Back to Journals » ImmunoTargets and Therapy » Volume 14
Integrative Spatial Transcriptomics and Experimental Validation Reveal UBC-Mediated EMT Associated with Immune Evasion in Hepatocellular Carcinoma
Authors Li X, Qin X, Shi K, Lu G, Tian G, Chen Y, Yao R ![]()
Received 15 September 2025
Accepted for publication 11 December 2025
Published 19 December 2025 Volume 2025:14 Pages 1437—1454
DOI https://doi.org/10.2147/ITT.S567643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Xiaosong Li,1,2,* Xian Qin,3,4,* Kezhi Shi,5 Guangrui Lu,1,2 Guodong Tian,1,2 Yue Chen,1,2 Rucheng Yao1,2
1The First College of Clinical Medical Science, China Three Gorges University, Yichang, Hubei, People’s Republic of China; 2Hepatopancreatobiliary Surgery, Yichang Central People’s Hospital, Yichang, Hubei, People’s Republic of China; 3Department of Hepatobiliary and Pancreatic Surgery, Zhongnan Hospital of Wuhan University, Wuhan, Hubei, People’s Republic of China; 4Clinical Medicine Research Center for Minimally Invasive Procedure of Hepatobiliary & Pancreatic Diseases of Hubei Province, Wuhan, Hubei, People’s Republic of China; 5Oncology Department, Yichang Central People’s Hospital, Yichang, Hubei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Rucheng Yao, The First College of Clinical Medical Science, China Three Gorges University, Yichang, Hubei, People’s Republic of China, Email [email protected]
Background: Hepatocellular carcinoma (HCC) exhibits pronounced spatial heterogeneity that limits therapeutic efficacy. The contribution of epithelial–mesenchymal transition (EMT) to regional tumor progression and immune evasion remains incompletely understood.
Methods: We integrated bulk transcriptomic datasets (TCGA, GSE14520), 34 single-cell RNA-seq samples, and 15 spatial transcriptomic datasets to delineate EMT activity across distinct HCC regions. Immune infiltration profiling, pathway enrichment, and multi-model machine learning were used to identify candidate EMT regulators. Functional validation was performed in Hep3B cells through wound healing, Transwell migration/invasion, and immunofluorescence assays.
Results: EMT activity was significantly elevated at tumor margins (fold change = 2.7, p < 0.001) and was associated with poorer overall survival (HR = 2.15, 95% CI: 1.41– 3.27, p < 0.001). Regions with high EMT signatures showed reduced CD8⁺ T-cell infiltration and increased immunosuppressive cells, including MDSCs, M2 macrophages, and Tregs, along with elevated expression of immune checkpoints (PDCD1, CTLA4, LAG3). Among candidate regulators, UBC was consistently ranked as a top EMT-associated gene across all models. Functional assays confirmed that UBC overexpression enhanced migration, invasion, and vimentin expression, whereas UBC knockdown reversed these effects.
Conclusion: Through integrative spatial multi-omics and experimental validation, we identify UBC as a key mediator of EMT and immune suppression at HCC margins. These findings provide mechanistic insight into spatial heterogeneity and suggest that targeting UBC could have translational potential for overcoming immune evasion in HCC.
Keywords: hepatocellular carcinoma, epithelial–mesenchymal transition, UBC, multi-omics integration, tumor microenvironment
Background
Epidemiological data indicate that hepatocellular carcinoma (HCC) remains a major global health burden, with approximately 841,000 new cases and 782,000 deaths annually, over half occurring in China.1,2 Despite therapeutic advances, patients with advanced HCC continue to experience poor outcomes due to high recurrence and limited treatment efficacy. A major obstacle lies in the pronounced spatial and temporal heterogeneity of HCC.3–5 Intertumoral diversity drives patient-specific treatment responses, whereas intratumoral heterogeneity fosters dynamic interactions between malignant subclones and the tumor microenvironment, promoting resistance and clonal evolution.6–8
The advent of single-cell and spatial transcriptomics has revolutionized the study of HCC heterogeneity.9 Single-cell RNA sequencing (scRNA-seq) dissects the transcriptional landscape at single-cell resolution, identifying malignant subpopulations and immune components.10 Spatial transcriptomics complements this by preserving tissue architecture, enabling investigation of regional signaling, niche formation, and microenvironmental reprogramming.11 Recent studies reveal spatially distinct malignant phenotypes;12,13 immunosuppressive remodeling via TAM polarization and endothelial interaction;14 and clonal continuity from cirrhosis to carcinoma.15 These insights underscore spatial heterogeneity as a key determinant of therapeutic resistance.
However, an important gap remains in understanding how epithelial–mesenchymal transition (EMT) contributes to spatial immune remodeling and immune evasion in HCC. EMT not only enhances cellular plasticity and invasiveness but also reprograms cytokine signaling and antigen presentation, facilitating immune suppression within the tumor microenvironment. Yet, the spatial relationship between EMT activity and regional immune landscapes has not been systematically explored in HCC.
Here, we hypothesize that UBC regulates EMT-mediated spatial immune remodeling in HCC. To test this, we integrated bulk, single-cell, and spatial transcriptomic datasets to delineate EMT activity across tumor regions and combined bioinformatic analyses with functional validation to uncover the molecular basis of spatial heterogeneity and immune evasion.
Method
Data Acquisition
This study integrated multiple publicly available HCC sequencing datasets, including bulk transcriptomic data from TCGA-LIHC (Supplementary Table 1) and GSE1452016 (Supplementary Table 2, single-cell transcriptomic data from 34 samples encompassing tumor core, invasive margin, and adjacent non-tumor tissues17 (Supplementary Table 3, and spatial transcriptomic data from 12 samples covering the same anatomical regions18 (Supplementary Table 4). All datasets were obtained from public repositories with corresponding clinical and histological annotations.
Transcriptome Data Processing
Bulk RNA-seq data were preprocessed and normalized using standard pipelines. Batch effects across cohorts (TCGA vs GEO) were corrected using the ComBat function in the sva R package (parameters: par.prior = TRUE, prior.plots = FALSE). The resulting normalized expression matrix was used for downstream analyses.
scRNA-Seq Data Processing
scRNA-seq data were processed using the Seurat V5 framework. A total of 59,302 high-quality cells passed quality control thresholds, including 300 < nFeature_RNA < 6000, 500 < nCount_RNA < 50,000, and mitochondrial gene ratio < 15%. Cells expressing hemoglobin genes (indicative of red blood cells) were removed prior to downstream analysis, ensuring data reliability and comparability across samples. The filtered expression matrix was normalized using methods appropriate for single-cell data (SCTransform), and highly variable genes were selected for downstream analyses. Dimensionality reduction was first performed via principal component analysis (PCA), followed by uniform manifold approximation and projection (UMAP) for low-dimensional visualization. Cell clusters were identified using a graph-based clustering algorithm (Louvain) and annotated based on marker gene expression, reference databases (SingleR), and manual curation. To integrate data from multiple samples or batches, an anchor-based integration strategy or batch correction method (Harmony) was applied to preserve biological variability while minimizing technical artifacts. For analyses of cellular lineage evolution or state transitions, trajectory inference tools such as Monocle and CellChat were utilized to reconstruct developmental trajectories and infer intercellular communication networks.
Spatial Transcriptomics Data Processing
Spatial transcriptomics data were generated using the 10x Genomics Visium platform, with a spatial resolution of 55 μm per spot. Reads were aligned to tissue section images, and gene expression counts were assigned to individual spatial spots. Spots with <200 detected genes or low UMI counts were filtered out. The processed data were analyzed in Seurat’s spatial framework, integrating histological images (H&E) for domain annotation. Normalization and dimensionality reduction followed SCTransform and PCA–UMAP workflows adapted from single-cell analyses.
The “border” was defined based on both histological and spatial transcriptomic information. Pathologists first identified the tumor–normal transition zone on H&E-stained sections, characterized by the presence of tumor nests adjacent to nonmalignant hepatocytes and stromal components. This visually defined boundary typically corresponded to a spatial distance of approximately 150–250 μm from the tumor core in Visium spatial maps (∼3–5 spot diameters). For quantitative consistency across samples, spots located within this boundary region were annotated as the border region.
Immune Cell Infiltration Analysis
Immune infiltration was quantified using multiple complementary algorithms.
For bulk transcriptomes, CIBERSORT (1000 permutations) estimated immune cell proportions, while ssGSEA assessed immune pathway activity. Immune and stromal scores were computed using the ESTIMATE algorithm. To ensure comparability, expression matrices from TCGA and GEO were batch-corrected prior to integration using ComBat. Cross-validation between algorithms ensured robustness of inferred immune composition.
Gene Set Enrichment and Module Score Analyses
Functional enrichment was performed using GSVA, with hallmark and KEGG gene sets from MSigDB. At the single-cell level, AUCell quantified the activation of selected pathways, complemented by Seurat::AddModuleScore for cluster-wise functional scoring. Adjusted p-values were computed using the Benjamini–Hochberg (FDR) method, with significance set at FDR < 0.05.
Machine Learning
In this study, we applied multiple machine learning approaches to perform feature selection on single-cell transcriptomic data derived from hepatocytes, with the aim of identifying key genes associated with EMT and tissue heterogeneity in HCC. Prior to analysis, the raw data underwent quality control and label preprocessing, followed by a random split into a training set (70%) and a test set (30%). To enhance model robustness, five-fold repeated cross-validation (repeatedCV) was implemented during training, and parallel computing was employed to expedite model fitting.
For feature selection, we systematically applied six built-in models from the caret package—random forest, gradient-boosted trees, Lasso regression, PLS, discriminant analysis, and decision trees—as well as two independently developed gradient-boosting frameworks, CatBoost and LightGBM. (CatBoost: models were trained with learning_rate = 0.1, depth = 6, and iterations = 1000 using the default loss function, and a fixed random seed for reproducibility. LightGBM: Models were trained with num_leaves = 31, learning_rate = 0.05, and n_estimators = 500, with a fixed random seed and default objective function). Following model training, permutational feature importance was computed using the DALEX framework, standardized to a 0–100 scale, and the top ten genes were extracted from each model. For CatBoost and LightGBM, dedicated interfaces were utilized to ensure optimal performance and interpretability, as these models are not natively integrated into the caret package.
Finally, feature selection results from all models were aggregated, and the frequency of gene selection and average importance across models were calculated. The results were visualized using bar charts, UpSet plots, and scatter plots. This multi-model integration strategy minimizes bias introduced by any single algorithm, identifies core genes consistently selected across models, and provides a solid foundation for subsequent biological validation.
Cell Lines and Culture Conditions
The human hepatocellular carcinoma Hep3B cell line was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2.
Cell Scratch Assay
A wound-healing assay was performed to evaluate the migratory capacity of cells. Briefly, cells were seeded into six-well plates and cultured until reaching approximately 90% confluence. A sterile 200 μL pipette tip was used to generate a linear scratch across the monolayer. Detached cells were removed by washing with phosphate-buffered saline (PBS), and fresh serum-free medium was added. Images of the wound area were captured at 0 h and at designated time points under an inverted microscope. The wound closure percentage was quantified using ImageJ software. All assays were performed in triplicate biological replicates.
Transwell Migration and Invasion Assay
Cell migration and invasion were assessed using Transwell chambers (8 μm pore size; Corning, USA). For the migration assay, cells suspended in serum-free medium were seeded into the upper chamber, while medium containing 10% fetal bovine serum (FBS) was added to the lower chamber as a chemoattractant. After incubation for 24 h, non-migrated cells on the upper surface of the membrane were removed with a cotton swab, whereas migrated cells on the lower surface were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. For invasion assays, the upper chamber was pre-coated with Matrigel (BD Biosciences), and otherwise the procedure was identical. Cells in five random microscopic fields were counted for quantification. All assays were performed in triplicate biological replicates.
Immunofluorescence Staining
Cells were grown on sterilized glass coverslips and fixed with 4% paraformaldehyde for 15 min at room temperature. After permeabilization with 0.2% Triton X-100 for 10 min and blocking with 5% bovine serum albumin (BSA) for 1 h, cells were incubated overnight at 4 °C with primary antibodies targeting the indicated proteins. After washing, cells were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature in the dark. Nuclei were counterstained with DAPI. Fluorescence images were obtained using a confocal laser scanning microscope (Leica Microsystems).
Statistical Analysis
Data were analyzed using R (v4.3.3). Two-group comparisons employed the Wilcoxon rank-sum test. Survival analyses were conducted using the Kaplan–Meier method with the Log rank test. Multiple testing correction was performed using the Benjamini–Hochberg FDR procedure. P < 0.05 or FDR < 0.05 was considered statistically significant. All code is provided in the Supplementary code.
Result
Enhanced EMT Activity at the Tumor Margin in HCC
The GSE189903 dataset includes single-cell sequencing data from 34 liver cancer tissue samples, encompassing regions such as the tumor core, tumor margin, and adjacent normal tissue. Following quality control, the 34 single-cell datasets were integrated and annotated (Figure 1A–D). Analysis demonstrated that liver cancer cell compositions vary not only across patients but also among different tumor regions within the same individual. To investigate these spatial differences, GSVA enrichment analysis was conducted on cells from distinct tumor regions across 13 tumor-associated phenotypes. The results indicated that EMT is significantly upregulated in the tumor margin, suggesting its potential role as a key driver of regional tumor heterogeneity (Figure 1E).
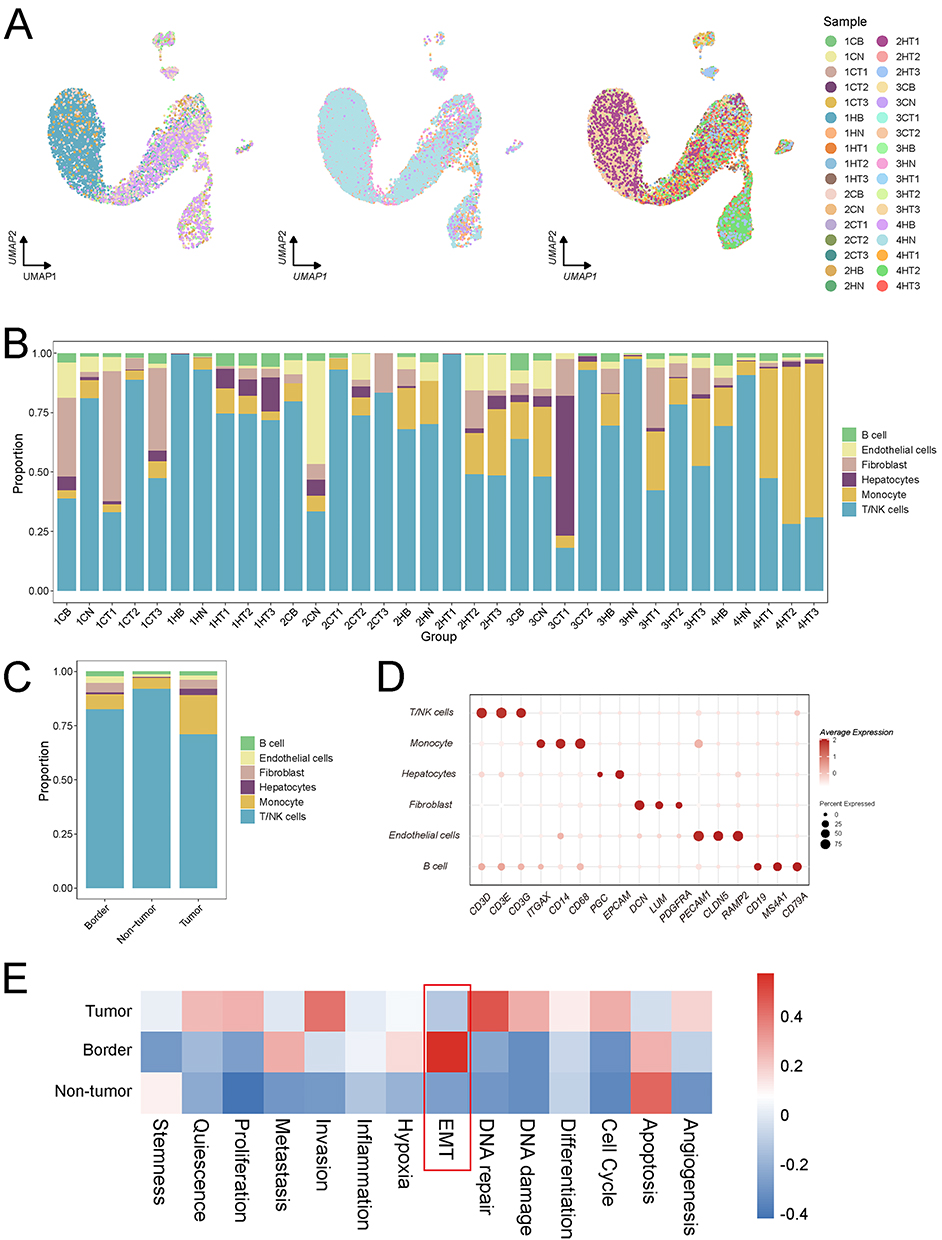 |
Figure 1 Analysis of the 34 single-cell datasets from the HRA000437 cohort revealed that EMT activity was significantly elevated in the HCC margin. (A) The Harmony method was employed to integrate the 34 single-cell datasets, with the integrated results visualized according to tissue origin. (B) Proportions of various cell types following annotation of the 34 single-cell datasets. (C) Distribution of cell types across the tumor core, tumor margin, and adjacent normal tissue. (D) A bubble plot depicting cell marker gene annotations. (E) GSVA enrichment analysis demonstrated that EMT activity was most pronounced in the tumor margin of hepatocellular carcinoma. The red-colored box highlights that UBC exhibits the strongest correlation with EMT at the tumor border. |
Prognostic and Immune Microenvironment Implications of EMT in HCC
Following the identification of the characteristic distribution of EMT in liver cancer tissues through single-cell analysis, we further examined large-scale transcriptomic data to evaluate the clinical and immunological implications of EMT in HCC patients. The results demonstrated that EMT serves as an independent prognostic factor in HCC: higher EMT scores were significantly associated with poorer patient survival (Figure 2A and B). Furthermore, advanced tumor stages correlated with elevated EMT scores, suggesting that EMT may promote malignant progression in HCC (Figure 2C). Analysis of the tumor microenvironment revealed a negative association between EMT scores and immune activity. In the high EMT score group, both the functional capacity and infiltration levels of CD8⁺ T cells were reduced compared to those in the low score group, whereas myeloid-derived suppressor cells (MDSCs), M2 macrophages, and regulatory T cells (Tregs) were markedly enriched (Figure 2D–F). Immune profiling further indicated that mesenchymal cells were predominant in the high EMT score group, with a relatively low abundance of immune cells. EMT scores showed positive correlations with the expression of immunosuppressive or tolerance-associated molecules, including PDCD1 (r = 0.41, FDR < 0.001), CTLA4 (r = 0.44, FDR < 0.001), and LAG3 (r = 0.25, FDR < 0.001), and negative correlations with immune-stimulatory molecules such as CD70 (r = −0.42, FDR < 0.001) and CD86 (r = −0.49, FDR < 0.001) (Figure 2G).
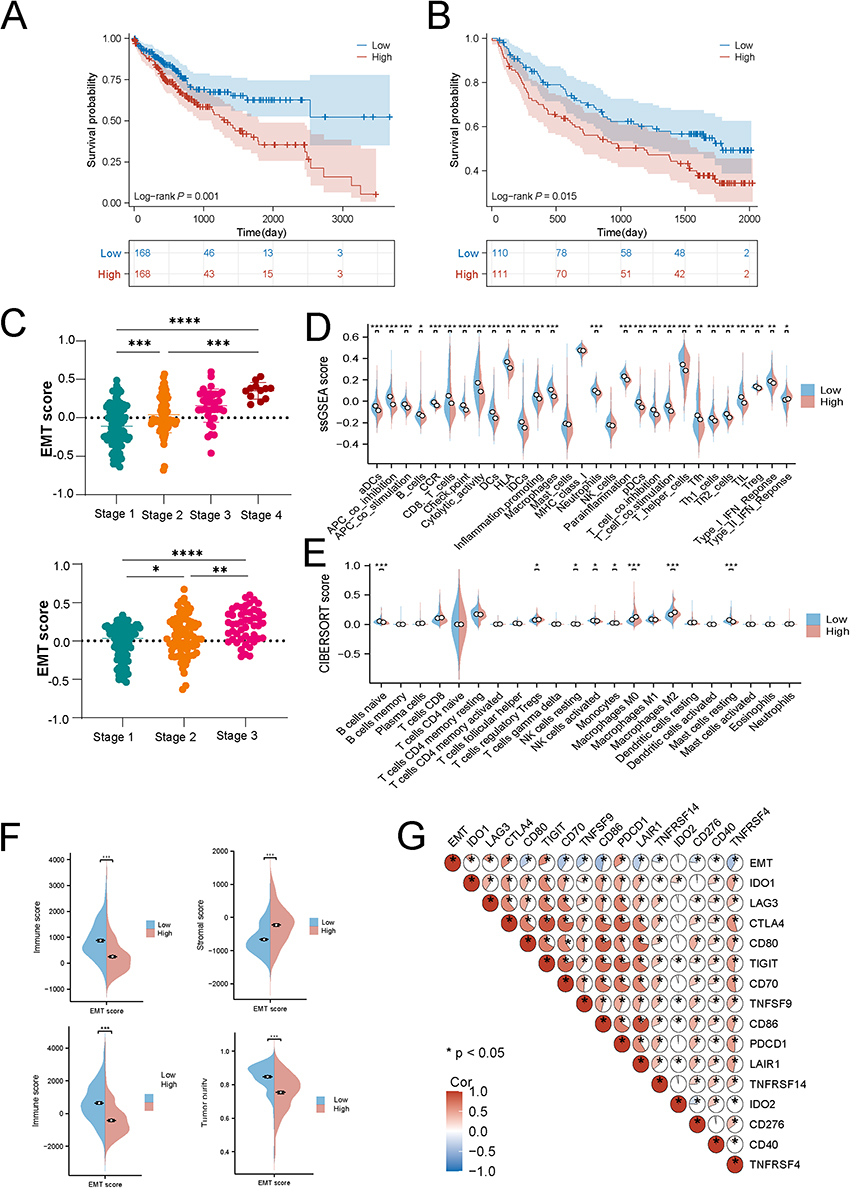 |
Figure 2 Large-scale transcriptomic data validate the association between epithelial-mesenchymal transition (EMT) scores and clinical outcomes, as well as immune profiles, in hepatocellular carcinoma patients. (A) Higher EMT scores predicted poorer survival in TCGA cohorts. (B) Higher EMT scores predicted poorer survival in GSE14520 cohorts. (C) Higher tumor grades in hepatocellular carcinoma are correlated with elevated EMT activity. (D) The CIBERSORT algorithm analysis indicates that patients with high EMT scores exhibit increased proportions of myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and M2 macrophages compared to those with low EMT scores. (E) The ssGSEA algorithm analysis indicates that patients with high EMT scores exhibit increased proportions of myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and M2 macrophages compared to those with low EMT scores. (F) Patients with high EMT scores display higher mesenchymal scores and lower immune scores. (G) EMT scores show a strong positive correlation with immune exhaustion markers, including LAG3, PDCD1, and CTLA4. (p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****)). |
Functional Features of EMT-Positive Cells
Based on these findings, we focused on liver epithelial cells to characterize the functional properties of EMT-positive cells. Re-clustering and functional enrichment analysis of epithelial cells revealed that cluster 0 exhibited the most significant differences across the three tumor regions and represented the predominant population of EMT-positive cells (r=0.19, FDR < 0.001) (Figure 3A–E). Functional enrichment analysis further demonstrated that this cluster was highly enriched in pathways associated with cell migration, invasion, and metastasis, with elevated expression of genes such as TLN1, MMP9, and LAMA1 (Figure 3F). Trajectory inference analysis indicated that tumor core cells corresponded to the terminal differentiation state, whereas tumor edge and normal cells represented earlier differentiation states. EMT-related genes showed dynamic expression patterns during epithelial cell differentiation, with the highest levels observed in tumor edge cells (Figure 4).
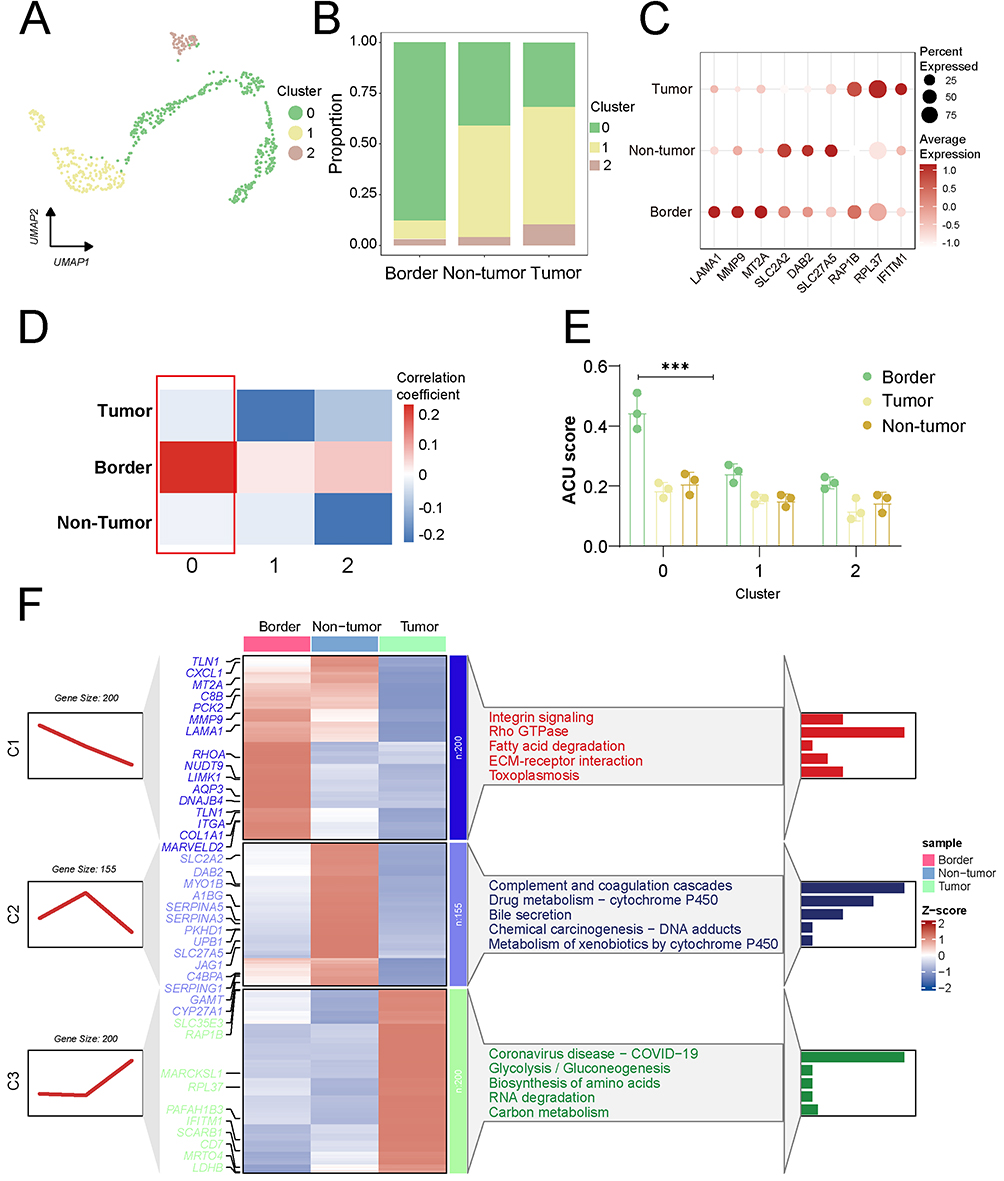 |
Figure 3 EMT-positive cells exhibit increased invasive activity. (A) Re-clustering analysis of liver epithelial cells. (B) The proportion of cells in clusters 0, 1, and 2 at the tumor border, tumor core, and non-tumor. (C) Bubble chart revealed increased expression of invasive genes in cluster 0. Red color represents the mean expression level. (D) Cluster 0 EMT activity was highest at the tumor margin. Red color represents the magnitude of the correlation coefficient. (E) The red-colored box indicates that cell cluster 0 shows the highest correlation with the EMT score at the tumor margin. (F) Invasive activity of cluster 0 cancer cells was significantly higher than that of other cell clusters. Red color represents the magnitude of the Z-score correlation coefficient. (p < 0.001 (***)). |
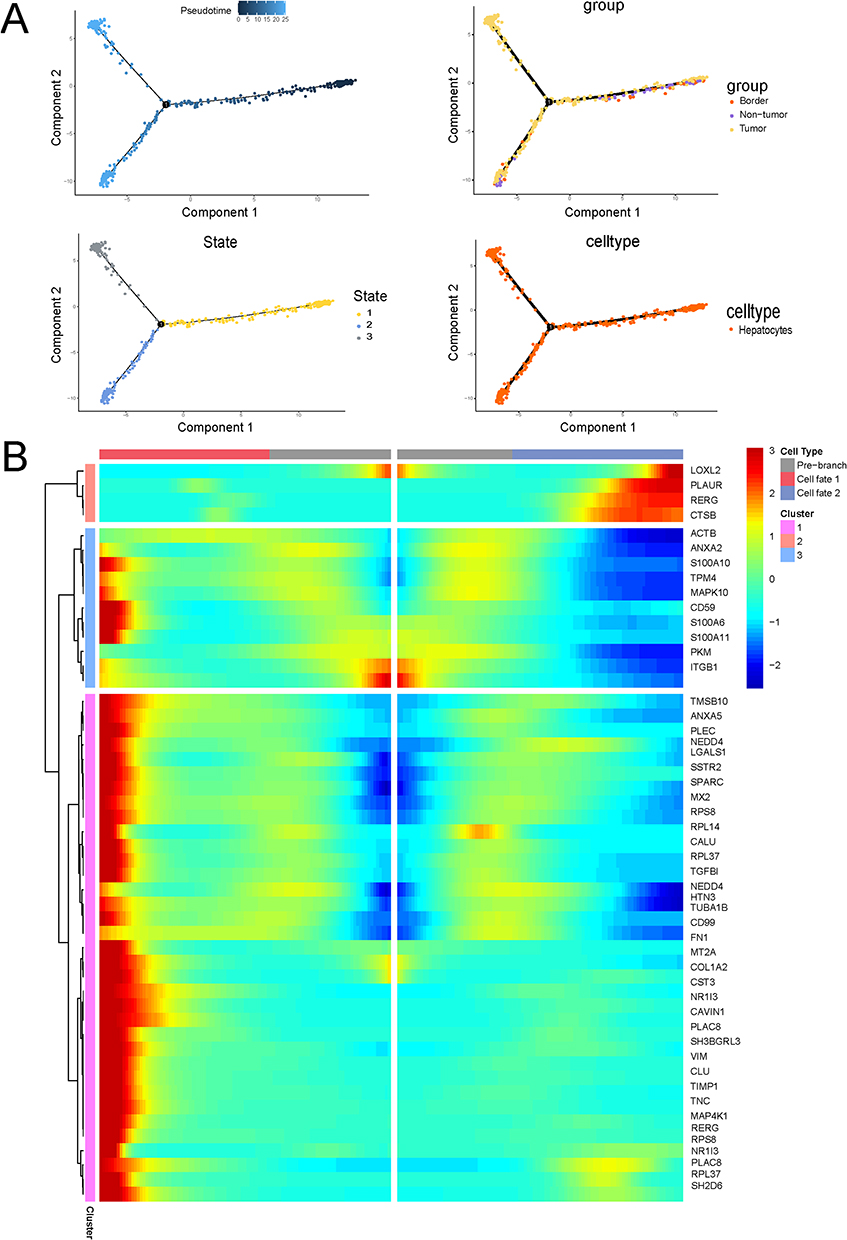 |
Figure 4 Single-cell trajectory analysis demonstrated that the EMT gene set exhibited the highest expression at the tumor margin. (A) Differentiation trajectories of liver epithelial cells across the tumor core, tumor margin, and cancer-adjacent regions. (B) The EMT gene set showed elevated expression levels in the tumor margin during the differentiation of liver epithelial cells. |
Single-Cell Transcriptomic Insights into EMT in HCC
We applied CellChat to analyze intercellular communication networks in single-cell transcriptomic data, investigating signaling interactions among distinct cell types within the tumor microenvironment. The results revealed extensive communication across cell populations, with tumor margin-associated populations displaying a more complex and interactive signaling network (Figure 5A and B). Notably, in the tumor margin, the interaction between tumor-associated fibroblasts and epithelial cells was significantly upregulated, mediated through key pathways such as EGFR and TGF-β. These findings underscore the pivotal role of EMT in the peripheral regions of HCC and suggest potential mechanisms underlying its dynamic crosstalk with the tumor microenvironment (Figure 5C).
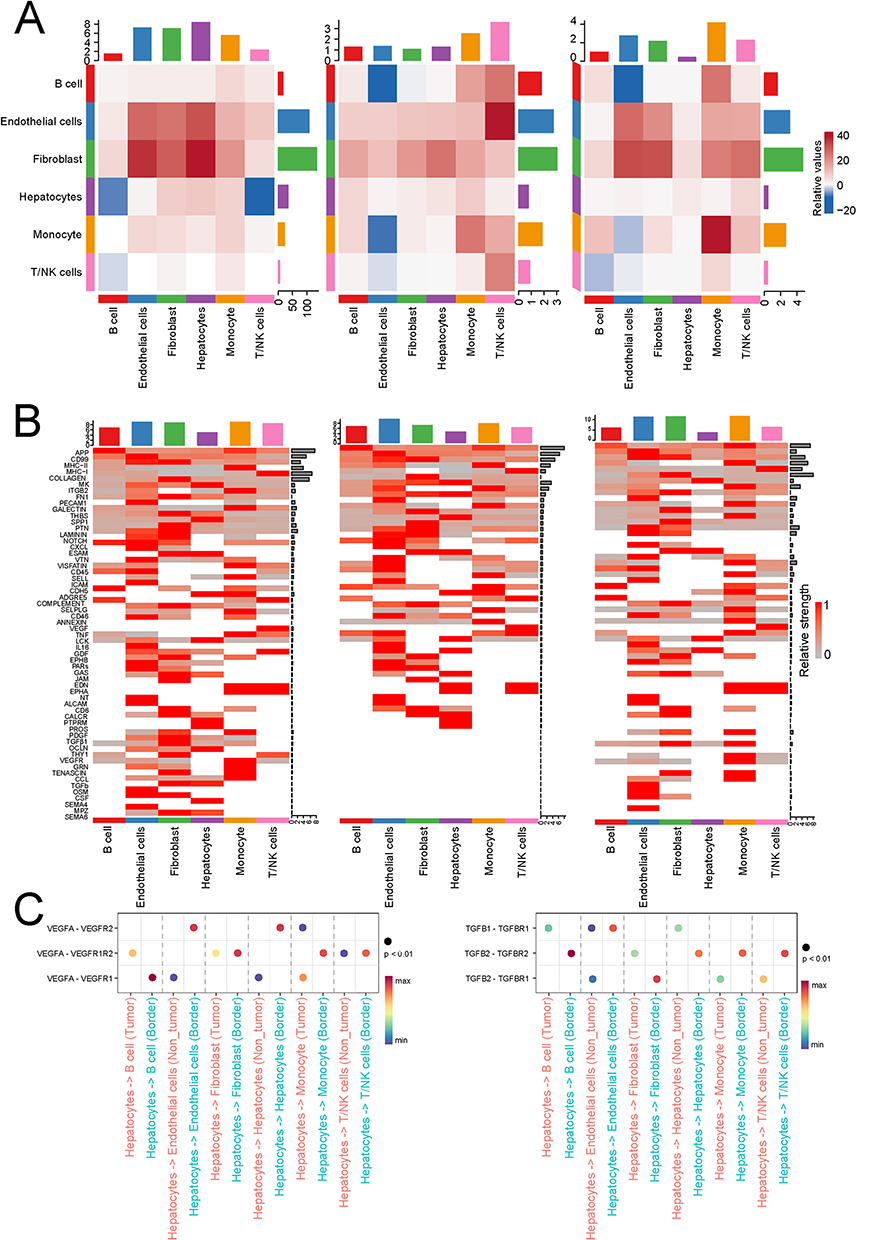 |
Figure 5 Cell cluster 0 showed significantly increased EGFR and TGF-β activity at the tumor margin. (A) The whole-cell view shows the interaction between cell clusters 0, 1, 2, and other cell types. (B) The heatmap displays gene expression trends within signaling pathways across three regions. (C) Results of ligand and receptor interactions revealed that VEGF and TGF-β activity was significantly higher in the tumor margin region than in the tumor core and adjacent tissue. |
Spatial Transcriptomics Validation
The HRA000437 dataset includes spatial transcriptomics data from four HCC tissues, encompassing regions such as the tumor core, periphery, and adjacent normal tissues. Analysis further confirmed the enriched activation of the EMT pathway in the tumor periphery (Figure 6A–C). EMT was predominantly observed in cell clusters with high EPCAM expression, and EMT scores in the tumor margin were significantly elevated compared to those in the tumor core and adjacent non-tumor regions. EPCAM expression exhibited a strong positive correlation with EMT scores, further supporting the notion that EMT activity at the tumor margin is closely linked to the epithelial cell state (Figure 6D and E).
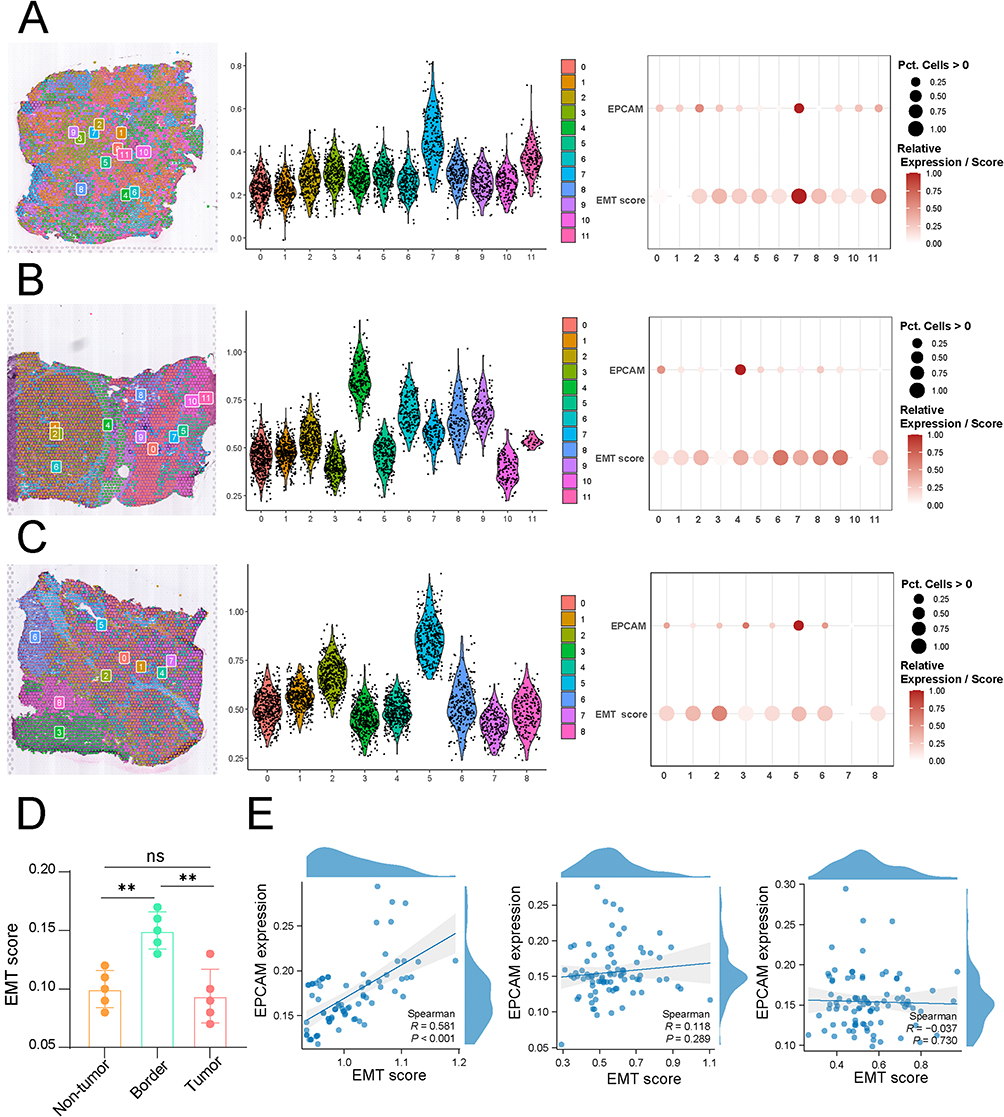 |
Figure 6 Spatial transcriptomics data revealed that EMT scores were positively correlated with the activity and malignant epithelial content at the tumor margin. (A) EMT scores in the tumor core, tumor margin, and adjacent non-tumor tissue in cohort 1. (B) EMT scores in the tumor core, tumor margin, and adjacent non-tumor tissue in cohort 2. (C) EMT scores in the tumor core, tumor margin, and adjacent non-tumor tissue in cohort 3. (D) EMT scores were highest at the tumor margin, and the proportion of EMT-positive epithelial cells was higher at the tumor margin than in the tumor core and adjacent non-tumor tissue. (E) Correlation between EMT scores and EPCAM expression levels in tumor core, tumor border, and non-tumor within single-cell cohorts. (p < 0.01 (**)). |
UBC as a Key Regulator of EMT at Tumor Margins
By screening for EMT-regulating genes in tumor margin regions using eight machine learning algorithms, UBC was identified as one of the top ten key genes across all models (For each model, genes were ranked based on normalized importance scores (0–100) derived from permutational importance using DALEX. Genes consistently appearing in the top 10% of ranked features across at least five of eight algorithms were defined as “consensus regulators”). UBC expression was elevated in the tumor margin regions of hepatocellular carcinoma and showed a strong positive correlation with EMT scores (Figure 7A–D). Analysis of TCGA and GSE14520 data revealed that patients with high UBC expression had poorer outcomes, and UBC expression remained significantly positively correlated with EMT scores, suggesting that UBC may be a critical driver of EMT activity in the tumor margin regions of hepatocellular carcinoma (r_TCGA = 0.366, FDR < 0.001; r_GSE14520 = 0.357, FDR < 0.001) (Figure 7E–G).
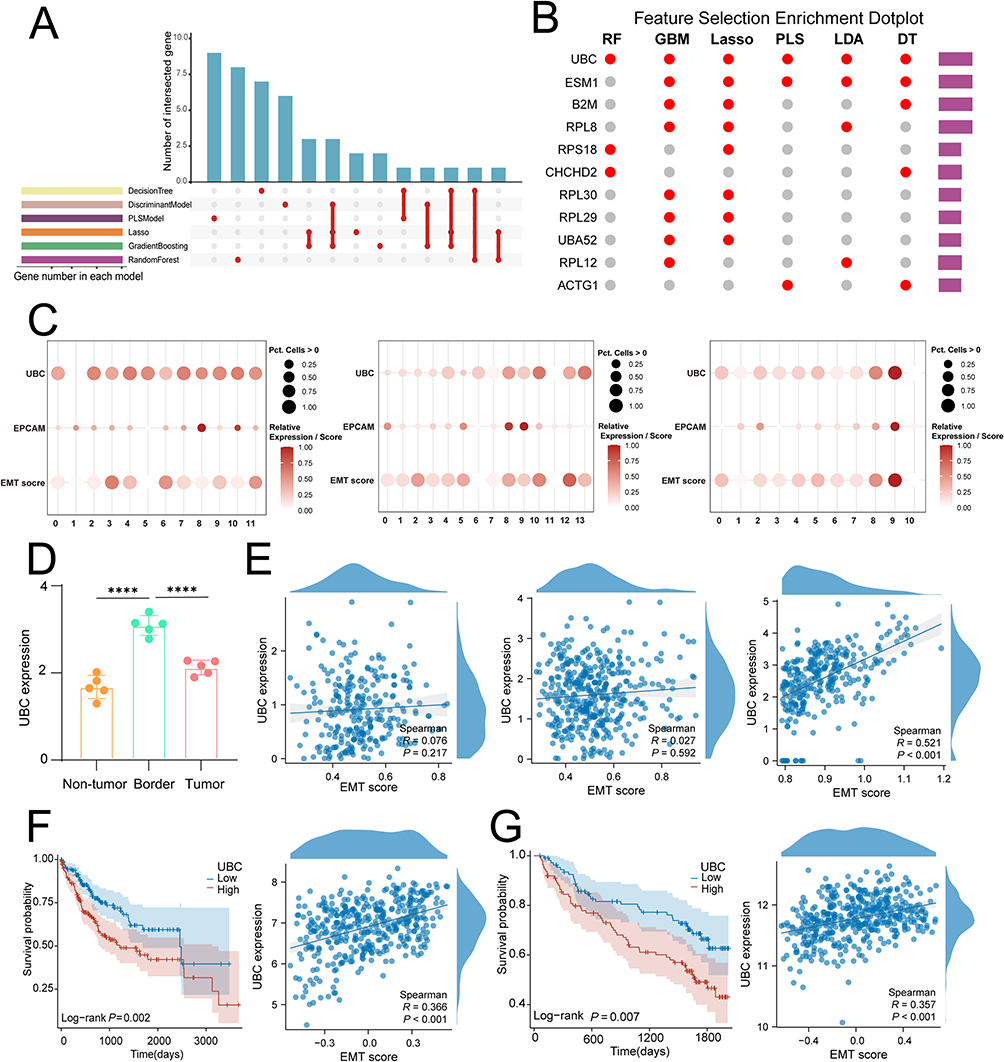 |
Figure 7 Machine learning algorithms reveal that the UBC gene is a key regulator of EMT. (A) Machine learning upset plot. (B) Bubble chart displaying genes selected by machine learning. (C) Spatial transcriptomics data indicate that UBC is highly expressed in epithelial cell clusters in the tumor margin region and shows a positive correlation with EMT scores. (D) UBC expression was highest at tumor border. (E) Spatial transcriptomics data analysis revealed that UBC expression was highest in the core of the tumor margin and showed a high positive correlation with EMT expression. (F) Validation using the TCGA hepatocellular carcinoma cohort showed that patients with high UBC expression had poorer outcomes, and EMT scores were highly positively correlated with UBC expression levels. (G) Validation using the GSE14520 dataset showed that patients with high UBC expression had poorer outcomes, and EMT scores were highly positively correlated with UBC expression levels. (p < 0.0001 (****)). |
UBC Facilitates EMT and Metastasis in vivo
To further validate the biological function of UBC in HCC, we manipulated UBC expression in HCC cell lines and examined associated EMT-related phenotypic changes. Wound healing assays showed that UBC overexpression enhanced wound closure (85% vs 93%), whereas short hairpin RNA (shRNA)-mediated UBC knockdown inhibited this process (92% vs 79%) (Figure 8A and B). Similarly, Transwell assays demonstrated that UBC overexpression promoted the metastatic capacity of HCC cells (450 cells vs 810 cells), while UBC silencing reduced it (840 cells vs 440 cells) (Figure 8C and D). At the molecular level, immunofluorescence staining confirmed UBC’s pro-EMT role: UBC upregulation led to decreased E-cadherin expression and increased N-cadherin expression (Figure 8E and F). Collectively, these in vitro findings identify UBC as a positive regulator of EMT in HCC.
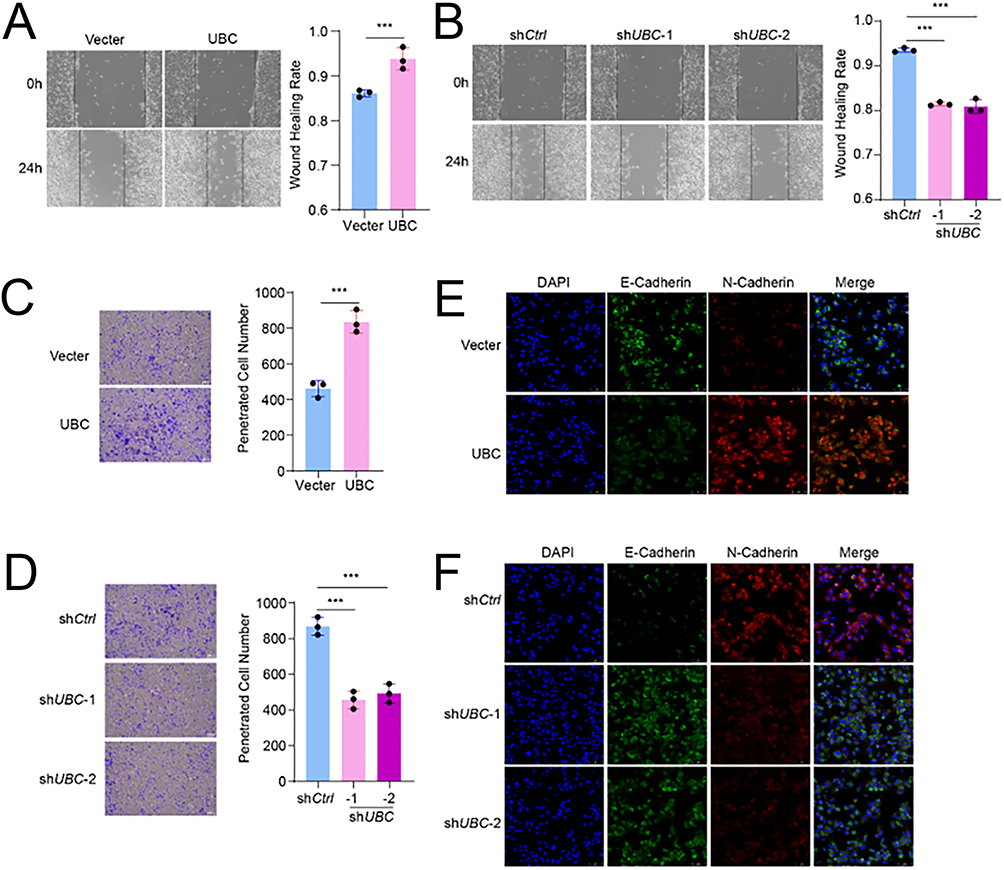 |
Figure 8 UBC facilitates HCC cells metastasis and EMT. (A) Wound healing images and wound healing rates quantification of HCC cells with over expression UBC. (B) Wound healing images and wound healing rates quantification of HCC cells with knock down UBC. (C) Transwell images and penetrated cell number quantification of HCC cells with over expression UBC. (D) Transwell images and penetrated cell number quantification of HCC cells with knock down UBC. (E) Immunofluorescence images of E-Cadherin and N-Cadherin in HCC cells with over expression UBC. (F) Immunofluorescence images of E-Cadherin and N-Cadherin in HCC cells with knock down UBC. (p < 0.001 (***)). |
Discussion
HCC is a highly prevalent and lethal malignancy worldwide, with major therapeutic challenges stemming from substantial molecular and spatial heterogeneity.19 In recent years, advances in single-cell sequencing and spatial transcriptomics have transformed the understanding of HCC from a tumor cell-centric view to a more comprehensive “tumor–microenvironment” framework.20 However, the precise role of EMT in the spatial progression of HCC and its interactions with the immune microenvironment remains incompletely characterized.21 This study integrates multi-omics approaches to uncover the spatial enrichment of EMT at the tumor margin and identifies UBC as a key regulatory gene. These findings not only corroborate existing knowledge of EMT but also expand our understanding of the spatial heterogeneity in liver cancer and its underlying mechanisms of immune evasion.
Numerous studies have established that EMT plays a central role in tumorigenesis, invasion, and metastasis.22,23 Classic EMT marker genes, such as VIM, SNAIL, and TWIST, have been associated with poor prognosis in various solid tumors. In HCC, previous studies have demonstrated that EMT activation is linked to tumor angiogenesis, metastatic potential, and drug resistance.24,25 However, most of these studies have predominantly relied on bulk RNA-seq data or in vitro experimental models, lacking precise spatial resolution of EMT activity.26 Our results reveal that EMT is significantly enriched in the peripheral regions of hepatocellular carcinoma and is closely associated with the epithelial cell state. This finding provides molecular evidence supporting the tumor periphery as an “invasion frontier”. Integrating our observations with recent spatial transcriptomics studies—which have shown that the peripheral regions of HCC are frequently enriched with immunosuppressive cell populations and angiogenesis-related signaling pathways—our conclusions suggest that EMT may serve as a central regulatory mechanism driving this spatial distribution pattern.
In recent years, EMT has been increasingly recognized as a key process associated with responses to immunotherapy.27,28 Studies have shown that EMT-activated tumor cells often exhibit impaired antigen presentation and suppress effector immune responses through the secretion of immunosuppressive factors such as TGF-β and IL-10.29–31 Our study further confirmed that patients with high EMT scores display prominent immunosuppressive features, including reduced CD8⁺ T cell infiltration, increased accumulation of MDSCs and M2-type macrophages, and elevated expression of immune checkpoint molecules such as PDCD1, CTLA4, and LAG3. These findings are consistent with the emerging concept of the “EMT–immune suppression axis”.32–34 For example, a study demonstrated that tumors with high EMT activity often exhibit an “immune-cold” phenotype and show limited responses to immune checkpoint inhibitors.35,36 Our study not only replicates this pattern in HCC but also reveals its significant spatial dependency: while EMT is activated at the tumor margin, the local microenvironment undergoes concurrent immunosuppressive remodeling. This observation provides novel insights into the mechanisms underlying immunotherapy resistance.
Single-cell trajectory analysis revealed that EMT-positive cells are predominantly localized at the tumor periphery and exist in a transitional state between epithelial and mesenchymal differentiation. This intermediate state exhibits dual epithelial and mesenchymal characteristics, marked by the activation of pathways involved in cell migration, invasion, and extracellular matrix remodeling. Recent studies have introduced the concept of “partial EMT”, wherein tumor cells remain in a partially transformed state, displaying enhanced plasticity and metastatic potential. Our findings are highly consistent with this concept and are further supported by spatial transcriptomics, which confirms a significant association between EPCAM⁺ cells and EMT activity. These results suggest that EMT-positive cells in the tumor periphery may function as a key “pioneer population” driving metastasis and dissemination in HCC.
The role of tumor microenvironment signals in inducing EMT has been extensively documented. Signaling pathways such as TGF-β, EGF, and HGF are all capable of driving EMT, with these signals predominantly originating from stromal cells, fibroblasts, or immune cells.37–40 In this study, CellChat analysis revealed that the communication between fibroblasts and epithelial cells at the tumor margin was significantly upregulated, mediated through key pathways including EGFR and TGF-β. This observation is consistent with previous studies on the involvement of cancer-associated fibroblasts in EMT regulation, indicating that EMT is not exclusively tumor cell-intrinsic but is instead shaped by tumor–stromal interactions.41 In recent years, targeting CAF–TGF-β signaling has emerged as a promising strategy in combination immunotherapy for cancer.42,43 Our findings offer novel spatial-level evidence supporting the therapeutic rationale for this approach in HCC.
This study consistently identified UBC as a key regulator of EMT in the tumor periphery using multiple machine learning algorithms. As a ubiquitin-conjugating enzyme, UBC participates in protein degradation and signal transduction processes. Previous studies have demonstrated that the ubiquitin system plays a critical role in EMT regulation, for instance, by modulating the stability of EMT transcription factors such as SNAIL and ZEB1, thereby influencing cellular phenotypes.44,45 However, the specific involvement of UBC in HCC remains largely uncharacterized.
In this study, we identified UBC as a key regulator of EMT in HCC, particularly at the tumor margin. Our results demonstrate that UBC expression correlates with higher EMT scores and poor prognosis, suggesting its critical role in EMT and tumor progression. However, while these findings provide important insights into UBC’s functional role, there is still a need for a deeper exploration of its upstream regulators and post-translational modifications (PTMs). UBC is an essential component of the ubiquitin-proteasome system, acting as a ubiquitin-conjugating enzyme (E2) in regulating protein degradation. Its expression can be influenced by various upstream signaling pathways, including NF-κB, which has been implicated in EMT and tumor progression. Additionally, miRNAs and long non-coding RNAs (lncRNAs) may regulate UBC at the post-transcriptional level, providing a layer of control that could further modulate EMT in HCC. Investigating these regulatory networks will enhance our understanding of how UBC contributes to tumor progression. Moreover, UBC is subject to a variety of post-translational modifications, including phosphorylation, acetylation, and SUMOylation, which could influence its activity and interactions with EMT-related proteins such as Snail, Slug, and Twist. These modifications may alter UBC’s ability to regulate key substrates involved in EMT and metastasis. Further studies exploring these PTMs and their impact on UBC’s function will be crucial for understanding its precise role in HCC.
We identified UBC as a key regulator of EMT in HCC, with significant clinical implications. Inhibition of UBC may block EMT signaling, potentially reducing tumor invasiveness and metastasis, particularly in advanced tumors with high EMT activity. Combining UBC inhibition with immune checkpoint inhibitors could restore immune function and improve patient prognosis. Therefore, UBC inhibitors hold promise as a novel therapeutic strategy for HCC.
It should be noted that this study has several limitations. First, the data analyzed were obtained from public databases with relatively small sample sizes, and variations in clinical backgrounds and experimental conditions across different cohorts may impact the generalizability of the findings. Despite efforts to minimize batch effects, the heterogeneity of the datasets—particularly in terms of sample types, sequencing depths, and data preprocessing methods—remains a critical challenge. This variability may introduce technical biases that could influence the robustness and reproducibility of our conclusions. Therefore, further validation in larger, more homogeneous cohorts is needed to confirm the findings and better assess their applicability in clinical settings.
We acknowledge that the ubiquitination system is essential for maintaining normal cellular homeostasis, and the broad involvement of UBC raises concerns about potential off-target effects. To address this, we examined publicly available transcriptomic datasets (GSE14520, TCGA) and found that UBC is ubiquitously expressed at low to moderate levels in normal hepatocytes, consistent with its housekeeping role. However, our data indicate that UBC expression is significantly elevated in tumor regions compared with adjacent normal tissues, suggesting a cancer-specific upregulation. Considering the indispensable role of UBC in normal cells, direct inhibition might lead to toxicity; thus, future therapeutic strategies should focus on selectively disrupting UBC-mediated interactions or downstream EMT-related signaling rather than global suppression of UBC expression.
Second, although we performed partial cellular experiments to support the computational predictions, comprehensive validation is still lacking—particularly regarding the direct functional role of UBC in regulating EMT. The molecular mechanisms underlying UBC’s regulation of EMT in HCC require further confirmation through in vitro and in vivo studies. Additionally, while we identified CAF-TGF-β signaling as a potential driver of EMT in the tumor microenvironment, the lack of experimental validation of this pathway is an important limitation. Future experiments should specifically address the role of CAF-TGF-β signaling in regulating EMT, either through genetic silencing or pathway inhibitors to confirm its contribution to the observed EMT processes.
Finally, the resolution of current spatial transcriptomics technologies remains limited, making it challenging to fully distinguish signals originating from adjacent cell populations. Although we used existing spatial transcriptomic data, the low resolution of the available technology restricted our ability to resolve fine-grained cellular interactions. Future research should incorporate higher-resolution spatial multi-omics approaches, such as single-cell spatial transcriptomics or spatial proteomics, to better dissect the cellular and molecular heterogeneity at the tumor margin and to address the spatial complexity of EMT.
Conclusion
This study systematically characterized the spatial enrichment patterns of EMT in the peripheral regions of HCC, revealing its association with the immunosuppressive tumor microenvironment. For the first time, we suggest that UBC may act as a key regulator of EMT. These findings provide new insights into spatial heterogeneity and immune evasion mechanisms in HCC and lay the groundwork for potential therapeutic strategies targeting the UBC–EMT axis. As spatial omics and computational methods continue to evolve, these discoveries hold promise for improving the precision of therapeutic interventions in HCC. Future research should focus on validating UBC as a therapeutic target and explore its role in combination therapies, particularly in overcoming immune suppression and metastasis.
Approval of the Research Protocol by an Institutional Reviewer Board
All transcriptomic and clinical data were obtained from publicly available repositories (TCGA, GEO, HRA). No human or animal subjects were directly enrolled, and thus no additional ethical approval was required. Cell line-based experiments did not involve human or animal subjects and thus did not require ethics approval. The study was approved by the Medical Ethics Committee of Yichang Central People’s Hospital (approval number 2024-08-01).
Abbreviations
HCC, Hepatocellular carcinoma; EMT, Epithelial–mesenchymal transition; UBC, Ubiquitin C; TAM, Tumor-associated macrophage; CAF, Cancer-associated fibroblast; TCGA, The Cancer Genome Atlas; GEO, Gene Expression Omnibus; GSVA, Gene Set Variation Analysis; UMAP, Uniform manifold approximation and projection; SsGSEA, Single-sample Gene Set Enrichment Analysis.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68(3):526–549. doi:10.1016/j.jhep.2017.09.016
2. Singal AG, Kanwal F, Llovet JM. Global trends in hepatocellular carcinoma epidemiology: implications for screening, prevention and therapy. Nat Rev Clin Oncol. 2023;20(12):864–884. doi:10.1038/s41571-023-00825-3
3. Pinheiro PS, Jones PD, Medina H, et al. Incidence of etiology-specific hepatocellular carcinoma: diverging trends and significant heterogeneity by race and ethnicity. Clin Gastroenterol Hepatol. 2024;22(3):562–571.e8. doi:10.1016/j.cgh.2023.08.016
4. Toh MR, Wong EYT, Wong SH, et al. Global epidemiology and genetics of hepatocellular carcinoma. Gastroenterology. 2023;164(5):766–782. doi:10.1053/j.gastro.2023.01.033
5. Zheng B, Zhu YJ, Wang HY, Chen L. Gender disparity in hepatocellular carcinoma (HCC): multiple underlying mechanisms. Sci China Life Sci. 2017;60(6):575–584. doi:10.1007/s11427-016-9043-9
6. Chan YT, Zhang C, Wu J, et al. Biomarkers for diagnosis and therapeutic options in hepatocellular carcinoma. Mol Cancer. 2024;23(1):189. doi:10.1186/s12943-024-02101-z
7. Dong LQ, Peng LH, Ma LJ, et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J Hepatol. 2020;72(5):896–908. doi:10.1016/j.jhep.2019.12.014
8. Yang F, Hilakivi-Clarke L, Shaha A, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78(5):1602–1624. doi:10.1097/hep.0000000000000005
9. Liu X, Zhang K, Kaya NA, et al. Tumor phylogeography reveals block-shaped spatial heterogeneity and the mode of evolution in hepatocellular carcinoma. Nat Commun. 2024;15(1):3169. doi:10.1038/s41467-024-47541-9
10. Song Y, Boerner T, Drill E, et al. A novel approach to quantify heterogeneity of intrahepatic cholangiocarcinoma: the hidden-genome classifier. Clin Cancer Res. 2024;30(16):3499–3511. doi:10.1158/1078-0432.Ccr-24-0657
11. Wang YF, Yuan SX, Jiang H, et al. Spatial maps of hepatocellular carcinoma transcriptomes reveal spatial expression patterns in tumor immune microenvironment. Theranostics. 2022;12(9):4163–4180. doi:10.7150/thno.71873
12. Kuratani A, Okamoto M, Kishida K, et al. Platelet factor 4-induced T(H)1-T(reg) polarization suppresses antitumor immunity. Science. 2024;386(6724):eadn8608. doi:10.1126/science.adn8608
13. Wang K, Jiang X, Jiang Y, et al. EZH2-H3K27me3-mediated silencing of mir-139-5p inhibits cellular senescence in hepatocellular carcinoma by activating TOP2A. J Exp Clin Cancer Res. 2023;42(1):320. doi:10.1186/s13046-023-02855-2
14. Wang W, Wei C. Advances in the early diagnosis of hepatocellular carcinoma. Genes Dis. 2020;7(3):308–319. doi:10.1016/j.gendis.2020.01.014
15. Lee PC, Wu CJ, Hung YW, et al. Distinct gut microbiota but common metabolomic signatures between viral and MASLD HCC contribute to outcomes of combination immunotherapy. Hepatology. 2025. doi:10.1097/hep.0000000000001446
16. Roessler S, Jia HL, Budhu A, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70(24):10202–10212. doi:10.1158/0008-5472.Can-10-2607
17. Ma L, Heinrich S, Wang L, et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat Commun. 2022;13(1):7533. doi:10.1038/s41467-022-35291-5
18. Wu R, Guo W, Qiu X, et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci Adv. 2021;7(51):eabg3750. doi:10.1126/sciadv.abg3750
19. Wu Y, Liu Z, Xu X. Molecular subtyping of hepatocellular carcinoma: a step toward precision medicine. Cancer Commun. 2020;40(12):681–693. doi:10.1002/cac2.12115
20. Sun Y, Wu L, Zhong Y, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184(2):404–421.e16. doi:10.1016/j.cell.2020.11.041
21. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65(4):798–808. doi:10.1016/j.jhep.2016.05.007
22. Dong Y, Zheng Q, Wang Z, et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J Hematol Oncol. 2019;12(1):112. doi:10.1186/s13045-019-0795-5
23. Jia L, Li J, Li P, et al. Site-specific glycoproteomic analysis revealing increased core-fucosylation on FOLR1 enhances folate uptake capacity of HCC cells to promote EMT. Theranostics. 2021;11(14):6905–6921. doi:10.7150/thno.56882
24. Jia G, He P, Dai T, et al. Spatial immune scoring system predicts hepatocellular carcinoma recurrence. Nature. 2025;640(8060):1031–1041. doi:10.1038/s41586-025-08668-x
25. Zhou ZJ, Dai Z, Zhou SL, et al. HNRNPAB induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by transcriptionally activating SNAIL. Cancer Res. 2014;74(10):2750–2762. doi:10.1158/0008-5472.Can-13-2509
26. Yuan J, Lv T, Yang J, et al. The lipid transporter HDLBP promotes hepatocellular carcinoma metastasis through BRAF-dependent epithelial-mesenchymal transition. Cancer Lett. 2022;549:215921. doi:10.1016/j.canlet.2022.215921
27. Huang Y, Hong W, Wei X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J Hematol Oncol. 2022;15(1):129. doi:10.1186/s13045-022-01347-8
28. Zhang F, Guo J, Zhang Z, et al. Mesenchymal stem cell-derived exosome: a tumor regulator and carrier for targeted tumor therapy. Cancer Lett. 2022;526:29–40. doi:10.1016/j.canlet.2021.11.015
29. Bourdon M, Santulli P, Jeljeli M, et al. Immunological changes associated with adenomyosis: a systematic review. Hum Reprod Update. 2021;27(1):108–129. doi:10.1093/humupd/dmaa038
30. Huang W, Solouki S, Koylass N, Zheng SG, August A. ITK signalling via the Ras/IRF4 pathway regulates the development and function of Tr1 cells. Nat Commun. 2017;8(1):15871. doi:10.1038/ncomms15871
31. Maimon A, Levi-Yahid V, Ben-Meir K, et al. Myeloid cell-derived PROS1 inhibits tumor metastasis by regulating inflammatory and immune responses via IL-10. J Clin Invest. 2021;131(10). doi:10.1172/jci126089
32. Bai F, Zhang LH, Liu X, et al. GATA3 functions downstream of BRCA1 to suppress EMT in breast cancer. Theranostics. 2021;11(17):8218–8233. doi:10.7150/thno.59280
33. Fu Z, Huang Z, Xu H, et al. IL-2-inducible T cell kinase deficiency sustains chimeric antigen receptor T cell therapy against tumor cells. J Clin Invest. 2024;135(4). doi:10.1172/jci178558
34. Song G, Yu X, Shi H, Sun B, Amateau S. miRNAs in HCC, pathogenesis, and targets. Hepatology. 2024. doi:10.1097/hep.0000000000001177
35. Cheng Y, Mo F, Li Q, et al. Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin. Mol Cancer. 2021;20(1):62. doi:10.1186/s12943-021-01355-1
36. Taki M, Abiko K, Ukita M, et al. Tumor immune microenvironment during epithelial-mesenchymal transition. Clin Cancer Res. 2021;27(17):4669–4679. doi:10.1158/1078-0432.Ccr-20-4459
37. Clapéron A, Mergey M, Nguyen Ho-Bouldoires TH, et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J Hepatol. 2014;61(2):325–332. doi:10.1016/j.jhep.2014.03.033
38. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21(1):104. doi:10.1186/s12943-022-01569-x
39. Wang X, Eichhorn PJA, Thiery JP. TGF-β, EMT, and resistance to anti-cancer treatment. Semin Cancer Biol. 2023;97:1–11. doi:10.1016/j.semcancer.2023.10.004
40. Zhao Y, Kaushik N, Kang JH, et al. A feedback loop comprising EGF/TGFα sustains TFCP2-mediated breast cancer progression. Cancer Res. 2020;80(11):2217–2229. doi:10.1158/0008-5472.Can-19-2908
41. Zheng Y, Jiang F, Wang C, et al. Regulation of Semaphorin3A in the process of cutaneous wound healing. Cell Death Differ. 2022;29(10):1941–1954. doi:10.1038/s41418-022-00981-6
42. Ebrahimi N, Manavi MS, Faghihkhorasani F, et al. Harnessing function of EMT in cancer drug resistance: a metastasis regulator determines chemotherapy response. Cancer Metastasis Rev. 2024;43(1):457–479. doi:10.1007/s10555-023-10162-7
43. Sheng W, Shi X, Lin Y, et al. Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin Cancer Res. 2020;39(1):16. doi:10.1186/s13046-020-1521-4
44. Wei CY, Zhu MX, Yang YW, et al. Downregulation of RNF128 activates Wnt/β-catenin signaling to induce cellular EMT and stemness via CD44 and CTTN ubiquitination in melanoma. J Hematol Oncol. 2019;12(1):21. doi:10.1186/s13045-019-0711-z
45. Yuan H, Li Q, Li L, et al. DDX39B K63-linked ubiquitination mediated by TRIM28 promotes NSCLC metastasis by enhancing ECAD lysosomal degradation. Signal Transduct Target Ther. 2025;10(1):221. doi:10.1038/s41392-025-02305-9
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.