Back to Journals » International Journal of General Medicine » Volume 17
Interstitial Lung Disease in Patients with Mixed Connective Tissue Disease: A Retrospective Study
Received 23 March 2024
Accepted for publication 1 May 2024
Published 13 May 2024 Volume 2024:17 Pages 2091—2099
DOI https://doi.org/10.2147/IJGM.S464704
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Woon-Man Kung
Xueyan Shan,1,2 Yongpeng Ge3
1Department of Rheumatology, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China; 2Postgraduate School, Beijing University of Chinese Medicine, Beijing, People’s Republic of China; 3Department of Rheumatology, The Key Laboratory of Myositis, China-Japan Friendship Hospital, Beijing, People’s Republic of China
Correspondence: Yongpeng Ge, Department of Rheumatology, The Key Laboratory of Myositis, China-Japan Friendship Hospital, Yinghua East Road, Chaoyang District, Beijing, 100029, People’s Republic of China, Email [email protected]
Objective: To investigate the clinical features, severity and prognosis of interstitial lung disease (ILD) in patients with mixed connective tissue disease (MCTD).
Methods: We performed a retrospective study on clinical data of MCTD patients admitted to China-Japan Friendship Hospital between October 2012 and October 2022. Data including long-term follow-up were retrieved from medical records. We compared MCTD patients with and without ILD in terms of clinical features, laboratory and imaging findings, severity and treatment response.
Results: A total of 59 patients were included, with a mean age of 46 years, among which 91.5% (n = 54) were females. Symptoms of pulmonary involvement were present in 44 patients (74.6%, 95% CI: 62.3– 84.9%). Based on lung high-resolution computed tomography (HRCT), ILD was diagnosed in 39 (66.1%) patients, among which 31 (79.5%) showed nonspecific interstitial pneumonia (NSIP) as the radiological pattern, 21 (53.9%) showed a reticulation pattern, while 24 (61.5%) showed ground glass opacity (GGO). Eight (13.6%) patients had pulmonary arterial hypertension (PAH), and 7 (11.9%) had pleural effusions. Based on pulmonary function tests (PFTs), 27 patients were divided into the mild 13 (48.1%) and moderate 14 (51.9%) groups. Multivariate analysis showed that gastroesophageal reflux (GER; OR=5.28, p=0.010) and cough (OR=4.61, p=0.043) were the predictive factors for ILD. With a median follow-up of 50 months, the mortality rate was 2.38%.
Conclusion: ILD is common in MCTD patients, with NSIP as the common imaging pattern. Patients with GER and cough are relevant factors in the development of ILD. The majority of MCTD patients with ILD are mild to moderate in severity.
Keywords: mixed connective tissue disease, interstitial lung disease, gastroesophageal reflux
Background
Mixed connective tissue disease (MCTD) is characterized by positive anti-U1RNP (nRNP) antibodies, with Raynaud’s phenomenon, synovitis and myositis, and first described in 1972 by Sharp et al.1 Patients suffering from MCTD have several clinical features of other connective tissue diseases (CTDs), such as systemic lupus erythematosus (SLE), systemic sclerosis (SSc) or polymyositis/dermatomyositis (PM/DM). Lung involvement in CTD significantly contributes to high mortality and poor outcomes.2 In MCTD, the main manifestation of lung involvement includes interstitial lung diseases (ILD) and pulmonary arterial hypertension (PAH). Previous studies on different cohorts reported the prevalence of ILD to range from 27.4% to 78%,2–4 and showed that ILD is also a major contributor to poor prognosis in MCTD.5
Many studies described the factors associated with ILD. Narula et al demonstrated that dysphagia and Raynaud’s phenomenon were associated with ILD,3 while another study conducted by Fagundes et al found patients with gastroesophageal reflux (GER) and esophageal dilatation to be more susceptible to ILD.4 However, only a few studies investigated the prevalence and clinical characteristics of ILD in Chinese MCTD patients. The factors associated with complications in this group remain unclear.
The objective of this study is to investigate the prevalence, clinical and radiological features as well as the severity of ILD in MCTD patients in China. We aim to identify associated risk factors and improve the recognition of pulmonary involvement in MCTD patients.
Materials and Methods
Patients
Patients admitted to the Department of Rheumatology in China-Japan Friendship Hospital between October 2012 and October 2022 were included in this study. We collected the clinical and laboratory data, including the age, clinical features, laboratory tests, pulmonary function tests (PFTs), high-resolution computed tomography (HRCT), treatment and follow-up from the medical records. In this study, pulmonary involvement of patients was recorded in detail including ILD, PAH, pleural effusion and pulmonary hypertension secondary to ILD (PH-ILD). The serological examinations included complete blood count, urinalysis, liver and renal function tests, electrolyte analysis, creatine kinase, anti-extractable nuclear antigen (ENA) spectrum, immunoglobulin levels, complement levels, rheumatoid factor, C-reactive protein, and tumor markers. Cardiac involvement (CI) was defined based on electrocardiography and transthoracic Doppler echocardiography abnormalities, including right ventricular hypertrophy, atrial enlargement and ventricular block. Neurological impairment (NI) included headache, limb numbness, and facial numbness as manifestations of trigeminal or peripheral neuropathy. GER was determined based on clinical manifestations, including gagging, panting, belching and dysphagia. In some cases, GER was confirmed by gastroscopy or esophagoscopy.
The inclusion criteria were as follows: (i) older than 18 years old; (ii) fulfilling either the Alarcón-Segovia or the Kasukawa criteria.6,7 The exclusion criteria included the following: (i) missing primary data; (ii) diagnosed with other CTDs at follow-up. Furthermore, patients who were diagnosed with ILD prior to their MCTD diagnosis were excluded to confirm that the ILD cases were directly attributable to MCTD.
The study was in line with the Declaration of Helsinki and was approved by the Research Review Committee (RRC) and Ethics Review Committee (ERC) of China-Japan Friendship Hospital (reference number: 2023-KY-345). All patient data were used anonymously, and the written informed consent for patients was waived for this retrospective study.
ILD Assessment
ILD was defined as having evidence of ground-glass attenuations, consolidations, reticulations and/or honeycombing noted on HRCT as well as restrictive impairments in the lung function examinations. All pulmonary HRCT for the diagnosis of ILD were independently reviewed by an experienced radiologist and a rheumatologist. HRCT presentations are classified into usual interstitial pneumonia (UIP), organizing pneumonia (OP), nonspecific interstitial pneumonia (NSIP), respiratory bronchiolitis (RB), desquamative interstitial pneumonia (DIP) and diffuse alveolar damage (DAD).8 As recommended by the American Thoracic Society/European Respiratory Society,9 the classification of ILD using CT imaging involves honeycombing, reticulation, ground glass opacity (GGO), traction bronchiectasis (TB), interlobular septal thickening (IST) and airspace consolidation. In some cases, lung biopsy results corroborating ILD, were also recorded.
Pulmonary Function Tests
PFTs were performed within 4 weeks after the corresponding HRCT. The tested variables included the forced vital capacity (FVC), forced expiratory volume in one second (FEV1), the ratio of FEV1 to FVC (FEV1/FVC) and the diffusing capacity of the lung for carbon monoxide (DLCO). According to a consensus from the American Thoracic Society (ATS) on Idiopathic Pulmonary Fibrosis,10 an absolute decline or increase in FVC of >5% or in DLCO of >10% within 1 year is considered as ILD progression or improvement, respectively. Conversely, a decrease or increase in FVC of <5% or in DLCO of <10% is defined as stable disease. Hence, the patients were divided into 3 groups based on the initial PFT values: mild group (FVC > 75% and DLCO > 55%), moderate group (FVC between 50–75% or DLCO between 35–55%) and severe group (FVC < 50% and DLCO < 35%).11,12
Transthoracic Doppler Echocardiography
Transthoracic Doppler echocardiography (TTDE) was conducted for the assessment of cardiac structure and function. According to previous studies and the 2022 ESC/ERS guidelines for pulmonary arterial hypertension,13,14 PAH is defined as a mean pulmonary artery pressure of > 20 mmHg at right heart catheterization or if TTDE shows pulmonary artery systolic pressure (PASP) > 40 mmHg.
PAH combined with ILD,15 also known as disproportionate pulmonary hypertension, is suspected when pulmonary hypertension is detected in patients with mild or normal respiratory function impairment (FVC > 70%) and minimal parenchymal involvement on CT imaging. Pulmonary hypertension arising from ILD itself is called PH-ILD.15 This form of pulmonary hypertension results from the effects of lung scarring or fibrosis on the pulmonary vasculature, causing increased pulmonary artery pressure. All patient subgroups are identified through detailed evaluation by specialized physicians.
Statistical Analysis
The features of MCTD patients with ILD were compared with those of MCTD patients without ILD using the Student’s test for age and chi-square or Fisher’s exact tests where appropriate for others. Univariate logistic regression identified significant variables with p-values below 0.05 for inclusion in the multivariate analysis. Using logistic regression backward stepwise elimination, variables with less statistical significance were gradually removed, resulting in a final model that retained only the most significant variables. All p-values were 2-sided, and a p-value less than 0.05 was considered statistically significant. The statistical analyses were performed using SPSS (version 26).
Results
General Information on MCTD Patients
As shown in Figure 1, 92 patients were initially enrolled, among which we excluded 29 patients who were later diagnosed with other CTDs and 4 patients with missing consecutive data. Eventually, 59 patients with MCTD were included, including 54 (91.5%) females and 5 (8.5%) males. The age of onset ranged from 18 to 76 years, with a mean age of 46 ± 14.5 years, and the median disease duration was 2 years.
 |
Figure 1 Flowchart of the screening process. |
At disease onset, the initial symptom in the majority of MCTD patients was Raynaud’s phenomenon (35.6%) or synovitis (25.4%), while shortness of breath (11.9%) or myositis (6.8%) was present in a minority of patients. However, most patients (n=44, 74.6%, 95% CI: 62.3–84.9%) developed pulmonary involvement during the follow-up period, including 39 (66.1%) patients with ILD, 8 (13.6%) with PAH and 7 (11.9%) with pleural effusions. Three of these patients had PAH combined with ILD. Of those with pleural effusions, 5 of 7 had ILD, while 1 had PAH. The patients gradually developed multiple manifestations. During the course of disease, 44 (74.6%) patients had Raynaud’s phenomenon, 47 (79.7%) had synovitis, 33 (55.9%) had typical GER symptoms, 40 (67.8%) had myositis, 22 (37.3%) suffered from NI and 2 (4.1%) had CI.
Notably, during the follow-up period, 17 (28.8%) patients suffered from different degrees of lung infection. Five were infected with bacteria, 4 with viruses, including herpes simplex virus (n=2), EB virus (n=2) and cytomegalovirus (n=3), 3 with fungi, including Pseudomonas albicans (n=2), Candida albicans (n=1) and Aspergillus flavus (n=1), and 5 with multiple pathogens.
All patients with MCTD were positive for anti-U1RNP and antinuclear antibodies (ANA). Among them, 46 (77.9%) patients showed granular nuclear staining and 10 (16.9%) granular cytoplasmic staining, while 2 showed homogeneous nuclei and 1 a speckled nucleus.
Comparison of MCTD Patients with and without ILD
All patients underwent HRCT at the time of their initial MCTD diagnosis. In total, 39 (66.1%) patients were diagnosed with ILD based on lung images or pulmonary histopathology. The radiological patterns of ILD included NSIP in 31 (79.5%) patients and UIP in 8 (20.5%). In most cases, abnormal shadows were mainly bilaterally distributed on the inferior lobe and subpleural areas. Honeycombing, reticulation, GGO, IST, pleural thickening, TB and airspace consolidations were observed in 7 (18.0%), 21 (53.9%), 24 (61.5%), 11 (28.2%), 13 (33.3%), 7 (18.0%) and 6 (15.4%) patients, respectively. Throughout the course of disease, 37 (62.8%) MCTD patients developed respiratory symptoms, including 21 (35.6%) with dry cough, 6 (10.2%) with dyspnea and 10 (17.0%) with other manifestations, while 9 (23.1%) patients in the ILD group did not have any respiratory symptoms. A total of 27 patients had available PFTs, which were as follows: FVC <75% in 11 (40.7%) cases, FEV1 <80% in 12 (44.4%) cases and DLCO <55% in 11 (40.7%) cases. All patients had FEV1/FVC ≥70%. Based on PFTs, 13 (48.1%) were included in the mild group, and 14 (51.9%) were included in the moderate group.
The characteristics of the patients at the time of their initial MCTD diagnosis were reviewed. The age at onset, sex, duration and smoking history did not show statistically significant differences in the ILD group compared with the non-ILD group (p>0.05; Table 1). Meanwhile, the frequency of cough, shortness of breath and GER manifestations were significantly higher in the ILD group than in the non-ILD group (p<0.05). As shown in Table 2, the IgG level (2157.0 ± 961.8 g/L vs 1647.1 ± 470.6 g/L, p=0.008) and RF positivity rate (54.1% vs 23.5%, p=0.036) were significantly higher in the ILD group compared with the non-ILD group. The levels of tumor markers, such as cytokeratin 19 fragment (CYFRA211), carcinoembryonic antigen (CEA) and carbohydrate antigen 199 (CA199), were significantly higher in ILD group than in the non-ILD group (p<0.05). In addition, the levels of serum Krebs von den lungen-6 (KL-6) were measured in 7 ILD patients and shown to be elevated with a mean value of 1161.7 U/mL.
 |
Table 1 Characteristics of MCTD Patients Combined with ILD |
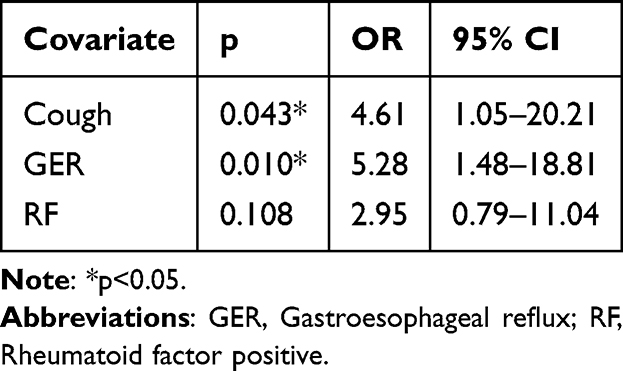 |
Table 2 Logistic Regression Analysis of MCTD Patients with ILD |
Comparing the PAH and Non-PAH Groups
The PAH group included 8 female patients, with a mean age of 47.5 ± 6.8 years and median duration of 4 years. The mean baseline PASP was 66.7 ± 22.5 mmHg. Among them, 4 (50%) patients had accelerated tricuspid valve flow velocity with a mean tricuspid regurgitant jet velocity (TRV) of 412.3 ± 61.8 cm/s. The tricuspid regurgitation was mild in 5 (72.5%) patients, moderate in 2 (25%) and severe in 1 (12.5%).
As shown in Supplementary Table 1, the frequency of anemia (50% vs 13.7%, respectively) and CI (25% vs 0%, respectively) were significantly higher in the PAH group compared with the non-PAH group (p<0.05). Meanwhile, the frequency of GER symptoms was significantly lower in the PAH group than in the non-PAH group (12.5% vs 62.7%, respectively, p=0.023). The levels of C-reactive protein (CRP; 47.4 ± 44.8 mg/L vs 1.3 ± 0.3 mg/L, respectively, p=0.002) and CA724 (10.5 ± 3.9 U/mL vs 1.5 ± 0.9 U/mL, respectively, p=0.01) were significantly higher in the PAH group than in non-PAH group.
Predictive Factors for ILD
The resulting variables from the univariate analysis were further analyzed with multivariate logistic regression. The results in Table 2 showed that GER (OR=5.28, p=0.010) and cough (OR=4.61, p=0.043) were significantly associated with ILD.
Treatment and Follow Up
Eight (13.6%) patients were treated only with glucocorticoid (GC), and 50 (84.7%) with GC combined with immunosuppressant, including hydroxychloroquine (25, 42.4%), cyclophosphamide (18, 30.5%), methotrexate (12, 20.3%), cyclosporine (8, 13.6%), mycophenolate mofetil (7, 11.9%), leflunomide (7, 11.9%), iguratimod (6, 10.2%) and azathioprine (3, 5.1%). Furthermore, herbal preparations, such as tripterygium glycosides (12, 20.3%) or total glucosides of paeony (6, 10.2%), were also used in combination with GC in the treatment of some patients. One patient (1.7%) was not treated with GC but with hydroxychloroquine and total glucosides of paeony.
We performed a follow-up over a period of 12–112 months on 42 patients, with a median follow-up time of 50 months. We followed up with all patients by telephone to track their outcomes. Among the 17 cases that were lost to follow-up, there were no fatalities, indicating that the lack of follow-up data was due to other reasons. During the follow-up period, one patient died, who had combined ILD and PAH with severe pulmonary infection, resulting in respiratory and circulatory failure; the mortality rate of MCTD patients was 2.38%. Two patients with combined ILD and PAH developed malignancy. Eighteen patients underwent HRCT review during the follow-up period, one MCTD patient developed ILD 5 years after disease onset.
During the follow-up period, 8 patients with ILD underwent HRCT and PFT, among which 7 had stable PFTs and 1 had progression. In addition, HRCT revealed stable findings in 5 patients, while 3 patients showed new abnormal shadows. The detailed results are shown in Supplementary Table 2.
Discussion
In this retrospective study, we found that pulmonary involvement, especially ILD, frequently occurred in patients with MCTD but was generally mild to moderate. GER and cough were found to be highly associated with ILD. After treatment, the prognosis of lung involvement was good.
Previous studies demonstrated that patients with MCTD were frequently females and had synovitis, Raynaud’s phenomenon and esophageal hypermobility.2,16 In our cohort, common clinical features among MCTD patients included Raynaud’s phenomenon, synovitis, shortness of breath, GER and myositis. Pulmonary involvement frequently occurred in MCTD patients, with the prevalence of PAH ranging from 6.9% to 17.8%. Previous studies reported varied prevalence of ILD, ranging from 27.4% to 78%.2–4 In our cohort, 66.1% of the patients had MCTD combined with ILD and 13.6% with PAH.
The majority of HRCT presentations in the cohort of ILD patients were distributed on the bilateral inferior lobe and subpleural areas, with reticulations and GGO being the most common, which is consistent with previous findings.17,18 ILD subtyping was predominantly NSIP, followed by UIP. However, subtyping might be less accurate in some patients due to the unavailability of bronchoscopy or lung biopsy. Decreased lung physiological parameters indicated the development and progression of ILD.17 Pulmonary function abnormalities could precede the clinical symptoms, characteristic alterations include restrictive ventilation disorders and diffusion dysfunction, and the most common abnormal manifestation is decreased DLCO.19 In this study, all patients with ILD had mild to moderate decreased DLCO.
In the present study, GER manifestations were significantly more frequent in the ILD group and strongly associated with ILD. The relationship between SSc-ILD and esophageal symptoms has been explained in previous studies.4,20 Briefly, gastroesophageal acid after GER tends to cause bronchoconstriction, while prolonged the microaspiration of gastric acid, repeatedly damaging the lung parenchyma and leading to fibroblast activation, which eventually predisposes to ILD.4,21 In this study, the prevalence of cough and shortness of breath was significantly higher in patients with combined ILD, and logistic regression analysis showed cough to be significantly associated with the development of ILD. This suggests that in clinical practice, respiratory symptoms in patients with MCTD may indicate concomitant ILD. The occurrence of infection, though more frequent among patients with combined ILD, is likely a consequence of MCTD treatment or complications arising from ILD.
Our results showed that MCTD-PAH patients had significantly higher prevalence of anemia and CI. Anemia can contribute to the onset and progression of PAH through mechanisms such as inflammation and disrupted iron homeostasis, leading to reduced oxygen delivery and increased cardiovascular stress.22 The pathogenesis of PAH is associated with the impaired secretion of vasoactive mediators, such as nitric oxide, and increased production of vasoconstrictor and proliferative factors, which affect the vascular tone and lead to vascular remodeling.23 The deoxygenated form of hemoglobin can reduce nitrite to nitric oxide, which promotes vasodilation of the pulmonary vascular bed. However, the reduced hemoglobin content in anemia may impair the reaction, leading to an increase in the pulmonary vascular pressure.24
RF and IgG levels were significantly higher in the ILD group, in consistence with the study of Narula et al,3 which reported RF and anti-Sm antibody positivity to be higher in ILD patients with MCTD. In addition, we found that the levels of CRP and ESR were significantly elevated in MCTD patients with PAH, which is consistent with previous findings on idiopathic PAH and SLE-PAH, especially since CRP is associated with the prognosis and adverse events in PAH.23,25 The mechanism behind this effect can be explained by the level of inflammatory infiltration in the perivascular region associated with pulmonary vascular remodeling.26
Tumor markers are synthesized and secreted by tumor cells and other cells in the tumor tissue, which can be classified into different groups according to their production source and biological properties. Bao et al found significant differences in the markers of CA199, CEA, CA153 and CYFRA211 between CTD-ILD patients and non-ILD patients.27 We also found that patients with MCTD-ILD had significantly higher levels of CA199, CEA and CYFRA211. The expression of CYFRA211 in respiratory fine bronchial and alveolar epithelial cells has indeed been identified as a possible marker of epithelial cell injury in ILD patients.28 The CA199 level is negatively correlated with DLCO and could be a diagnostic indicator of RA-ILD.29 As for CEA, it reflects the proliferation and secretion of epithelial cells, which may be potentially linked to the mechanism of ILD (persistent damage of epithelial cells).30 Our findings may provide new evidence on the relationship between elevated tumor markers and MCTD-ILD. However, two MCTD-ILD patients with elevated tumor markers had detectable tumors during follow-up. So, we should follow up the occurrence of cancer and the influence of therapeutic drugs on the tumor marker in all patients in the following years.
In the study of Chan et al on CTD-ILD, they found the mortality rate to be 23.8% at a median follow-up time of 4 years,31 while the study of Li et al showed the overall survival rates of CTD-PAH patients at 1, 3 and 5 years to be 98%, 78% and 59%, respectively.32 Although the pulmonary involvement is a common cause of death in patients with CTD, one patient in our cohort died of respiratory and complicative circulatory failure. Although a high prevalence of ILD was observed in MCTD patients, it was mostly mild to moderate, and the disease progression was relatively slow after early and aggressive treatment.
This study has some limitations. Firstly, only a relatively small number of patients was included and the follow-up time was relatively short in some patients. Furthermore, because this was a retrospective study, a complete set of clinical data was not available for every patient. This study included only hospitalized patients, excluding those admitted on an outpatient basis. As hospitalized patients may represent a more severe subgroup, this could affect the observed prevalence of ILD. Finally, the gold standard for the diagnosis of PAH is right heart catheterization, and due to its invasive nature, we relied on non-invasive echocardiographic findings as a screening criterion, which may result in false positive or negative cases.
Conclusion
In summary, ILD occurs frequently in MCTD patients. The imaging pattern of MCTD-ILD is mostly NSIP, and it is generally less severe. Our findings show that GER and cough are predictive factors of ILD. Early identification of ILD and PAH may help to improve the prognosis of patients.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Acknowledgments
The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Funding
This study was supported by the National High Level Hospital Clinical Research Funding (2022-NHLHCRF-YS-02) and Elite Medical Professionals Project of China-Japan Friendship Hospital (NO. ZRJY2023-GG02).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease--an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52(2):148–159. doi:10.1016/0002-9343(72)90064-2
2. Alves MR, Isenberg DA. “Mixed connective tissue disease”: a condition in search of an identity. Clin Exp Med. 2020;20(2):159–166. doi:10.1007/s10238-020-00606-7
3. Narula N, Narula T, Mira-Avendano I, Wang B, Abril A. Interstitial lung disease in patients with mixed connective tissue disease: pilot study on predictors of lung involvement. Clin Exp Rheumatol. 2018;36(4):648–651.
4. Fagundes MN, Caleiro MT, Navarro-Rodriguez T, et al. Esophageal involvement and interstitial lung disease in mixed connective tissue disease. Respir Med. 2009;103(6):854–860. doi:10.1016/j.rmed.2008.12.018
5. Hyldgaard C, Bendstrup E, Pedersen AB, Pedersen L, Ellingsen T. Interstitial lung disease in connective tissue diseases: survival patterns in a population-based cohort. J Clin Med. 2021;10(21):4830. doi:10.3390/jcm10214830
6. D. Alarcón-Segovia and M. Villareal. Classification and diagnostic criteria for mixed connective tissue disease. In: Mixed Connective Tissue Disease and Antinuclear Antibodies. Amsterdam: Elsevier; 1987:3340.
7. R. Kasukawa, T. Tojo, and S. Miyawaki. Preliminary diagnostic criteria for classification of mixed connective tissue disease. In: Mixed Connective Tissue Disease and Antinuclear Antibodies. Amsterdam: Elsevier; 1987:41–47.
8. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. doi:10.1164/rccm.201308-1483ST
9. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68. doi:10.1164/rccm.201807-1255ST
10. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an Update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47. doi:10.1164/rccm.202202-0399ST
11. Aiko N, Yamakawa H, Iwasawa T, et al. Clinical, radiological, and pathological features of anti-asparaginyl tRNA synthetase antibody-related interstitial lung disease. Respir Investig. 2020;58(3):196–203. doi:10.1016/j.resinv.2019.12.003
12. Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
13. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731. doi:10.1093/eurheartj/ehac237
14. Humbert M, Kovacs G, Hoeper MM, et al. Corrigendum to: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur Heart J. 2023;44(15):1312. doi:10.1093/eurheartj/ehad005
15. Waxman AB, Elia D, Adir Y, Humbert M, Harari S. Recent advances in the management of pulmonary hypertension with interstitial lung disease. Eur Respir Rev. 2022;31(165):210220. doi:10.1183/16000617.0220-2021
16. Hajas A, Szodoray P, Nakken B, et al. Clinical course, prognosis, and causes of death in mixed connective tissue disease. J Rheumatol. 2013;40(7):1134–1142. doi:10.3899/jrheum.121272
17. Perelas A, Arrossi AV, Highland KB. Pulmonary manifestations of systemic sclerosis and mixed connective tissue disease. Clin Chest Med. 2019;40(3):501–518. doi:10.1016/j.ccm.2019.05.001
18. Gunnarsson R, Aaløkken TM, Molberg Ø, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Ann Rheum Dis. 2012;71(12):1966–1972. doi:10.1136/annrheumdis-2011-201253
19. Hetlevik SO, Flatø B, Aaløkken TM, et al. Pulmonary manifestations and progression of lung disease in juvenile-onset mixed connective tissue disease. J Rheumatol. 2019;46(1):93–100. doi:10.3899/jrheum.180019
20. Salaffi F, Di Carlo M, Carotti M, Fraticelli P, Gabrielli A, Giovagnoni A. Relationship between interstitial lung disease and oesophageal dilatation on chest high-resolution computed tomography in patients with systemic sclerosis: a cross-sectional study. Radiol med. 2018;123(9):655–663. doi:10.1007/s11547-018-0894-3
21. Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27(1):136–142. doi:10.1183/09031936.06.00037005
22. Sonnweber T, Pizzini A, Tancevski I, Löffler-Ragg J, Weiss G. Anaemia, iron homeostasis and pulmonary hypertension: a review. Intern Emerg Med. 2020;15(4):573–585. doi:10.1007/s11739-020-02288-1
23. Zanatta E, Polito P, Famoso G, et al. Pulmonary arterial hypertension in connective tissue disorders: pathophysiology and treatment. Exp Biol Med. 2019;244(2):120–131. doi:10.1177/1535370218824101
24. Crawford JH, Isbell TS, Huang Z, et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107(2):566–574. doi:10.1182/blood-2005-07-2668
25. Huang C, Li M, Liu Y, et al. Baseline characteristics and risk factors of pulmonary arterial hypertension in systemic lupus erythematosus patients. Medicine. 2016;95(10):e2761.
26. Cerik IB, Dindas F, Koyun E, et al. New prognostic markers in pulmonary arterial hypertension: CRP to albumin ratio and uric acid. Clin Biochem. 2022;100:22–28. doi:10.1016/j.clinbiochem.2021.11.004
27. Bao Y, Zhang W, Shi D, Bai W, He D, Wang D. Correlation between serum tumor marker levels and connective tissue disease-related interstitial lung disease. Int J Gen Med. 2021;14:2553–2560. doi:10.2147/IJGM.S310917
28. Vercauteren IM, Verleden SE, McDonough JE, et al. CYFRA 21.1 in bronchoalveolar lavage of idiopathic pulmonary fibrosis patients. Exp Lung Res. 2015;41(8):459–465. doi:10.3109/01902148.2015.1073407
29. Zheng M, Lou A, Zhang H, Zhu S, Yang M, Lai W. Serum KL-6, CA19-9, CA125 and CEA are diagnostic biomarkers for rheumatoid arthritis-associated interstitial lung disease in the Chinese Population. Rheumatol Ther. 2021;8(1):517–527. doi:10.1007/s40744-021-00288-x
30. Strieter RM, Mehrad B. New mechanisms of pulmonary fibrosis. Chest. 2009;136(5):1364–1370. doi:10.1378/chest.09-0510
31. Chan C, Ryerson CJ, Dunne JV, Wilcox PG. Demographic and clinical predictors of progression and mortality in connective tissue disease-associated interstitial lung disease: a retrospective cohort study. BMC Pulm Med. 2019;19(1):192. doi:10.1186/s12890-019-0943-2
32. Li X, Sun X, Huang Y, et al. Simplified risk stratification for pulmonary arterial hypertension associated with connective tissue disease. Clin Rheumatol. 2019;38(12):3619–3626. doi:10.1007/s10067-019-04690-3
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.