Back to Journals » Drug Design, Development and Therapy » Volume 11
Justification of disintegration testing beyond current FDA criteria using in vitro and in silico models
Authors Uebbing L ![]() , Klumpp L, Webster GK, Löbenberg R
, Klumpp L, Webster GK, Löbenberg R ![]()
Received 28 December 2016
Accepted for publication 21 February 2017
Published 11 April 2017 Volume 2017:11 Pages 1163—1174
DOI https://doi.org/10.2147/DDDT.S131213
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Lukas Uebbing,1,2,* Lukas Klumpp,1,3,* Gregory K Webster,4 Raimar Löbenberg1
1Faculty of Pharmacy and Pharmaceutical Sciences, Katz Group-Rexall Centre for Pharmacy and Health Research, University of Alberta, Edmonton, Canada; 2Institute of Pharmacy and Biochemistry, Johannes Gutenberg University, Mainz, 3Institute of Pharmaceutical Technology, Goethe University Frankfurt, Frankfurt, Germany; 4Global Research and Development, AbbVie Inc., North Chicago, IL, USA
*These authors contributed equally to this work
Abstract: Drug product performance testing is an important part of quality-by-design approaches, but this process often lacks the underlying mechanistic understanding of the complex interactions between the disintegration and dissolution processes involved. Whereas a recent draft guideline by the US Food and Drug Administration (FDA) has allowed the replacement of dissolution testing with disintegration testing, the mentioned criteria are not globally accepted. This study provides scientific justification for using disintegration testing rather than dissolution testing as a quality control method for certain immediate release (IR) formulations. A mechanistic approach, which is beyond the current FDA criteria, is presented. Dissolution testing via United States Pharmacopeial Convention Apparatus II at various paddle speeds was performed for immediate and extended release formulations of metronidazole. Dissolution profile fitting via DDSolver and dissolution profile predictions via DDDPlus™ were performed. The results showed that Fickian diffusion and drug particle properties (DPP) were responsible for the dissolution of the IR tablets, and that formulation factors (eg, coning) impacted dissolution only at lower rotation speeds. Dissolution was completely formulation controlled if extended release tablets were tested and DPP were not important. To demonstrate that disintegration is the most important dosage form attribute when dissolution is DPP controlled, disintegration, intrinsic dissolution and dissolution testing were performed in conventional and disintegration impacting media (DIM). Tablet disintegration was affected by DIM and model fitting to the Korsmeyer–Peppas equation showed a growing effect of the formulation in DIM. DDDPlus was able to predict tablet dissolution and the intrinsic dissolution profiles in conventional media and DIM. The study showed that disintegration has to occur before DPP-dependent dissolution can happen. The study suggests that disintegration can be used as performance test of rapidly disintegrating tablets beyond the FDA criteria. The scientific criteria and justification is that dissolution has to be DPP dependent, originated from active pharmaceutical ingredient characteristics and formulations factors have to be negligible.
Keywords: API, dissolution, disintegration, DDDPlus, quality-by-design, product specification, model fitting
Introduction
Quality-by-design (QbD) approaches aim to utilize the most appropriate performance or quality control tests for a drug product.1,2 Still, the critical quality attributes (CQAs) are often more based on empirical values and guidelines, instead of understanding mechanistic processes and excipient-active pharmaceutical ingredient (API) interactions.
The ICH Guideline Q6A “Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances” outlines acceptance criteria for different dosage forms and routes of administration. The guidance document contains decision tree #7.1, which allows disintegration instead of dissolution testing to be used as a performance/quality control test for rapidly dissolving dosage forms (Q>80% in 15 minutes) containing highly soluble drugs (BCS class I/III), if a relationship between dissolution and disintegration has been established.3
The new United States Pharmacopeial Convention (USP) Chapter <2> “Oral drug products – Product quality test”4 follows the ICH guidance criteria and states under specific tests for tablets (excerpt of the original text):
The disintegration test, if included, is used only as a quality control test and not as a product performance test and should conform with the specifications in the monograph.4
The US Food and Drug Administration (FDA) draft guidance on “Specification Criteria for Immediate-Release Solid Oral Dosage Forms Containing Biopharmaceutics Classification System (BCS) Class 1 and 3 Drugs” allows the use of disintegration testing as a surrogate for routine release and stability dissolution testing for rapidly dissolving BCS class I/III drug products (Q=80% in 15 minutes). The acceptance criterion is disintegration within 5 minutes in 0.01M HCl.5
The current FDA guidance on “Dissolution Testing of Immediate Release Dosage Forms”, which the new draft will supersede, suggests a single-point dissolution tests specification of “Q=80% in 60 minutes” as a replacement for dissolution testing for rapidly dissolving BCS class I/III drugs.6
In industry, there is little consensus on how to apply these guidelines. Many companies simply default to dissolution testing rather than justify disintegration as the easiest path forward for a global product. However, with a better mechanistic understanding and knowledge of critical parameters in the dissolution process, this aversion can be avoided. A modeling-based approach is needed to justify important product specifications and support CQA beyond guideline assumptions. This will enable globally operating companies to justify their product specifications beyond sometimes contradicting national guidances.
In this study, metronidazole, a BCS class I drug, was chosen as the model API.7 Four different formulations were tested, with two being fast disintegrating, immediate release (IR#1 and IR#2) tablets, and the other two formulated for a slow erosion and drug release (slow eroding tablet [SET] and granulated tablet [GT]). API and excipient interactions were investigated using model fitting and computer simulations of the obtained dissolution profiles. The influence of disintegration on API-controlled dissolution was studied using new disintegration impacting media (DIM).
This study mechanistically investigated disintegration and dissolution behavior of different formulations. Model fitting was utilized in order to differentiate between API and formulation controlled drug release. This approach enables formulation scientists to identify CQAs for IR tablets. Disintegration testing might be used if drug particle properties (DPP) control dissolution. This was confirmed by simulations using DDDPlus software. Dissolution testing is required if the formulation significantly controls the dissolution process. This approach goes beyond the current FDA criteria for IR tablets and provides scientific justification for using disintegration instead of dissolution testing in a QbD environment.
Materials and methods
Materials
Metronidazole (for tablets and quantification standard) was purchased from Medisca® (Richmond, BC, Canada; LOT 601124/C). Microcrystalline cellulose (Avicel® PH-102 NF; for IR#2 formulation) was obtained from FMC Biopolymer (Philadelphia, PA, USA). Microcrystalline cellulose NF, dicalcium phosphate dihydrate NF (for IR#1 formulation) and croscarmellose sodium were purchased from PCCA Canada (London, ON, Canada). Galen IQ™ 801 was obtained from BENEO-Palatinit GmbH (Mannheim, Germany) and magnesium stearate from H.L. Blachford Ltd (Mississauga, ON, Canada; IR#1) and Street Chemicals & Co (Montreal, QC, Canada; IR#2). Mannitol was purchased from EM Science (Gibbstown, NJ, USA), Starch 1500 from Colorcon (Indianapolis, IN, USA), Povidone K30 “Kollidon® 30” from BASF (Mt Olive, NJ, USA) and sodium carbonate (anhydrous) was obtained from BDH Inc. (Toronto, ON, Canada). Ethanol was obtained from GreenField Specialty Alcohols Inc. (Brampton, ON, Canada).
Buffer media for dissolution testing were prepared according to USP specifications for acetate buffer pH 4.5 and SGFsp (simulated gastric fluid sine pepsin).4 Sodium acetate trihydrate was purchased from Caledon Laboratories Ltd (Georgetown, ON, Canada) and glacial acetic acid USP, hydrochloric acid NF and sodium chloride USP were purchased from Fisher Scientific (Fair Lawn, NJ, USA). For the sugar solutions, Rogers Granulated Sugar from Lantic Inc. (Montreal, QC, Canada) was used. High-performance liquid chromatography (HPLC) grade water and water for the dissolution and disintegration test media were generated in an Elgastat Maxima UF and an Elgastat Option 3B water purifier by ELGA Laboratories Ltd. (Mississauga, ON, Canada) and filtered through a 0.45 μm membrane MCE filter by Fisher Scientific (Pittsburgh, PA, USA; for immersion media) and a Durapore® 0.22 μm GV filter by Millipore Canada Ltd. (Etobicoke, ON, Canada; for HPLC mobile phase), respectively. Acetonitrile for the HPLC mobile phase was purchased from VWR International LLC. (Radnor, PA, USA) and filtered through a Durapore 0.45 μm HV filter by Millipore Canada Ltd. (Etobicoke, ON, Canada).
Methods
Tablets
Tablets were pressed with a Carver Laboratory Press by Fred S Carver Inc. Hydraulic Equipment (Manomonee Falls, WI, USA). Direct compression IR tablets (IR#1 and IR#2) were pressed at 1 metric ton pressure for 30 seconds, after blending the formulation ingredients (Tables 1 and 2) in a rotating blender by Erweka GmbH (Heusenstamm, Germany) for 30 minutes. These parameters had previously been established as a starting point that usually provided tablets of adequate quality in this group.8 Direct compression SETs (Table 3) were pressed for 1 minute at 1 metric ton pressure. The granulate for the GTs (Table 4) was prepared by adding ~5 mL of 70% ethanol to about 11 g of intragranular formulation mix, granulating through a No 20 sieve, drying in vacuum at 45°C and sieving the granulate through a No 20 sieve, onto a No 40 sieve. Magnesium stearate was then added as lubricant and the tablets were pressed for at 1 metric ton pressure for 30 seconds.
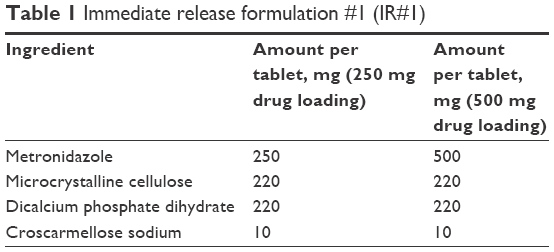 | Table 1 Immediate release formulation #1 (IR#1) |
 | Table 2 Immediate release formulation #2 (IR#2) |
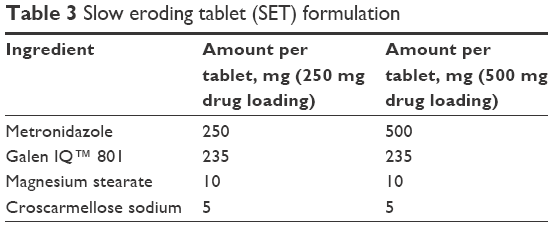 | Table 3 Slow eroding tablet (SET) formulation |
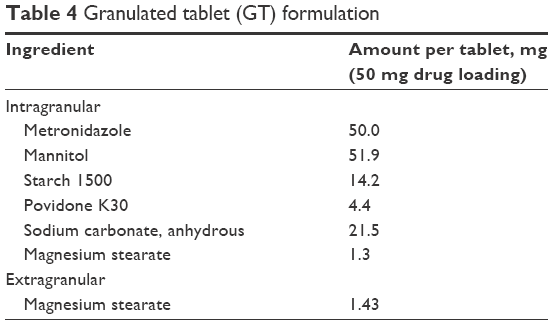 | Table 4 Granulated tablet (GT) formulation |
Dissolution and disintegration testing
The pH of the dissolution and disintegration media was measured using an accumet® XL 20 pH-meter by Fisher Scientific (Fair Lawn, NJ, USA). Dissolution testing was performed using a VK 7020 system from Varian Inc. (Cary, NC, USA) equipped with 70 μm Full Flow™ Filters (Varian Inc.), since smaller pore sizes proved to be problematic with the more viscous DIM, and a VK 8000 auto sampler (Varian Inc.). All tests were performed with USP Apparatus 2 and 900 mL dissolution medium (SGFsp, acetate buffer USP pH 4.5, 10%/20%/30% sucrose solution). SGFsp and acetate buffer were deaerated by filtration, ultrasound and vacuum. Samples (1.0 mL) were withdrawn without replacement at each time point (5, 10, 15, 20, 30, 45 and 60 minutes) and were transferred into 2.5 mL vials for HPLC analysis. The drug concentration in the vessel was adjusted by calculation.
Disintegration testing was performed in an ED-2L disintegration tester by Electrolab India Pvt. Ltd. (Navi-Mumbai, India) in the same media as the dissolution tests according to USP standards.4
Intrinsic dissolution testing was performed in the same media as the dissolution tests using a modified version of the rotating disk apparatus described in USP Chapter <1087>.4 Approximately 160–170 mg were pressed into the die to be used as a rotating disk apparatus using the Carver Laboratory Press at 2 metric tons pressure for 90 seconds. The apparatus was mounted in a type RZR50 stirrer by Caframo Ltd. (Wiarton, ON, Canada) and immersed in a beaker containing 100 mL dissolution medium, with the temperature being kept constant at 37°C±0.5°C by a hot water bath. The test was performed at 100 rpm and 1.0 mL samples were drawn using a BD 1 mL syringe (Franklin Lakes, NJ, USA) and transferred into HPLC vials for analysis by filtering them through a 13 mm syringe filter with a 0.2 μm PTFE membrane by VWR International LLC (Radnor, PA, USA). SGFsp and acetate buffer were deaerated by filtration, ultrasound and vacuum.
HPLC
The standard solution for HPLC quantification of metronidazole was prepared by dissolving metronidazole in the respective medium, using a Branson 3800 ultrasonic bath from Emerson Industrial Automation (Ferguson, MO, USA), and the calibration curve was prepared for a range from 3.75% to 120% of the expected maximum drug concentrations. Quantification of metronidazole was performed via a slightly modified version of a previously published HPLC method.7 A VP-class Shimadzu Scientific Instruments (Kyoto, Japan) liquid chromatograph, equipped with a Lichrospher® 60 RP Select B column (5 μm, 12.5×4 mm, by Merck Darmstadt, Germany) with a matching guard column and connected to a CBM-20A system controller, two LC-10AS pumps, an SIL-10ADVP auto sampler and a SPD-M10AVP diode array detector, was used. The system was controlled using the data acquisition software “EZ Start 7.4” (Shimadzu). The mobile phase was deaerated before use, using a combination of vacuum filtration, and ultrasound and the flow rate was set to 1 mL/minute, using a 70:30 mix of water and acetonitrile. A sample volume of 10 μL was directly injected without dilution and the retention time for metronidazole was ~2 minutes with a total run time of 3 minutes. For all dissolution tests, the correlation coefficient (r2) for the calibration curve was ≥0.998.
DDDPlus™ simulation software
DDDPlus (Dose Disintegration and Dissolution Software) version 5.0.0011 by Simulations Plus, Inc. (Lancaster, CA, USA) was used to simulate dissolution behavior of different formulations by defining excipients and test conditions.9 The software is divided into three main tabs – formulation, dissolution method and simulation. The formulation tab lets the user chose from eight different dosage forms – such as IR tablet, coated tablet, powder or capsule – and provide manufacturing properties, such as compression force or tablet diameter, as well as the formulation composition. The latter can be made up from excipients in the included database or user-defined ingredients. The role of the ingredient (API, disintegrant, polymer, etc.) can be set by the user; the physicochemical properties, such as solubility, pKa, diffusion coefficient or logD and the particle size distribution can be defined and a dissolution model (eg, mass transfer, Nernst-Brunner, intrinsic dissolution) is chosen. An excipient-specific coefficient which represents the influence of the excipient on the formulation, as well as a calibration coefficient, can be defined. The parameters which were used for the simulations in this study can be found in Tables 5 and 6 and were either measured, collected from literature, or part of the default excipient database in DDDPlus. A solubility vs pH profile was established using the “pKa Table” dialog and literature values, which were also used in the biowaiver monograph for metronidazole, and an “.spd” (solubility-pH data) file was created.10,11
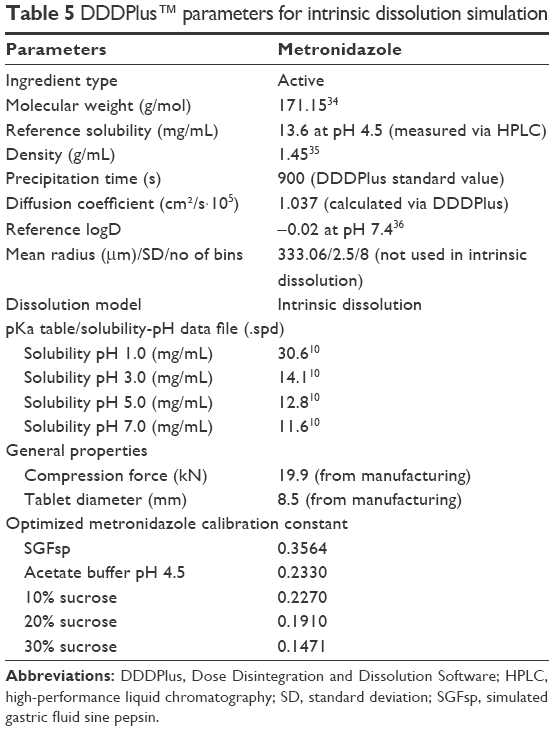 | Table 5 DDDPlus™ parameters for intrinsic dissolution simulation |
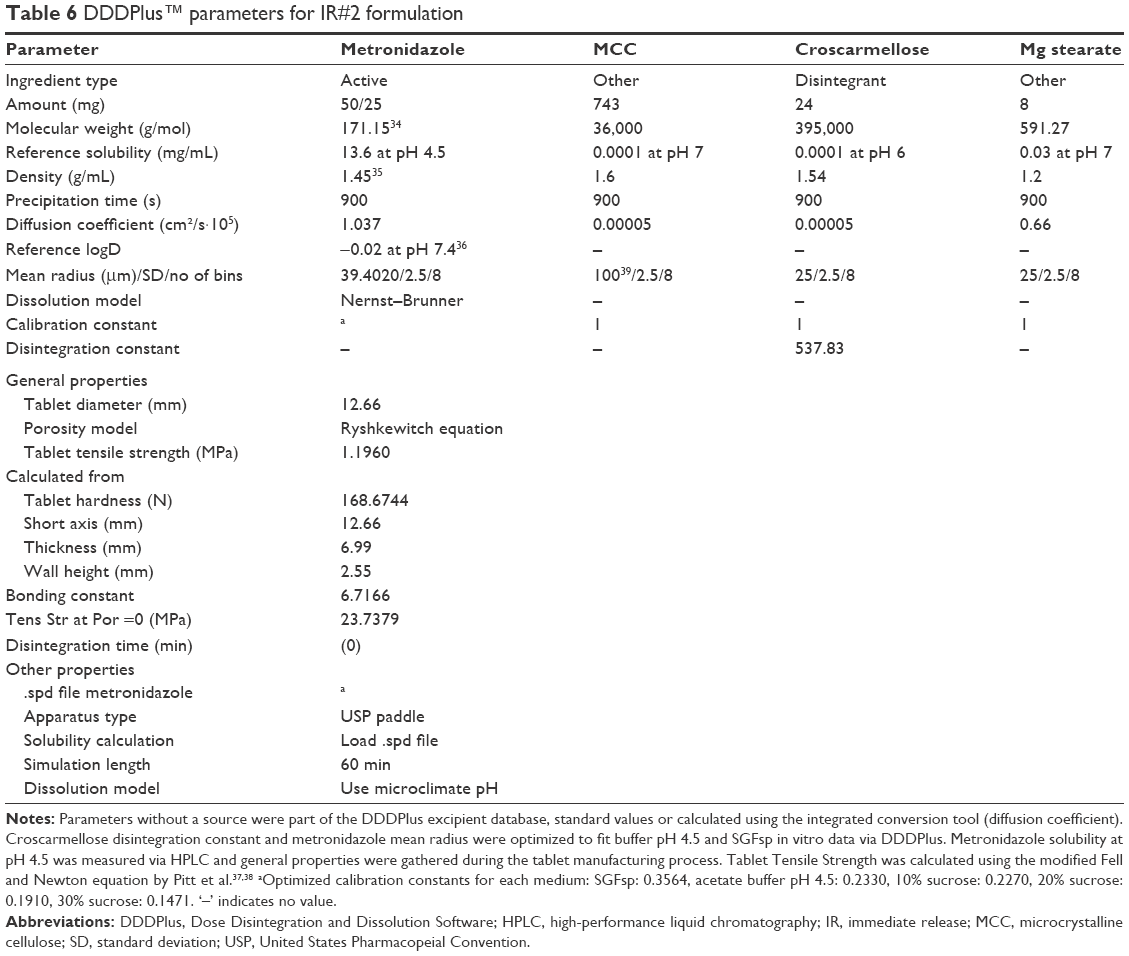 | Table 6 DDDPlus™ parameters for IR#2 formulation |
Simulation test conditions were chosen to be identical to the actual dissolution and intrinsic dissolution test conditions of the in vitro tests performed, using 900 mL medium and 100 mL medium, respectively, as well as paddle speeds of 25, 50 and 75 rpm (basket speed: 100 rpm) and a 60 minutes (25 minutes) simulation length. New media were defined using DDDPlus integrated medium composition tool, with the pH being the measured pH and dynamic viscosity at 37°C being estimated values based on inter- and extrapolation of literature data.12 The chosen parameters for the simulations can be found in Table 7.
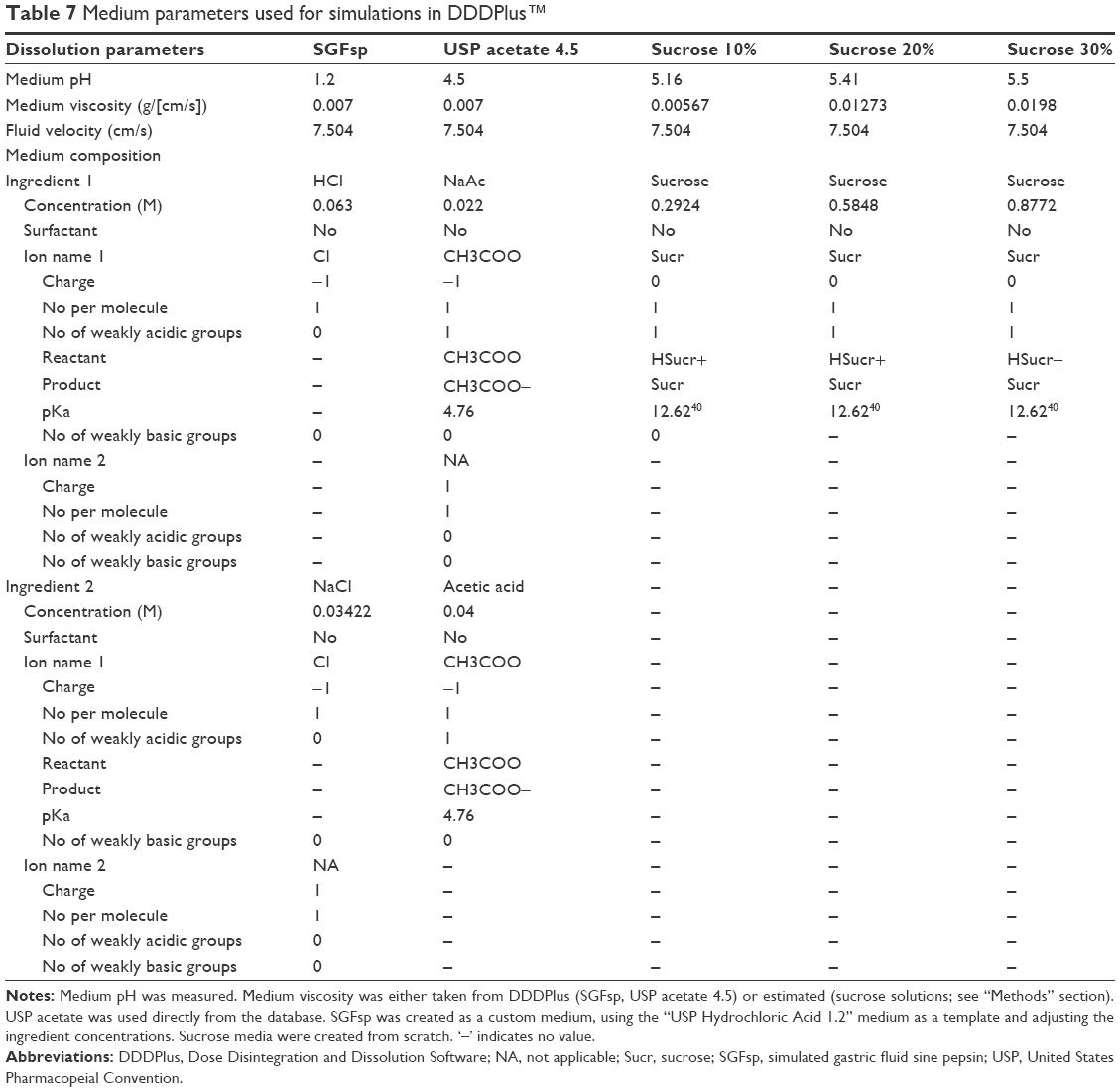 | Table 7 Medium parameters used for simulations in DDDPlus™ |
The third tab in the software is used to simulate either a single simulation, a parameter sensitivity analysis, a virtual trial, parameter optimization using provided in vitro data, or compare the simulation results to the provided in vitro data using f1/f2-testing. The results can be displayed graphically for each ingredient and can be exported for further use.
Data analysis
All data analysis was performed via either DDDPlus or using Microsoft Excel™ with DDSolver. Dissolution and intrinsic dissolution tests were graphically plotted in Microsoft Excel and statistically evaluated using DDSolver, a free excel plugin designed for dissolution profile data analysis, like profile comparison or modeling.13,14 Profiles showing a higher dissolution rate than 100%, due to calibration inaccuracy, were normalized to a maximum of 100% for better compatibility with DDDPlus. This was achieved by defining the highest fraction dissolved value as 100% and multiplying all the other profiles in the plot figure by the same factor in order to maintain comparability. Pairwise dissolution data comparison was performed via f2 statistics for both in vitro and in vitro data, as well as for evaluating in silico predictions. The coefficient of determination (R2) for evaluating in silico data correlation to in vitro data was obtained from DDDPlus. Model fitting in DDSolver was used to determine drug release mechanisms of the different tablet formulation, by using zero order, first order, Gompertz, Weibull and Hopfenberg functions, as well as the Korsmeyer–Peppas equation.15–17 Korsmeyer-Peppas modeling requires Q-values of <65%. If this was not possible, the lowest amount of points (three points) that DDSolver accepts was used. For zero-order curve fitting, only the portion of the dissolution profile with Q-values <100% was modeled because only these values represent the actual dissolution process. The adjusted coefficient of determination (R2adj), the Akaike information criterion and the model selection criterion were used to determine the most appropriate release model and are all included in DDSolver.13,18
Results and discussion
The results from the dissolution tests for the IR#1 and IR#2, the SET formulation and the GT formulation in USP pH 4.5 acetate buffer are shown in Figures 1–7.
 | Figure 1 Dissolution of IR#1 formulation (250 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds. |
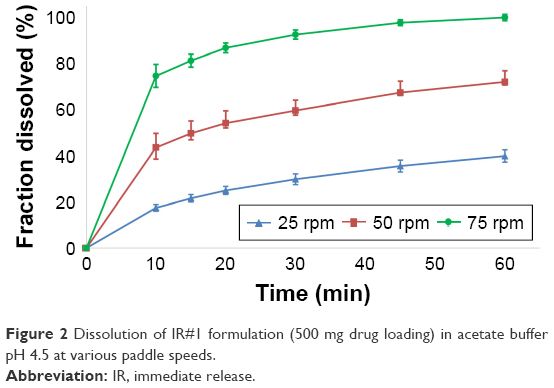 | Figure 2 Dissolution of IR#1 formulation (500 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds. |
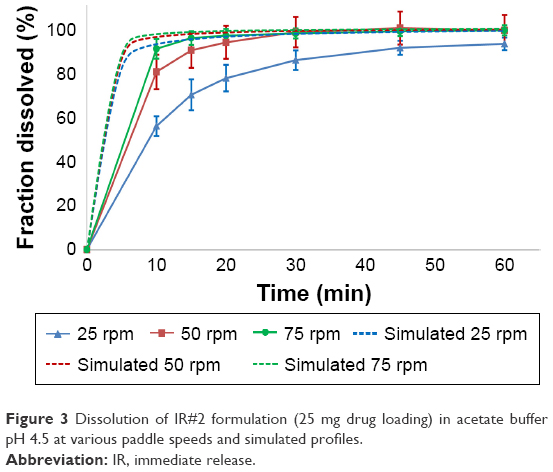 | Figure 3 Dissolution of IR#2 formulation (25 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds and simulated profiles. |
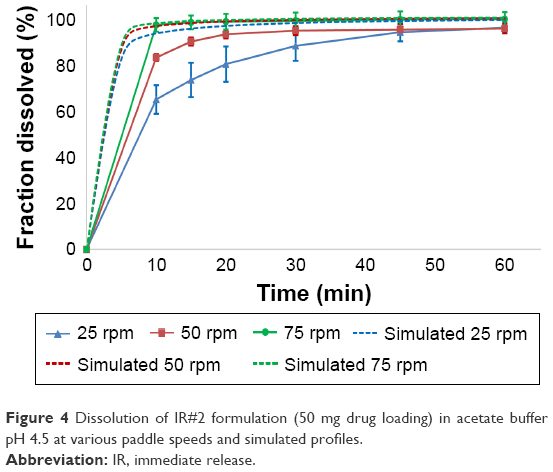 | Figure 4 Dissolution of IR#2 formulation (50 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds and simulated profiles. |
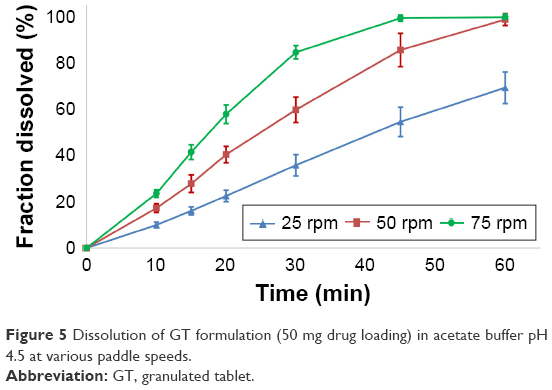 | Figure 5 Dissolution of GT formulation (50 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds. |
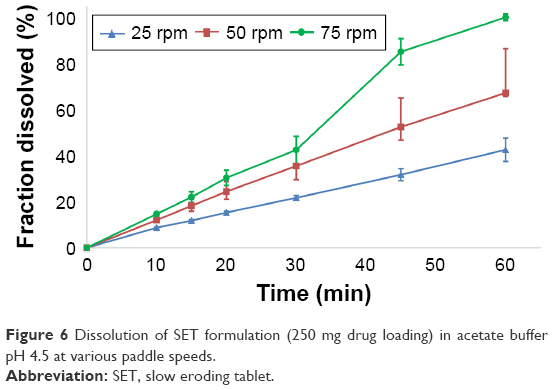 | Figure 6 Dissolution of SET formulation (250 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds. |
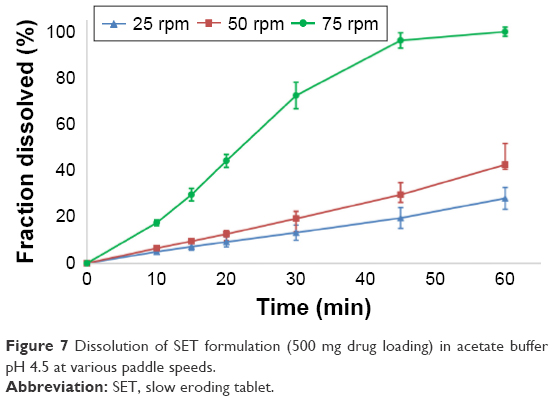 | Figure 7 Dissolution of SET formulation (500 mg drug loading) in acetate buffer pH 4.5 at various paddle speeds. |
Both IR#1 and IR#2 showed rapid disintegration and all reached the FDA requirement for 80% dissolution within 15 minutes, as long as the tests were performed at a paddle speed of 75 rpm.5 At lower paddle speeds coning was observed and the dissolution rates declined. This resulted in IR#1 failing the FDA criteria at paddle speeds of ≤50 rpm and IR#2 failing at 25 rpm. Coning is an in vitro phenomenon, where a loose powder bed is formed by undissolved drug and excipients under the paddle after the tablet has partially or completely disintegrated. It is not relevant to the in vivo performance of tablets.19,20 In general, the effect was less noticeable in the dissolution profiles for IR#2, which could be due to the lower drug loading of this formulation. At 25 rpm, the IR#2 profiles still failed the FDA criteria and were not considered similar to the 75 rpm profiles (f2-test, 25 mg: 30; 50 mg: 32). Although the FDA criteria were met at 50 rpm, the profiles were close to being considered not similar to the 75 rpm values (f2-test, 25 mg: 56; 50 mg: 50).
Model fitting via DDSolver produced very good fits (R2adj ≥0.992) for the Gompertz and Weibull models, as well as good fits (R2adj =0.848–0.993 for 250 mg and 0.798–0.985 for 500 mg) to a first-order release for IR#1. The models showed weaker correlations at lower paddle speeds. The usefulness of the Weibull model was shown by Langenbucher to model a variety of dissolution curves which could not be modeled using only exponential or log-normal models. The Gompertz model was successfully used to model the dissolution behavior of methyldopa tablets. Both the Weibull and Gompertz models are frequently used to describe the release from IR tablets.13,15,21–23 In the Korsmeyer–Peppas equation, n-values ≤0.43 suggest an only diffusion-controlled dissolution (only DPP controlled), whereas values ≥1.00 represent zero-order release kinetics, with anomalous (non-Fickian) diffusion for the values in between.24 Applying the Korsmeyer–Peppas equation to the in vitro data resulted in n-values of 0.183/0.336/0.434 (250 mg) and 0.209/0.286/0.460 (500 mg) for 75, 50 and 25 rpm, respectively. This shows a shift away from DPP controlled dissolution to an increasing formulation effect for the IR#1 samples. For the IR#2 tablets, model fitting provided good correlations for both the Gompertz (R2adj =0.827–0.996) and Weibull (R2adj =0.795–0.998) functions. Korsmeyer–Peppas modeling produced n-values of 0.128/0.283/0.558 (25 mg) and 0.027/0.196/0.301 (50 mg) for 75, 50 and 25 rpm, respectively. Thus, diffusion was the main mechanism for dissolution. As with IR#1, an increasing formulation effect on dissolution could be observed at lower paddle speeds, due to coning.
To better demonstrate the effect of the formulation, without having to rely on uncommon paddle speeds and the in vitro phenomenon of coning, two formulations with a more controlled release (SET and GT) were produced.
The SET formulation did not show immediate disintegration and completely disintegrated only at 75 rpm. There was no coning occurring at lower paddle speeds, since only magnesium stearate and the disintegrant (croscarmellose sodium) were insoluble and the formulation only contained small amounts of these excipients. The tablets containing a drug loading of 250 mg broke in half after ~40 minutes, resulting in a higher surface area and therefore a higher dissolution rate. Overall, these tablets showed a controlled release rate, which was supported by a good correlation to a zero-order model (R2adj =0.936–0.998), as well as n-values of 0.887/0.976/0.979 (250 mg; 25/50/75 rpm) and 0.947/1.045/1.338 (500 mg) for the Korsmeyer–Peppas equation. Weibull and Hopfenberg modeling produced good correlations (R2adj =0.915–1.000). The Hopfenberg equation was developed for erodible tablets in various shapes.25
The results for the GT formulation were similar to the modeling outcomes for the SET formulation. Here, a slow erosion of the tablet instead of a fast disintegration could be observed as well. Again, a correlation to zero order (R2adj =0.980–0.997), Weibull (R2adj =0.990–1.000) and Hopfenberg (R2adj =0.978–0.996) models could be established, with higher correlations at lower paddle speeds. A look at the n-values of 1.137/1.145/1.253 (25–75 rpm) for Korsmeyer–Peppas suggests a zero order (case II) transport for the drug dissolution process for this formulation too.
These results show that dissolution in rapidly disintegrating, IR formulations is DPP-controlled after the tablet has disintegrated. Thus, the dissolution of the API can be described by basic mechanistic mass-transport equations such as Nernst–Brunner.15 The advantage of these mechanistic models is that they can predict drug dissolution, and depend only on very basic parameters such as particle size and particle size distribution, solubility, density, diffusion coefficient, diffusion layer thickness, bulk/micro-climate pH, and experimental conditions.26,27 Such parameters are characterized for an API in a QbD approach and dissolution can be predicted for different media. For IR tablet formulations, where API dissolution is known and controlled by DPP, disintegration is the most critical and clinically relevant dosage form parameter. If disintegration changes, the corresponding drug product performance will change. However, if disintegration occurs fast and complete, then dissolution depends only on DPP, and the excipient and formulation factors are not important for clinical performance. In this case the disintegration test is the most appropriate performance and quality control test for the final formulation because the API dissolution behavior is not significantly impacted by the formulation.
To demonstrate the importance of the disintegration step in more detail, DIM were introduced to evaluate changes in dissolution behavior due to different disintegration times. Our group had shown previously, how different media can impact disintegration of different dosage forms.28 SGFsp and acetate buffer pH 4.5 USP were used as conventional disintegration media, while also preparing 10%, 20% and 30% sucrose solutions as DIM. Sucrose solutions have been shown to impact tablet disintegration.29 The results from the disintegration test for the IR#2 (50 mg) formulation can be seen in Figure 8 and show a clear increase in disintegration time for the DIM compared to the conventional media, with longer disintegration times for higher sugar concentrations.
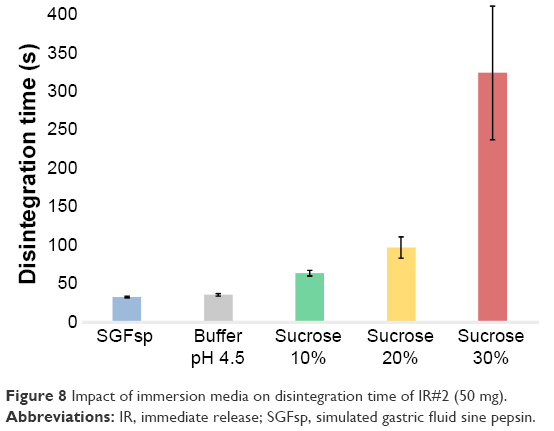 | Figure 8 Impact of immersion media on disintegration time of IR#2 (50 mg). |
This can be explained as follows: disintegration mechanisms can be divided into surface erosion, tablet fraction, dissolution of API and excipients and particle de-agglomeration by swelling of polymers, with the first two being hydrodynamics-dependent, dissolution being both hydrodynamics-dependent and hydrodynamics-independent and particle deagglomeration being hydrodynamically independent. These mechanisms can also be divided into dynamic and static tablet disintegration.30,31 Sucrose solutions in particular have been shown to affect tablet disintegration both by influencing water uptake (static and dynamic disintegration) and hydrodynamics (dynamic disintegration) because of their higher viscosity, and by causing a profound depression of water diffusivity – and therefore lower water uptake – by molecular interactions (eg, hydrogen bonding) between water and sucrose.29,31
After showing that DIM did indeed affect disintegration, intrinsic dissolution testing was performed in the same media to study their impact on the API alone. The results from these intrinsic dissolution tests are shown in Figure 9. Metronidazole dissolved fastest in SGFsp, where it has its highest solubility due to its pKa of 3.09. The other profiles showed slower dissolution rates as a result of the increasing sucrose concentrations. All dissolution profiles showed good (R2adj =0.940–1.000) correlation to a zero-order release model, as expected.
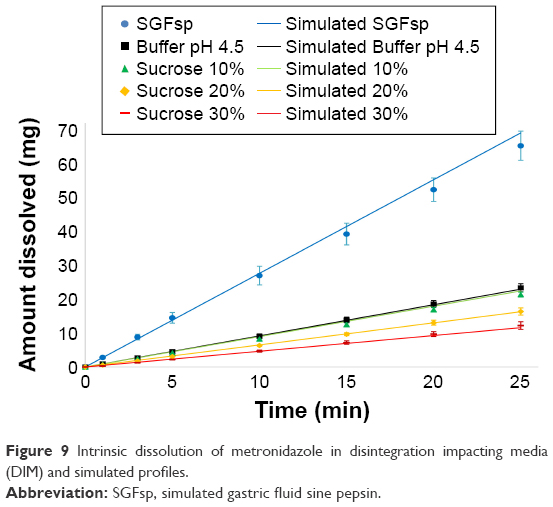 | Figure 9 Intrinsic dissolution of metronidazole in disintegration impacting media (DIM) and simulated profiles. |
Finally, dissolution testing for the IR#2 (50 mg) formulation was performed in conventional media and DIM – the results can be seen in Figure 10. Model fitting to the Korsmeyer–Peppas equation showed a growing impact of the formulation on dissolution, with rising n-values (0.037/0.095/0.126/0.201/0.502) suggesting a move away from DPP-controlled dissolution into anomalous dissolution mechanisms caused by the interaction of the API with the formulation and DIM. This identifies disintegration as the CQA of these tablets besides API properties.
 | Figure 10 Dissolution of IR#2 (50 mg) at 75 rpm in disintegration impacting media and simulated profiles. |
While the recent FDA draft guidance uses an empirical criterion to allow dissolution testing to be interchanged with disintegration testing, that criterion is not based on any mechanistic knowledge about the drug dissolution process or API-formulation-interactions. This can result in drug products meeting the FDA criteria for being tested via disintegration testing, even though their mechanism is not dependent on disintegration alone (as can be seen here in 30% sucrose solution) and dissolution testing would be appropriate. On the other hand, drug products might not be allowed to be tested via disintegration testing, because they do not meet the 80% dissolution in 15 minutes criterion – although their dissolution is only DPP dependent and the only influence the formulation has is disintegration.
Current guidelines ask for a correlation as a condition for replacing dissolution with disintegration testing. However, in a study published by Gupta et al, no such correlation between disintegration and dissolution could be shown.32 Instead, the extensive study showed the complexity of excipient effects and manufacturing parameters on disintegration times and dissolution rates of 24 different IR tablets. Since this very thorough study showed that no correlation exists, one may ask: which scientific data could justify disintegration testing as a surrogate for dissolution testing beyond the FDA criteria?
This study mechanistically supports that if dissolution occurs due to DPP, and disintegration has no significant impact on API dissolution, disintegration is the most relevant performance test for this dosage form.
A second goal of this study was to evaluate the possibility of predicting the impact of disintegration and the resulting changes in dissolution by using in silico methods. DDDPlus was used to simulate the previously observed in vitro data. First, simulations of the intrinsic dissolution process were performed in order to optimize a calibration parameter for the API in each medium, (Figure 10). This was used to account for influences of viscosity and osmolality on the dissolution of the API in the different media. The intrinsic dissolution parameters were used for the simulation of the predictions of tablet formulation (IR#2). The disintegration constant for croscarmellose sodium, as well as the particle size of metronidazole were optimized from the dissolution data using pH 4.5 acetate buffer and SGFsp. The resulting parameters were then used to simulate dissolution in DIM and at various paddle speeds. The workflow process is shown in Figure 11. Comparison via f2-test (built-in tool in DDDPlus) and R2 (for intrinsic dissolution; built-in tool in DDDPlus) were used to evaluate the accuracy of these simulations. The simulations can be found in the same figures as their corresponding in vitro data, and the results of the f2-tests are listed in Table 8.
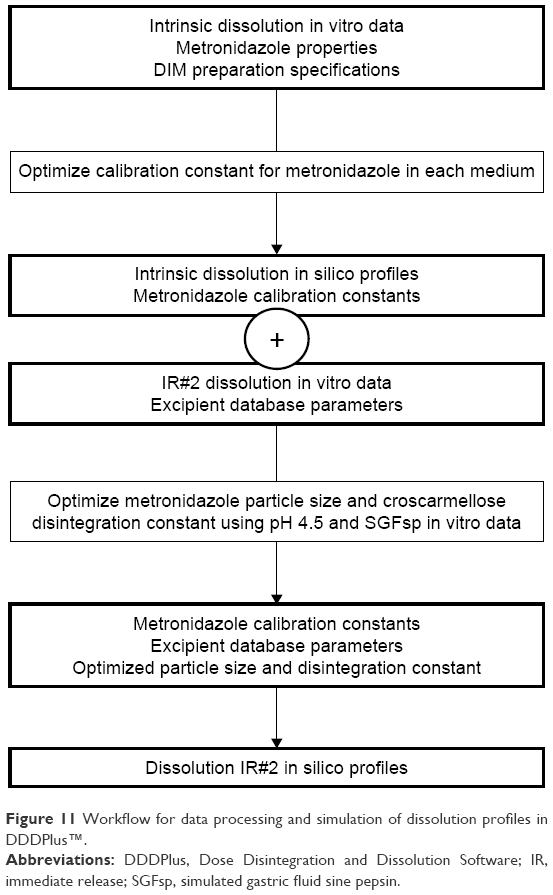 | Figure 11 Workflow for data processing and simulation of dissolution profiles in DDDPlus™. |
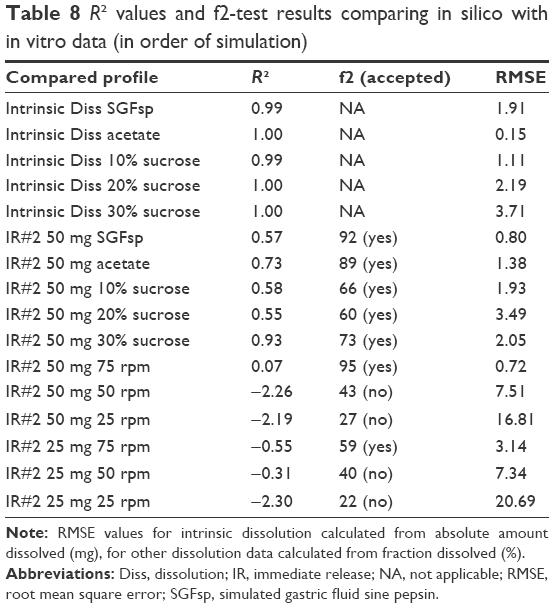 | Table 8 R2 values and f2-test results comparing in silico with in vitro data (in order of simulation) |
Simulation of the intrinsic dissolution process in DDDPlus worked very well. The in silico dissolution profiles of IR#2 in different media, that were obtained using the previously described optimization process, were all considered similar to their in vitro data according to f2-testing at 75 rpm. For lower rotation speeds, a vast overestimation of the fraction dissolved was observed, due to the previously described issue of coning (Figures 3 and 4). The software does not account for this artificial in vitro phenomenon and assumes normal hydrodynamic conditions throughout the dissolution vessel, resulting in the observed overestimation.
This study shows that in silico methods can be utilized in order to predict the impact of disintegration on the dissolution of certain IR formulations. It also shows that the current software capabilities are not yet suitable to predict all aspects of dosage form and dissolution behavior. The software was good at predicting dissolution behavior when DPP governed the dissolution process and no artificial coning appeared. However, for formulations where API excipient interactions occur, dissolution data was needed to optimize dissolution predictions and to determine the appropriate interaction coefficients. Further work is needed to build a library of excipient parameters. Then the software might be used for formulation design, even before any in vitro testing is performed.
Conclusion
The goal in QbD approaches during pharmaceutical development is to have all critical processes and quality attributes sufficiently described and controlled. Today, statistical approaches and simulations can be used to enable rational product design and to assess possible process or product risks, based on a thorough understanding of product and process performance.33
This study shows the current capabilities and limitations of DDDPlus to predict API and tablet dissolution behavior in different media and rotation speeds. The dissolution model fitting results via DDSolver were able to differentiate between DPP and formulation influenced tablet dissolution behavior.
This study mechanistically investigated disintegration and dissolution behavior of IR tablets. It was found that disintegration and dissolution can be sequential or parallel processes, or both. If disintegration occurs first, DPP dependent dissolution can happen and disintegration can be used as performance test of rapidly disintegrating tablets beyond the current FDA criteria – the scientific data needed for this justification being, that dissolution has to be DPP dependent, originated from API characteristics, and formulation factors have to be negligible for the dissolution process. However, dissolution should be used as the quality control method if the formulation significantly influences dissolution behavior.
This approach will enable globally operating pharmaceutical companies to scientifically justify their product specifications for disintegration independent from national – sometimes contradicting – regulatory guidance documents.
Acknowledgments
The authors thank Simulations Plus, the Drug Development and Innovation Centre of the University of Alberta and AbbVie for their support. AbbVie jointly participated in the interpretation of data, writing, reviewing and approving this publication. The study contains no proprietary AbbVie data.
Parts of the data were presented at the 2016 AAPS annual meeting as poster presentation.
Disclosure
Gregory K Webster is an employee of AbbVie. The other authors report no conflicts of interest in this work.
References
Yu LX. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. 2008;25(4):781–791. | ||
Almukainzi M, Salehi M, Chacra NAB, Löbenberg R. Comparison of the rupture and disintegration tests for soft-shell capsules. Dissolution Technol. 2011;18(1):21–25. | ||
European Medicines Agency. ICH Topic Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. EMEA; 2000. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002823.pdf. Accessed May 6, 2016. | ||
The United States Pharmacopeia 2016: USP 39; The national formulary: NF 34. Rockville, MD: United States Pharmacopeial Convention; 2016. | ||
Center for Drug Evaluation and Research (CDER). Guidance for industry: specification criteria for immediate-release solid oral dosage forms containing biopharmaceutics classification system class 1 and 3 Drugs. Food and Drug Administration; 2015. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM456594.pdf. Accessed May 9, 2016. | ||
Center for Drug Evaluation and Research (CDER). Guidance for industry: dissolution testing of immediate release solid oral dosage forms. Food and Drug Administration; 1997. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070237.pdf. Accessed October 3, 2016. | ||
Löbenberg R, Chacra NB, Stippler ES, et al. Toward global standards for comparator pharmaceutical products: case studies of amoxicillin, metronidazole, and zidovudine in the Americas. AAPS J. 2012;14(3):462–472. | ||
Schmid K, Löbenberg R. Influence of the changed USP specifications on disintegration test performance. Dissolution Technol. 2010;17(1):6–10. | ||
Almukainzi M, Okumu A, Wei H, Lobenberg R. Simulation of in vitro dissolution behavior using DDDPlus. AAPS PharmSciTech. 2015;16(1):217–221. | ||
Ogata H, Aoyagi N, Kaniwa N, Ejima A. Effect of food on bioavailability of metronidazole from sugar-coated tablets having different dissolution rates in subjects with low gastric acidity. Int J Clin Pharmacol Ther Toxicol. 1986;24(6):279–282. | ||
Rediguieri CF, Porta V, G Nunes DS, et al. Biowaiver monographs for immediate release solid oral dosage forms: metronidazole. J Pharm Sci. 2011;100(5):1618–1627. | ||
Asadi M. Beet-Sugar Handbook: Hoboken NJ: John Wiley & Sons, Inc. Available from: http://onlinelibrary.wiley.com/doi/10.1002/9780471790990.oth1/pdf. Accessed March 14, 2017. | ||
Zhang Y, Huo M, Zhou J, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–271. | ||
Zuo J, Gao Y, Bou-Chacra N, Löbenberg R. Evaluation of the DDSolver software applications. Biomed Res Int. 2014;2014:204925. | ||
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. | ||
Korsmeyer RW, von Meerwall E, Peppas NA. Solute and penetrant diffusion in swellable polymers II. Verification of theoretical models. J Polym Sci B Polym Phys. 1986;24(2):409–434. | ||
Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv Drug Deliv Rev. 2001;48(2–3):139–157. | ||
Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;19(6):716–723. | ||
Armenante P. Inherent method variability in dissolution testing: the effect of hydrodynamics in the USP II apparatus. A Technical Report Submitted to the Food and Drug Administration; 2005. Available from: http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4187B1_01_04-Effect-Hydrodynamics.pdf. Accessed October 8, 2016. | ||
Bai G, Armenante PM. Velocity distribution and shear rate variability resulting from changes in the impeller location in the USP dissolution testing apparatus II. Pharm Res. 2008;25(2):320–336. | ||
Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–981. | ||
Labastie M, Nacco R, Cumps J. Tablet dissolution parameters: a statistical evaluation. J Pharm Biomed Anal. 1992;10(10–12):1105–1108. | ||
Adams E, Coomans D, Smeyers-Verbeke J, Massart D. Non-linear mixed effects models for the evaluation of dissolution profiles. Int J Pharm. 2002;240(1–2):37–53. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5(1):23–36. | ||
Hopfenberg HB. Controlled release from erodible slabs, cylinders, and spheres. In: Paul DR, Harris FW, editors. Controlled Release Polymeric Formulations. Washington: American Chemical Society. 1976;33:26–32. | ||
Agata Y, Iwao Y, Miyagishima A, Itai S. Novel mathematical model for predicting the dissolution profile of spherical particles under non-sink Conditions. Chem Pharm Bull. 2010;58(4):511–515. | ||
Simulations Plus Inc. DDDPlus User Manual. 2016. | ||
Zuo J, Gao Y, Almukainzi M, Löbenberg R. Investigation of the disintegration behavior of dietary supplements in different beverages. Dissolution Technol. 2013;20(4):6–9. | ||
Radwan A, Ebert S, Amar A, et al. Mechanistic understanding of food effects: water diffusivity in gastrointestinal tract is an important parameter for the prediction of disintegration of solid oral dosage forms. Mol Pharm. 2013;10(6):2283–2290. | ||
Kindgen S, Wachtel H, Abrahamsson B, Langguth P. Computational fluid dynamics simulation of hydrodynamics and stresses in the PhEur/USP disintegration tester under fed and fasted fluid characteristics. J Pharm Sci. 2015;104(9):2956–2968. | ||
Radwan A, Wagner M, Amidon GL, Langguth P. Bio-predictive tablet disintegration: effect of water diffusivity, fluid flow, food composition and test conditions. Eur J Pharm Sci. 2014;57:273–279. | ||
Gupta A, Hunt RL, Shah RB, Sayeed VA, Khan MA. Disintegration of highly soluble immediate release tablets: a surrogate for dissolution. AAPS PharmSciTech. 2009;10(2):495–499. | ||
Kesisoglou F, Mitra A. Application of absorption modeling in rational design of drug product under quality-by-design paradigm. AAPS J. 2015;17(5):1224–1236. | ||
DrugBank [homepage on the internet]. Metronidazole; http://www.drugbank.ca/drugs/DB00916. Accessed June 17, 2016. | ||
Guidechem.com: 443-48-1|C6H9N3O3 Metronidazole Density, Molecular Structure, Formula, Synonyms, Boiling Point, Flash Point, Risk Codes, Storage Temperature. [homepage on the internet]. http://www.guidechem.com/dictionary/en/443-48-1.html. Accessed June 17, 2016. | ||
Bao JJ, Liu X, Zhang Y, Li Y. The development of a high-throughput measurement method of octanol/water distribution coefficient based on hollow fiber membrane solvent microextraction technique. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;967:183–189. | ||
Pitt KG, Newton JM, Stanley P. Effects of compaction variables on porosity and material tensile strength of convex-faced aspirin tablets. J Pharm Pharmacol. 1991;43(4):219–225. | ||
Gamlen Tableting Ltd. The importance of normalisation when comparing tablet properties. Available from: http://www.gamlentableting.com/images/The%20importance%20of%20normalization%20when%20analysing%20tablet%20hardness.pdf. Accessed September 30, 2016. | ||
FMC Health and Nutrition. [homepage on the internet]. Avicel for solid dose forms. Available from: http://www.fmcbiopolymer.com/Pharmaceutical/Products/Avicelforsoliddoseforms.aspx Accessed June 17, 2016. | ||
Bhattacharyya L, Rohrer JS. Applications of ion chromatography for pharmaceutical and biological products. Hoboken NJ: John Wiley & Sons; 2012. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.