Back to Journals » Journal of Pain Research » Volume 15
Interactions Among Non-Coding RNAs and mRNAs in the Trigeminal Ganglion Associated with Neuropathic Pain
Authors Fang ZH ![]() , Liao HL, Tang QF, Liu YJ, Zhang YY, Lin J, Yu HP
, Liao HL, Tang QF, Liu YJ, Zhang YY, Lin J, Yu HP ![]() , Zhou C, Li CJ, Liu F, Shen JF
, Zhou C, Li CJ, Liu F, Shen JF
Received 23 July 2022
Accepted for publication 13 September 2022
Published 22 September 2022 Volume 2022:15 Pages 2967—2988
DOI https://doi.org/10.2147/JPR.S382692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qi Fang
Zhong-Han Fang,1,2 Hong-Lin Liao,1,2 Qing-Feng Tang,1,2 Ya-Jing Liu,1,2 Yan-Yan Zhang,1,2 Jiu Lin,1,2 Hao-Peng Yu,3 Cheng Zhou,4 Chun-Jie Li,1,5 Fei Liu,1,2,* Jie-Fei Shen1,2,*
1State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, National Center for Stomatology, West China School of Stomatology, Sichuan University, Chengdu, People’s Republic of China; 2Department of Prosthodontics, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China; 3West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 4Laboratory of Anesthesia and Critical Care Medicine, Translational Neuroscience Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 5Department of Head and Neck Oncology, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fei Liu; Jie-Fei Shen, Department of Prosthodontics, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China, Email [email protected]; [email protected]
Background: Recent studies have demonstrated the contribution of non-coding RNAs (ncRNAs) to neuropathic pain. However, the expression profile of ncRNAs in the trigeminal ganglion (TG) and their functional mechanism underlying trigeminal neuropathic pain are still unclear.
Methods: In the present study, the trigeminal neuropathic pain model induced by chronic constriction injury of the infraorbital nerve (CCI-ION) was used to study the expression profile and potential regulatory mechanism of miRNAs, lncRNAs, circRNAs, and mRNAs in the TG by RNA-sequencing (RNA-seq) and bioinformatics analysis. CCI-ION mice suffered from mechanical allodynia from 3 days to 28 days after surgery.
Results: The RNA-seq results discovered 67 miRNAs, 216 lncRNAs, 14 circRNAs, 595 mRNAs, and 421 genes were differentially expressed (DE) in the TG of CCI-ION mice 7 days after surgery. And 39 DEGs were known pain genes. Besides, 5 and 35 pain-related DE mRNAs could be targeted by 6 DE miRNAs and 107 DE lncRNAs, respectively. And 23 pain-related DEGs had protein–protein interactions (PPI) with each other. GO analysis indicated membrane-related cell components and binding-related molecular functions were significantly enriched. KEGG analysis showed that nociception-related signaling pathways were significantly enriched for DE ncRNAs and DEGs. Finally, the competing endogenous RNA (ceRNA) regulatory network of DE lncRNA/DE circRNA-DE miRNA-DE mRNA existed in the TG of mice with trigeminal neuropathic pain.
Conclusion: Our findings demonstrate ncRNAs are involved in the development of trigeminal neuropathic pain, possibly through the ceRNA mechanism, which brings a new bright into the study of trigeminal neuropathic pain and the development of novel treatments targeting ncRNAs.
Keywords: trigeminal neuropathic pain, non-coding RNAs, RNA-sequencing, trigeminal ganglion, trigeminal nerve injury, bioinformatics
Introduction
Trigeminal neuropathic pain is a severe health problem and is difficult to treat because of its complex mechanisms. It often occurs after injuries to the trigeminal sensory fibers induced by trauma and tooth iatrogenic injuries from dental treatments, such as local anesthetic injections, root canal therapies, extractions, oral surgery, dental implants, orthognathic surgery, and other invasive procedures.1,2 Although lots of potential intervention molecular targets in the trigeminal ganglion (TG) for treating trigeminal neuropathic pain have been found,3 for instance, multiple groups of molecules (ATP, glutamate, SP, IL-1β, and TNF-α), receptors (P2X, NMDAR, NK1, and GPCRs), ion channels (voltage-gated potassium channels and voltage-gated calcium channels), and intracellular signaling pathways (MAPKs, PKC, PKA),3–5 consistently effective treatment options are still not available.3,6 Therefore, more preclinical research is expected to elucidate the molecular mechanism underlying trigeminal neuropathic pain and identify novel therapeutic targets to develop a more effective treatment for trigeminal neuropathic pain.
In recent years, emerging evidence suggests that non-coding ribose nucleic acids (ncRNAs) play a vital role in the pathophysiological process of neuropathic pain,7,8 which brings novel landscapes for the diagnosis and treatment of trigeminal neuropathic pain. According to their size, ncRNAs mainly consist of micro RNAs (miRNAs), long ncRNAs (lncRNAs), circular RNAs (circRNAs), and small nucleolar RNAs (snoRNAs), which typically do not encode proteins but functionally regulate protein expression.9,10 miRNAs in the TG have been proved to play an essential role in oral cancer pain and orofacial inflammatory pain.11,12 In addition, emerging data indicate that lncRNAs and circRNAs interact with miRNAs to regulate mRNA expression in the progression of neuropathic pain.8,13 Notably, recent studies have reported that lncRNAs in the TG also contribute to trigeminal neuropathic pain and chronic inflammatory temporomandibular joint pain.14,15 These data support that ncRNAs in the TG may be associated with trigeminal neuropathic pain. However, the regulatory functions of ncRNAs in the TG under trigeminal neuropathic pain and their functional mechanisms have not been systematically described.
Chronic constriction injury of the infraorbital nerve (CCI-ION) is a classic animal model of trigeminal neuropathic pain,16,17 resulting in long last orofacial pain status. In this study, a whole-transcriptome sequencing technique was performed to systematically analyze ncRNAs and mRNA expression signatures in the TG of mice with or without trigeminal neuropathic pain induced by CCI-ION. Furthermore, bioinformatics analysis interpreted the potential biological functions of differentially expressed (DE) ncRNAs, DE mRNAs, and DE genes (DEGs) related to pain and analyzed the possible regulatory mechanisms of ncRNAs in trigeminal neuropathic pain.
Methods
Animals
Male and female C57BL/6 mice (7 weeks) were used in the present study. All animal procedures performed in this study were reviewed and approved by the Research Ethics Committee of West China Hospital of Stomatology and were conducted according to the International Association for the Study of Pain guidelines. Animals were habituated for one week before experiments and maintained on a 12:12 light-dark cycle at a room temperature of 22 ± 1 °C with free access to food and water.
Trigeminal Neuropathic Pain Model Induced by CCI-ION
Twenty-four male mice were randomly divided into CCI-ION (n = 12) and sham (n = 12) groups. In addition, six female mice were randomly divided into CCI-ION (n = 3) and sham groups (n = 3). Chronic constriction injury of the infraorbital nerve (CCI-ION) was carried out to establish orofacial neuropathic pain.17 Animals were anesthetized with 4% pentobarbital sodium (50 mg/kg, i.p.) and laid on their back. Intraoral surgery was performed under aseptic conditions. Briefly, a 3-mm-long incision was made along the gingivobuccal margin in the buccal mucosa, beginning next to the maxillary first left molar. The distal segment of ION outside the infraorbital foramen was isolated from surrounding connective tissue and clearly visualized from the infraorbital foramen. Then, it was loosely tied with two chromic gut (4-0) ligatures at an interval of 2 mm. Finally, the mucosa incision was closed by 5-0 silk sutures. Sham-operated mice received only unilateral nerve exposure without ligature.
Behavioral Test
Nociceptive responses of Sham (n = 6) and CCI-ION (n = 6) male mice to mechanical stimuli were measured on 1 day before and 3, 7, 10, 14, 21, and 28 days after surgery. A series of 8 von Frey filaments (0.04 g, 0.16 g, 0.4 g, 0.6 g, 1 g, 1.4 g, 2 g, and 4 g, Stoelting Company, Wood Dale, USA) were applied to the left orofacial skin to examine the head withdrawal threshold (HWT) of mice in sham and CCI-ION groups in the up-down method described previously.18 The mouse was placed in a metal mesh cage (8 cm × 8 cm × 8 cm). After it calmed down, the hairs were presented at the left whisker pad, of which the hair was cut before the test. The starting filament was the middle of the series. Each filament was tested at an interval of several seconds. A positive response was noted if the head was sharply withdrawn. Once a negative response followed by a positive response or vice versa was approached, another four von Frey presentations were done according to the up-down rules. The mechanical pain threshold was quantified as EF50 (the response frequency), which was calculated by combining the value of the factor based on the response pattern.18 When continuous positive or negative responses were observed to the exhaustion of the stimulus set, values of 4 g and 0.04 g were assigned, respectively. The data of behavioral tests were presented as the means ± SD. The results of the Sham and CCI-ION groups on the same day were statistically analyzed using a t-test. The time tendency of behavioral testing data was detected with repeated measure ANOVA. Significance was established at P < 0.05.
RNA Extraction, Library Preparation, and RNA Sequencing
On 7 days after the operation, the left TG was collected only from six male mice (3 mice in sham group and 3 mice in CCI-ION group) after being deeply anesthetized with isoflurane. Total RNA was extracted from the isolated TG using TRIZOL (Invitrogen, Carlsbad) according to the manufacturer’s instructions. RNA degradation and contamination were monitored on 1% agarose gels. RNA purity and concentration were examined using the NanoPhotomet ® spectrophotometer (IMPLEN, CA, USA) and Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Library construction and sequencing were conducted by Novogene (Beijing, China). For lncRNA, circRNA, and mRNA sequencing, about 3 µg of total RNA from each sample was used to deplete ribosomal RNA according to the Epicenter Ribo-zeroTM rRNA Removal Kit (Epicenter, USA) following the manufacturer’s instruction. After the rRNA-free residue was cleaned up by ethanol precipitation, RNA was then fragmented into 250~300 bp fragments. Fragmented RNAs were reverse-transcribed to create the first-strand cDNA, which was used to synthesize U-labeled second strand DNAs with DNA polymerase I, RNase H, and dNTPs followed by adapter ligation and PCR amplification. For small RNA sequencing, about 3 μg total RNA per sample was used to generate a small RNA library using NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA) following the manufacturer’s recommendations. Briefly, after 3’ end of small RNAs was ligated by NEB 3’ SR Adaptor, the SR RT Primers were used to transform the single-stranded DNA into a double-stranded DNA. Then, PCR amplification was performed using LongAmp Taq 2X Master Mix, SR Primer for illumina and index (X) primer. The RNA fragments from the above two libraries were finally sequenced on an Illumina NovaSeq 6000 platform, and 50 bp single-end reads for the small RNAs were generated, and 150 bp paired-end reads for the library of lncRNAs, circRNAs, and mRNAs were generated. The raw and clean reads number of the library for miRNAs for the samples were 13,077,884–16,706,212 and 12,891,853–16,477,632, respectively. The raw and clean reads number of the library for lncRNAs, circRNAs, and mRNAs for the samples were 79,399,034–105,087,144 and 77,718,888–103,727,040, respectively. The raw and clean bases of the library for miRNAs for the samples were, respectively, 0.654G–0.835G and 0.645G–0.824 G. The raw and clean bases of the library for lncRNAs, circRNAs, and mRNAs for the samples were, respectively, 11.91G–15.76G and 11.66G–15.56G. The error rate of the library for miRNAs and library of lncRNAs, circRNAs, and mRNAs for the samples were, respectively, <0.01 and <0.03%. The raw data of RNA-seq for miRNAs (SRA ID: SUB10791226, BioProject ID: PRJNA788681) and lncRNAs, circRNAs, and mRNAs (SRA ID: SUB10816303, BioProject: PRJNA788734) have been published in the NCBI SRA database.
Differential Analysis of ncRNAs and mRNAs
The small RNA tags were mapped to the reference sequence by Bowtie with 1 mismatch to analyze their expression and distribution. Clean reads with a length of 18–35 nt were screened as miRNA. miRBase 22 was used as a reference to look for the known miRNA. Modified software mirdeep2 and srna-tools-cli were used to obtain the potential miRNA and draw the secondary structures.19 Only miRNAs with Fold Change 1.4 and P value < 0.05 were classified as DE miRNAs. All transcriptomes from all samples were merged to reconstruct a comprehensive transcriptome using Perl scripts. Hisat220 was used to map reads to the genome of mice. LncRNAs were identified from the assembled transcripts following steps: (1) removal of short transcripts < 200 bp and < 2 exons; (2) removal of the transcripts with protein-coding capability determined using CNCI (Coding-Non-Coding-Index),21 Pfam and CPC (Coding Potential Calculator)22 database; Those remaining were considered to be lncRNAs. Novel lncRNAs were named following the rules of HGNC (The HUGO Gene Nomenclature Committee). StringTie23 was used to perform the expression level of mRNAs and lncRNA by calculating FPKM (fragments per kilobase of transcript per million mapped reads). The differential expression analysis was performed using the edgeR package (1.10.1). The resulting P values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. The adjusted P value < 0.05 and |log2 (fold change)| > 1 were defined as the cutoff criteria for differentially expressed mRNAs (DE mRNAs) and DE Genes (DEGs) encoding proteins. The DE lncRNAs were selected with adjusted P-value < 0.05 and |log2 (fold change)| > 1. Find_circ and CIRI224,25 were used to de novo assemble the unmapped reads to circRNAs. The raw counts were first normalized using TPM (Read Count*1,000,000)/libsize). DE circRNAs were extracted with |log2 (fold change)| >1 and with statistical significance (P value < 0.05).
ncRNA Target Analysis
The target gene of miRNAs was performed by miRanda and RNAhybrid.26,27 The intersection result of the above two software was considered the target of miRNAs. The target mRNA analysis of DE lncRNAs was performed with two methods: co-location and co-expression analysis by bedtools and R package cor.test, respectively. Co-location analysis is based on the Cis-acting theory that lncRNAs are acting on the nearby protein-coding genes. The threshold of co-location was set within 100 kb upstream and downstream of lncRNAs. For co-expression analysis (Trans role of target gene prediction), target gene prediction is based on the expression correlation of lncRNAs and mRNAs. The absolute value of correlation was >0.95. For circRNA target analysis, some source genes of circRNAs can also be normally transcribed into linear mRNAs.28 Therefore, the expression level of circRNA may also affect the expression level of mRNA from the same source gene. The correlation analysis between the expression level of DE circRNA source genes and DE mRNA source genes was performed in this study. In order to predict the role of DE ncRNAs and DEGs with trigeminal neuropathic pain, the list of pain genes (Supplementary Table 1) was constructed based on PainNetworks,29 PainGene databases,30 and literature curation.31 The visualization of the regulatory network of DE ncRNAs and targeted DE mRNAs were represented by Cytoscape 3.9.0.
Go Annotations, KEGG Pathways, and PPI Analysis
Gene Ontology (GO) enrichment analysis and KEGG pathway analysis were performed to predict the function of DE ncRNAs and DEGs encoding proteins in the condition of trigeminal neuropathic pain. GO analysis was used to elucidate genetic regulatory networks of the DE ncRNAs and DEGs according to the biological process (BP), cellular component (CC), and molecular function (MF). GOseq R package-based Wallenius non-central hypergeometric distribution was implemented for GO enrichment analysis.32 The number of genes was statistically analyzed with significant enrichment of each GO term and displayed in a histogram. KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database resource for understanding high-level functions and utilities of the biological system from molecular-level information.33 KOBAS software was used to test the statistical enrichment of the target gene candidates in KEGG pathways.34 A protein–protein interaction (PPI) network of DEGs was drawn using Cytoscape 3.9.0 based on the reported 1002 PPIs involved in pain.35 Single edges not connected to the main network were removed. The STRING was used to analyze the PPI of the DEGs related to the known pain genes. Nociceptor-related DEGs were identified in the whole TG, referencing the reported results of single-cell RNA-seq analysis of DEGs in DRG sensory neurons during SNT injury in mice.36
Construction and Analysis of ceRNAs Regulatory Network
To reveal the role and interactions among ncRNAs and mRNAs in trigeminal neuropathic pain pathogenesis, competing endogenous RNA (ceRNA) regulatory networks were constructed for DE lncRNAs, DE circRNAs, DE miRNAs, and DE mRNAs with miRanda software. The interaction network was built and visually displayed using Cytoscape 3.9.0 based on the screening of DE lncRNA-DE miRNA-DE mRNA pairs and DE circRNA-DE miRNA-DE mRNA pairs. In the ceRNA regulation networks, DE lncRNAs/DE circRNAs were determined as a decoy, DE miRNAs as the center, DE mRNAs as the target. The size of the node was in direct proportion to the degree.
Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Totally, six male mice (3 mice in sham group and 3 mice in CCI-ION group) and six female mice (3 mice in sham group and 3 mice in CCI-ION group) were used for testing the randomly chosen DE ncRNAs and DE mRNA in the TG by RT-qPCR. RNA Extraction Kit (Takara, China) was used to extract the total RNA from TGs. The RNA was reversely transcribed into the first-strand cDNA with Prime Script™ RT reagent Kit (Takara, China). The Mir-X miRNA First-Strand Synthesis Kit (Cat. No. 638313, Takara, China) was used for converting miRNAs. The sequences of polymerase chain reaction (PCR) primers (Tsingke Biotechnology, China) are shown in Supplementary Table 2. With an ABI QuantStudio 7 (Applied Biosystems, Waltham, MA), the PCR amplification was performed at parameters: Step 1 (95°C for 30s); Step 2 (40 cycles: 95°C for 5s, 60°C for 34s); Step 3 (Melt Curve). Calculation of the relative expression of lncRNAs and circRNAs was consistent with mRNA using the 2−ΔΔCt method. The internal control was Gapdh. The expression levels of miRNAs were normalized to the U6 level in each sample. The data of the behavioral test and RT-qPCR were presented as the means ± SD. The RT-qPCR results were analyzed using a t-test. Significance was established at P < 0.05.
Results
CCI-ION Mice Suffer from Trigeminal Neuropathic Pain
The HWT of male Sham and CCI-ION mice were recorded on day 1 before and day 3, 7, 10, 14, 21, and 28 after surgery using the von Frey filament test. The behavioral test results showed that CCI-ION mice suffered from facial allodynia from day 3 after surgery compared with the Sham group (Figure 1). In addition, from day 7 after surgery, the HWT of CCI-ION mice reached a low level and lasted to at least day 28 after surgery. Moreover, compared with the baseline, there were no significant changes in the HWT of the Sham group. However, the HWT of CCI-ION mice began to decrease significantly from day 3 after surgery. These results indicated that CCI-ION treated mice suffered from trigeminal neuropathic pain from day 3 to day 28 after surgery.
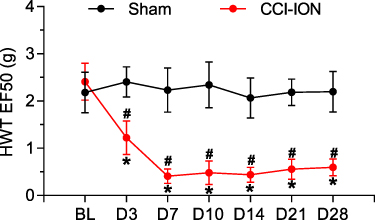 |
Figure 1 Orofacial mechanical allodynia induced by CCI-ION. The results of von Frey filaments indicated that the head withdrawal threshold (HWT) of CCI-ION mice began to decrease from 3 days and lasted at least 28 days after surgery compared to the baseline (n = 6, *P < 0.05). CCI-ION mice almost reached the significantly lowest HWT on day 7 after surgery compared to the Sham group (n = 6, Sham vs CCI, 2.32 ± 0.47 g vs 0.41 ± 0.16 g, #P < 0.05). |
CCI-ION Regulates the Expression of ncRNAs and mRNAs in the TG of Mice
Information on the DE miRNAs, DE lncRNAs, DE circRNAs, DE mRNAs, and DEGs in the TG of male CCI-ION mice compared with the male Sham group on 7 days after surgery are listed in Supplementary Tables 3–7, respectively. Totally, 1325 miRNAs were found in the TG of Sham and CCI-ION groups (Figure 2A). And 67 miRNAs (66 annotated miRNAs and 1 novel miRNAs) were differentially expressed compared with the Sham group (Figure 2B and C, Supplementary Table 3), including 36 up-regulation and 31 down-regulation (P value < 0.05, absolute log2FC > 0.485). For lncRNA, 43586 lncRNAs were detected in the TG of Sham and CCI-ION groups (Figure 3A). And 216 lncRNAs (165 annotated lncRNAs and 51 novel lncRNAs) were differentially expressed (adjusted P value < 0.05, absolute log2FC >1), including 144 up-regulation and 72 down-regulation (Figure 3B and C, Supplementary Table 4). For circRNAs, 2627 circRNAs were detected in Sham and CCI-ION groups (Figure 4A). And 14 circRNAs (5 annotated circRNAs and 9 novel circRNAs) were significantly regulated, including 9 up-regulation and 5 down-regulation (P value < 0.05, absolute log2FC >1) (Figure 4B and C, Supplementary Table 5). For mRNA analysis at the transcription level, 43695 mRNAs were detected in Sham and CCI-ION groups (Figure 5A). And 595 DE mRNA transcripts (adjusted P value < 0.05, absolute log2FC > 1) from 539 host genes were determined in the TG of CCI-ION mice compared with the Sham group, including 402 up-regulation and 193 downregulation (Figure 5B and C, Supplementary Table 6). For the analysis of DEGs encoding proteins, 18303 genes encoding proteins were determined, and 421 DEGs (adjusted P value < 0.05, absolute log2FC > 1, 388 up-regulation and 33 down-regulation) were determined in the TG of CCI-ION mice (Supplementary Figure 1A–C, Supplementary Table 7). Figure 6A shows the summary histogram of DE miRNAs, DE lncRNAs, DE circRNAs, DE mRNAs, and DEGs. To validate the reliability of the sequencing results, the change of ncRNAs and mRNAs expression 7 days after surgery were analyzed. One of DE miRNAs (mmu-miR-134-3p), DE lncRNAs (A630023A22Rik-202), DE circRNAs (mmu_circ_0000668), and DE mRNAs (Anxa10) were analyzed by RT-qPCR. All of the validated ncRNAs and mRNA were consistent with the results of RNA sequencing (Figure 6B and C). In addition, to investigate the changes in the expression of the above randomly chosen DE miRNA, DE lncRNA, DE circRNA, and DE mRNA in the TG after trigeminal nerve injury, RT-qPCR was also performed in female mice. The results showed that the changes in the expression of A630023A22Rik-202, mmu_circ_0000668, andAnxa10 mRNA were the same as that of male mice, while mmu-miR-134-3p has no significant change (Supplementary Figure 2A–D).
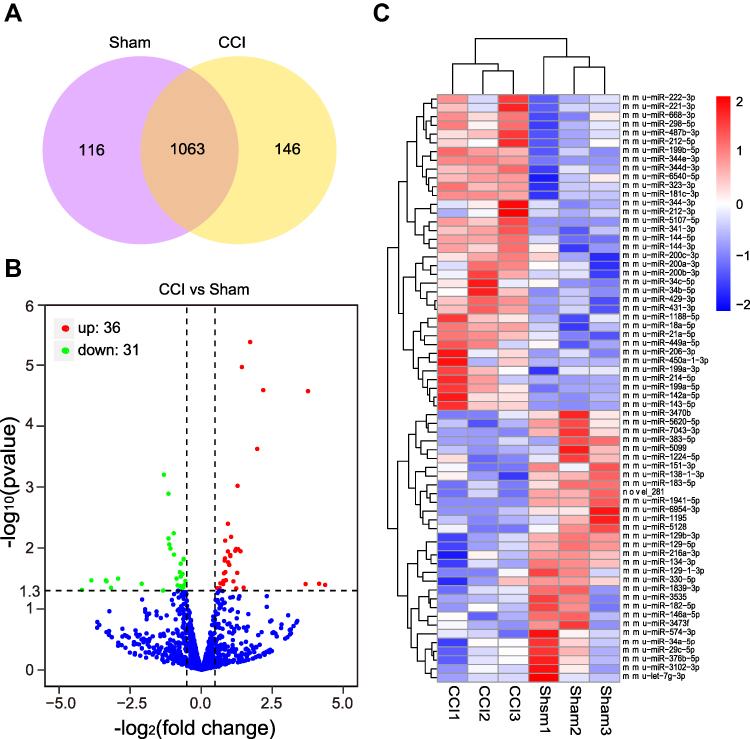 |
Figure 2 The expression profiling changes of miRNAs in the TG of CCI-ION mice (n = 3). (A) The Venn diagram shows the number of overlap miRNAs in the CCI-ION group compared with the Sham group. (B) The Volcano plot displays the DE miRNAs in the CCI-ION group compared with the Sham group. (C) The clustering map shows the top 20 up and down-regulated DE miRNAs in each sample TG of CCI-ION and Sham groups. |
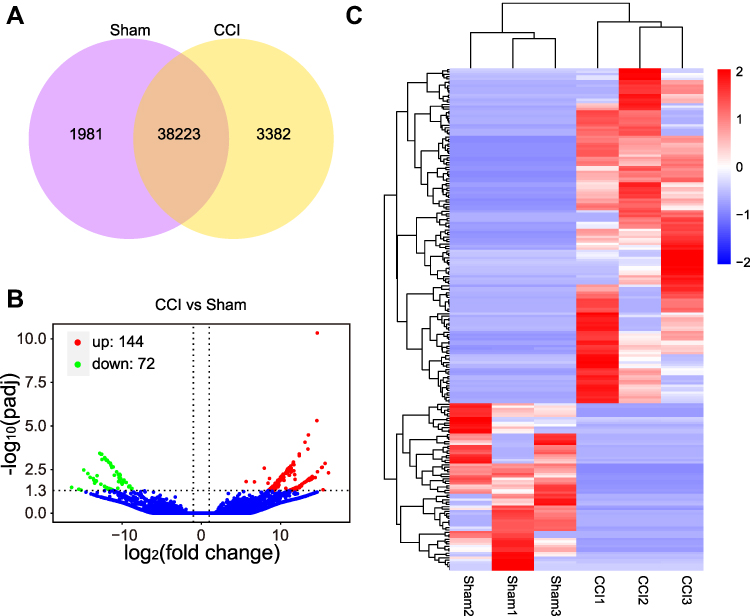 |
Figure 3 The expression profiling changes of lncRNAs in the TG of CCI-ION mice (n = 3). (A) The Venn diagram shows the number of overlap lncRNAs in the CCI-ION group compared with the Sham group. (B) The Volcano plot displays the DE lncRNAs in the CCI-ION group compared with the Sham group. (C) The clustering map shows the DE lncRNAs in each sample TG of CCI-ION and Sham groups. |
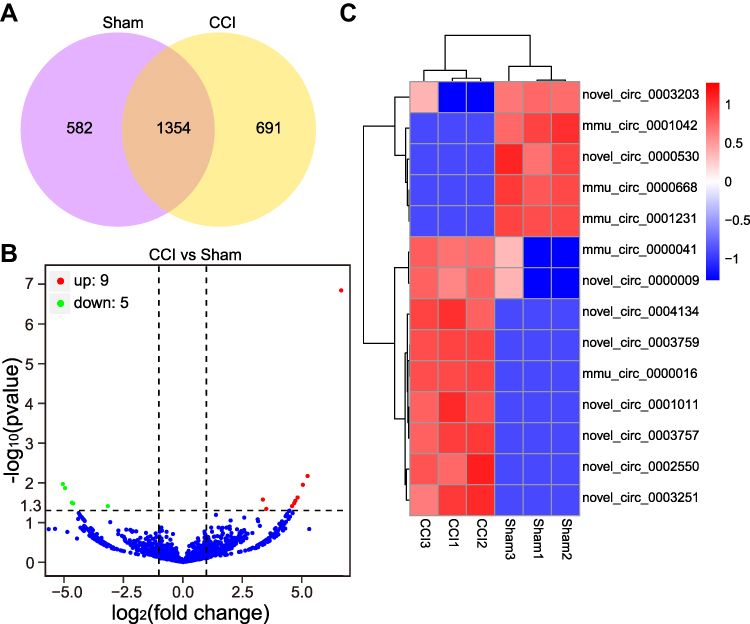 |
Figure 4 The expression profiling changes of circRNAs in the TG of CCI-ION mice (n = 3). (A) The Venn diagram shows the number of overlap circRNAs in the CCI-ION group compared with the Sham group. (B) The Volcano plot displays the DE circRNAs in the CCI-ION group compared with the Sham group. (C) The clustering map shows the DE circRNAs in each sample TG of CCI-ION and Sham groups. |
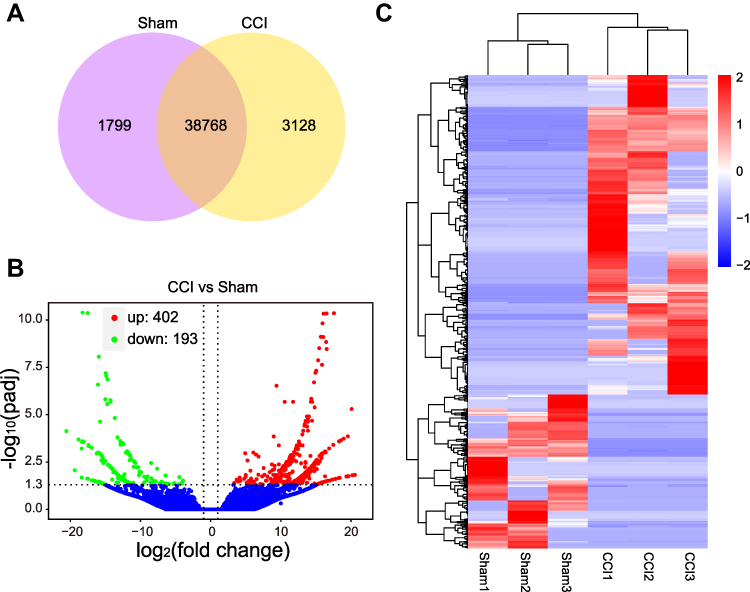 |
Figure 5 The expression profiling changes of mRNAs in the TG of CCI-ION mice (n = 3). (A) The Venn diagram shows the number of overlap mRNAs in the CCI-ION group compared with the Sham group. (B) The Volcano plot displays the DE mRNAs in the CCI-ION group compared with the Sham group. (C) The clustering map shows the DE mRNAs in each sample TG of CCI-ION and Sham groups. |
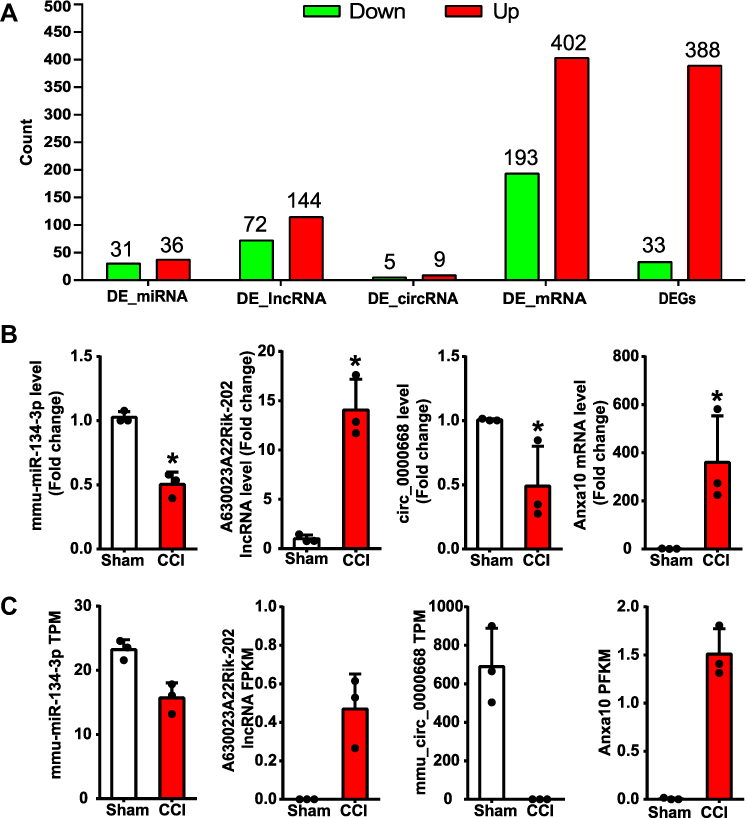 |
Figure 6 Summary of DE ncRNAs, DE mRNAs, and DEGs in the TG of CCI-ION mice and RT-qPCR validation of RNA-seq results. (A) The histogram shows the number of down and up-regulated lncRNAs, circRNAs, miRNAs, and genes encoding mRNAs. (B) RT-qPCR validation of the expression level of randomly chosen DE miRNA, DE lncRNA, DE circRNA, and DE mRNA in the TG of male mice, *P< 0.05, n = 3. (C) The expression level of chosen DE miRNA, DE lncRNA, DE circRNA, and DE mRNA by RNA sequencing. |
Some Pain-Related DE mRNAs Could Be Targeted by DE miRNAs and DE lncRNAs
ncRNAs can regulate the expression of mRNAs to exact biological functions. In order to predict DE mRNAs regulated by ncRNAs, Venn diagram analysis was performed between the targeted mRNAs of DE ncRNAs (miRNAs, lncRNAs, and circRNAs) and DE mRNAs (Figure 7A–C). Firstly, we searched the pain-related studies of the 67 DE miRNAs in PubMed and found that 18 DE miRNAs have been reported to be involved in pain (Supplementary Table 8), including neuropathic pain, inflammatory pain, diabetic neuropathic pain, and cancer pain. MiRNAs can degrade mRNAs or inhibit translation by acting on specific sequences in the 3` untranslated region (UTR) of target mRNAs.7 The intersection of the targeted mRNAs of DE miRNAs and DE mRNAs indicated that 125 DE mRNAs were the same as the targeted mRNAs of 37 DE miRNAs (Supplementary Table 9, Figure 7A). Among the 37 DE miRNAs, 9 DE miRNAs have been reported to be associated with pain (Supplementary Table 9). Meanwhile, among the 125 targeted DE mRNAs, 5 DE mRNAs belonging to the known pain genes (Cacna1a, Grm1, Il1rl1, Lmx1b, and Ctsd) were targeted by 6 DE miRNAs (Supplementary Table 9). Moreover, 5 proteins encoded by DE mRNAs (Cacna1a, Pik3cd, Il1rl1, Cga, and Spr) targeted by 5 DE miRNAs were involved with the 8 pain-specific PPI relations (Supplementary Table 10). Finally, the negative regulation network of DE miRNA and DE mRNA showed that 30 DE miRNAs were predicted to negatively target 80 DE mRNAs (Figure 8, Supplementary Table 11). In the network, 7 DE miRNAs have been reported to be associated with pain, and 4 DE mRNAs (Cacna1a, Il1rl1, Lmx1b, and Ctsd) were related to the known pain genes.
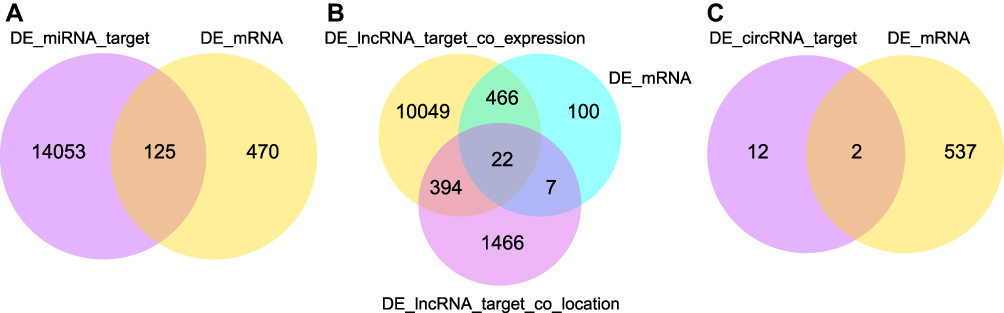 |
Figure 7 Intersection analysis of DE mRNAs and DE ncRNAs targeting mRNAs. (A) Venn diagram showing the overlap number of the targeted mRNAs of DE miRNAs and DE mRNAs. (B) Venn diagram displaying the overlap number of co-expression and co-location targeted mRNAs of DE lncRNAs with DE mRNAs. (C) The Venn diagram shows the overlap number of targeted mRNAs of DE circRNAs and DE mRNAs. |
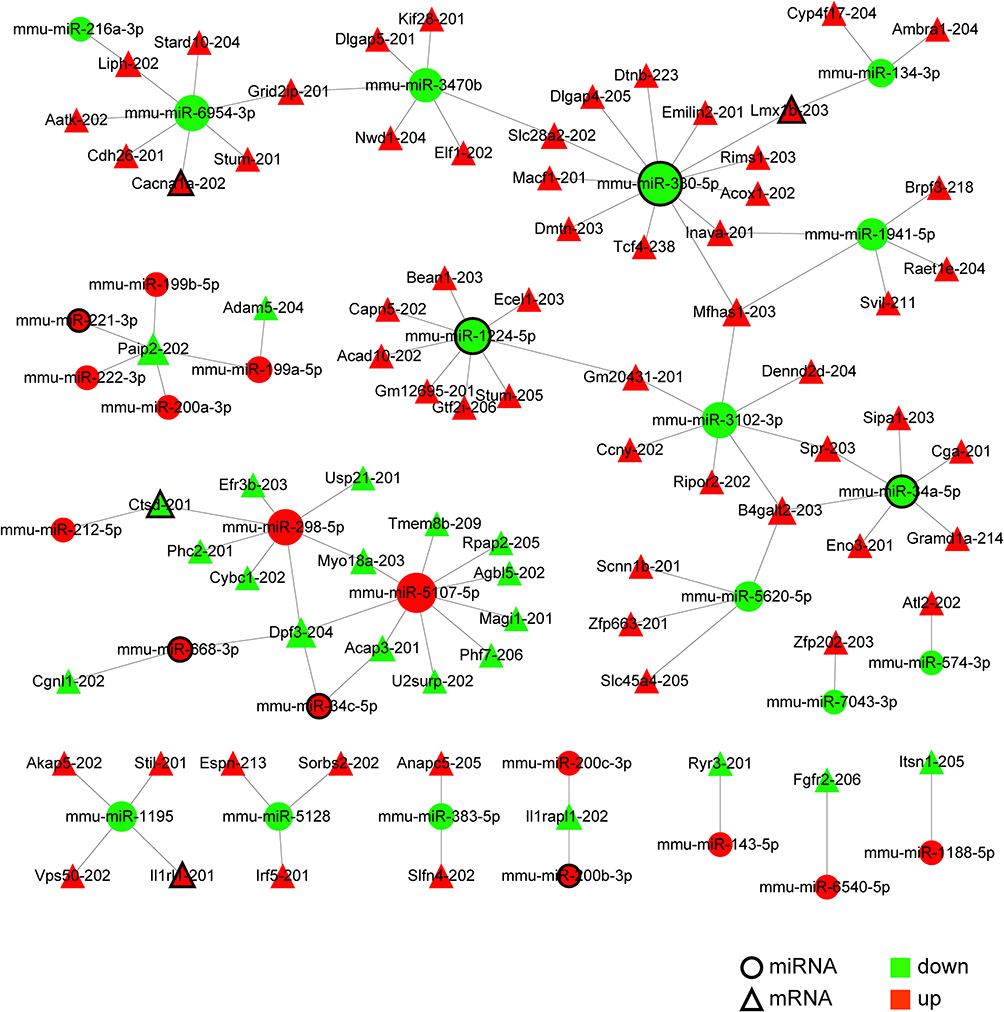 |
Figure 8 The regulatory network of DE miRNAs negatively targeting DE mRNAs. Totally, 30 DE miRNAs were predicted to target 80 DE mRNAs negatively. In the network, 7 DE miRNAs (circles with black border) have been associated with pain, and 4 DE mRNAs (triangles with black border) were related to the known pain genes. The node size is directly proportional to the number of their corresponding partners. |
The Venn diagram shows the interaction between DE lncRNA targeted mRNAs by co-location or co-expression with DE mRNAs (Figure 7B). Co-expression analysis demonstrated that 139 DE lncRNAs were predicted to target 488 DE mRNAs (82% DE mRNAs). Figure 9 shows the regulatory network of 107 DE lncRNAs targeting 35 DE mRNAs related to known pain genes according to the list of pain genes constructed based on PainNetworks, PainGene databases, and previous literature.29–31 The detailed information on the 107 DE lncRNAs and 35 pain-related DE mRNAs is listed in Supplementary Table 12. Co-location analysis suggested that 24 DE lncRNAs were predicted to target 29 DE mRNAs (Supplementary Table 13, Supplementary Figure 3), but no DE mRNA was related to the known pain genes. Furthermore, 22 DE mRNAs were predicted to be regulated by DE lncRNAs both through co-expression and co-location. However, only 3 DE mRNAs (Ly6a-202, Lrp8-201, and Esd-202) were predicted to be regulated by the same DE lncRNAs (Gm20275-201, 4933424M12Rik-202, and TCONS_00087827) through co-expression and co-location, respectively (Supplementary Figure 4), which are more associated with trigeminal neuropathic pain. CircRNAs may regulate the expression of mRNAs from the same host gene.28 Intersection analysis of DE circRNA host genes and DE mRNA host genes indicated the potential regulatory relationship between DE circRNAs and DE mRNAs (Figure 7C). Notably, 2 DE circRNAs (mmu_circ_0000016 and mmu_circ_0000041) were from the same source genes as 2 DE mRNAs (Rims1 and Hecw2), respectively.
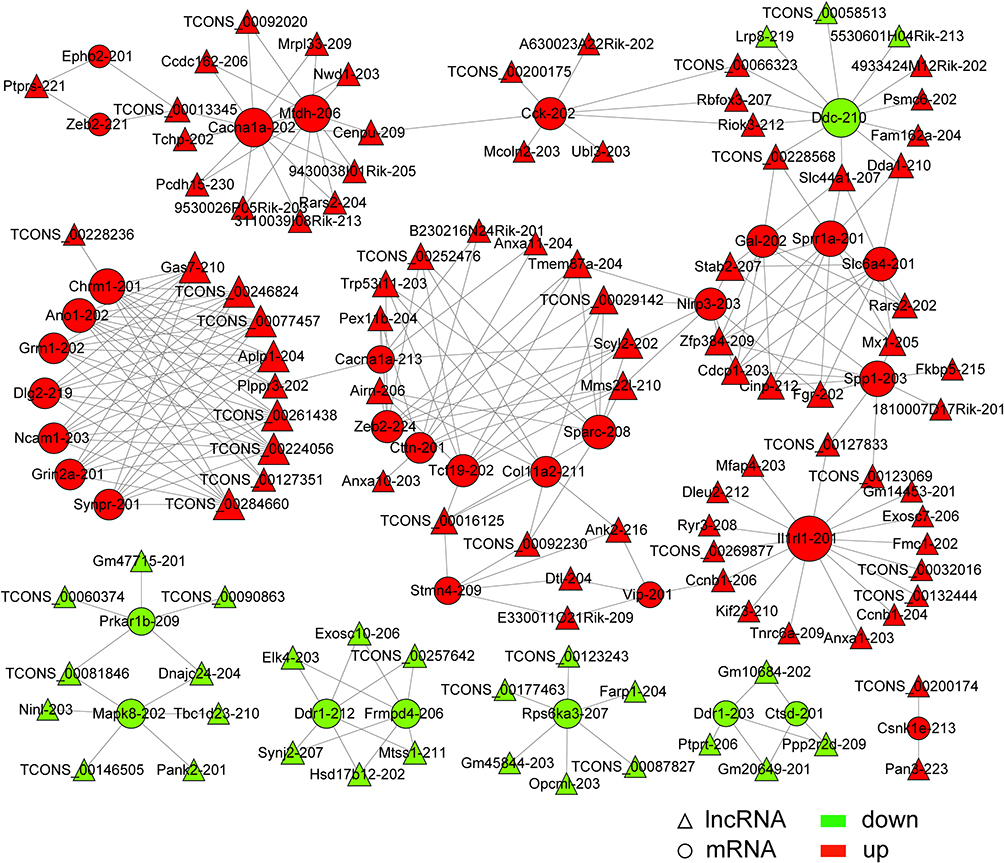 |
Figure 9 The regulatory network of DE lncRNAs targeting DE mRNAs related to the known pain genes by co-expression. Totally, 107 DE lncRNAs were predicted to target 35 DE mRNAs associated with pain by co-expression. The size of the node is in direct proportion to the degree. |
Nociception-Related Signaling Pathways are Enriched for DE ncRNAs Targeting Genes
GO and KEGG pathway analyses were performed to predict the gene function of DE ncRNAs in the condition of trigeminal neuropathic pain. According to the distribution of predicted target genes in the Gene Ontology, the target gene function of DE ncRNAs was clarified. According to the GO analysis of target genes of DE miRNAs, the most significant biological processes (BP) were cellular component organization or biogenesis, metabolic process, organelle organization, etc. The most significantly enriched cellular components (CC) were related to the intracellular part, cytoplasm, organelle, etc. The most significantly enriched molecular function (MF) were protein binding, binding, catalytic activity, etc. (Figure 10A). Based on the GO analysis of the co-localization and co-expression genes of DE lncRNAs, the most significantly enriched GO terms were similar to the enriched GO terms of target genes of DE miRNAs. Significantly activated BP of these genes included cellular component organization, cellular component organization or biogenesis, positive regulation of the biological process, etc. The most noteworthy enriched CC was related to the intracellular part, cytoplasm, organelle, etc. The most significantly enriched MF were protein binding, binding, ion binding, etc. (Figure 10B). Based on the GO analysis of predicted target genes of DE circRNAs, the most significantly enriched BP were positive regulation of peptidyl-threonine phosphorylation, regulation of peptidyl-threonine phosphorylation, and intracellular protein transport, etc. The most significantly enriched CC was mitotic spindle, adherens junction, anchoring junction, etc. The most significantly enriched MF were protein binding, ankyrin repeat binding, acetyltransferase activator activity, etc. (Figure 10C).
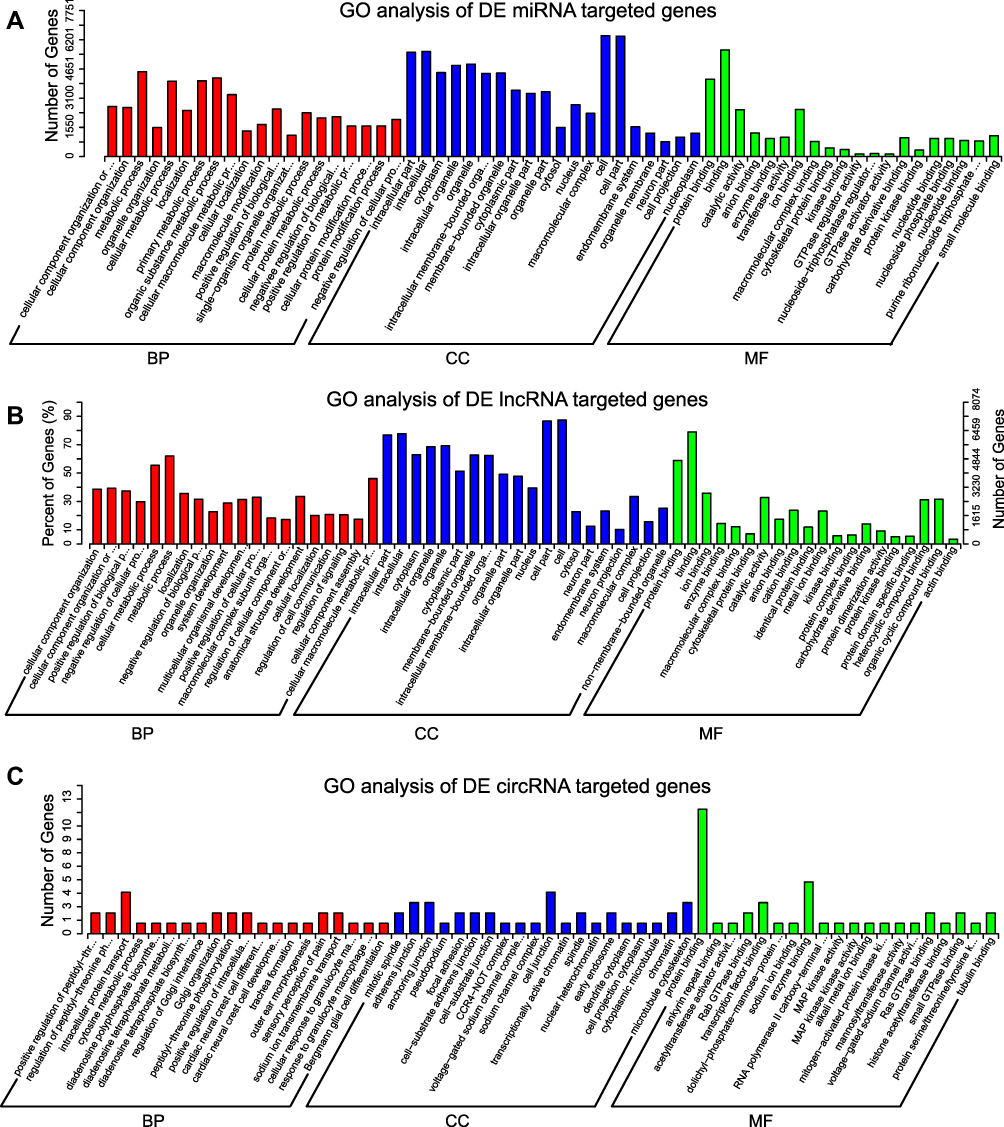 |
Figure 10 GO analysis of the target genes of DE miRNAs, DE lncRNAs, and DE circRNAs. The top 20 significant items in the biological process (BP), cellular component (CC), molecular function (MF) by GO analysis of the target genes of DE miRNAs (A), DE lncRNAs (B), and DE circRNAs (C). |
KEGG analysis of predicted genes of DE miRNAs in the TG of CCI-ION mice demonstrated the Rap1 signaling pathway, oxytocin signaling pathway, cAMP signaling pathway, MAPK signaling pathway, and lysosome were significantly enriched (Figure 11A), which were related to hyperalgesia.37,38 The KEGG analysis of predicted genes of DE lncRNAs in the TG of CCI-ION mice indicated that the most significantly enriched pathways were focal adhesion. It was noteworthy that MAPK signaling pathway, PI3K-Akt signaling pathway, ECM-receptor interaction, and gap junction were also significantly enriched (Figure 11B), which were involved in the process of nociceptive transmission. For the host genes of 9 DE circRNAs determined in the TG of CCI-ION mice, only retrograde endocannabinoid signaling pathways were significantly enriched (Supplementary Figure 5).
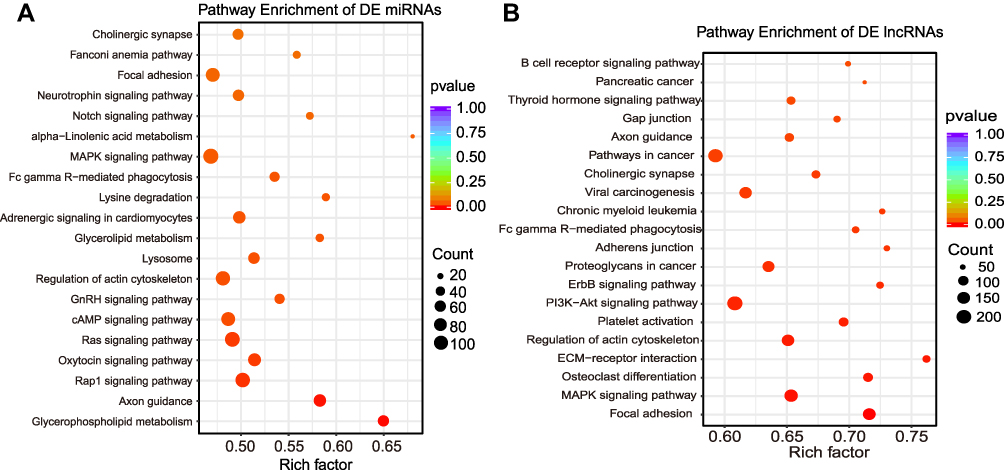 |
Figure 11 KEGG analysis of the target genes of DE lncRNAs and DE miRNAs. (A) The scatter dot graph shows the top 20 enriched pathways related to the target genes of DE miRNAs; (B) The scatter dot graph shows the top 20 enriched pathways related to the target genes of DE lncRNAs (P < 0.05). |
Some DE Genes Encoding Proteins are Related to Pain
Among the DEGs encoding proteins (421 genes from Supplementary Table 7) in the TG of CCI-ION mice, Barx1 was expressed specifically in the TG compared to the DRG.39 In order to characterize the changes in the functional properties of DEGs, GO, KEGG pathway, and PPI analysis were performed. Based on the GO analysis, the most noteworthy enriched BP were defense response, immune system process, response to external stimulus, and response to stress. The extracellular region, extracellular space, extracellular region part, and plasma membrane part in the category of the CC were enriched. The most noteworthy enriched MF were binding, ion binding, anion binding, and carbohydrate derivative binding (Supplementary Figure 6A–D). In the KEGG analysis of DEGs, the most significantly involved pathways in CCI-ION pathogenesis were PI3K−Akt signaling pathway, focal adhesion, ECM−receptor interaction, and protein digestion and absorption (Supplementary Figure 7).
To explore the DEGs in the TG induced by CCI-ION involved in the pain-related PPI network, the previously published pain interactome, a PPI network of 611 interconnected pain-related proteins, was used.35 Totally, 30 up-regulated DEGs and 66 partner genes were involved in the pain-specific PPI network without down-regulated DEGs, including 107 PPIs (Figure 12, Supplementary Table 14). Several up-regulated hub genes (Prkcg, Grin2b, Gal, Npy, Vip, and Mmp9) were obviously centered around by other genes. These genes were related to nerve injury and regeneration (Gal and Npy),40,41 pro-nociceptive signaling transmission (Prckg, Grin2b, Vip, and Mmp9),42–45 and anti-nociceptive signaling transmission (Npy).46 To further determine the DEGs related to the known pain genes in the TG of CCI-ION mice, an intersection analysis of DEGs and the known 804 pain genes (Supplementary Table 1) according to previous databases and literature was performed.29–31 The results showed that 39 DEGs (38 up-regulation and 1 down-regulation) were known pain genes (eg, top 10 up regulated genes: Prok2, Sprr1a, Vip, Npy, Slc6a4, Mpo, Kcnj5, Col9a1, Lcn2, Grin2b, and the down-regulated Nmu) (Supplementary Table 15). The STRING network analysis of the 39 DEGs related to the known pain genes showed an interconnected network of 23 DEGs and 5 partner genes (Tnf, Cd79a, Cyba, Ncf1, and Dlg4) (Figure 13). The cluster of Atf3, Lcn2, Mmp9, Mpo, Serpinb1a, Spp1, Tnf, and Tnfrsf1b was significantly enriched in the IL-17 signaling pathway, TNF signaling pathway, and immune system. The cluster of Gal, Npy, Nts, and Vip was enriched in neuropeptide signaling pathway and peptide hormone binding. The cluster of Dlg4, Grin2a, Grin2b, and Shisa9 was enriched in glutamate-gated calcium ion channel activity and long-term potentiation. The cluster of Cd79a, Itpr1, Lyn, and Prkcg was significantly enriched in long-term depression and potentiation. The cluster of Cckar and Cckbr was implicated in peptide hormone binding and calcium signaling pathway. Moreover, the network included nerve injury-related genes Atf3, Tnf, Mmp9, and Anxa1. Finally, according to the lists of genes related to small nonpeptidergic (NP) nociceptors, medium peptidergic (PEP) nociceptors, and large myelinated (LM) sensory neurons,36 73 DEGs (17% DEGs) were related to nociceptor (NP or PEP), 72 DEGs were related to NP nociceptors, 26 DEGs were related to medium PEP nociceptors, and 18 DEGs were related to LM sensory neurons (Supplementary Table 16).
 |
Figure 12 The pain-related protein–protein intersection (PPI) network of the DEGs encoding proteins in the TG of CCI-ION mice. The network was built based on the pain-specific PPI network. Using DEGs in the TG of CCI-ION mice and partner genes from the pain-specific PPI network (DEGs and non-DEGs). The colored arrows represent the type and direction of interactions; The colored nodes mark the expression changes. Totally, 30 up-regulated genes were involved in the pain-specific PPI network, and no down-regulated DEGs were involved in the pain-specific PPI network. DEG, differentially expressed gene; PPI, protein–protein interaction. The size of the node is in direct proportion to the degree. |
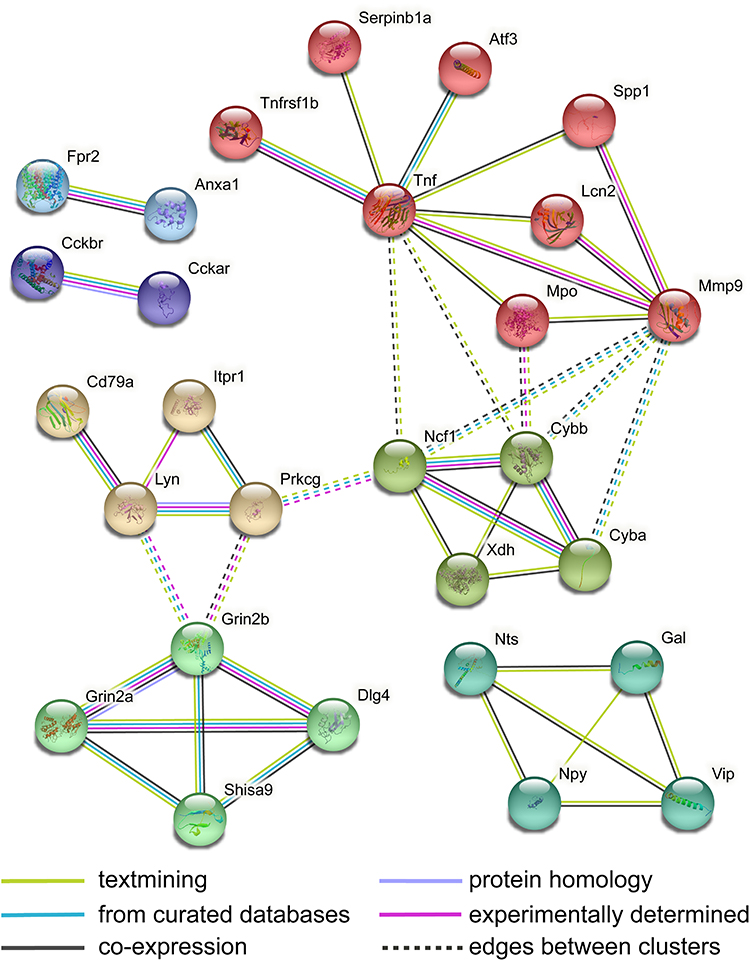 |
Figure 13 PPI analysis of biological interactions of the pain-related DEGs. The analysis was performed with STRING, using high confidence of interactions (score 0.7) and not more than 5 interactors in the first shell. The nodes were clustered using the MCL algorithm. The disconnected genes in the network are not shown. |
CeRNA Regulatory Networks Involved with Pain Exist in the TG of CCI-ION Mice
It is well known that lncRNAs and circRNAs can sponge miRNAs to regulate the expression of mRNAs, which is proven to be involved in the development and maintenance of neuropathic pain.7,8 Therefore, to further study the molecular mechanisms in ncRNAs involved in trigeminal neuropathic pain, the lncRNA/circRNA-miRNA-mRNA regulatory networks based on the ceRNA regulatory theory were analyzed. The ceRNA regulation network of DE lncRNAs, DE miRNAs, and DE mRNAs demonstrated that 42 down-regulated lncRNAs were predicted to regulate the expression of 21 down-regulated mRNAs by sponging 15 up-regulated miRNAs in the TG of CCI-ION mice (Figure 14A). Among the DE mRNAs in the above down-up-down regulatory network, Ctsd was related to the known pain gene. The network in Figure 14B displayed that 96 up-regulated lncRNAs were predicted to sponge 15 down-regulated miRNA to regulate the expression of 59 up-regulated mRNAs in the TG of CCI-ION mice. Among the DE mRNAs in the above up-down-up regulatory network, 3 up-regulated mRNAs (Cacna1a, Il1rl1, and Lmx1b) were related to the known pain genes.29–31 In Figure 15, the ceRNA regulatory network of DE circRNAs, DE miRNAs, and DE mRNAs showed that 3 down-regulated circRNAs were predicted to regulate the expression of 45 up-regulated mRNAs by sponging 5 down-regulated miRNAs. Besides, 6 up-regulated circRNAs were predicted to regulate the expression of 20 down-regulated mRNAs through sponging 11 up-regulated miRNAs. In the above ceRNA regulation network of DE circRNA-DE miRNA-DE mRNA, the up-regulated Ctsd and the down-regulated Cacna1a and Lmx1b were associated with the known pain genes.29–31
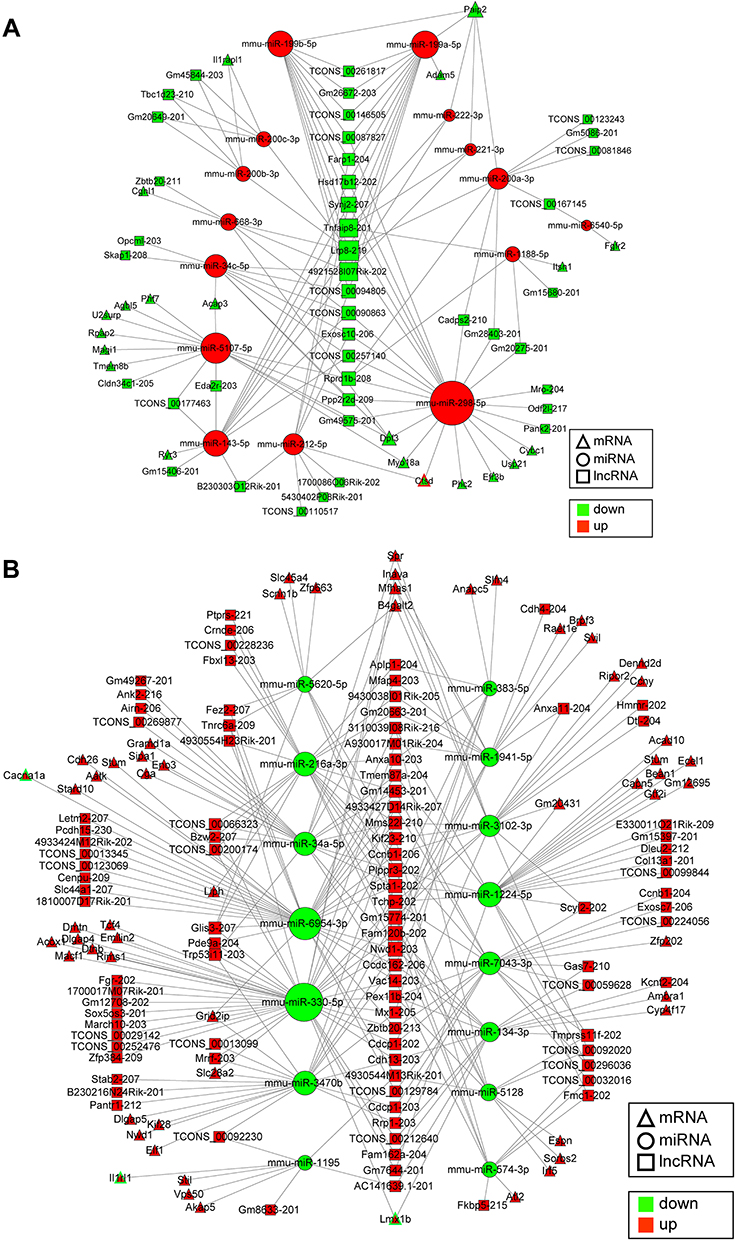 |
Figure 14 Regulatory network analysis of DE lncRNA-DE miRNA-DE mRNA in the TG of CCI-ION mice based on the ceRNA regulation theory. (A) The down-regulated DE lncRNAs were predicted to regulate the expression of the down-regulated DE mRNAs by sponging the up-regulated DE miRNAs in the TG of CCI-ION mice. The triangle with a red border (Ctsd) represents the DE mRNA related to pain. (B) The up-regulated lncRNAs were predicted to regulate the expression of the up-regulated mRNAs by sponging the down-regulated miRNAs in the TG of CCI-ION mice, the triangle with a green border (Cacna1a, Il1rl1, and Lmx1b) represents the DE mRNA related to pain. The size of the node is in direct proportion to the degree. |
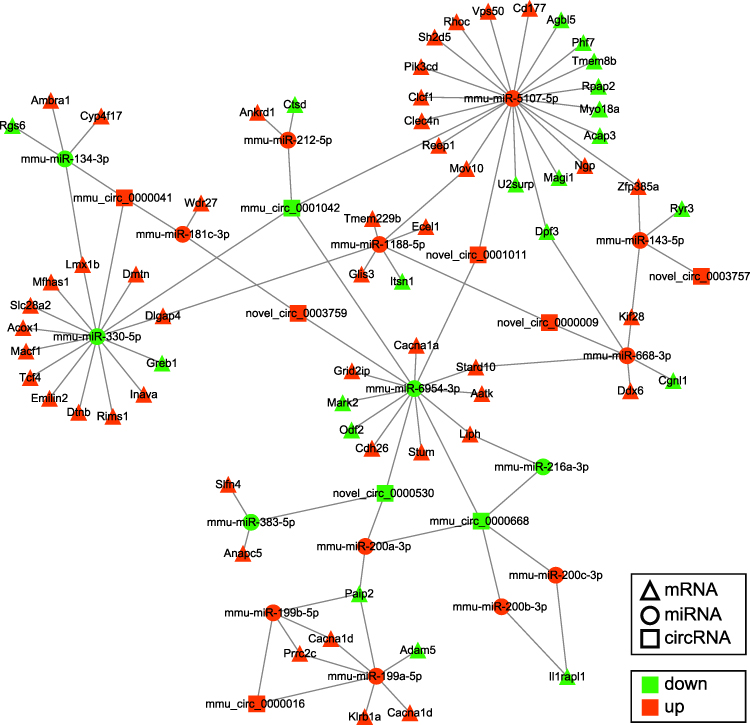 |
Figure 15 Regulatory network analysis of circRNA-miRNA-mRNA in the TG of CCI-ION mice according to the ceRNA mechanism. The up-regulated circRNAs and down-regulated circRNAs were predicted to regulate the expression of DE mRNAs by sponging DE miRNAs in the TG of CCI-ION mice. The triangles with a black border represent the DE mRNAs related to the known pain genes. The size of the node is in direct proportion to the degree. |
Based on the GO analysis of target mRNAs in the ceRNA regulatory network of DE lncRNAs, DE miRNAs, and DE mRNAs, the noteworthy enriched BP were macromolecule localization, secretion, and regulation of cellular localization, etc. The most noteworthy enriched CC were cell projection, membrane region, and dendrite. The most noteworthy enriched MF were binding, protein binding, ion binding, etc. (Supplementary Figure 8A). Based on the GO analysis of target mRNAs of the regulatory network of DE circRNAs, DE miRNAs, and DE mRNAs according to the theory of ceRNA, the most significantly enriched BP were cell–cell junction organization, regulation of neuron differentiation, and regulation of neurogenesis, etc. The most noteworthy enriched CC were cell projection, cell projection part, and dendrite, which were associated with neurons. The most noteworthy MF were binding, protein binding, and ion binding (Supplementary Figure 8B). When the data were KEGG-analyzed with the predicted mRNAs in the DE lncRNA-DE miRNA-DE mRNA regulatory network based on the theory of ceRNA under the pathogenesis of CCI-ION induced neuropathic pain, the most noteworthy involved pathways were retrograde endocannabinoid signaling, progesterone-mediated oocyte maturation, and aldosterone-regulated sodium reabsorption (Supplementary Figure 9A). The KEGG analysis of the DE circRNA-DE miRNA-DE mRNA regulatory network indicated that the most noteworthy KEGG pathways were retrograde endocannabinoid signaling, cholinergic synapse, carbohydrate digestion and absorption, and synaptic vesicle cycle (Supplementary Figure 9B). These results illustrate the regulatory relationship between ncRNAs and mRNAs in the mechanism of trigeminal neuropathic pain. However, the regulatory role of ncRNAs in trigeminal neuropathic pain was complicated. It needs studies furthermore to elucidate their role in the development and maintenance of trigeminal neuropathic pain.
Discussion
Trigeminal neuropathic pain belongs to orofacial pain that is attributed to lesions or diseases of cranial nerves according to the International Classification of Orofacial Pain (ICOP), 1st edition.1 However, highly effective treatments or drugs for trigeminal neuropathic pain are still unavailable because of the lack of a complete understanding of its mechanisms.3,6 Although increasing data demonstrate that ncRNAs (miRNAs, lncRNAs, and circRNAs) play crucial roles in neuropathic pain, little is known about the expression profile and roles of ncRNAs in the TG under the condition of trigeminal neuropathic pain. As far as we know, this study was the first to study the expression profile of miRNAs, lncRNAs, and circRNAs in the TG of CCI-ION mice (a classical trigeminal neuropathic pain model) by whole-transcriptome sequencing and bioinformatics analysis. The results of this study support that miRNA, lncRNA, and circRNA contribute to the development of trigeminal neuropathic pain.
miRNA is a group of small non-coding RNAs consisting of 18~23 nucleotide sequences. Since miRNA was proved to be involved in orofacial inflammatory pain through regulating the cytokine mRNA expression and had an analgesic action at the spinal cord level towards an anti-inflammatory phenotype,47–49 the role of miRNAs in pain regulation has attracted increasing attention. In this study, 18 DE miRNAs have been reported to be involved in pain (Supplementary Table 8). miRNA with the “activated” RNA-induced silencing complex (RISC) attaches to the 3’ UTR of a target mRNA, resulting in the degradation of target mRNAs (full complementarity) or translational repression (incomplete complementarity).7 According to the above mechanism, 37 DE miRNAs were predicted to target 125 DE mRNAs negatively, and 4 DE target mRNAs (Ctsd, Lmx1b, Il1rl1, and Cacnala) of DE miRNAs were related to the known pain genes. These results suggest that miRNAs may play a role in trigeminal neuropathic pain by targeting the known pain genes. Furthermore, several nociception-related signaling pathways were significantly enriched in the KEGG analysis, including Rap1 signaling pathway, oxytocin signaling pathway, cAMP signaling pathway, MAPK signaling pathway, lysosome, etc.37,50,51 For instance, protein kinase A is involved in neuropathic pain by activating the p38 MAPK pathway to mediate spinal cord cell apoptosis.52 The above results provide a theoretical basis for using miRNAs as therapeutic targets in trigeminal neuropathic pain.
LncRNAs with lengths exceeding 200 nucleotides are involved in many physiological and pathological processes including embryonic development, cancer, inflammation, cardiovascular and neurobiological diseases, etc.9,53,54 Recently, the role of lncRNAs has been quickly attracted in neuropathic pain.55 For instance, Zhou et al discovered 126 DE lncRNAs in the spinal cord of rats using RAN-sequencing 7 days after SNI surgery.56 Most recently, evidence also has shown that lncRNAs (Gm14461 and uc.48) in the TG play crucial roles in trigeminal neuralgia.14,57 In the present study, 216 DE lncRNAs were determined in the TG of mice treated with CCI-ION. LncRNAs can modulate gene expression through trans (co-expression) and cis (co-location) regulation.58 Intersection analysis of the DE mRNAs and the target mRNAs of DE lncRNA by co-expression demonstrated that 139 DE lncRNAs were predicted to target 488 DE mRNAs, including 35 DE mRNAs transcribed from the known pain genes.29–31 Furthermore, 3 DE mRNAs (Ly6a, Lrp8, and Esd) were predicted to be regulated by the same DE lncRNAs (Gm20275-201, 4933424M12Rik-202, and TCONS_00087827) through co-expression and co-location, respectively. Notably, Lrp8 is involved in the reelin signaling pathways that contribute to nociceptive processing.59 KEGG analysis suggested MAPK signaling pathway, PI3K-Akt signaling pathway, ECM-receptor interaction, and gap junction were enriched, which were related to neuropathic pain.52,60,61
CircRNAs are non-coding RNAs with covalently closed circular structures with 5’ and 3’ ends. The sequences of circRNAs are conserved among mammalian species and have higher stability than liner mRNAs because the particular ring structure enables them to escape from RNase degradation.28,62 Dysregulation of circRNAs is associated with tumors, cardiovascular diseases, endocrine diseases, immune system diseases, and neurodegenerative diseases.28,62 Recently, emerging studies found that circRNAs play important roles in developing and maintaining neuropathic pain.13,63 For instance, Zhang et al found 22 DE circRNAs in rat spinal cord on days 7 and 14 following SNL. And they proved that the up-regulated CircAnks1a in the spinal cord of rats contributes to the hypersensitivity in the paw induced by SNL injury.13 Besides, Zhou et al determined 188 DE circRNAs in the rat spinal cord on day 14 after SNI was controlled to the sham group with RNA-seq and bioinformatic analysis.10 In the present study, 9 DE circRNAs were determined in the TG of CCI-ION mice. Intersectional analysis showed that 2 DE circRNAs (mmu_circ_0000016 and mmu_circ_0000041) were from the same source genes as 2 DE mRNAs (Rims1 and Hecw2), respectively. RIMS1 can regulate neurotransmitter release at the presynaptic active zones.64,65 Besides, mmu_circ_0000041 (circRNA HECW2) engaged in the endothelial-mesenchymal transition related to the damage of the blood–brain barrier.66 KEGG analysis showed that only retrograde endocannabinoid signaling pathways were significantly enriched for the target genes of DE circRNAs. Tissue injury can activate retrograde endocannabinoid signaling,67,68 which is involved in multiple pathophysiological functions, such as anti-nociception, cognition and memory, endocrine function, nausea, and vomiting, inflammation, immune recognition, and modulation of synaptic transmission and plasticity.69,70
The best-known mechanism underlying the functional regulation of ncRNAs is that lncRNA/circRNA acts as a ceRNA to sponge miRNAs, reducing the number of available miRNAs and helping to improve the translation of target mRNAs.8 For example, increased circAnks1a sponged miR-324-3p to promote the VEGFB expression in the spinal cord of rats after SNL, resulting in neuropathic pain.13 Among the target DE mRNAs in the ceRNA regulatory network of DE lncRNAs, Ctsd, Cacna1a, Il1rl1, and Lmx1b are related to the known pain genes.29–31 Among the target DE mRNAs in the ceRNA regulatory network of DE circRNA, the up-regulated Ctsd and the down-regulated Cacna1a and Lmx1b were associated with the known pain-specific genes.29–31 These results support that lncRNAs/circRNAs in the TG of CCI-ION mice may play an essential role in the development of trigeminal neuropathic pain through ceRNA regulation.
Trigeminal nerve damage or compression of the trigeminal nerve root could cause apparent changes in the expression of various genes in the TG, which contribute to the initiation and sustainability of trigeminal neuropathic pain.3,6 In the present study, a total of 421 DEGs encoding proteins were found in the TG of mice on 7 days following CCI-ION surgery. KEGG pathway analysis showed PI3K−Akt signaling pathway, focal adhesion, ECM−receptor interaction, protein digestion and absorption and so on were significantly enriched, which was similar to the results of pathway enrichment of DEGs in the spinal cord of rats treated with SNI.10 Based on a previously reported pain-related PPI network,35 PPI analysis indicated several up-regulated hub genes (Prkcg, Grin2b, Gal, Npy, Vip, and Mmp9) were involved in the pain-specific PPI network. These genes were related to nerve injury and regeneration and nociception regulation (Gal and Npy),40,41 pro-nociceptive signaling transmission (Prckg, Grin2b, Vip, and Mmp9),42–45 and anti-nociceptive signaling transmission (Npy).46 In addition, 39 DEGs were known pain-specific genes.29–31 TG mainly consists of trigeminal ganglion neurons (TGNs), satellite glial cells (SGCs), Schwann cells, and macrophages.71 In the present study, we examined the expression profile of ncRNAs and mRNAs in the whole TG while not determining the changes in the expression of ncRNAs and mRNAs in different types of cells in the TG. In order to predict the cell-type origin of the identified DEGs, previously published data of single-cell RNA-seq analysis of DEGs in the mouse nonpeptidergic nociceptors (NP), peptidergic nociceptors (PEP), and large myelinated (LM) sensory neurons under the status of neuropathic pain caused by sciatic nerve transection (SNT) was used.36 The results indicated 73 DEGs (17%) were nociceptor (NP or PEP)-related, and 18 DEGs (4%) were related to LM sensory neurons. These results suggest the nociceptive TGNs are involved in the trigeminal neuropathic pain and that some DEGs may be from other types of cells in the TG, contributing to trigeminal neuropathic pain.
Moreover, the sex difference is a well-known factor in the pathology of chronic orofacial pain. For instance, females have a greater susceptibility to neuropathic pain, temporomandibular joint pain and migraine.72,73 To investigate the changes in the expression of randomly chosen DE miRNA (mmu-miR-134-3p), DE lncRNA (A630023A22Rik-202), DE circRNA (mmu_circ_0000668), and DE mRNA (Anxa10) in both male and female mice after trigeminal nerve injury, RT-qPCR was performed. The results showed that the changes in the expression of A630023A22Rik-202, mmu_circ_0000668, and Anxa10 mRNA were the same as the results of RNA-sequencing in both male and female mice. Only mmu-miR-134-3p in the female mice showed no significant change. The above difference in miRNA expression between male and female mice may result from sex differences, however, very few studies have examined whether there are potential sex-specific effects on the expression of ncRNAs in the pathogenesis of trigeminal neuropathic pain. In an animal model of posttraumatic widespread pain (PTWP),74 microRNA-19b was found expressed in a sex-dependent manner in dorsal root ganglia (DRG), which was thought to be affected by sex hormones. Therefore, the sex difference should be considered when we research the role of ncRNAs in the development and maintenance of trigeminal neuropathic pain in the following-up studies.
Conclusion
The results of the present study suggest that ncRNAs (including miRNAs, lncRNAs, and circRNAs) may play a vital role in the development of trigeminal neuropathic pain as well as mRNAs by mediating the pain-related genes and nociceptive signaling pathways through ceRNA regulation of circRNA-miRNA-mRNA and lncRNA-miRNA-mRNA. In addition to the known DE ncRNAs and pain genes in the TG of CCI-ION mice, the detailed roles of the novel DE ncRNAs and DEGs currently unrelated to pain also deserve to study in the future. Therefore, the results of this study bring novel insight into the mechanisms and potential treatment targets of trigeminal neuropathic pain.
Ethics Approval
This study was approved by the West China Hospital of Stomatology Institutional Review Board (Animal Protocol Number: WCHSIRB-D-2021-146).
Acknowledgments
The authors thank Ms. Da-Qing Liao and Ms. Xiang-Li Kong for their help with this study.
The study was funded by the National Natural Science Foundation of China (Grant Numbers: 81870800, 82071149), the Department of Science and Technology of Sichuan Province (Grant Numbers: 2021JDRC0166, 2020YJ0224), and the China Postdoctoral Science Foundation (Grant Number: 2020M683329).
Disclosure
The authors declare no conflicts of interest in relation to this work and that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Orofacial Pain Classification Committee. International classification of orofacial pain, 1st edition (ICOP). Cephalalgia. 2020;40(2):129–221. doi:10.1177/0333102419893823
2. Kohli D, Katzmann G, Benoliel R, Korczeniewska OA. Diagnosis and management of persistent posttraumatic trigeminal neuropathic pain secondary to implant therapy: a review. J Am Dent Assoc. 2021;152(6):483–490. doi:10.1016/j.adaj.2020.08.017
3. Nagakura Y, Nagaoka S, Kurose T. Potential molecular targets for treating neuropathic orofacial pain based on current findings in animal models. Int J Mol Sci. 2021;22(12):6406. doi:10.3390/ijms22126406
4. Nakajima A, Tsuboi Y, Suzuki I, et al. PKCgamma in Vc and C1/C2 is involved in trigeminal neuropathic pain. J Dent Res. 2011;90(6):777–781. doi:10.1177/0022034511401406
5. Jiang BC, Zhang J, Wu B, et al. G protein-coupled receptor GPR151 is involved in trigeminal neuropathic pain through the induction of Gβγ/extracellular signal-regulated kinase-mediated neuroinflammation in the trigeminal ganglion. Pain. 2021;162(5):1434–1448. doi:10.1097/j.pain.0000000000002156
6. Baad-Hansen L, Benoliel R. Neuropathic orofacial pain: facts and fiction. Cephalalgia. 2017;37(7):670–679. doi:10.1177/0333102417706310
7. Kalpachidou T, Kummer KK, Kress M. Non-coding RNAs in neuropathic pain. Neuronal Signal. 2020;4(1):NS20190099. doi:10.1042/NS20190099
8. Song G, Yang Z, Guo J, Zheng Y, Su X, Wang X. Interactions among lncRNAs/circRNAs, miRNAs, and mRNAs in neuropathic pain. Neurotherapeutics. 2020;17(3):917–931. doi:10.1007/s13311-020-00881-y
9. Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127(3):761–771. doi:10.1172/JCI84424
10. Zhou J, Xiong Q, Chen H, Yang C, Fan Y. Identification of the spinal expression profile of non-coding RNAs involved in neuropathic pain following spared nerve injury by sequence analysis. Front Mol Neurosci. 2017;10:91. doi:10.3389/fnmol.2017.00091
11. Dong Y, Li P, Ni Y, Zhao J, Liu Z. Decreased microRNA-125a-3p contributes to upregulation of p38 MAPK in rat trigeminal ganglions with orofacial inflammatory pain. PLoS One. 2014;9(11):e111594. doi:10.1371/journal.pone.0111594
12. Pereira CM, Sehnem D, da Fonseca EO, et al. miRNAs: important targets for oral cancer pain research. Biomed Res Int. 2017;2017:4043516. doi:10.1155/2017/4043516
13. Zhang SB, Lin SY, Liu M, et al. CircAnks1a in the spinal cord regulates hypersensitivity in a rodent model of neuropathic pain. Nat Commun. 2019;10(1):4119. doi:10.1038/s41467-019-12049-0
14. Xu M, Yan Y, Zhu M, Wang Z, Zhang X, Zhang D. Effects of long non-coding RNA Gm14461 on pain transmission in trigeminal neuralgia. J Inflamm. 2020;17:1. doi:10.1186/s12950-019-0231-1
15. Dong T, Si H, Li Z, Bai Q, Tao F. Transcriptomic analysis of trigeminal ganglion and spinal trigeminal nucleus caudalis in mice with inflammatory temporomandibular joint pain. J Pain Res. 2022;15:1487–1502. doi:10.2147/JPR.S364887
16. Vos BP, Strassman AM, Maciewicz RJ. Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat’s infraorbital nerve. J Neurosci. 1994;14(5 Pt 1):2708–2723. doi:10.1523/JNEUROSCI.14-05-02708.1994
17. Ding W, You Z, Shen S, et al. An improved rodent model of trigeminal neuropathic pain by unilateral chronic constriction injury of distal infraorbital nerve. J Pain. 2017;18(8):899–907. doi:10.1016/j.jpain.2017.02.427
18. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi:10.1016/0165-0270(94)90144-9
19. Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. doi:10.1093/nar/gkr688
20. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi:10.1038/nmeth.3317
21. Sun L, Luo H, Bu D, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41(17):e166. doi:10.1093/nar/gkt646
22. Kong L, Zhang Y, Ye ZQ, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(WebServer issue):W345–W349. doi:10.1093/nar/gkm391
23. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. doi:10.1038/nbt.3122
24. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. doi:10.1038/nature11928
25. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19(5):803–810. doi:10.1093/bib/bbx014
26. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1. doi:10.1186/gb-2003-5-1-r1
27. Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34(WebServer issue):W451–454. doi:10.1093/nar/gkl243
28. Chen LL. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21(8):475–490. doi:10.1038/s41580-020-0243-y
29. Perkins JR, Lees J, Antunes-Martins A, et al. PainNetworks: a web-based resource for the visualisation of pain-related genes in the context of their network associations. Pain. 2013;154(12):2586e2581–2586e2512. doi:10.1016/j.pain.2013.09.003
30. Lacroix-Fralish ML, Ledoux JB, Mogil JS. The pain genes database: an interactive web browser of pain-related transgenic knockout studies. Pain. 2007;131(1–2):3 e1–4. doi:10.1016/j.pain.2007.04.041
31. Pokhilko A, Nash A, Cader MZ. Common transcriptional signatures of neuropathic pain. Pain. 2020;161(7):1542–1554. doi:10.1097/j.pain.0000000000001847
32. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. doi:10.1186/gb-2010-11-2-r14
33. Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Databaseissue):D480–484. doi:10.1093/nar/gkm882
34. Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–3793. doi:10.1093/bioinformatics/bti430
35. Jamieson DG, Moss A, Kennedy M, et al. The pain interactome: connecting pain-specific protein interactions. Pain. 2014;155(11):2243–2252. doi:10.1016/j.pain.2014.06.020
36. Hu G, Huang K, Hu Y, et al. Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci Rep. 2016;6:31851. doi:10.1038/srep31851
37. Eijkelkamp N, Wang H, Garza-Carbajal A, et al. Low nociceptor GRK2 prolongs prostaglandin E2 hyperalgesia via biased cAMP signaling to Epac/Rap1, protein kinase Cepsilon, and MEK/ERK. J Neurosci. 2010;30(38):12806–12815. doi:10.1523/JNEUROSCI.3142-10.2010
38. Huang CL, Liu F, Zhang YY, et al. Activation of oxytocin receptor in the trigeminal ganglion attenuates orofacial ectopic pain attributed to inferior alveolar nerve injury. J Neurophysiol. 2021;125(1):223–231. doi:10.1152/jn.00646.2020
39. Kogelman LJA, Christensen RE, Pedersen SH, et al. Whole transcriptome expression of trigeminal ganglia compared to dorsal root ganglia in Rattus Norvegicus. Neuroscience. 2017;350:169–179. doi:10.1016/j.neuroscience.2017.03.027
40. Liu F, Yajima T, Wang M, Shen JF, Ichikawa H, Sato T. Effects of trigeminal nerve injury on the expression of galanin and its receptors in the rat trigeminal ganglion. Neuropeptides. 2020;84:102098. doi:10.1016/j.npep.2020.102098
41. Oztürk G, Tonge DA. Effects of leukemia inhibitory factor on galanin expression and on axonal growth in adult dorsal root ganglion neurons in vitro. Exp Neurol. 2001;169(2):376–385. doi:10.1006/exnr.2001.7667
42. Gabra BH, Kessler FK, Ritter JK, Dewey WL, Smith FL. Decrease in N-methyl-D-aspartic acid receptor-NR2B subunit levels by intrathecal short-hairpin RNA blocks group I metabotropic glutamate receptor-mediated hyperalgesia. J Pharmacol Exp Ther. 2007;322(1):186–194. doi:10.1124/jpet.107.120071
43. McDougall JJ, Watkins L, Li Z. Vasoactive intestinal peptide (VIP) is a modulator of joint pain in a rat model of osteoarthritis. Pain. 2006;123(1–2):98–105. doi:10.1016/j.pain.2006.02.015
44. Peirs C, Williams SG, Zhao X, et al. Mechanical allodynia circuitry in the dorsal horn is defined by the nature of the injury. Neuron. 2021;109(1):73–90.e77. doi:10.1016/j.neuron.2020.10.027
45. Escolano-Lozano F, Gries E, Schlereth T, et al. Local and systemic expression pattern of MMP-2 and MMP-9 in complex regional pain syndrome. J Pain. 2021;22(10):1294–1302. doi:10.1016/j.jpain.2021.04.002
46. Intondi AB, Dahlgren MN, Eilers MA, Taylor BK. Intrathecal neuropeptide Y reduces behavioral and molecular markers of inflammatory or neuropathic pain. Pain. 2008;137(2):352–365. doi:10.1016/j.pain.2007.09.016
47. Bai G, Ambalavanar R, Wei D, Dessem D. Downregulation of selective microRNAs in trigeminal ganglion neurons following inflammatory muscle pain. Mol Pain. 2007;3:15. doi:10.1186/1744-8069-3-15
48. Palanisamy V, Jakymiw A, Van Tubergen EA, D’Silva NJ, Kirkwood KL. Control of cytokine mRNA expression by RNA-binding proteins and microRNAs. J Dent Res. 2012;91(7):651–658. doi:10.1177/0022034512437372
49. Willemen HL, Huo XJ, Mao-Ying QL, Zijlstra J, Heijnen CJ, Kavelaars A. MicroRNA-124 as a novel treatment for persistent hyperalgesia. J Neuroinflammation. 2012;9:143. doi:10.1186/1742-2094-9-143
50. Hung HY, Chow LH, Kotlinska JH, et al. LVV-hemorphin-7 (LVV-H7) plays a role in antinociception in a rat model of alcohol-induced pain disorders. Peptides. 2021;136:170455. doi:10.1016/j.peptides.2020.170455
51. Wang ZB, Liu YD, Wang S, Zhao P. High-frequency spinal cord stimulation produces long-lasting analgesic effects by restoring lysosomal function and autophagic flux in the spinal dorsal horn. Neural Regener Res. 2022;17(2):370–377. doi:10.4103/1673-5374.317989
52. Deng Y, Yang L, Xie Q, et al. Protein kinase A is involved in neuropathic pain by activating the p38MAPK pathway to mediate spinal cord cell apoptosis. Mediators Inflamm. 2020;2020:6420425. doi:10.1155/2020/6420425
53. Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol. 2017;1008:1–46.
54. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi:10.1016/j.cell.2013.06.020
55. Wu S, Bono J, Tao YX. Long noncoding RNA (lncRNA): a target in neuropathic pain. Expert Opin Ther Targets. 2019;23(1):15–20. doi:10.1080/14728222.2019.1550075
56. Zhou J, Fan Y, Chen H. Analyses of long non-coding RNA and mRNA profiles in the spinal cord of rats using RNA sequencing during the progression of neuropathic pain in an SNI model. RNA Biol. 2017;14(12):1810–1826. doi:10.1080/15476286.2017.1371400
57. Xiong W, Tan M, Tong Z, et al. Effects of long non-coding RNA uc.48+ on pain transmission in trigeminal neuralgia. Brain Res Bull. 2019;147:92–100. doi:10.1016/j.brainresbull.2019.02.009
58. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393–407. doi:10.1016/j.cell.2018.01.011
59. Akopians AL, Babayan AH, Beffert U, Herz J, Basbaum AI, Phelps PE. Contribution of the Reelin signaling pathways to nociceptive processing. Eur J Neurosci. 2008;27(3):523–537. doi:10.1111/j.1460-9568.2008.06056.x
60. Okerman T, Jurgenson T, Moore M, Klein AH. Inhibition of the phosphoinositide 3-kinase-AKT-cyclic GMP-c-Jun N-terminal kinase signaling pathway attenuates the development of morphine tolerance in a mouse model of neuropathic pain. Mol Pain. 2021;17:17448069211003375. doi:10.1177/17448069211003375
61. Spray DC, Hanani M. Gap junctions, pannexins and pain. Neurosci Lett. 2019;695:46–52. doi:10.1016/j.neulet.2017.06.035
62. Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(3):428–442. doi:10.1016/j.molcel.2018.06.034
63. Xu D, Ma X, Sun C, et al. Emerging roles of circular RNAs in neuropathic pain. Cell Prolif. 2021;54:e13139. doi:10.1111/cpr.13139
64. Acuna C, Liu X, Südhof TC. How to make an active zone: unexpected universal functional redundancy between RIMs and RIM-BPs. Neuron. 2016;91(4):792–807. doi:10.1016/j.neuron.2016.07.042
65. Abrahamsson T, Chou CYC, Li SY, et al. Differential regulation of evoked and spontaneous release by presynaptic NMDA receptors. Neuron. 2017;96(4):839–855.e835. doi:10.1016/j.neuron.2017.09.030
66. Yang L, Han B, Zhang Y, et al. Engagement of circular RNA HECW2 in the nonautophagic role of ATG5 implicated in the endothelial-mesenchymal transition. Autophagy. 2018;14(3):404–418. doi:10.1080/15548627.2017.1414755
67. Hillard CJ, Weinlander KM, Stuhr KL. Contributions of endocannabinoid signaling to psychiatric disorders in humans: genetic and biochemical evidence. Neuroscience. 2012;204:207–229. doi:10.1016/j.neuroscience.2011.11.020
68. de Vries M, van Rijckevorsel DC, Wilder-Smith OH, van Goor H. Dronabinol and chronic pain: importance of mechanistic considerations. Expert Opin Pharmacother. 2014;15(11):1525–1534. doi:10.1517/14656566.2014.918102
69. Ohno-Shosaku T, Tanimura A, Hashimotodani Y, Kano M. Endocannabinoids and retrograde modulation of synaptic transmission. Neuroscientist. 2012;18(2):119–132. doi:10.1177/1073858410397377
70. Basavarajappa BS. Neuropharmacology of the endocannabinoid signaling system-molecular mechanisms, biological actions and synaptic plasticity. Curr Neuropharmacol. 2007;5(2):81–97. doi:10.2174/157015907780866910
71. Batbold D, Shinoda M, Honda K, et al. Macrophages in trigeminal ganglion contribute to ectopic mechanical hypersensitivity following inferior alveolar nerve injury in rats. J Neuroinflammation. 2017;14(1):249. doi:10.1186/s12974-017-1022-3
72. Sannajust S, Imbert I, Eaton V, et al. Females have greater susceptibility to develop ongoing pain and central sensitization in a rat model of temporomandibular joint pain. Pain. 2019;160(9):2036–2049. doi:10.1097/j.pain.0000000000001598
73. Vetvik KG, MacGregor EA. Sex differences in the epidemiology, clinical features, and pathophysiology of migraine. Lancet Neurol. 2017;16(1):76–87. doi:10.1016/S1474-4422(16)30293-9
74. Linnstaedt SD, Rueckeis CA, Riker KD, et al. MicroRNA-19b predicts widespread pain and posttraumatic stress symptom risk in a sex-dependent manner following trauma exposure. Pain. 2020;161(1):47–60. doi:10.1097/j.pain.0000000000001709
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024