Back to Journals » International Journal of General Medicine » Volume 17
JAG1 Variants Confer Genetic Susceptibility to Thyroid Dysgenesis and Thyroid Dyshormonogenesis in 813 Congenital Hypothyroidism in China
Authors Li M, Wang X, Wang F, Wang F, Zhao D, Liu S
Received 19 October 2023
Accepted for publication 5 February 2024
Published 7 March 2024 Volume 2024:17 Pages 885—894
DOI https://doi.org/10.2147/IJGM.S445557
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Miaomiao Li,1,2 Xiaoyu Wang,1,2 Fang Wang,3 Fengqi Wang,1,2 Dehua Zhao,4 Shiguo Liu1,2
1Department of Medical Genetic, the Affiliated Hospital of Qingdao University, Qingdao, People’s Republic of China; 2Prenatal Diagnosis Center, the Affiliated Hospital of Qingdao University, Qingdao, People’s Republic of China; 3Endocrinology Department, the Affiliated Hospital of Qingdao University, Qingdao, People’s Republic of China; 4Neonatal Screening Center, the Third Affiliated Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China
Correspondence: Dehua Zhao, The Third Affiliated Hospital of Zhengzhou University, Neonatal Screening Center, Zhengzhou, Henan, 450052, People’s Republic of China, Email [email protected] Shiguo Liu, The Affiliated Hospital of Qingdao University, Department of Medical Genetic, Prenatal Diagnosis Center, Qingdao, Shandong, 266003, People’s Republic of China, Email [email protected]
Background and Objective: Congenital hypothyroidism (CH) is indeed a prevalent neonatal endocrine disorder, affecting approximately 1 in 2000– 3000 newborns worldwide, and 1 in 2400 newborns in China. Despite its high incidence, the genetic causes of CH, particularly those related to thyroid dysgenesis (TD), are still not well understood. However, previous studies have suggested that JAG1 may be a potential susceptibility gene for congenital thyroid defects. To explore the association between JAG1 and CH, we screened JAG1 variants in a large cohort of 813 CH patients.
Methods: We performed genetic analysis of JAG1 using next-generation sequencing in 813 CH cases. The pathogenicity of the variants was assessed by bioinformatics softwares, protein sequence conservation analysis, and hydrophobic analysis. Further genetic analysis was conducted targeting 20 CH-related genes in these 25 JAG1 variant carriers.
Results: We identified 10 pathogenic missense mutations (p.V45L, p.V272I, p.P552L, p.G610E, p.G852D, p.A891T, p.E1030K, p.R1060W, p.A1131T, p.P1174L) carried by 25 patients, the mutation rate of JAG1 in CH was 3.08%. Among these 25 patients, 16 with 1 variant, 6 with 2 variants, and the other 3 with 3 variants. Our findings indicated that JAG1 variants confer genetic susceptibility to both TD and DH, but with different inheritance models. JAG1 variants lead to TD mainly through monogenic model, while for DH cases, both monogenic mechanisms and oligogenic mechanisms play a pivotal role. Oligogenicity may contribute to the disease severity of DH.
Conclusion: JAG1 is a shared genetic factor in TD and DH, with a detection rate of 3.08% in Chinese individuals with CH. A comparison between the oligogenic and monogenic groups suggests a gene dosage effect in CH. Patients with the same JAG1 mutation exhibit diverse clinical phenotypes, indicating complex mechanisms underlying phenotypic heterogeneity.
Keywords: congenital hypothyroidism, JAG1, pathogenic variant, inheritance model, phenotypic heterogeneity
Introduction
Congenital hypothyroidism (CH) characterized by primary thyroid hormone deficiency, is the most common and preventable neonatal endocrine disorder, with an incidence of 1 in 2000–3000 newborns worldwide, and 1 in 2400 in China.1,2 Extensive research in human and experimental models has confirmed the genetic origin of CH.3 CH can be classified into two groups based on the pathogenesis: dyshormonogenesis (DH) and thyroid dysgenesis (TD). DH is associated with genes such as DUOX2, DUOXA2, DUOX1, IYD, SLC5A5, TG, TPO, SLC26A4, and SLC26A7, which are inherited in an autosomal-recessive manner.4 In contrast, the pathogenesis of TD is more complex, with well-established pathogenic genes including NKX2.1, PAX8, TSHR, and FOXE1. Next-generation sequencing has led to the identification of additional candidate genes for TD, such as NKX2.5, GLIS3, CDCA8, NTN1, TUBB1, THRB, NNT, DUOX2, and JAG1.5,6 TD caused by these genetic defects can occur in isolation or in conjunction with extrathyroidal abnormalities, but the risk is significantly higher in the context of syndromes. Therefore, exploring TD pathogenesis from the perspective of syndromic TD pathogenic genes may be a more efficient strategy.
JAG1 (NM_000214.3) is located on human chromosome 20p12.2 and encodes a ligand of the Notch signaling pathway. It is expressed on the cell surface and activates Notch signaling by interacting with receptors on adjacent cells. Defects in JAG1 have been identified as the main cause of Alagille syndrome (ALGS; OMIM 118450), a multisystemic disorder that affects the liver, heart, skeleton, eyes, and facial features.7 JAG1 is known to play a role in the development of various organ systems, including the thyroid. Luca Persani et al confirmed the importance of the jagged1-Notch signal in thyroid development and function using zebrafish models. They found that loss of jag1 function resulted in thyroid hypoplasia.8 Subsequently, they screened 100 cases of CH and identified four carriers of JAG1 variants, three of which had TD, suggesting that JAG1 is a novel susceptibility gene for congenital thyroid defects. Subsequently, they screened 100 cases of CH and identified four carriers of JAG1 variants, three of which had TD, suggesting that JAG1 is a novel susceptibility gene for congenital thyroid defects.9 However, further clinical and experimental data are needed to explore the association between JAG1 and CH.
In this study, we performed JAG1 analysis in a larger cohort of 813 CH patients using next-generation sequencing, and identified 10 likely pathogenic mutations carried by 25 patients. We further analyzed 20 CH-related genes in these 25 carriers, assessed the pathogenicity of variants and the association between genotype and phenotype of JAG1 variants. Our findings revealed that the JAG1 variants may confer genetic susceptibility to both TD and DH, but with different inheritance model.
Method
Patients
Eight hundred and thirteen CH patients in Henan Province and Shandong Province from January 2012 to Mar 2019 were included in the study. Newborns with TSH levels of >10 μIU/mL were recalled for the re‑examination of serum TSH and FT4 levels by electrochemiluminescence assay. A diagnosis of CH was made on the basis of elevated TSH (TSH ≥ 10 mIU/l) level and decreased free T4 (fT4 < 12 pmol/l) level. To ensure the accuracy of the study, patients with other congenital malformations were excluded from the analysis. Additionally, all patients underwent thyroid ultrasonography and technetium thyroid scans for classification purposes. Generally, after 2–3 years of L-T4 replacement therapy, patients were assessed as permanent congenital hypothyroidism (PCH) or transient congenital hypothyroidism (TCH) by retesting TSH and FT4 one month after drug withdrawal. This study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University (QDFY WZLL 28515) and the Third Affiliated Hospital of Zhengzhou University, and complied with the Declaration of Helsinki. Written informed consent had been obtained from the parents of all participants.
Next-Generation Sequencing and Data Analysis
We extracted the genomic DNA from peripheral blood leukocytes using the Tiangen DNA extraction kit (TIANGEN, Beijing, China). A total of 10 ng qualified DNA per sample was randomly fragmented into 180 ~ 250 bp by Sonication (Covaris S2, Covaris, USA) for the DNA libraries preparation. The exons and 10 bp exon–intron boundaries of JAG1 were selected by a gene capture strategy, using the GenCap custom enrichment kit (MyGenostics, Beijing, China) for mutational screening. Sequencing of amplicon libraries was carried out using the MiSeq system (Illumina, CA, USA). Surecall software (V3.0.1.4, Agilent) was used for data analysis and variant annotation.
We detected 111 variants in JAG1 in 813 CH patients. Variants located in introns or synonymous variants were excluded. Forty-one variants with MAF < 0.01 in all database (1000 Genome, ExAC, esp6500siv2, PopFreqMax) and located in exons and splicing site were retained. And then the functional impact of variants were tested by 4 online programs (SIFT, Polyphen2_HDIV, MutationTaster), CADD (threshold≥20), we regard the variants predicted to be possibly damaging by ≥2 programs as pathogenic variants. In the end, a total of 12 pathogenic variants were identified through screening, and out of those, 10 pathogenic variants were confirmed through Sanger sequencing in 25 patients.
For these 25 JAG1 variant carriers, we performed further genetic analysis targeting 20 CH-related genes (TG, TPO, DUOX1, DUOXA1, DUOX2, DUOXA2, IYD, SLC26A4, SLC26A5, TSHR, NKX2.1 FOXE1, PAX8, HHEX, THRB, GLIS3, CDCA8, NKX2.5, NTN1, TUBB1). With the same method of variant detection and filtering, we finally validated 7 pathogenic variants in DUOX1, 1 in DUOXA2 in 9 patients.
Sanger Sequencing
2 × Rapid Taq Master Mix (Vazyme, Nanjing, China) was used for PCR amplification. BigDye® Terminator Cycle Sequencing Kit and automated sequencer ABI 3730XL were used for sequencing reaction of the PCR products. The sequencing results were interpreted using Chromas software V.2.5.
Statistical Analysis
The statistical significance was assessed using the Mann–Whitney U-test and chi-square test. Differences were considered significant at P <0.05. All analyses were performed using SPSS.
Results
Clinical Characteristics of Patients
The present study enrolled a total of 813 CH patients consisting of 224 TD and 589 DH, the ratio of male to female was 1:1.07. Except for 306 patients without classification, the other 296 patients were diagnosed as PCH and 211 were TCH. We summarized the clinical characteristics of all subjects and made a comparison between all subjects and JAG1 variant carriers, indicating that the carriers had lower levels of T3, T4, fT3 and fT4 in diagnostic serum, but only the fT4 level was statistically different (p<0.05). Based on European Society for Paediatric Endocrinology criteria, serum fT4 concentration correlates with CH severity, which can be classified into mild (>10 pmol/L), moderate (5 to < 10 pmol/L), and severe (<5 pmol/L). The JAG1 variant carriers appear to have a higher distribution in severe phenotypes (56% vs 41%). Table 1 records additional symptoms associated with CH, including prolonged jaundice, abdominal distension, umbilical hernia, low heart sounds, hoarse cry, and drowsiness, among others. Among the patients, 12 had a family history of CH, while the remaining cases were sporadic.
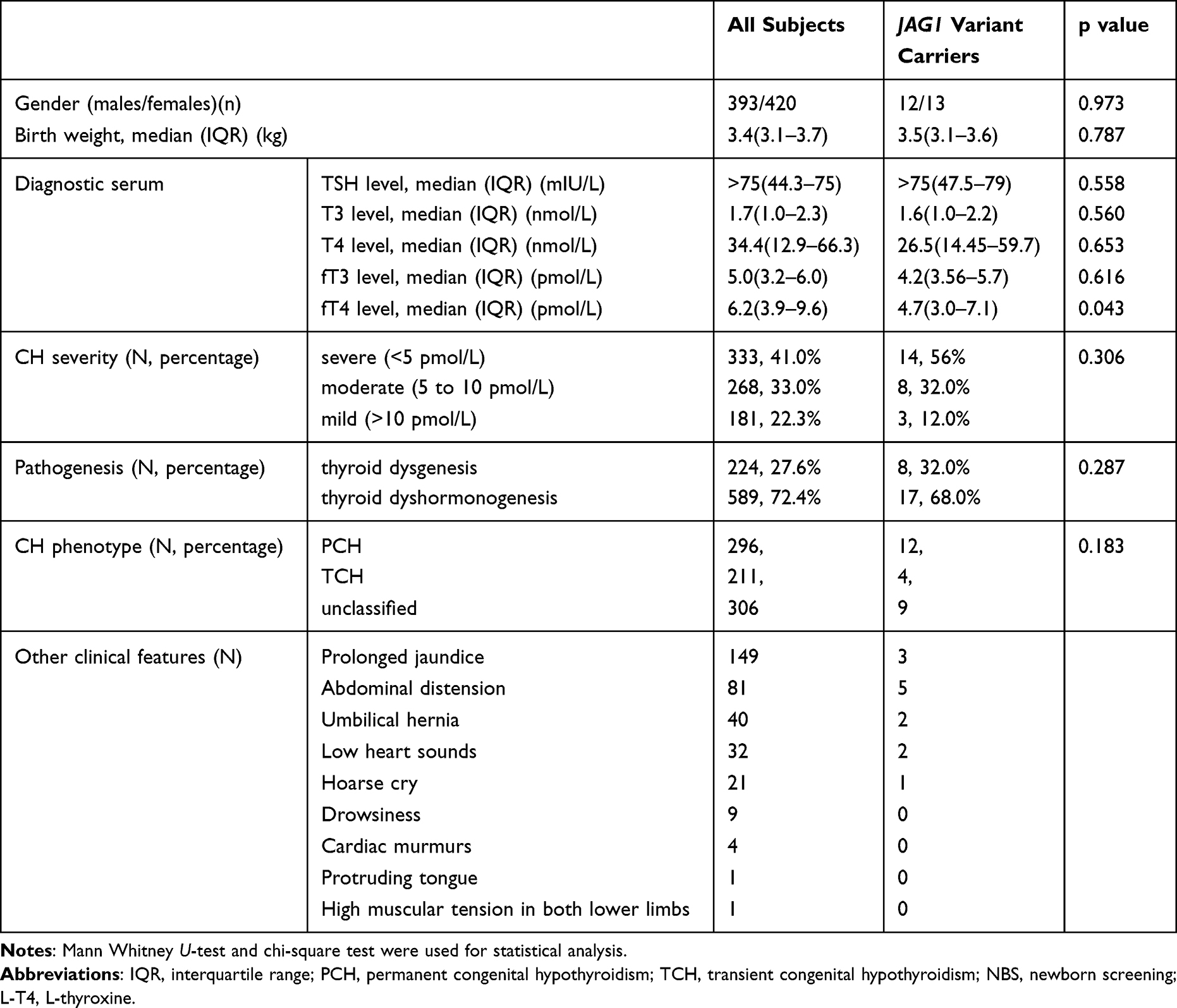 |
Table 1 The Clinical Characteristics of All CH Subjects and JAG1 Variant Carriers |
Variant Detection and Mutation Spectrum
After data analysis and verification by Sanger sequencing, we identified 10 JAG1 pathogenic variants in 25 patients, the mutation rate is 3.08%. The mutant p.V45L was the most common variant, accounting for 8 cases, followed by mutant p.P552L (5 cases), p.A1131T (3 cases), p.G610E (2 cases), p.P1174L (2 cases) and the others (p.V272I, p. G852D, p.A891T, p.E1030K, p.R1060W). All of the identified variants were missense mutations. Among them, the p.G852D and p.E1030K variants were first reported by your research team, while the remaining variants were potential disease-causing mutations previously identified in patients with ALGS or pituitary stalk interruption syndrome. Currently, only the mutant p.P1174L was described in hypothyroidism. In addition, we screened 20 CH associated genes (TG, TPO, DUOX1, DUOXA1, DUOX2, DUOXA2, IYD, SLC26A4, SLC26A5, TSHR, NKX2.1 FOXE1, PAX8, HHEX, THRB, GLIS3, CDCA8, NKX2.5, NTN1, TUBB1) in these 25 patients in the same way, and found that 1 with DUOXA2 monoallelic variant, 5 with DUOX2 monoallelic variant, and 3 with DUOX2 biallelic variants (Table 2). Specifically, we screened out 7 pathogenic variants in DUOX2 and 1 in DUOXA2, all these variants had been reported in previous study of CH (Supplementary Figure 1, Supplementary Table 1). It is worth noting that all the pathogenic variants identified in this study were found to be heterozygous. Detailed information regarding these variants, as well as relevant data, can be found in Supplementary Table 1.
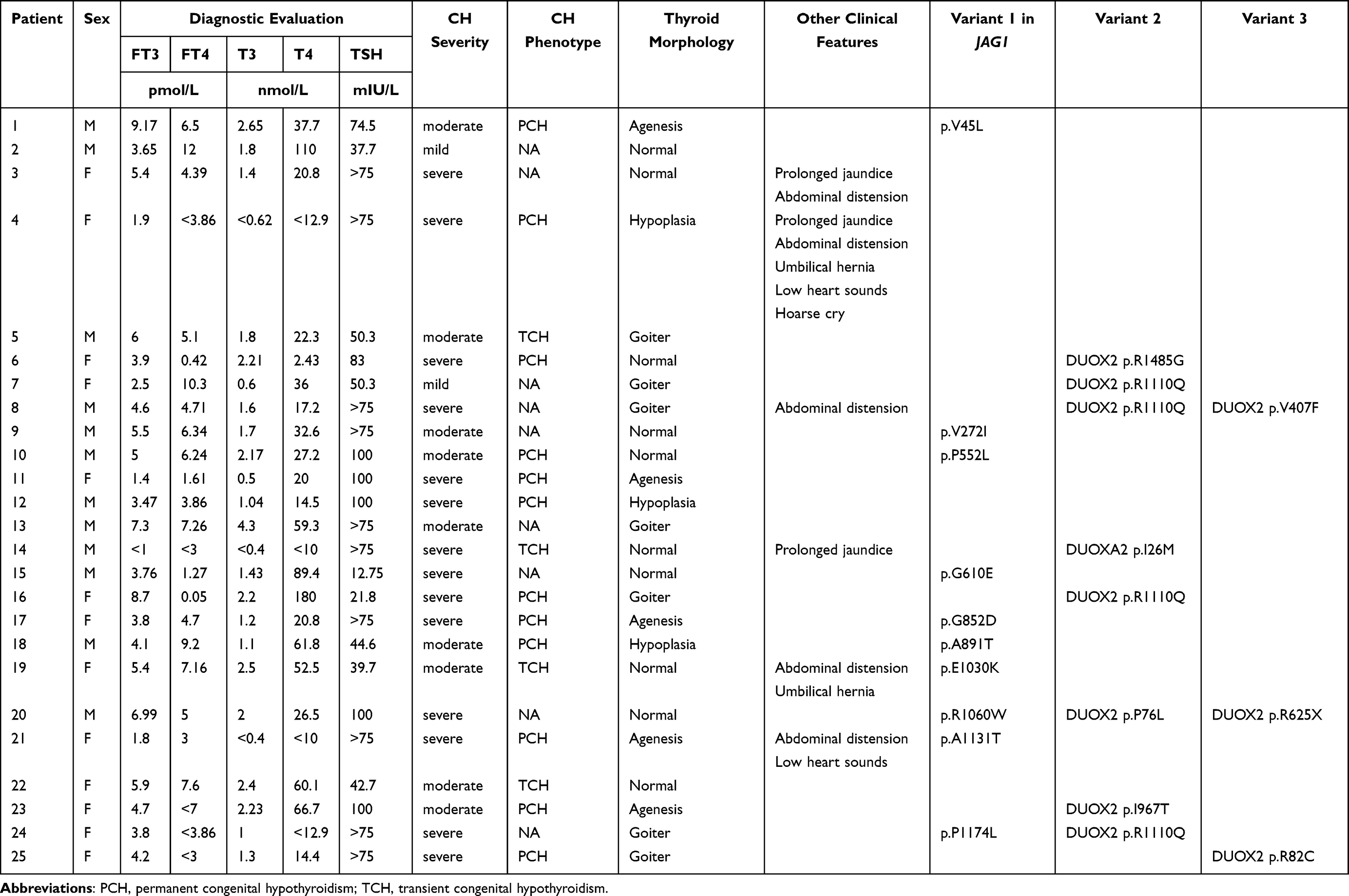 |
Table 2 Clinical Characteristics of Patients with Potential Pathological Variants |
Pathogenicity Assessment
We assessed all variants pathogenicity using SIFT, PolyPhen2, Mutation Taster and CADD (threshold≥20), the pathogenic variants were predicted to be deleterious by at least 2 bioinformatics software. To assess the conservation of the amino acid site where the missense mutation is located, a multiple sequence alignment was performed using ClustalW2 across eight different species (Homo sapiens, Mus musculus, Rattus norvegicus, Sus scrofa, Pan troglodytes, Bos Taurus, Macaca mulatta and Felis catus). The results revealed that all the amino acid sites were highly conserved except for the 1174 site of JAG1, 407 and 967 site of DUOX2 (Figure 1). Considering the effect of amino acid hydrophobicity on protein structure and stability, we analyzed the changes of hydrophobic parameters caused by amino acid substitution, most of these substitution lead to significant increase (R>C, P>L, R>G, R>W) or decrease (I>T, G>D, G>E, I>M, V>F) of hydrophobic parameters, a few with minor changes (R>Q, V>I, V>L, E>K) (Figure 2). According to American College of Medical Genetics and Genomics (ACMG) guidelines, 1 variants were classified as pathogenic, 6 were likely pathogenic, and the other 11 were variants of uncertain significant (VUS) (Supplementary Table 1).
 |
Figure 1 Multiple sequences alignment. Multiple sequences alignment were conducted using ClustalW2 among Homo sapiens, Mus musculus, Rattus norvegicus, Sus scrofa, Pan troglodytes, Bos Taurus, Macaca mulatta and Felis catus. (A) Multiple sequences alignment of JAG1; (B) Multiple sequences alignment of DUOX2; (C) Multiple sequences alignment of DUOXA2. The mutation sites were marked by red arrows. |
 |
Figure 2 The changes of hydrophobic parameters caused by amino acid substitution. Wild-type amino acids were marked with black, substituted amino acids were marked with red; “+” means increased hydrophobicity, “-” means decreased hydrophobicity. |
Genotype and Phenotype Relationship
Among these 25 patients, 16 carried with 1 variant, 6 with 2 variants, and the other 3 with 3 variants (Table 2). The distribution of the number of variants carried by each patients in TD and DH groups showed that TD is mainly caused by monogenic mutation (7/8), while for DH, monogenic mutation (9/17) and multiple mutations (8/17) account for about half of the cases respectively (Figure 3A). In this study, the patients were divided into two groups based on the number of variants they carried: the monogenic group and the oligogenic group. The concentration of fT4 was then compared between these two groups to assess any differences. The results showed that the fT4 concentration in oligogenic group was lower than that in monogenic group, though it was not statistically significant (P=0.192) (Figure 3B). Further analysis was conducted on the basis of CH classification. For TD patients, only one case in oligogenic group, which cannot be used for statistical analysis. For DH patients, the concentration of fT4 in oligogenic group was significantly lower than that in monogenic group (P=0.043) (Figure 3C). Compared with DH cases, TD cases showed a more severe phenotype. In monogenic group, the level of fT4 in TD was lower (P=0.185) (Figure 3D), and in subjects with classification, all TD patients were diagnosed as PCH (Table 2). We conducted the burden testing of the JAG1 variants we identified using publicly available data in GnomAD (n=60,146) as control, the results indicated the association between JAG1 variants and CH (OR = 4.003, p = 0.000). Although the pathogenic mechanisms and clinical manifestations of JAG1 variant in TD and DH are different, there is no difference in its distribution in TD and DH patients (P=0.287) (Table 1).
 |
Figure 3 Analysis of genotype and phenotype relationship (A) The distribution of the number of variants carried by each patients; (B) The comparison of fT4 concentration between monogenic group and oligogenic group; (C) The comparison of fT4 concentration between monogenic DH and oligogenic DH; (D) The comparison of fT4 concentration between monogenic TD and monogenic DH. “ns” means P>0.05, “*” means P<0.05. |
 |
Figure 4 Clustering analysis of JAG1 variants in CH. Distance in nucleotides between missense mutations within exons 1–21 and exons 22–26. Statistical significance was calculated using an unpaired, two-tailed t test, “*” means P<0.05. |
Discussion
In this study, we performed NGS targeting JAG1 in a large cohort of 813 subjects with CH, and then identified 10 pathogenic missense mutations (p.V45L, p.V272I, p.P552L, p.G610E, p.G852D, p.A891T, p.E1030K, p.R1060W, p.A1131T, p.P1174L) carried by 25 patients, the mutation rate of JAG1 in CH was 3.08%.
JAG1 is expressed widely during mammalian development and plays a crucial role in the development of organ systems associated with Alagille syndrome (ALGS) and the thyroid.7,10 In the context of ALGS, JAG1 mutations have been characterized, with frameshift mutations being the most common (37%), followed by nonsense mutations (22%), large gene deletions (13%), missense mutations (13%), splice site mutations (12%), and other types (<3%).11 Missense variants in JAG1 tend to cluster within the first 6 exons of the gene, and approximately 25% of these variants result in the gain or loss of a cysteine within the EGF-like domain, which is statistically significant.11 In the genetic screening of JAG1 variants in cases of CH, Filippis et al identified seven missense variants (p.P476T, p.F509L, p.Y663C, p.R744Q, p.R937Q, p.I1021T, p.R1094W).9,12 In your study, 10 missense variants were found in CH patients. Notably, all the JAG1 variants detected in CH were missense mutations, and they were significantly concentrated in exons 22–26 (p=0.049) (Figure 4), which differs from the pattern observed in ALGS.11 Phenotypic effects of JAG1 mutations are highly penetrant, but there is considerable variability in expressivity, which is consistent with the findings of your research. Patients harboring the same pathogenic variant can exhibit a high degree of variable expressivity. This observation led to the hypothesis that a second gene could act as a modifier of the phenotype. Candidate genetic modifiers, such as glycosyltransferases (Lunatic Fringe, Radical Fringe, Manic Fringe, and POGLUT1) and THBS2, have been reported.13–15 However, whether the hypothesis is applicable for the pathogenic mechanism of JAG1 in CH still needs further verification.
Among these 813 CH patients we enrolled, 224 were classified as TD and the other 589 were DH. Our data showed once again that DH is a more common cause of CH than TD in China. Combined with the genetic screening of 20 genes associated with CH in these 25 JAG1 variants carriers, we further analyzed the correlation between genotype and phenotype, the results revealed that the JAG1 variants confer genetic susceptibility to both TD and DH, but with different inheritance model.
In TD cases, 7 of 8 were monogenic JAG1 variant carriers, the other 1 with heterozygous variant in JAG1 and DUOX2, indicating that JAG1 variants lead to TD mainly through monogenic model. Currently, most knowledge on TD genetics has been accumulated on monogenetic forms of TD, which concern mutations in genes TSHR, NKX2-1, PAX8, FOXE1 and NKX2-5. However, as causative mutations have been identified in less than 5% of cases, and the genetics of TD do not follow simple Mendelian patterns, the transmission mode of TD remains a topic of debate. Polygenic and epigenetic mechanisms have been proposed alongside the monogenic model.16,17 The polygenic origin of TD was initially supported by mouse models, as Nkx2-1 +/− and Pax8 +/− mice displayed normal thyroid gland, while [Nkx2-1, Pax8] +/− mice showed hypoplasia or hemiagenesis.18 Polygenic variants were also identified in very few TD patients, the combination of variants in genes reported in previous studies were TSHR and TPO/DUOX2/GANS/PAX8/JAG1, PAX8 and NKX2-5, TG and PAX8/TPO/JAG1. Notably, one patient with variants in both JAG1 (p.R937Q) and TSHR (p.S526G) exhibited thyroid hypoplasia, and another patient carried JAG1 (p.R744Q) and biallelic TSHR variants (p.R1250L, p.R1136Q) and was diagnosed with thyroid ectopy.12,17,19 Additionally, a JAG1 variant in combination with a DUOX2 variant was found in a CH patient without further classification.20 In this study, only 1 patient with agenesis was detected with digenic variants in JAG1 (p.A1131T) and DUOX2 (p.I967T), the low incidence of polygenic mutations indicating it possess a marginal role in TD.
In DH cases, both monogenic mechanisms (9/17, 52.9%) and oligogenic mechanisms (8/17, 47.1%) play a pivotal role. It is worth noting that DH caused by monogenic variants of JAG1 is not inherited in the classic autosomal dominant manner, as all parents of affected individuals were euthyroid. Although biallelic and monogenic mutations are considered the most common causes of DH, oligogenicity has been found in 20–43.5% of patients with CH, particularly in association with DUOX2 and DUOXA2.20,21 DUOX2 is the primary gene responsible for DH, and the detection rate of DUOX2 mutations in China is as high as 28%-44%,22 which is consistent with the detection rate obtained in this study (44%). In our patients with DH, 2 out of 9 cases in monogenic group were severe CH, while for oligogenic group, 7 out of 8 cases were severe CH. Pearson chi-square analysis showed that oligogenic inheritance was significantly associated with severe CH (P=0.007), and the finding is supported by previous study.23 To examine the correlation between variant dosage and severity, the study compared the free thyroxine (fT4) levels in the monogenic and oligogenic groups. The results indicated that oligogenicity may contribute to the disease severity of DH, which is consistent with the findings of a study by Takeshi et al.20 Therefore, the oligogenic model may partly account for the differences in the penetrance and expressivity of CH.
The role of JAG1 in thyroid development has been investigated in zebrafish, where it was found to affect the proliferation and survival of thyroid follicular cells, possibly through a permissive effect on the TSH signal. In the biosynthesis of thyroid hormone, JAG1 may play a role by regulating SLC5A5 through the Notch effector HES1.8,10 Interestingly, there is an overlap of genetic causes between TD and thyroid DH, as identified in many studies. Patients with the same mutation can exhibit a wide range of clinical phenotypes.12,20,24 This suggests that the genetics of CH are complex, and the mechanisms involved in its phenotypic heterogeneity remain unclear.
In conclusion, we screened out 25 JAG1 variant carriers in 813 CH patients, the mutation rate is 3.08%. Further analysis revealed that the JAG1 variants confer genetic susceptibility to both TD and DH, but with different inheritance model. The comparation between oligogenic group and monogenic group indicated that CH may exhibit a gene dosage effect. There are two limitations in the present study should be noted. First, patients with other congenital malformations were excluded when collecting samples, as a result, we could not evaluate the effects of these JAG1 variants on other organ systems. Second, we did not carry out functional studies of these JAG1 variants.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Acknowledgments
We thank all the patients and team members for participating in the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The present work was supported by grants from the National Natural Science Foundation of China (82270829, 82071683, 82201914).
Disclosure
The authors declare no potential conflicts of interest in this work.
References
1. van Trotsenburg P, Stoupa A, Leger J, et al. Congenital Hypothyroidism: a 2020-2021 Consensus Guidelines Update-An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid. 2021;31(3):387–419. doi:10.1089/thy.2020.0333
2. Deng K, He C, Zhu J, et al. Incidence of congenital hypothyroidism in China: data from the national newborn screening program, 2013-2015. J Pediatr Endocrinol Metab. 2018;31(6):601–608. doi:10.1515/jpem-2017-0361
3. Persani L, Rurale G, de Filippis T, Galazzi E, Muzza M, Fugazzola L. Genetics and management of congenital hypothyroidism. Best Pract Res Clin Endocrinol Metab. 2018;32(4):387–396. doi:10.1016/j.beem.2018.05.002
4. Kostopoulou E, Miliordos K, Spiliotis B. Genetics of primary congenital hypothyroidism-a review. Hormones. 2021;20(2):225–236. doi:10.1007/s42000-020-00267-x
5. Mio C, Grani G, Durante C, Damante G. Molecular defects in thyroid dysgenesis. Clin Genet. 2020;97(1):222–231. doi:10.1111/cge.13627
6. Kizys MML, Louzada RA, Mitne-Neto M, et al. DUOX2 Mutations Are Associated With Congenital Hypothyroidism With Ectopic Thyroid Gland. J Clin Endocrinol Metab. 2017;102(11):4060–4071. doi:10.1210/jc.2017-00832
7. Grochowski CM, Loomes KM, Spinner NB. Jagged1 (JAG1): structure, expression, and disease associations. Gene. 2016;576(1 Pt 3):381–384. doi:10.1016/j.gene.2015.10.065
8. Porazzi P, Marelli F, Benato F, et al. Disruptions of global and JAGGED1-mediated notch signaling affect thyroid morphogenesis in the zebrafish. Endocrinology. 2012;153(11):5645–5658. doi:10.1210/en.2011-1888
9. de Filippis T, Marelli F, Nebbia G, et al. JAG1 Loss-Of-Function Variations as a Novel Predisposing Event in the Pathogenesis of Congenital Thyroid Defects. J Clin Endocr Metab. 2016;101(3):861–870. doi:10.1210/jc.2015-3403
10. Marelli F, Persani L. Role of Jagged1-Notch pathway in thyroid development. J Endocrinol Invest. 2018;41(1):75–81. doi:10.1007/s40618-017-0715-x
11. Gilbert MA, Bauer RC, Rajagopalan R, et al. Alagille syndrome mutation update: comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum Mutat. 2019;40(12):2197–2220. doi:10.1002/humu.23879
12. de Filippis T, Gelmini G, Paraboschi E, et al. A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet. 2017;26(13):2507–2514. doi:10.1093/hmg/ddx145
13. Ryan MJ, Bales C, Nelson A, et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology. 2008;48(6):1989–1997. doi:10.1002/hep.22538
14. Thakurdas SM, Lopez MF, Kakuda S, et al. Jagged1 heterozygosity in mice results in a congenital cholangiopathy which is reversed by concomitant deletion of one copy of Poglut1 (Rumi). Hepatology. 2016;63(2):550–565. doi:10.1002/hep.28024
15. Tsai EA, Gilbert MA, Grochowski CM, et al. THBS2 Is a Candidate Modifier of Liver Disease Severity in Alagille Syndrome. Cell Mol Gastroenterol Hepatol. 2016;2(5):663–675 e662. doi:10.1016/j.jcmgh.2016.05.013
16. Stoupa A, Kariyawasam D, Carre A, Polak M. Update of Thyroid Developmental Genes. Endocrinol Metab Clin North Am. 2016;45(2):243–254. doi:10.1016/j.ecl.2016.01.007
17. Szinnai G. Genetics of normal and abnormal thyroid development in humans. Best Pract Res Clin Endocrinol Metab. 2014;28(2):133–150. doi:10.1016/j.beem.2013.08.005
18. Amendola E, De Luca P, Macchia PE, et al. A mouse model demonstrates a multigenic origin of congenital hypothyroidism. Endocrinology. 2005;146(12):5038–5047. doi:10.1210/en.2005-0882
19. Makretskaya N, Bezlepkina O, Kolodkina A, et al. High frequency of mutations in ‘dyshormonogenesis genes’ in severe congenital hypothyroidism. PLoS One. 2018;13(9):e0204323. doi:10.1371/journal.pone.0204323
20. Yamaguchi T, Nakamura A, Nakayama K, et al. Targeted Next-Generation Sequencing for Congenital Hypothyroidism With Positive Neonatal TSH Screening. J Clin Endocrinol Metab. 2020;105(8):e2825–e2833. doi:10.1210/clinem/dgaa308
21. Wang H, Kong X, Pei Y, et al. Mutation spectrum analysis of 29 causative genes in 43 Chinese patients with congenital hypothyroidism. Mol Med Rep. 2020;22(1):297–309. doi:10.3892/mmr.2020.11078
22. Huang M, Lu X, Dong G, et al. Analysis of Mutation Spectra of 28 Pathogenic Genes Associated With Congenital Hypothyroidism in the Chinese Han Population. Front Endocrinol. 2021;12:695426. doi:10.3389/fendo.2021.695426
23. Oliver-Petit I, Edouard T, Jacques V, et al. Next-Generation Sequencing Analysis Reveals Frequent Familial Origin and Oligogenism in Congenital Hypothyroidism With Dyshormonogenesis. Front Endocrinol. 2021;12:657913. doi:10.3389/fendo.2021.657913
24. Stoupa A, Kariyawasam D, Muzza M, et al. New genetics in congenital hypothyroidism. Endocrine. 2021;71(3):696–705. doi:10.1007/s12020-021-02646-9
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.