TRAM-34 (10 μM) or PDTC (1000 μM) or AG490 (10 μM) for 30 min, and then stimulated by LCWE (1 μg/mL) for 12 h. Subsequently, MCAECs in 6-well plates were incubated with RAW264.7 cells-conditioned medium for 12 h to establish a MCAECs injury model.
Isolation, Culture and Treatment of Mouse Primary Peritoneal Macrophage
Mouse primary peritoneal macrophages (PMΦs) were harvested as previously described by I-Chun Lin et al.24 Briefly, 4-6-week-old male C57BL/6 mice were intraperitoneally injected with 2 mL 3% thioglycolate medium (Merck, Darmstadt, Germany) for 3 days. Peritoneal macrophages were isolated by washing the peritoneal cavity with 5 mL sterile PBS for 3 times. The peritoneal fluid of each mouse was collected separately. After 5 min centrifugation at 1200 rpm, the supernatant was discarded and the cell pellet was suspended in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were seeded into 6-well plates and allowed to adhere the culture plates overnight at 37 °C under a 5% CO2 atmosphere. Non-adherent cells were removed using PBS, and the remaining adherent cells were treated with TRAM-34 (10 μΜ) for 30 min, and then stimulated by LCWE (1 μg/mL) for 12 h.
Cell Viability
Cell viability was measured by cell counting kit (CCK) 8 assay. Briefly, RAW264.7 cells or MCAECs were seeded into 96-well plates at a density of 5×103 cells per well. RAW264.7 cells were treated with TRAM-34 at different concentrations (0, 1, 5, 10, and 15 μΜ) in the presence or absence of LCWE (1 μg/mL). MCAECs were incubated with RAW-CM from different groups as mentioned above. Following by 12 h of incubation, the medium was added with 10 μL CCK (YeaSen, Shanghai, China) for 3 h. The optical density at a wavelength of 450 nm was measured by using a spectrophotometer (Merinton, Beijing, China).
Quantitative Realtime-Polymerase Chain Reaction (qRT-PCR)
Total RNA in cultured cells was extracted by TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) followed a standard chloroform extraction method. The RNA was converted to cDNA with PrimeScript TM RT Master Mix (Takara, Dalian, China) as described in the manufacturer’s instructions. qRT-PCR was performed using the Applied Biosystems 7500 real-time PCR system. The condition of qRT-PCR was 95 °C for 2 min and 40 cycles of 15 s at 95 °C and 34 s at 60 °C. The primers for qRT-PCR were listed in Table 1. The relative mRNA level was calculated using the 2(−ΔΔ Ct) method.
 |
Table 1 Primers Used for qRT-PCR |
Nitric Oxide (NO) Assay
NO production was detected by Griess reagent (Beyotime Biotechnology, Shanghai, China). RAW264.7 cells seeded in 24-well plates were pretreated with TRAM-34 (1, 5, 10 μM) or PDTC (100, 200, 500, 1000 μM) or AG490 (1, 5, 10 μM) for 30 min prior to the addition of LCWE (1 μg/mL) for 12 h. The 100 μL supernatant was transferred and mixed with 100 μL Griess reagent in 96-well plates. After 10-min incubation at 37 °C in the dark, the absorbance was measured at 540 nm by using a spectrophotometer (Merinton, Beijing, China)
Western Blotting
Total proteins were extracted by RIPA lysis buffer containing protease and phosphatase inhibitors (Beyotime Biotechnology, Shanghai, China). Cytoplasmic and nuclear proteins were obtained using a nuclear and cytoplasmic protein extraction kit (Beyotime Biotechnology, Shanghai, China). The concentration of protein was determined by a BCA protein assay kit (Thermo Fisher, Waltham, MA, USA). Equal protein amounts were separated by 4–20% SDS-PAGE gels (GenScript, Nanjing, China) and then transferred to polyvinylidene fluoride (PVDF) membranes (Merck-Millipore, Darmstadt, Germany). The membranes were blocked in QuickBlock buffer (Beyotime Biotechnology, Shanghai, China) for 15 min at room temperature, then were incubated with the primary antibodies at 4 °C overnight. The membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. The blots were visualized by a chemiluminescence reagent (Thermo Fisher, Waltham, MA, USA). The optical density of each band was quantified with ImageJ software (NIH, Bethesda, MD, USA).
Statistical Analysis
Data were expressed as mean ± SD and analyzed with Graph Pad Prism7 software (La Jolla, CA, USA). A two-tailed unpaired Student’s t-test was used to compare two groups, and a comparison of more than two groups was carried out using single-factor ANOVA. Differences with a p value less than 0.05 were considered as statistically significant. All experiments were performed at least three times.
Results
LCWE Increases KCa3.1 Level in RAW264.7 Cells
KCa3.1, a pro-inflammatory ion channel, is significantly increased in pro-inflammatory M1 macrophages. LCWE has been reported to be a strong inducer for macrophage inflammation. Hence, we first investigated whether LCWE upregulated KCa3.1 level in RAW264.7 macrophages. As expected, the result of Western blot showed that KCa3.1 protein expression was increased when RAW264.7 macrophages were stimulated by LCWE (Figure 1A).
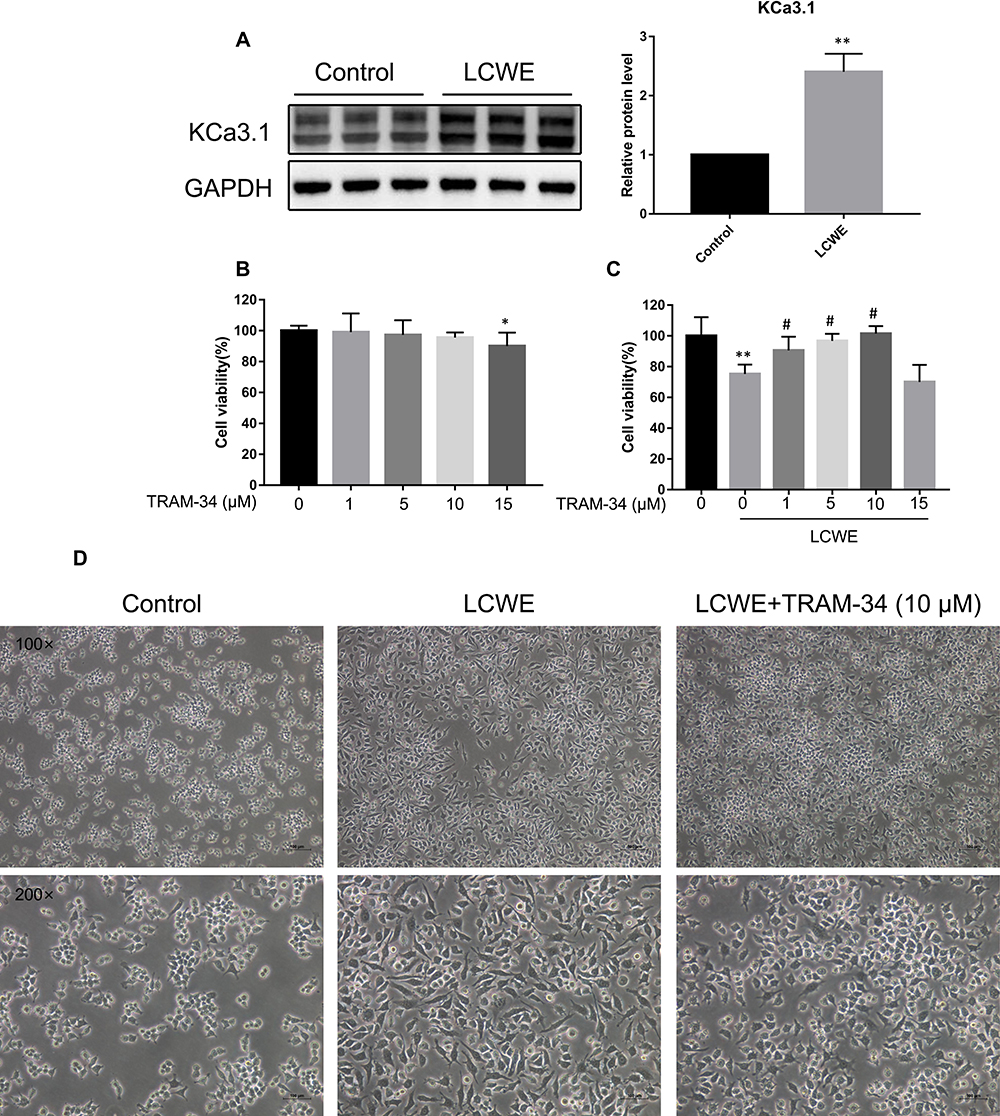 |
Figure 1 Effects of KCa3.1 inhibition by TRAM-34 on LCWE-induced cytotoxicity and activation in RAW264.7 cells. (A) RAW264.7 cells were stimulated with LCWE (1 μg/mL) for 12 h. The protein level of KCa3.1 was detected by Western blot. Results are expressed as the mean ± SD. **p < 0.01 versus control group. (B, C) Cytotoxicity of RAW264.7 cells treated by TRAM-34 (0, 1, 5, 10, 15 μM) with or without LCWE (1 μg/mL) was determined by CCK8 assay. Results (n = 5) are expressed as the mean ± SD, * p < 0.05, **p < 0.01 versus untreated group, # p < 0.05 versus LCWE-treated group. (D) The representative microphotographs showed phenotypic changes of RAW 264.7 cells in responsive to different stimuli (LCWE (1 μg/mL), TRAM-34 (10 μM)) for 12 h. Magnification = 100× or 200×. Scale bar = 100 µm. |
Effects of KCa3.1 Inhibitor TRAM-34 on RAW264.7 Cells Viability and Activation
The effect of TRAM-34 on RAW264.7 cell viability was evaluated by CCK8 assay. TRAM-34 has no significant toxicity at a concentration of ≤ 10 µM with or without LCWE treatment (Figure 1B and C). According to this result, TRAM-34 at concentrations of 1, 5, 10 µM was used in subsequent experiments. To examine the inhibitory effect of TRAM-34 on macrophage activation, optical microscopy was performed on RAW264.7 cells stimulated by LCWE with or without TRAM-34. Compared with the control cells, LCWE-stimulated RAW cells had irregular shapes, elongated pseudopodia and increased intercellular space. These morphological changes were much less in the TRAM-34-pretreated cells (Figure 1D). These findings indicate that KCa3.1 blockade by TRAM-34 inhibits LCWE-mediated macrophages activation.
KCa3.1 Blockade Suppresses Pro-Inflammatory Molecule Expression in LCWE-Stimulated RAW264.7 Cells
Inflammatory mediators, such as cytokines and chemokines, have been reported to promote the development of vascular lesions in KD.1 Blocking KCa3.1 with TRAM-34 impairs inflammatory gene expression during macrophage is polarized into pro-inflammatory M1 phenotype. Thus, we examined whether TRAM-34 could inhibit LCWE-mediated inflammatory molecule expression in RAW264.7 cells. To explore the inhibitory effect of TRAM-34 on LCWE induced pro-inflammatory gene expression, the mRNA levels of IL-1β, IL-6, tumor necrosis factor (TNF)-α and monocyte chemotactic protein (MCP)-1 were detected by qRT-PCR, respectively. The results showed that LCWE significantly increased the mRNA levels of IL-1β, IL-6, TNF-α and MCP-1 in RAW264.7 cells, and pretreatment with TRAM-34 decreased the mRNA levels of IL-1β, IL-6, TNF-α and MCP-1 in a dose-dependent manner (Figure 2A–D). To further confirm the anti-inflammatory role of TRAM-34, we examined the levels of MMP-9, iNOS and NO, which were responsible for the production of pro-inflammatory mediators and contributed to the vascular injury of LCWE-induced KD model. As shown in Figure 2E–I, TRAM-34 pretreatment drastically decreased the expression of MMP-9 as well as iNOS and NO production in LCWE-stimulated RAW264.7 cells in a dose-dependent manner. These results suggest KCa3.1 inhibition reduces the expression of pro-inflammatory mediators in LCWE-stimulated RAW264.7 macrophages.
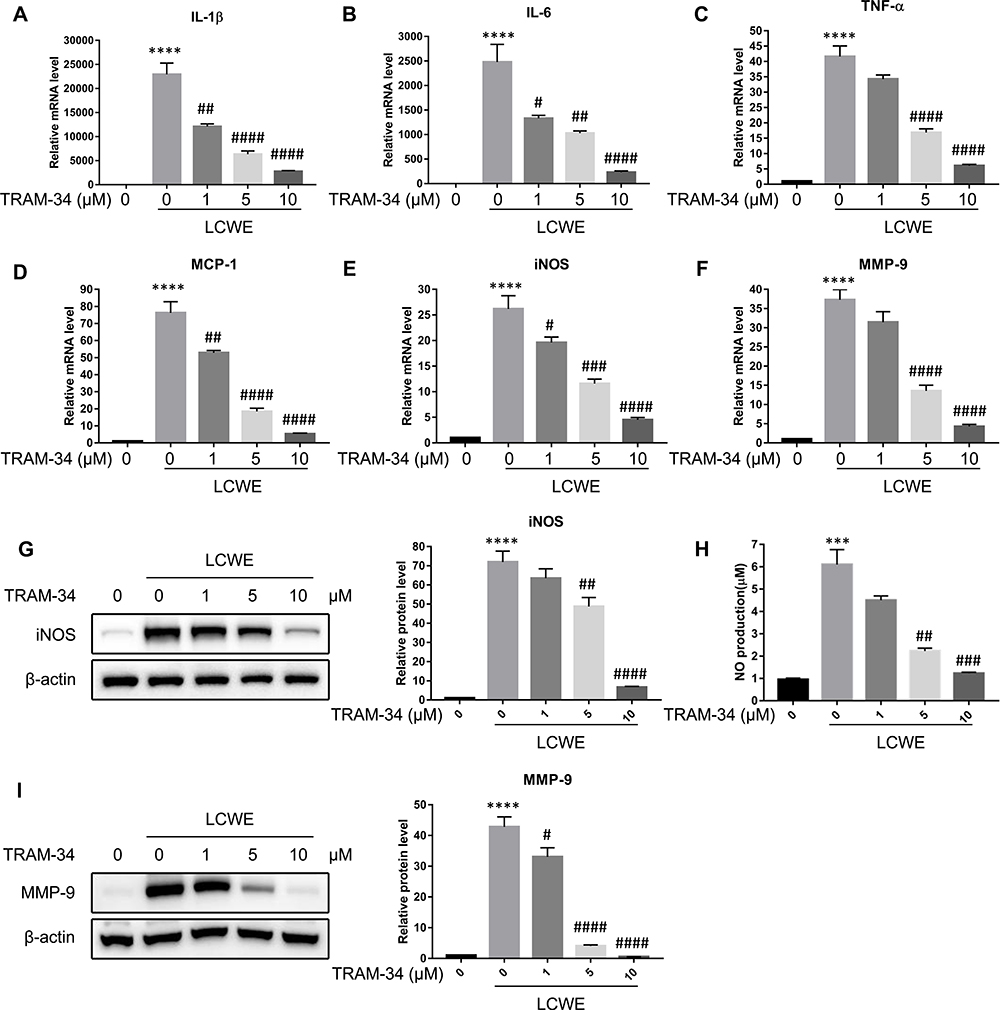 |
Figure 2 Blocking KCa3.1 with TRAM-34 inhibits LCWE-induced inflammatory response in RAW264.7 cells. RAW264.7 cells were pretreated with TRAM-34 (0, 1, 5, 10 μM) 30 min, then exposed to LCWE (1 μg/mL) for 12 h. The mRNA expressions of IL-1β (A), IL-6 (B), TNF-α (C), MCP-1 (D), iNOS (E) and MMP-9 (F) were assayed by qRT-PCR, respectively. The protein levels of iNOS (G) as well as MMP-9 (I) were detected using Western blot. (H) Culture supernatants of RAW264.7 cells were collected for NO detection by Griess reagent. All results are expressed as the mean ± SD. ***p < 0.001, ****p < 0.0001 versus untreated group; # p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus LCWE group. |
KCa3.1 Inhibition Attenuates Inflammatory Reaction in LCWE-Stimulated Mouse Peritoneal Macrophages
LCWE-induced upregulation of inflammatory mediators in PMΦs is closely associated with vascular lesions in KD murine model.24,33 Therefore, we tested whether inhibition of KCa3.1 by TRAM-34 could affect LCWE-mediated inflammatory gene expression in PMΦs. Similarly, TRAM-34 also reduced the mRNA expression of IL-1β, IL-6, TNF-α and MCP-1 in LCWE-stimulated PMΦs (Figure 3A–D). We then evaluated the role of TRAM-34 in LCWE-induced MMP-9 and iNOS expression in PMΦs. As expected, pretreatment with TRAM-34 markedly downregulated LCWE-induced MMP-9 and iNOS expression at both the mRNA and protein levels (Figure 3E–H). Moreover, as shown in Supplementary Figure 1, the phenotypic changes of LCWE stimulated-PMΦs were reversed by TRAM-34 pretreatment. These results indicate that inhibition of KCa3.1 attenuates LCWE-mediated inflammatory reaction in mouse primary peritoneal macrophages.
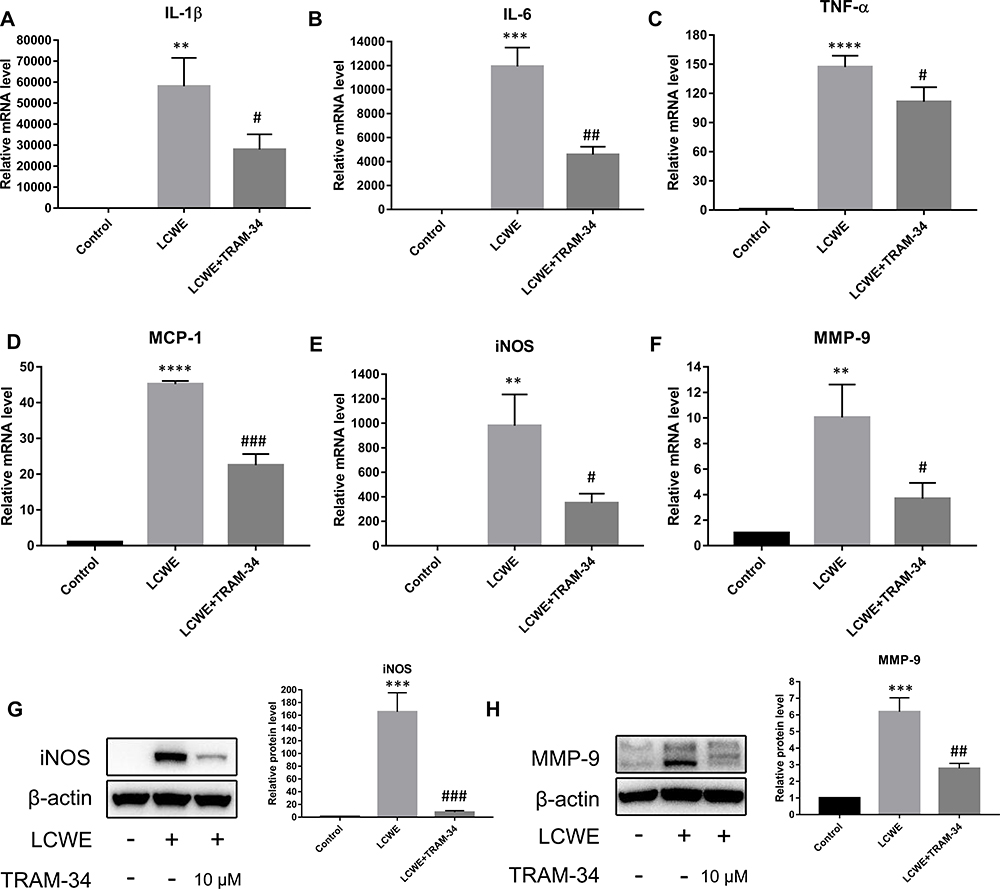 |
Figure 3 KCa3.1 inhibition by TRAM-34 suppresses inflammatory reaction in LCWE-stimulated mouse peritoneal macrophages. Mouse peritoneal macrophages were pretreated with TRAM-34 (10 μM) 30 min, then stimulated with LCWE (1 μg/mL) for 12 h. The expressions of IL-1β (A), IL-6 (B), TNF-α (C), MCP-1 (D), iNOS (E) and MMP-9 (F) at the mRNA levels were measured using qRT-PCR, respectively. Effects of TRAM-34 pretreatment on the protein expressions of iNOS (G) and MMP-9 (H) were analyzed by Western blot. All results are expressed as the mean ± SD. **p < 0.01, ***p < 0.001, ****p < 0.0001 versus control group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus LCWE group. |
Inhibition of KCa3.1 Blocks LCWE-Induced NF-κB and STAT3 Activation in RAW264.7 Cells
NF-κB/STAT3/p38 signaling plays a critical role in the transcription of those inflammatory mediators mentioned above. Thus, we investigated the effect of KCa3.1 inhibition on activation of NF-κB, STAT3 and p38 pathways in LCWE-induced RAW264.7 cells by Western blot. Western blotting analysis showed that LCWE increased nuclear protein levels of NF-κB p65 and reduced its cytoplasmic protein levels compared with the control group. This phenomenon was significantly attenuated by TRAM-34 pretreatment (Figure 4A). Meanwhile, as shown in Figure 4B and C, the phosphorylation of STAT3 induced by LCWE was decreased by TRAM-34 in a concentration-dependent manner, whereas the phosphorylation of p38 induced by LCWE was not altered. These findings suggest that inhibition of KCa3.1 suppresses LCWE-induced inflammatory response in macrophages by blocking NF-κB nuclear translocation and phosphorylation activation of STAT3.
 |
Figure 4 KCa3.1 blockade by TRAM-34 inhibits the activation of NF-κB and STAT3 caused by LCWE stimulation in RAW264.7 cells. RAW264.7 cells were stimulated with LCWE (1 μg/mL) for 12 h after 30 min pretreatment of TRAM-34 (0, 1, 5, 10 μM). (A) The nuclear or cytoplasmic protein levels of NF-κB p65 were detected by Western blot. (B) The expressions of p-STAT3 and STAT3 were examined via Western blot. (C) The protein levels of p-p38 and p38 were assayed by Western blot. All results are expressed as the mean ± SD. *p < 0.01, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus untreated group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus LCWE group. |
NF-κB and STAT3 Pathways are Involved in LCWE-Induced Inflammatory Response in RAW264.7 Cells
To further determine whether the NF-κB and STAT3 signaling pathways mediate LCWE-induced inflammatory response in RAW264.7 cells, cultures were co-incubated with NF-κB inhibitor PDTC or STAT3 inhibitor AG490. PDTC effectively inhibited the expressions of inflammatory genes by hampering nuclear translocation of NF-κB p65 in LCWE-stimulated RAW264.7 cells (Figure 5). Similarly, LCWE-induced the expressions of pro-inflammatory mediators in RAW264.7 cells were reduced by AG490 in a dose-dependent manner (Figure 6). These results imply that the signaling via NF-κB and STAT3 pathways are indispensable for LCWE-mediated inflammatory response in RAW264.7 cells.
 |
Figure 5 NF-κB signal pathway is necessary for LCWE-induced inflammatory reaction in RAW264.7 cells. RAW264.7 cells were pretreated with PDTC (0, 100, 200, 500, 1000 μM) 30 min before stimulation with LCWE (1 μg/mL) for 12 h. qRT-PCR was performed to test the mRNA expressions of IL-1β (A), IL-6 (B), TNF-α (C), MCP-1 (D), iNOS (E) and MMP-9 (F) in RAW264.7 cells, respectively. The protein expressions of iNOS (G), MMP-9 (I) and NF-κB p65 at cytoplasm or nucleus (J) were analyzed by Western blot, respectively. (H) NO production was detected by Griess reagent. All results are expressed as the mean ± SD. **p < 0.01, ***p < 0.001, ****p < 0.0001 versus untreated group; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus LCWE group. |
 |
Figure 6 STAT3 signal pathway is involved in LCWE-induced inflammatory response in RAW264.7 cells. RAW264.7 cells were pretreated with AG490 (0, 1, 5, 10 μM) 30 min prior to the stimulation with LCWE (1 μg/mL) for 12 h. qRT-PCR was conducted to analyze the relative mRNA levels of IL-1β (A), IL-6 (B), TNF-α (C), MCP-1 (D), iNOS (E) and MMP-9 (F) in RAW264.7 cells, respectively. The relative protein levels of iNOS (G), MMP-9 (I), p-STAT3 and STAT3 (J) were examined by Western blot, respectively. (H) NO production was detected by Griess reagent. All results are expressed as the mean ± SD. ***p < 0.001, ****p < 0.0001 versus untreated group; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus LCWE group. |
KCa3.1 Inhibition of Macrophages Abates MCAECs Inflammation Induced by LCWE-Treated RAW-CM
As for the LCWE disease model, mouse vascular endothelial damage is mainly mediated by cytokine factors produced by macrophages.34 Thus, to re-create the inflammatory environment surrounding endothelial cells in KD model, we utilized LCWE-treated RAW-CM to stimulate MCAECs to establish an in vitro model of endothelial cell injury. We first examined whether KCa3.1 inhibition of macrophages by TRAM-34 suppressed inflammatory reaction in MCAECs induced by LCWE-treated RAW-CM. Results of qRT-PCR demonstrated that the mRNA levels of IL-1β, TNF-α and MCP-1 in MCAECs were significantly upregulated by LCWE-treated RAW-CM and reduced by TRAM-34 or PDTC or AG490-treated RAW-CM (Figure 7). These results indicate that inhibition of KCa3.1 in macrophages alleviates the expressions of inflammatory mediators at the transcriptional level in MCAECs.
 |
Figure 7 KCa3.1 inhibition of macrophages alleviates MCAECs inflammation induced by LCWE-treated RAW-CM. MCAECs were incubated with RAW264.7 cells-conditioned medium from various groups (Control, LCWE, LCWE+TRAM-34, LCWE+PDTC, LCWE+AG490) for 12 h. Expressions of IL-1β (A), TNF-α (B), MCP-1 (C) at the mRNA levels was examined by qRT-PCR. All results are expressed as the mean ± SD. ***p < 0.001, ****p < 0.0001 versus control group; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus LCWE group. |
KCa3.1 Inhibition of Macrophages Protects MCAECs Against LCWE-Treated RAW-CM-Induced Injury
Next, we investigated the effects of KCa3.1 blockade in macrophages by TRAM-34 on LCWE-treated RAW-CM-mediated MCAECs injury. Compared with the control group, LCWE-treated RAW-CM induced a reduction in cell viability of MCAECs, which was reversed by TRAM-34 or PDTC or AG490-treated RAW-CM (Figure 8A). Significant markers of mouse vascular lesion in the LCWE-induced KD model,34,35 including VCAM-1, MMP-9 and IL-1β, were increased in MCAECs after exposure to LCWE-treated RAW-CM, while these MCAECs injury-associated molecules were decreased by TRAM-34 or PDTC or AG490-treated RAW-CM (Figure 8B). KCa3.1 protein level in MCAECs was upregulated by LCWE-treated RAW-CM and reduced in TRAM-34 or PDTC or AG490-treated RAW-CM group (Figure 8B), which further certified that KCa3.1 is a pro-inflammatory ion channel. Furthermore, we examined whether NF-κB and STAT3 pathways mediated MCAECs damage in this KD model. As shown in Figure 8C and D, phosphorylation of NF-κB and STAT3 was markedly induced by stimulation with LCWE-treated RAW-CM, which was notably attenuated by treatment with TRAM-34 or PDTC or AG490-treated RAW-CM. These results identify that the protective effects of KCa3.1 inhibition against LCWE-treated RAW-CM-induced injury are linked with the inactivation of NF-κB and STAT3 pathways.
 |
Figure 8 KCa3.1 inhibition of macrophages attenuates MCAECs injury mediated by LCWE-treated RAW-CM. MCAECs were incubated with RAW264.7 cells-conditioned medium from various groups (Control, LCWE, LCWE+TRAM-34, LCWE+PDTC, LCWE+AG490) 12 h for the detection of cell viability, MCAECs injury-related protein and STAT3 phosphorylation, or 1 h for the detection of phosphorylation of NF-κB p65. (A) Cell survival of MCAECs was determined by using CCK8 assay (n=5). (B) Expressions of VCAM-1, MMP-9, IL-1β and KCa3.1 were analyzed by Western blot. (C) Expressions of p-p65 and p65 at protein levels were measured by Western blot. (D) The protein levels of p-STAT3 and STAT3 were detected by Western blot. All results are expressed as the mean ± SD. ***p < 0.001, ****p < 0.0001 versus control group; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus LCWE group. |
Discussion
In this study, we first established a vascular injury model of KD using co-culture of LCWE-treated RAW264.7 cells-conditioned medium with the mouse coronary artery endothelial cells to investigate the role of KCa3.1 in the pathogenesis of KD. KCa3.1 channel has been reported to be involved in the inflammatory response in vitro and in vivo.36 However, the potential role of KCa3.1 to modulate the inflammation on KD-related model is not clear. Our results revealed that inhibition of KCa3.1 suppressed inflammatory reaction via targeting NF-κB and STAT3 pathways in LCWE-stimulated macrophages. Subsequently, KCa3.1 inhibition of RAW264.7 macrophages by TRAM-34 attenuated mouse vascular inflammation and damage in this model of KD induced by LCWE-treated RAW-CM. Taken together, it is reasonable to use KCa3.1 as a promising molecular target for KD treatment.
Macrophage activation has been observed both in the peripheral circulation and the pathological lesions of KD patients with coronary artery lesions.14,37 Similar to the results in human patients with KD, murine macrophage activation was also seen both in vivo and in vitro after LCWE stimulation.38 Thus, immune activation of macrophage involves in local inflammatory damage of the coronary arteries in the LCWE-induced murine model of KD. In the current in vitro study, enhanced protein level of KCa3.1 which is necessary for macrophage activation was observed in LCWE-treated RAW264.7 macrophages. Meanwhile, we found that LCWE-stimulated RAW264.7 cells exhibited typical morphological changes and this phenotype was inhibited by treatment with KCa3.1 inhibitor TRAM-34, which indicated that the involvement of KCa3.1 in LCWE-induced macrophage activation. Collectively, our study not only showed that KCa3.1 upregulation is a possible indicator of macrophage activation by LCWE but also provided fundamental evidence for the therapeutic effect of KCa3.1 inhibition with TRAM-34 on LCWE-induced immune activation.
Elevated serum levels of inflammatory cytokines, such as IL-1β, IL-6, TNF-α and MCP-1, are observed in KD patients at acute phase,20,39–41 suggesting that these cytokine factors produced by macrophages may be implicated in the pathogenesis of KD. Similarly, LCWE-induced mouse coronary arterial lesions are associated with inflammatory cytokines by macrophages in this murine model of KD.33 Moreover, several cytokine antagonists have been used to improve coronary outcome in the model of KD induced by LCWE.23,42,43 Thus, treatments aimed at suppressing inflammatory mediators by macrophages have a potential therapeutic strategy for the therapy of KD. Our results confirmed that LCWE could strongly induce the expression of IL-1β, IL-6, TNF-α and MCP-1, whereas KCa3.1 blockade by TRAM-34 remarkably reduced the upregulation of IL-1β, IL-6, TNF-α and MCP-1 in LCWE-stimulated macrophages (RAW264.7 cells and mouse primary peritoneal macrophages). These findings provide the evidence to support the theory of the anti-inflammatory effects of KCa3.1 inhibition on KD model.
MMP-9, a member of matrix metalloproteases (MMPs) family, has been found to be involved in tissue destruction in KD.44 In an LCWE-induced animal model of KD, the production of MMP-9 results in elastin breakdown and aneurysm formation and its inhibition improves coronary outcome.45 NO is known to maintain normal vascular tension. However, high concentrations of NO mediated by iNOS enzyme in macrophages could result in vessel wall dilatation and lesions in acute KD.46 Likewise, NO elaborated by activated macrophages could initiate coronary arteriolar wall damage in the LCWE-treated mouse model.47 The previous study has shown that blockade of iNOS could prevent cerebral aneurysm formation through reducing NO release.48 These studies imply that agents with the ability to inhibit both the expression of MMP-9 and NO production offer a promising therapeutic strategy for KD. Interestingly, inhibition of KCa3.1 with TRAM-34 has an inhibitory effect on MMP-9 expression in TNF-α-stimulated vascular smooth muscle cells49 and LPS-induced iNOS expression and NO production in microglia are strongly inhibited by blocking KCa3.1 channels.28 The inhibitory effects of KCa3.1 inhibition by TRAM-34 on the levels of both MMP-9, iNOS and NO in LCWE-stimulated macrophages remain unknown yet. We found that TRAM-34 notably suppressed the upregulation of both MMP-9, iNOS and NO in macrophages induced by LCWE, suggesting that KCa3.1 inhibitor TRAM-34 might be investigated as a possible agent for KD therapy.
Findings of several studies suggest that activation of NF-κB is crucial in the pathological development of vasculitis during KD through regulation of inflammatory factor expression.14,15,50 NF-κB activation occurs in CD14+ monocytes/macrophages in peripheral blood mononuclear cells (PBMCs) from children with KD at acute stage.14 Furthermore, NF-κB inhibitor PDTC significantly reduces the secretion of inflammatory cytokines in human coronary artery endothelial cells induced by KD patient-extracted PBMCs-conditioned supernatants.51 LCWE is a strong inducer of NF-κB activation, while NF-κB controls the transcription of inflammatory factors mentioned above.22 KCa3.1-mediated Ca2+ influx results in an increase in cytoplasmic Ca2+ concentration, which is important to the activation of NF-κB in rat peritoneal macrophages.52 Moreover, a recent study has shown that KCa3.1 mediates the activation of the NF-κB signaling pathway in astrocytes.53 Thus, we examined the effects of KCa3.1 inhibition by TRAM-34 on LCWE-induced NF-κB activation. Our results revealed that LCWE increased the nuclear translocation of the NF-κB p65, which was alleviated by TRAM-34. Moreover, the involvement of NF-κB signal pathway in LCWE-mediated inflammatory response was further confirmed by using NF-κB inhibitor PDTC. These results suggest that the anti-inflammatory effect of KCa3.1 blockade is mainly dependent on the activation of NF-κB pathway.
It has been found that STAT3 signaling pathway mediates LPS induced inflammation in Raw264.7 macrophages.54–56 Also, targeting STAT3 pathway by its inhibitor AG490 attenuated rheumatoid arthritis57 and the anti-atherogenesis effect is partially attributed to the inhibition of STAT3 pathway.58 Meanwhile, several studies have revealed that STAT3 signaling pathway is partially involved in KD-induced vasculitis through regulating inflammatory response in endothelial cells.59,60 Accordingly, the modulation of the STAT3 signaling pathway may provide an effective therapeutic strategy for the treatment of inflammation-associated diseases. Our results showed that LCWE upregulated pro-inflammatory molecule expression in Raw264.7 macrophages through the involvement of STAT3 pathway. Meanwhile, LCWE-induced phosphorylation of STAT3 was reduced by blocking KCa3.1 channel by TRAM-34 in a dose-dependent manner. Our study implies that the effects of KCa3.1 inhibition on inflammatory reaction are regulated, at least in part, by suppressing the STAT3 signal pathway in LCWE-stimulated RAW264.7 macrophages.
As mentioned above, mouse endothelial cells are exposed to inflammatory conditions by macrophages in LCWE-induced KD model. Therefore, MCAECs were incubated with LCWE-treated RAW-CM to re-create the inflammatory environment surrounding endothelial cells in this in vitro model of KD. Previous studies have reported that endothelial cell damage and inflammation are the two pivotal pathological mechanisms for KD. Furthermore, VCAM-1, MMP-9 and IL-1β are regarded as MCAECs injury-associated molecules in LCWE-induced KD model.34,35 Hence, we examined whether KCa3.1 inhibition of RAW264.7 macrophages alleviated inflammation reaction and endothelial cell injury in this in vitro model induced by LCWE-treated RAW-CM. Since inhibition of KCa3.1 by TRAM-34 significantly suppressed LCWE-induced inflammatory response in RAW264.7 cells, it was not surprising to see that the levels of both inflammatory cytokines and endothelial cells injury-associated molecules were decreased in the TRAM-34-treated RAW-CM group compare to the LCWE-treated RAW-CM group. Meanwhile, our results also showed that MCAECs’ survival was increased in the TRAM-34-treated RAW-CM group. In addition, the involvement of both NF-κB and STAT3 pathways in mouse coronary arterial lesions was demonstrated by the PDTC and AG490-treated RAW-CM groups. These findings put forward the possibility that KCa3.1 inhibition could be used as an effective strategy in treating KD-mediated vasculitis.
In summary, our findings demonstrated that KCa3.1 blockade inhibited LCWE-induced pro-inflammatory molecules mediating vascular injury via the suppression of the NF-κB and STAT3 pathways and provided evidence for KCa3.1 as an attractive pharmacological target for KD. Of course, there are several limitations in this work. Whether inhibition of KCa3.1 is capacity of improving coronary outcomes in LCWE-induced murine model of KD in vivo remains uncertain, and requires further investigation. In addition, our study cannot exclude that other Ca2+-associated signaling pathways, such as NLRP3 inflammasome and NFAT signaling, are also involved in this in vitro model of KD. Nevertheless, our work presents the evidence that firmly demonstrates the KCa3.1 as a novel molecular target for KD treatment.
Acknowledgments
This work is supported, in part, by grants from the National Natural Science Foundation of China (No. 81670251, 81970434).
Author Contributions
All authors made substantial contributions to the conception, study design and execution, acquisition and analysis of data; took part in drafting, revising or critically reviewing this manuscript; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of this work.
Disclosure
The authors declare no conflict of interest in this work.
References
1. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of kawasaki disease: a scientific statement for health professionals from the American heart association. Circulation. 2017;135(17):17. doi:10.1161/CIR.0000000000000484
2. Newburger JW, Takahashi M, Beiser AS, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324(23):1633–1639. doi:10.1056/NEJM199106063242305
3. Kato H, Sugimura T, Akagi T, et al. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation. 1996;94(6):1379–1385. doi:10.1161/01.CIR.94.6.1379
4. Nakamura Y, Yashiro M, Uehara R, et al. Epidemiologic features of Kawasaki disease in Japan: results of the 2007–2008 nationwide survey. J Epidemiol. 2010;20(4):302–307. doi:10.2188/jea.JE20090180
5. Sundel RP, Burns JC, Baker A, Beiser AS, Newburger JW. Gamma globulin re-treatment in Kawasaki disease. J Pediatr. 1993;123(4):657–659.
6. Al-Mayouf SM. The use of corticosteroid therapy in refractory Kawasaki patients. Clin Rheumatol. 2004;23(1):11–13.
7. Furukawa T, Kishiro M, Akimoto K, Nagata S, Shimizu T, Yamashiro Y. Effects of steroid pulse therapy on immunoglobulin-resistant Kawasaki disease. Arch Dis Child. 2008;93(2):142–146.
8. Burns JC, Best BM, Mejias A, et al. Infliximab treatment of intravenous immunoglobulin-resistant Kawasaki disease. J Pediatr. 2008;153(6):833–838.
9. Nagatomo Y, Muneuchi J, Nakashima Y, et al. Effective infliximab therapy for the early regression of coronary artery aneurysm in Kawasaki disease. Int J Cardiol. 2018;271:317–321.
10. Suzuki H, Terai M, Hamada H, et al. Cyclosporin A treatment for kawasaki disease refractory to initial and additional intravenous immunoglobulin. Pediatr Infect Dis J. 2011;30(10):871–876. doi:10.1097/INF.0b013e318220c3cf
11. Hamada H, Suzuki H, Onouchi Y, et al. Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, Phase 3 trial. Lancet. 2019;393(10176):1128–1137.
12. Imagawa T, Mori M, Miyamae T, et al. Plasma exchange for refractory Kawasaki disease. Eur J Pediatr. 2004;163(4–5):263–264. doi:10.1007/s00431-003-1267-y
13. Koga M, Ishihara T, Takahashi M, Umezawa Y, Furukawa S. Activation of peripheral blood monocytes and macrophages in Kawasaki disease: ultrastructural and immunocytochemical investigation. Pathol Int. 1998;48(7):512–517. doi:10.1111/j.1440-1827.1998.tb03942.x
14. Ichiyama T, Yoshitomi T, Nishikawa M, et al. NF-kappaB activation in peripheral blood monocytes/macrophages and T cells during acute Kawasaki disease. Clin Immunol. 2001;99(3):373–377. doi:10.1006/clim.2001.5026
15. Matsubara T, Ichiyama T, Furukawa S. Immunological profile of peripheral blood lymphocytes and monocytes/macrophages in Kawasaki disease. Clin Exp Immunol. 2005;141(3):381–387. doi:10.1111/j.1365-2249.2005.02821.x
16. Hara T, Nakashima Y, Sakai Y, Nishio H, Motomura Y, Yamasaki S. Kawasaki disease: a matter of innate immunity. Clin Exp Immunol. 2016;186(2):134–143. doi:10.1111/cei.12832
17. Ohashi R, Fukazawa R, Shimizu A, et al. M1 macrophage is the predominant phenotype in coronary artery lesions following Kawasaki disease. Vasc Med. 2019;24(6):484–492. doi:10.1177/1358863X19878495
18. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27(1):451–483. doi:10.1146/annurev.immunol.021908.132532
19. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–185. doi:10.1002/path.4133
20. Furukawa S, Matsubara T, Jujoh K, et al. Peripheral blood monocyte/macrophages and serum tumor necrosis factor in Kawasaki disease. Clin Immunol Immunopathol. 1988;48(2):247–251. doi:10.1016/0090-1229(88)90088-8
21. Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. 1985;28(6):652–659. doi:10.1002/art.1780280609
22. Rosenkranz ME, Schulte DJ, Agle LMA, et al. TLR2 and MyD88 Contribute to Lactobacillus casei extract–induced focal coronary arteritis in a mouse model of kawasaki disease. Circulation. 2005;112(19):2966–2973. doi:10.1161/CIRCULATIONAHA.105.537530
23. Lee Y, Schulte DJ, Shimada K, et al. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–1550. doi:10.1161/CIRCULATIONAHA.111.072769
24. Lin IC, Suen JL, Huang SK, et al. Dectin-1/Syk signaling is involved in Lactobacillus casei cell wall extract-induced mouse model of Kawasaki disease. Immunobiology. 2013;218(2):201–212. doi:10.1016/j.imbio.2012.04.004
25. Sforna L, Megaro A, Pessia M, Franciolini F, Catacuzzeno L. Structure, Gating and Basic Functions of the Ca2+-activated K Channel of Intermediate Conductance. Curr Neuropharmacol. 2018;16(5):608–617. doi:10.2174/1570159X15666170830122402
26. Xu R, Li C, Wu Y, et al. Role of KCa3.1 channels in macrophage polarization and its relevance in atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2017;37(2):226–236. doi:10.1161/ATVBAHA.116.308461
27. Ma X-Z, Pang Z-D, Wang J-H, et al. The role and mechanism of KCa3.1 channels in human monocyte migration induced by palmitic acid. Exp Cell Res. 2018;369(2):208–217. doi:10.1016/j.yexcr.2018.05.020
28. Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27(1):234–244. doi:10.1523/JNEUROSCI.3593-06.2007
29. Kang H, Kerloc’h A, Rotival M, et al. Kcnn4 is a regulator of macrophage multinucleation in bone homeostasis and inflammatory disease. Cell Rep. 2014;8(4):1210–1224. doi:10.1016/j.celrep.2014.07.032
30. Hua X, Deuse T, Chen Y-J, et al. The potassium channel KCa3.1 as new therapeutic target for the prevention of obliterative airway disease. Transplantation. 2013;95(2):285–292. doi:10.1097/TP.0b013e318275a2f4
31. Chen Y-J, Raman G, Bodendiek S, O’Donnell ME, Wulff H. The KCa3.1 Blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cerebral Blood Flow Metabo. 2011;31(12):2363–2374. doi:10.1038/jcbfm.2011.101
32. Toyama K, Wulff H, Chandy KG, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118(9):3025–3037. doi:10.1172/JCI30836
33. Okitsu-Negishi S, Nakano I, Suzuki K, Hashira S, Abe T, Yoshino K. The induction of cardioangitis by Lactobacillus casei cell wall in mice. I. The cytokine production from murine macrophages by Lactobacillus casei cell wall extract. Clin Immunol Immunopathol. 1996;78(1):30–40. doi:10.1006/clin.1996.0005
34. Noval Rivas M, Arditi M. Kawasaki disease: pathophysiology and insights from mouse models. Nat Rev Rheumatol. 2020;16(7):391–405.
35. Gao L, Fu S, Wang W, Xie C, Zhang Y, Gong F. Notch4 signaling pathway in a Kawasaki disease mouse model induced by Lactobacillus casei cell wall extract. Exp Ther Med. 2017;13(6):3438–3442. doi:10.3892/etm.2017.4434
36. Wulff H, Castle NA. Therapeutic potential of KCa3.1 blockers: recent advances and promising trends. Expert Rev Clin Pharmacol. 2010;3(3):385–396. doi:10.1586/ecp.10.11
37. Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. 2001;184(7):940–943. doi:10.1086/323155
38. Schulte DJ, Yilmaz A, Shimada K, et al. Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking Kawasaki disease. J Immunol. 2009;183(8):5311–5318. doi:10.4049/jimmunol.0901395
39. Ueno Y, Takano N, Kanegane H, et al. The acute phase nature of interleukin 6: studies in Kawasaki disease and other febrile illnesses. Clin Exp Immunol. 1989;76(3):337–342.
40. Maury CP, Salo E, Pelkonen P. Circulating interleukin-1 beta in patients with Kawasaki disease. N Engl J Med. 1988;319(25):1670–1671.
41. Wong M, Silverman ED, Fish EN. Evidence for RANTES, monocyte chemotactic protein-1, and macrophage inflammatory protein-1 beta expression in Kawasaki disease. J Rheumatol. 1997;24(6):1179–1185.
42. Hui-Yuen JS, Duong TT, Yeung RS. TNF-alpha is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J Immunol. 2006;176(10):6294–6301. doi:10.4049/jimmunol.176.10.6294
43. Wakita D, Kurashima Y, Crother TR, et al. Role of interleukin-1 signaling in a mouse model of kawasaki disease-associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2016;36(5):886–897. doi:10.1161/ATVBAHA.115.307072
44. Senzaki H. The pathophysiology of coronary artery aneurysms in Kawasaki disease: role of matrix metalloproteinases. Arch Dis Child. 2006;91(10):847–851. doi:10.1136/adc.2005.087437
45. Lau AC, Duong TT, Ito S, Wilson GJ, Yeung RSM. Inhibition of matrix metalloproteinase-9 activity improves coronary outcome in an animal model of Kawasaki disease. Clin Exp Immunol. 2009;157(2):300–309. doi:10.1111/j.1365-2249.2009.03949.x
46. Yu X, Hirono K-I, Ichida F, et al. Enhanced iNOS expression in leukocytes and circulating endothelial cells is associated with the progression of coronary artery lesions in acute Kawasaki disease. Pediatr Res. 2004;55(4):688–694.
47. Adewuya O, Irie Y, Bian K, Onigu-Otite E, Murad F. Mechanism of vasculitis and aneurysms in Kawasaki disease: role of nitric oxide. Nitric Oxide. 2003;8(1):15–25.
48. Fukuda S, Hashimoto N, Naritomi H, et al. Prevention of rat cerebral aneurysm formation by inhibition of nitric oxide synthase. Circulation. 2000;101(21):2532–2538.
49. Freise C, Querfeld U. Inhibition of vascular calcification by block of intermediate conductance calcium-activated potassium channels with TRAM-34. Pharmacological Research. 2014;85:6–14.
50. Yin W, Wang X, Ding Y, et al. Expression of nuclear factor -κBp65 in mononuclear cells in Kawasaki disease and its relation to coronary artery lesions. Indian J Pediatr. 2011;78(11):1378–1382.
51. Tian J, An X, Niu L. Correlation between NF-kappaB signal pathway-mediated caspase-4 activation and Kawasaki disease. Exp Ther Med. 2017;13(6):3333–3336.
52. Zhou X, Yang W, Ca LJ. 2+- and protein kinase C-dependent signaling pathway for nuclear factor-kappaB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-alpha production in lipopolysaccharide-stimulated rat peritoneal macrophages. J Biol Chem. 2006;281(42):31337–31347.
53. Wei T, Wang Y, Xu W, Liu Y, Chen H, Yu Z. KCa3.1 deficiency attenuates neuroinflammation by regulating an astrocyte phenotype switch involving the PI3K/AKT/GSK3β pathway. Neurobiol Dis. 2019;132:10487.
54. Kim SH, Park SY, Park YL, Myung DS, Rew JS, Joo YE. Chlorogenic acid suppresses lipopolysaccharide-induced nitric oxide and interleukin1beta expression by inhibiting JAK2/STAT3 activation in RAW264.7 cells. Mol Med Rep. 2017;16(6):9224–9232.
55. Qi Z, Qi S, Ling L, Lv J, Feng Z. Salidroside attenuates inflammatory response via suppressing JAK2-STAT3 pathway activation and preventing STAT3 transfer into nucleus. Int Immunopharmacol. 2016;35:265–271.
56. Li R, Hong P, Zheng X. beta-carotene attenuates lipopolysaccharide-induced inflammation via inhibition of the NF-kappaB, JAK2/STAT3 and JNK/p38 MAPK signaling pathways in macrophages. Anim Sci J. 2019;90(1):140–148.
57. Park JS, Lee J, Lim MA, et al. JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J Immunol. 2014;192(9):4417–4424.
58. Zhang X, Li J, Qin JJ, et al. Oncostatin M receptor β deficiency attenuates atherogenesis by inhibiting JAK2/STAT3 signaling in macrophages. J Lipid Res. 2017;58(5):895–906.
59. Qi XL, Chen LL, Sun XG, Li XM, Zhao LH, Kong DJ. 1,25-Dihydroxyvitamin D3 regulates T lymphocyte proliferation through activation of P53 and inhibition of ERK1/2 signaling pathway in children with Kawasaki disease. Eur Rev Med Pharmacol Sci. 2017;21(16):3714–3722.
60. Wang X, Ding YY, Chen Y, et al. MiR-223-3p alleviates vascular endothelial injury by targeting IL6ST in kawasaki disease. Front Pediatr. 2019;7:288.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.