Back to Journals » OncoTargets and Therapy » Volume 13
Integrative Genomic Analysis Reveals Cancer-Associated Gene Mutations in Chronic Myeloid Leukemia Patients with Resistance or Intolerance to Tyrosine Kinase Inhibitor
Authors Wu W ![]() , Xu N, Zhou X, Liu L, Tan Y
, Xu N, Zhou X, Liu L, Tan Y ![]() , Luo J, Huang J, Qin J
, Luo J, Huang J, Qin J ![]() , Wang J, Li Z, Yin C, Zhou L, Liu X
, Wang J, Li Z, Yin C, Zhou L, Liu X
Received 10 April 2020
Accepted for publication 30 July 2020
Published 25 August 2020 Volume 2020:13 Pages 8581—8591
DOI https://doi.org/10.2147/OTT.S257661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr XuYu Yang
Waner Wu,1,* Na Xu,1,* Xuan Zhou,1 Liang Liu,1 Yaxian Tan,1 Jie Luo,1 Jixian Huang,2 Jiayue Qin,3 Juan Wang,3 Zhimin Li,3 Changxin Yin,1 Lingling Zhou,1 Xiaoli Liu1
1Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, Guangdong, People’s Republic of China; 2Department of Hematology, Yuebei People’s Hospital, Shantou University, Shaoguan 512025, Guangdong, People’s Republic of China; 3Yiwu Cancer Research Center, Fudan University Shanghai Cancer Center, Yiwu, Zhejiang 322000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoli Liu Email [email protected]
Introduction: While the acquisition of mutations in the ABL1 kinase domain (KD) has been identified as a common mechanism behind tyrosine kinase inhibitor (TKI) resistance, recent genetic studies have revealed that patients with TKI resistance or intolerance frequently harbor one or more genetic alterations implicated in myeloid malignancies. This suggests that additional mutations other than ABL1 KD mutations might contribute to disease progression.
Methods: We performed targeted-capture sequencing of 127 known and putative cancer-related genes of 63 patients with CML using next-generation sequencing (NGS), including 42 patients with TKI resistance and 21 with TKI intolerance.
Results: The differences in the number of mutations between groups had no statistical significance. This could be explained in part by not all of the patients having achieved major molecular remission in the early period as expected. Overall, 66 mutations were identified in 96.8% of the patients, most frequently in the KTM2C (31.82%), ABL1 (31.82%), FAT1 (25.76%), and ASXL1 (22.73%) genes. CUX1, KIT, and GATA2 were associated with TKI intolerance, and two of them (CUX1, GATA2) are transcription factors in which mutations were identified in 82.61% of patients with TKI intolerance. ASXL1 mutations were found more frequently in patients with ABL1 KD mutations (38.1% vs 15.21%, P=0.041). Although the number of mutations was low, pairwise interaction between mutated genes showed that ABL1 KD mutations cooccurred with SH2B3 mutations (P< 0.05). In Kaplan–Meier analyses, only TET2 mutations were associated with shorter progression-free survival (P=0.026).
Conclusion: Our data suggested that the CUX1, KIT, and GATA2 genes may play important roles in TKI intolerance. ASXL1 and TET2 mutations may be associated with poor patient prognosis. NGS helps improving the clinical risk stratification, which enables the identification of patients with TKI resistance or intolerance in the era of TKI therapy.
Keywords: chronic myeloid leukemia, mutations, tyrosine kinase inhibitor, intolerance, resistance
Introduction
The cytogenetic hallmark of chronic myeloid leukemia (CML) is the chromosomal translocation t(9;22)(q34;q11), yielding the Philadelphia (Ph) chromosome and generating a fusion gene that encodes BCR-ABL1, a constitutively active protein tyrosine kinase. Tyrosine kinase inhibitors (TKIs) targeting BCR-ABL1 have substantially improved the prognosis of patients with CML, from prolonging survival time to achieving treatment-free remission. However, some patients still exhibit resistance or intolerance to TKIs, leading to treatment failure. Although the acquisition of a point mutation in the BCR-ABL1 kinase domain (KD) has been noted as an important mechanism of drug resistance,1 recent studies2–4 have reported patients with poor prognosis bearing one or more somatic mutations associated with myeloid malignancies, which means that additional mutations may contribute to disease progression. In the last decade, a variety of somatic mutations besides those in BCR-ABL1 have been found in myeloid malignancy, which have different frequencies and combinations and often overlap in different diseases.5 In studies on transcription factors and signal-transduction genes, mutations were found in genes conferring a growth advantage, which caused cancers.6,7 Over time, other pathways such as epigenetic modification, abnormalities in RNA splicing, and adhesion protein complexes have also been found to be associated with myeloid leukemia.8–12 The purpose of this study was to identify the difference of additional CML-related mutations between patients resistant or intolerant to TKIs, and to understand the possible effects of these mutations on disease progression.
Materials and Methods
Study Design and Subjects
A total of 63 CML patients admitted to Nanfang Hospital from August 2018 to October 2019 who were resistant or intolerant to TKIs were analyzed retrospectively. All cases had clear evidence regarding bone marrow cytology and genetics and met the diagnostic criteria of CML in the hematopoietic and lymphoid tissue tumor classification revised by the WHO in 2016. The group consisted of 42 patients with TKI resistance and 21 with TKI intolerance. The clinical data of the patients are displayed in Table 1 (details in Supplementary file 1). There was no significant correlation between the age of all CML patients and the number of detected gene mutations (Spearman’s rho correlation coefficient 0.047, P = 0.703).
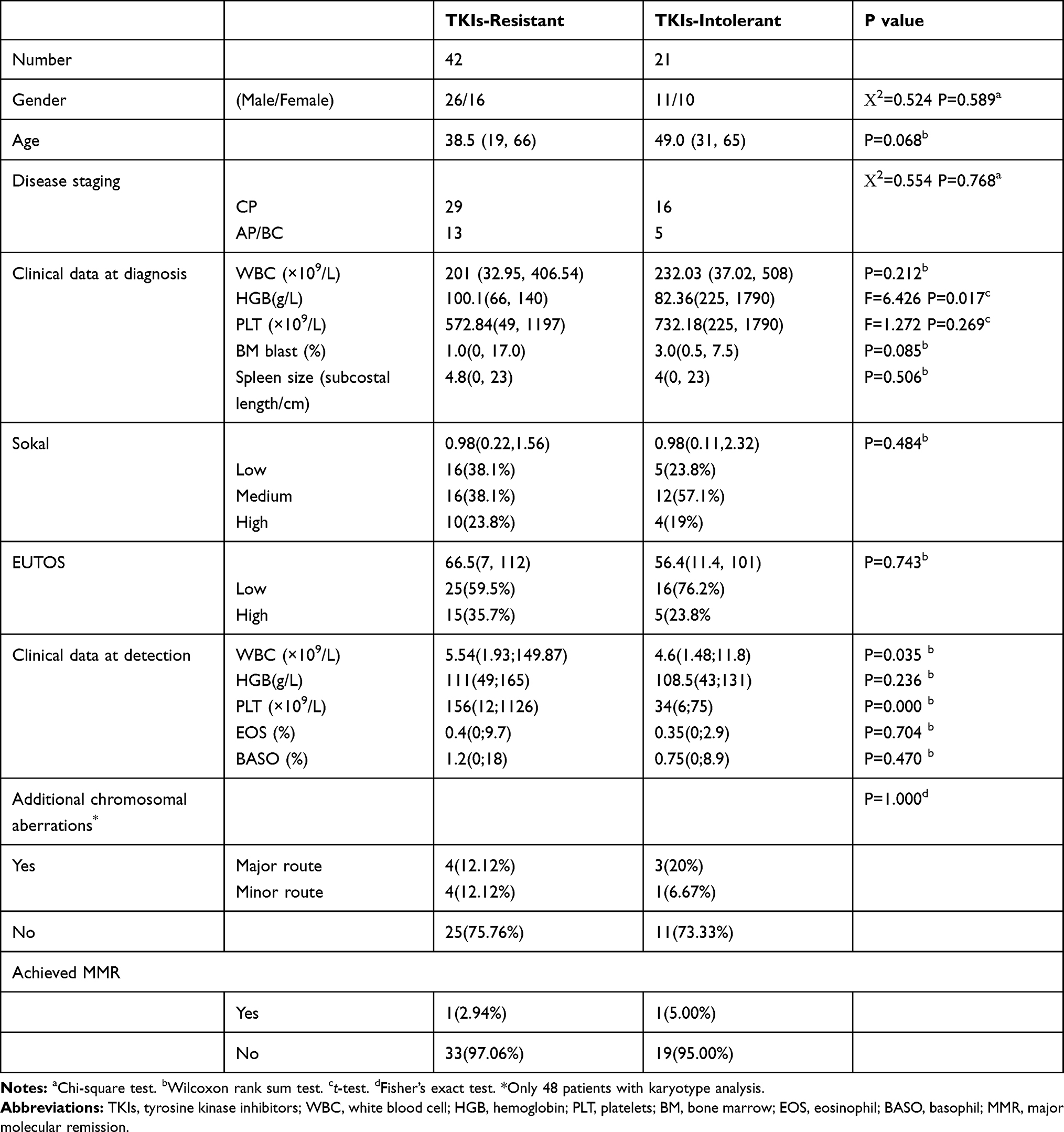 |
Table 1 Clinical Data of the Patients |
Inclusion Criteria
TKI resistance13 was defined as the lack of a complete hematologic response (CHR) after 3 months of TKI treatment, the lack of any cytogenetic response after 6 months of treatment, the lack of minor cytogenetic response (MCyR) (Ph-positive cells >35%) after 12 months of treatment, an increase of white blood cell (WBC) count in at least two consecutive samplings (with a doubling of the count from the nadir to ≥20×109/L or an absolute increase of ≥50×109/L), or a relapse after a CHR or MCyR. TKI intolerance13,14 was defined as at least grade 3 nonhematologic toxicity or grade 4 hematologic toxicity persisting for more than 7 days, related to TKIs at any dose.
Next-Generation Sequencing
All the patients in our group had recent q-PCR quantitative results of BCR-ABL1 fusion gene before the second-generation sequencing, and the enrollment standard was BCR-ABL1 fusion gene (IS) ≥10%, which make sure the proportion of Ph-positive cells will not be too low.
Using next-generation sequencing (NGS), we analyzed 127 genes (Table 2). After collecting peripheral blood of the patients, we extracted gDNA from the qualified samples using an automatic nucleic acid extractor for quality inspection. After passing the quality inspection, an Illumina standard library (Illumina, Inc.), was constructed to create a library suitable for high-throughput sequencing. The library peak map was detected by Agilent2100 (Agilent, Inc.), and the main peak size of the library was required to be about 350 bp. After qualification, the Roche NimbleGen hybridization capture chip (Roche, Inc.) was used to capture the target sequence of 127 genes related or putatively related to hematological disease. After the captured exon library passed the library quality inspection and achieved accurate qPCR quantification, PE75 sequencing was carried out on Illumina NextSeq 550AR (Illumina, Inc.). Bioinformatic analysis was carried out on the off-machine data qualified for quality control, including point mutations, insertions and deletions, internal tandem duplication, partial tandem duplication, and other types of variation. The Burrows‑Wheeler Alignment algorithm version 0.7.1215 was used to compare the sequence data with the human genome (version: GRCh37), and Picard version 1.115 (https://github.com/broadinstitute/picard) was used to mark the polymerase chain reaction duplicates. We corrected the quality value of the sequence alignment results by means of BaseRecalibrator in Genome Analysis Toolkit version 3.5.16 The MuTect2 version 3.5 software16 was employed for mutation detection, and all test results were annotated in the Annovar version 0722 software.17
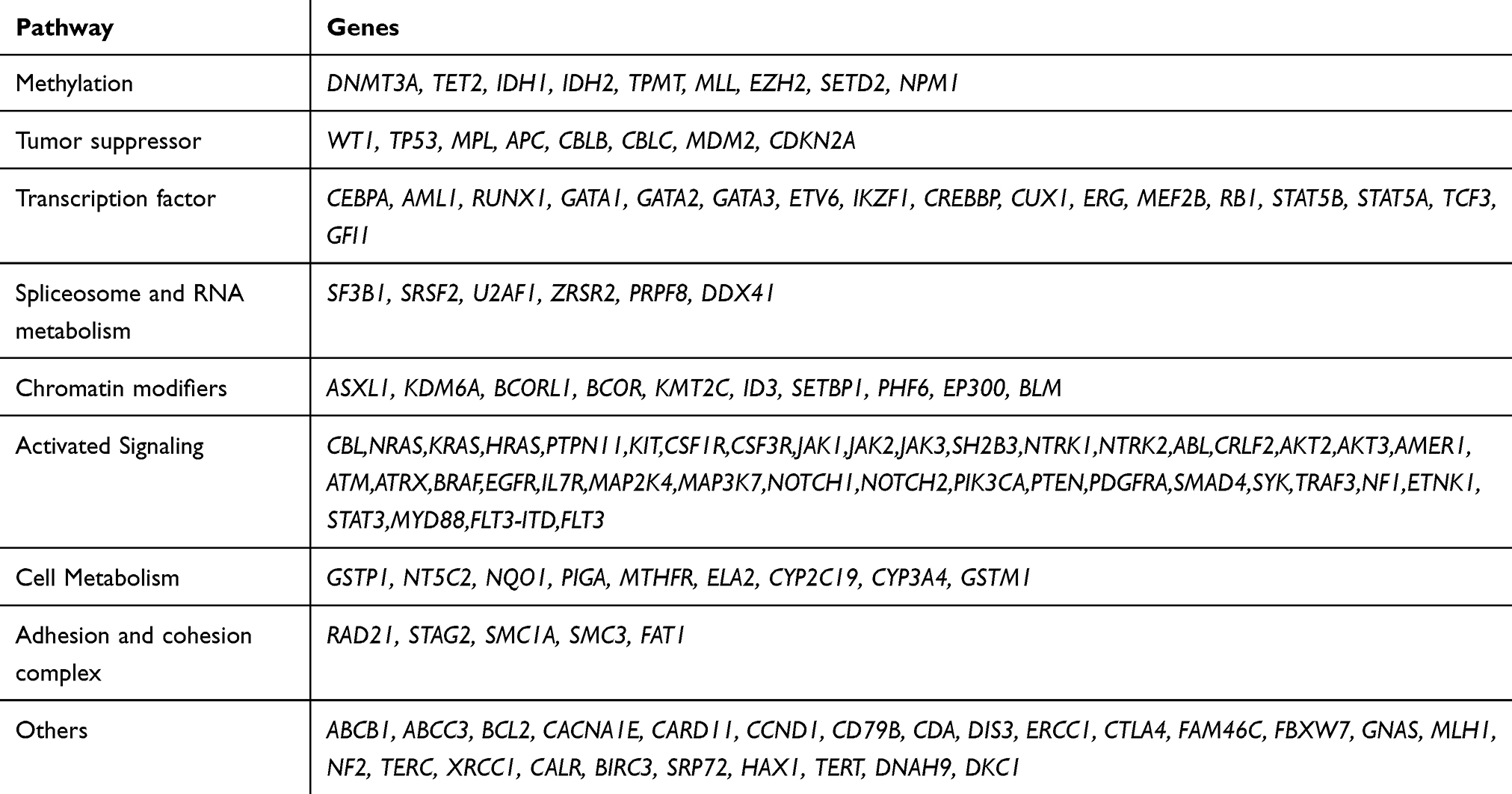 |
Table 2 Detected Genes |
Statistical Analysis
The Kaplan-Meier survival curve was analyzed using a Log rank test to compare progression-free survival between the TET2-positive and TET2-negative groups (GraphPad Prism; GraphPad Software, Inc.). For all other analyses, significant differences between two groups were examined using two‑sample Student’s t‑tests, Chi-square test or Fisher’s exact test depending on types of data. The odds ratio was calculated as the ratio of mutation frequency between two different groups. P<0.05 was considered to indicate a statistically significant difference.
Result
The results of the analysis of the 127 genes of CML patients with intolerance or resistance to TKIs confirmed that there were 66 individual somatic mutations in 63 patients. The number of mutations per patient ranged from 0 to 13, with a median of 3. There was no significant correlation between the number of mutations and the Sokal score or EUTOS score of the patients (Spearman’s rho correlation coefficient between Sokal and the number of mutations 0.174, P=0.171; Spearman’s rho correlation coefficient between EUTOS and the number of mutations 0.149, P=0.245).
Summary of Mutations in 127 Genes Detected
Among the total mutation rates, ABL1 KD showed the highest value (31%), followed by KMT2C, FAT1, and ASXL1 (Figure 1A). The coexistence relationship among genes is complicated (Figure 1B). In addition, we identified de novo mutations in CML patients with TKI intolerance, including in KIT1, CUX1, and GATA2 (Figure 1C, details in Supplementary file 2).
 |
Figure 1 Summary of mutations (A) Most of the 68 individual somatic mutations were missense mutations, while multiple hit mutations accounted for 38% of KMT2C mutations, and frameshift mutations also accounted for 46.67% of ASXL1. Among the total mutation rates, the highest was ABL1 KD (31%), followed by that of KMT2C, FAT1 and ASXL1, which were 31%, 25% and 22%, respectively. (B) The coexistence relationship among genes is complicated. (C) The TKIs-resistant group accounted for the majority of the mutations in most genes, while the number of mutations in the TKIs-intolerant group in the CUX1, GATA2 and KIT genes was higher than those in the TKIs resistant group. Abbreviations: KD, kinase domain; TKIs, tyrosine kinase inhibitors. |
There was no significant difference in the number of mutations between the TKI-resistant group and the TKI-intolerant group (3.10 vs. 3.79, P=0.123). There was also no significant difference in the ABL1 KD mutation rate between the TKI-resistant group and the TKI-intolerant group (34.09% vs. 26.09%, P=0.351).
Mutational Heterogeneity Between TKI-Resistant and TKI-Intolerant Groups
Myelodysplastic Syndrome (MDS)-Related Mutations Found in TKI-Intolerant Group
We analyzed 20 driving genes related to MDS in 63 patients and found that CUX1, GATA2, and KIT had higher mutation rates in patients with TKI intolerance, while AMER1 mutations were only found in drug-resistant patients (Table 3). Although the mutation rates of ASXL1 and RUNX1 did not show any difference between the two groups, the mutation rates of these two genes remained high in the two groups, indicating that their existence may lead to a poor response to TKIs.
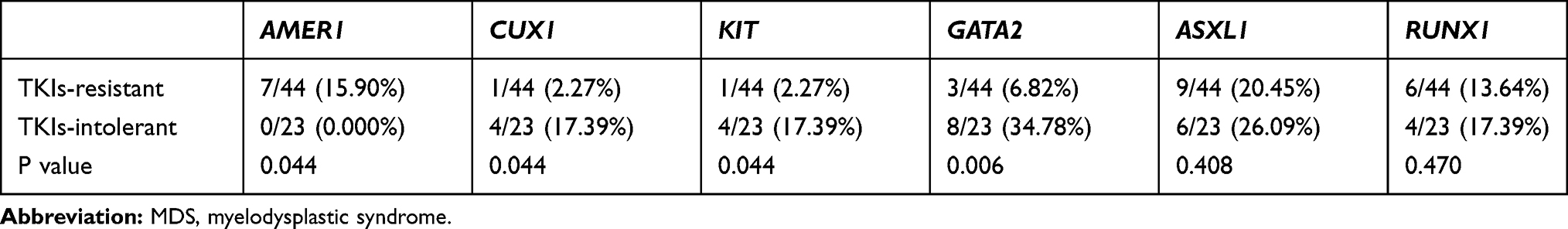 |
Table 3 Mutations Rates of Genes Related to MDS |
The distribution of mutations in these 20 genes was also different in the two groups (Figure 2). The median number of mutations of these 20 genes in the TKI-intolerant group was higher than that in the TKI-resistant group (2.0 vs. 1.0, P=0.029). This suggests that MDS-related genes may play an important role in TKI intolerance.
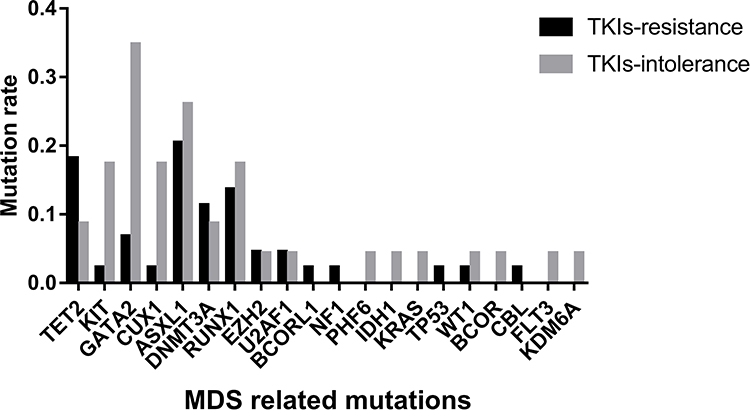 |
Figure 2 Mutation rates of MDS related genes. Mutation rates of 20 genes related to MDS; Mutation rates of KIT, GATA2 and CUX1 in patients with TKIs intolerance were significantly higher than in TKIs-resistant group. Abbreviation: MDS, myelodysplastic syndrome. |
Heterogeneity of Mutant Genes’ Function in TKI-Resistant or -Intolerant Patients
The number of mutations in genes encoding transcription factors was significantly higher in patients with TKI intolerance than that in patients with TKI resistance (0.83 vs. 0.27, P=0.014). GATA2 and CUX1, both of which encode transcription factors, have higher mutation rates in patients intolerant to TKIs (Figure 3).
 |
Figure 3 Mutation rates of genes with different functions. According to the function of genes, they were divided into chromatin modification (86.57%, 58/67), activated signaling (58.70%, 40/67), transcription factor (47.76%, 32/67), methylation (44.78%, 30/67), other (34.33%, 23/67), adhesion and cohesion complex (26.87%, 18/67), cell metabolism (16.42%, 11/67), tumor suppressor (8.96%, 6/67), spliceosome and RNA metabolism (5.97%, 4/67). It can be seen that the mutations rate in transcription factor is higher in patients intolerant to TKIs (82.60% vs 27.27%, P=0.016). |
Effect of Additional Gene Mutations on CML Progression
We also analyzed the effect of additional gene mutations on the progression of CML. In this paper, the term “additional gene mutations” is used to indicate the existence of somatic mutations besides those in ABL1 KD in patients with CML.
Coexistence of Additional Gene Mutations and ABL1 KD Mutations
The patients in this study were divided into two groups: those positive and negative for ABL1 KD mutation. Although the mutation rates of AXSL1, FAT1, and RUNX1 in patients with ABL1 KD mutation were higher than in those without it, this was statistically significant only for the ASXL1 gene mutation rate (Table 4). Moreover, the mutation rate of KIT in patients without ABL1 KD mutation was higher, but the difference did not reach statistical significance, which may have been due to the insufficient number of patients in this study.
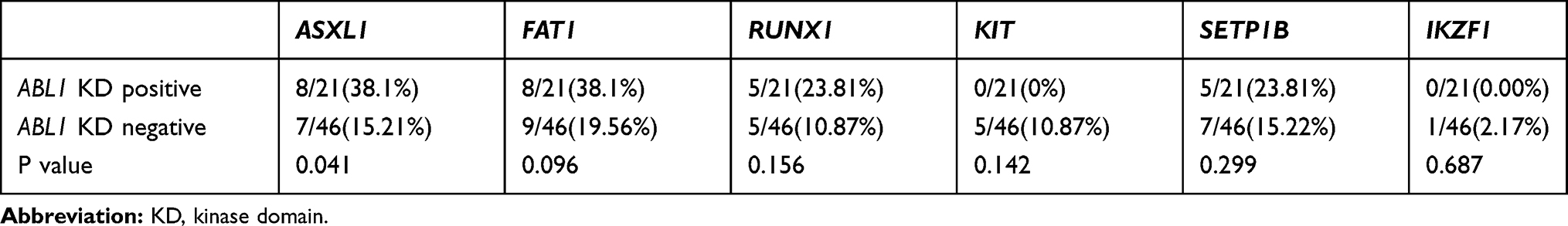 |
Table 4 Statistics on Mutation Rates of Additional Genes Associated with ABL KD |
The clinical features of patients with ASXL1 mutation also differed from those in patients without such mutation. Patients with ASXL1 mutation had a higher proportion of basophils in peripheral blood and a larger spleen at initial diagnosis (Table 5). Although Sokal score and EUTOS score did not show a difference between the two groups, ASXL1 mutation may increase the risk of disease progression. Analysis of the other mutated genes and clinical features at initial diagnosis was presented in Table S1. In the analysis of the existence of other mutations in combination with ABL1 KD mutation, the results showed that SH2B3 gene mutation significantly coexisted with it (Figure S1).
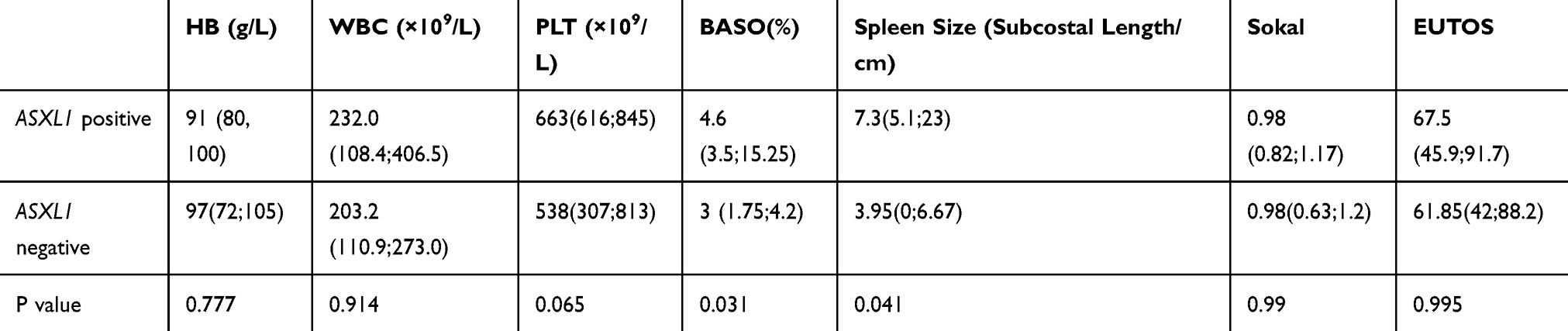 |
Table 5 Relationship Between ASXL1 Mutation and Clinical Features at Initial Diagnosis |
Additional Gene Mutations May Play an Important Role in CML Progression
The 63 patients were divided into a chronic phase (CP) group and an accelerated phase (AP)/blast crisis (BC) group. There was no difference in the number of gene mutations between the two groups (3.0 vs. 3.0). This may have been due to the fact that the patients in the CP group did not achieve early molecular remission or the optimal therapeutic response, in which additional gene mutations may play an important role. However, the rate of ABL1 KD mutations in the AP/BC group was higher than that in the CP group (38.89% vs. 28.57%, P=0.301), and similar trends were found for TET2 (38.89% vs. 6.12%, P=0.003) and FAT1 (33.33% vs. 22.45%, P=0.272). Similarly, patients with mutations in TET2 showed a poorer prognosis and had a shorter PFS than those without them (P=0.026) (Figure 4). Analysis of PFS for the other mutated genes was presented in Table S2. It was shown that patients with TET2 mutation had higher ETOUS score than those without, while there was no statistical difference in Sokal score between the two groups, which may because the number of patients with TET2 mutation was not enough (Table S3).
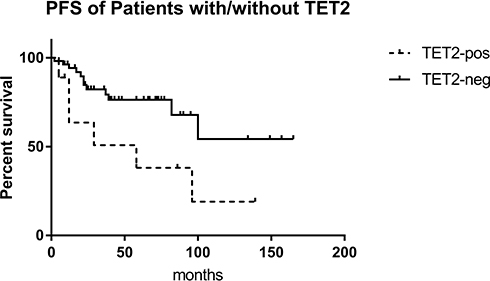 |
Figure 4 EFS of patients with/without TET2 mutation. In Kaplan-Meier analysis, the EFS of patients with TET2 mutation was significantly lower than those without (P=0.039). |
We defined disease-related events as ABL1 KD mutations or development to accelerated phase or blast crisis in CML patients. There were 35 patients (35/63) with disease-related events and 28 (28/63) in a relatively stable condition. It was found that patients with disease-related events carried more mutations than those in a relatively stable condition (3.5 vs. 2.0, P=0.024).
Discussion
TKI drug resistance and intolerance have remained major clinical challenges in the TKIs era. Although ponatinib, a third-generation TKI, improves outcomes for patients with BCR-ABL1-dependent mechanisms of resistance, including T315I mutation, a proportion of patients may have or develop BCR-ABL1-independent resistance and suffer from ponatinib treatment failure.18 The most frequent mechanism of BCR-ABL1-dependent resistance is point mutation in the ABL1 KD, which directly or indirectly impairs appropriate TKI binding to the kinase pocket either by altering BCR-ABL1 conformation with reduced affinity to the specific inhibitor or by interfering with a TKI-binding site.18 The causes of BCR-ABL1-independent resistance are complex, including an insufficient concentration of TKIs caused by their pharmacokinetics,19,20 activation of alternative signal pathways,21 and persistence of leukemia stem cells independent of BCR-ABL1 activity.22,23
In recent years, research on CML has greatly benefited from the development of NGS technology, which enables the profiling of patients with poor TKI response at the genetic level, so that we can better understand the potential mechanisms of drug resistance.
Relationship Between ABL1 KD Mutation and Additional Mutations
At the 2019 European Hematology Association, Branford et al2 mentioned that frequently mutated genes of CML patients with a poor prognosis at diagnosis were ASXL1, IKZF1, and RUNX1 and that the methyltransferase SETD1B was a novel repeatedly mutated gene. They also found that no patient with MLL fusion had an ABL1 KD mutation. Conversely, ABL1 KD mutations were frequently observed in patients with IKZF1 variants. In our research, ABL1 KD mutations were often observed in patients with AXSL1 variants.
However, in the analysis of gene interaction with ABL1 KD mutation, SH2B3 gene mutation was found to significantly coexist with it. It has been proven that LNK, a lymphocyte adapter protein encoded by the SH2B3 gene, restricts the expansion of hematopoietic stem cells and progenitor cells by binding to JAK2, which inhibits the activity of JAK2 kinase. In mice, LNK dysfunction may promote the development of hematological malignant tumors, and LNK deficiency can cooperate with BCR-ABL1 oncogenes in the development of CML.24
MDS-Related Gene Mutations are Common in Patients with TKI Intolerance
Schmidt et al25 found that 43% of CML patients with Ph-negative clonal cytogenetic abnormalities (CCAs) bear gene mutations, and most of these mutations are similar to those found in MDS patients. Bejar et al26 conducted a comprehensive analysis of 439 patients with MDS based on NGS and determined that three genes (TET2, DMNT3A, TP53) were independently associated with a decline in overall survival. Among the additional gene mutations that we detected, the mutation rates of additional genes related to MDS were different in the two groups of CML patients. The number of mutations in the TKI-intolerant patients was significantly higher than that in the TKI-resistant patients. In addition, the mutation rates of CUX1, GATA2, and KIT were all increased in the TKI-intolerant group, while the rate of BCR-ABL1 KD mutation was lower than that in the TKI-resistant group (26.09% vs. 34.09%), indicating that the acquisition of BCR-ABL1 KD mutation plays a major role in drug resistance to TKIs. However, the mutation of additional MDS-related genes mainly affects patients’ intolerance to TKIs.
In our data, 17.39% CUX1 mutation detected in patients who were intolerance to TKIs, two patients with CUX1 mutation accompanied with ACAs, including one with loss of chromosome 7. In the study of CUX1 in leukemia, it was found that CUX1 mutation was the most differentially expressed gene among the common deletion gene fragments,27 and it was haploinsufficient in leukemia patients with −7q or −7.28,29 The role of CUX1 gene in maintaining hematopoietic stem cell (HSC) quiescence through the PI3K/AKT signaling pathway has been confirmed in human CD34+ cells. Knockdown of the CUX1 gene promotes PI3K signal transduction and induces HSCs to shift from a quiescent to a proliferative state, leading to HSC exhaustion.29 In our patient cohort, mutations in the CUX1 gene were mainly missense and frameshift mutations (p.P1271fs). According to InterPro’s prediction, there are many DNA binding sites in the segment of 1256 to 1313 amino acids of the protein translated by the CUX1 gene, while frameshift mutation of the CUX1 gene in our patient cohort causes the 1271st and subsequent amino acids to change, which may change the original DNA binding site of the protein, leading to abnormal function.
We found 80% patients with KIT gene mutation intolerance to first- and second-generation TKIs. c-KIT is one of the targets of dasatinib,30 which can induce the death of acute myeloid leukemia cells (HEL92.1.7, Kasumi-1, and bone marrow cells of patients with c-KIT+).31 In our cohort, five patients carried KIT gene mutations, three of whom intolerance to dasatinib. Repeated dose reduction or even withdrawal of treatment due to thrombocytopenia caused treatment failure, and two patients progressed to blast crisis. We think that KIT gene mutation may attribute to the patients’ intolerance to dasatinib, but need larger sample to verify.
In addition, in this study the mutation rate of GATA2 was also increased in patients with TKI intolerance. Previous reports have shown that GATA2 haploinsufficiency causes hemocytopenia and immunodeficiency.32,33 This is consistent with the observation of decreased proliferation and increased apoptosis of hematopoietic stem cells in mouse models of GATA2 haploinsufficiency and conditioned GATA2 inactivation.34 In contrast, the overexpression of GATA2 inhibited the output and erythroid differentiation of HSCs,35 indicating that irrespective of whether the expression of GATA2 was decreased or increased, it would affect the proliferation and differentiation of HSCs. However, the mechanism of its role in CML is not completely clear. In our cohort, the mutation rate in patients with TKI intolerance is increased, which may be related to the decreased hemogram in patients with TKI intolerance during TKI treatment.
Among the three genes (CUX1, GATA2, KIT) closely related to TKI intolerance identified in this study, two of them (CUX1, GATA2) encode transcription factors. After analyzing the functions of 66 somatic mutations detected, it was shown that the number of transcription factor gene mutations in patients with TKI intolerance is greater than that in patients with TKI resistance (82.60% vs. 27.27%, P=0.016). Single HSC dysplasia can lead to the occurrence of malignant tumors such as leukemia, and transcription factors are essential for regulating nodes in the genetic map of cell development. Hamey et al36 described the genetic map of mouse LT-HSCs developing to progenitor cells by single-cell sequencing, simulated the transcriptional regulatory network, and confirmed that GATA2 regulates the transcription of many key factors. It can be seen that the distribution of mutations in transcription factor genes is very different in patients with drug resistance or intolerance to TKIs. In addition, the imbalance of the transcriptional regulatory network is severer in patients with TKI intolerance, which may suggest that it plays an important role in the mechanism of TKI intolerance.
Relationship Between Age-Related Clonal Hematopoiesis and CML Progression
In our study, patients with mutations in TET2 showed a poorer prognosis and had a shorter PFS than those without them (P=0.026). However, this may have been due to the insufficient number of patients included here, as its variant allele frequency (VAF) does not show a correlation with the progression of CML disease.
ASXL1 gene mutation showed an unusual relationship with CML in our study. First of all, these mutations were more common in ABL1 KD-mutant patients. Additionally, patients with ASXL1 mutation had a higher proportion of basophils in peripheral blood and a larger spleen at initial diagnosis. Although there were no differences in Sokal and EUTOS scores, ASXL1 mutation with increased the risk of disease progression. In MDS/MPN patients, ASXL1 mutation concentrated in exon 12. It has been reported37 that frameshift or nonsense mutation in exon 12 of ASXL1 leads to truncation and removal of the C-terminal of the protein, which contains a PHD finger, a structural motif found in nuclear proteins related to transcriptional regulation, chromatin modification, and histone demethylation. In this study, besides those in the ASXL1 gene, the main other gene mutations were missense mutations, but frameshift mutations in exon 12 of the ASXL1 gene accounted for 46.67% of the total, resulting in protein truncation, loss of PHD finger, and altered function of downstream proteins.
DNMT3A and TET2 genes were previously considered to be drivers of myeloid malignancies,38,39 but recent studies40 have shown that these two genes were most commonly mutated in age-related clonal hematopoiesis (ARCH). ARCH increases not only the risk of hematological malignancies in humans,40 but also the risk of nonhematological diseases,41–43 for which age is the main risk factor. However, Sasaki44 suggested that the VAFs of ASXL1, DNMT3A, JAK2, TET2, and TP53 mutations are closely related to the progression of AML and can be used as prognostic factors for overall survival. At present, there are different opinions on the correlation between such mutations and the disease progression of hematological malignancies. Stronger evidence is needed to explain their relationships.
Conclusion
In summary, we found that ABL1 KD mutations play dominant roles in drug resistance to TKIs, while CUX1, KIT, and GATA2 mutations may contributors to TKI intolerance. Although some mutations associated with ARCH, their associations with poor prognosis of CML patients cannot be ignored. We believe that genomic risk assessment can improve the risk stratification in clinical diagnosis of CML, which enables us to accurately identify CML patients who are resistant or intolerant to TKIs at an early stage in the era of TKI treatment.
Abbreviations
TKIs, tyrosine kinase inhibitors; CML, chronic myeloid leukemia; KD, kinase domain; NGS, next-generation sequencing; Ph chromosome, Philadelphia chromosome; CHR, complete hematologic response; MCyR, major cytogenetic response; WBC, white blood cell; MDS, myelodysplastic syndrome; CP, chronic phase; AP, accelerated phase; BC, blast crisis; CCAs, clonal cytogenetic abnormalities; HSC, hematopoietic stem cell; ARCH, age-related clonal hematopoiesis; VAF, variant allele frequency.
Data Sharing Statement
All data and materials analyzed during the current study are included in this published article.
Ethics Approval and Consent to Participate
This study was approved by the Medical Ethics Committee of Nanfang Hospital and carried out in according to the Declaration of Helsinki. All patients provided written informed consent.
Acknowledgment
Waner Wu and Na Xu contributed equally to this work and should be considered as co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Georgoulia PS, Todde G, Bjelic S, Friedman R. The catalytic activity of Abl1 single and compound mutations: implications for the mechanism of drug resistance mutations in chronic myeloid leukaemia. Biochim Biophys Acta Gen Subj. 2019;1863(4):732–741. doi:10.1016/j.bbagen.2019.01.011
2. Branford S, Wang P, Yeung DT, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132(9):948–961. doi:10.1182/blood-2018-02-832253
3. Kok CH, Yeung DT, Lu L, et al. Gene expression signature that predicts early molecular response failure in chronic-phase CML patients on frontline imatinib. Blood Adv. 2019;3(10):1610–1621. doi:10.1182/bloodadvances.2019000195
4. Kim T, Tyndel MS, Kim HJ, et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood. 2017;129(1):38–47. doi:10.1182/blood-2016-04-708560
5. Branford S, Kim DDH, Apperley JF, et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia. 2019;33(8):1835–1850. doi:10.1038/s41375-019-0512-y
6. Primon M, Hunter KD, Pandha HS, Morgan R. Kinase regulation of HOX transcription factors. Cancers. 2019;11(4):4. doi:10.3390/cancers11040508
7. Ngeow KC, Friedrichsen HJ, Li L, et al. BRAF/MAPK and GSK3 signaling converges to control MITF nuclear export. Proc Natl Acad Sci U S A. 2018;115(37):E8668–E8677. doi:10.1073/pnas.1810498115
8. Patel U, Luthra R, Medeiros LJ, Patel KP. Diagnostic, prognostic, and predictive utility of recurrent somatic mutations in myeloid neoplasms. Clin Lymphoma Myeloma Leuk. 2017;17:S62–S74. doi:10.1016/j.clml.2017.02.015
9. Ordoñez R, Martínez-Calle N, Agirre X, Prosper F. DNA methylation of enhancer elements in myeloid neoplasms: think outside the promoters? Cancers. 2019;11:10. doi:10.3390/cancers11101424
10. Holm F, Hellqvist E, Mason CN, et al. Reversion to an embryonic alternative splicing program enhances leukemia stem cell self-renewal. Proc Natl Acad Sci U S A. 2015;112(50):15444–15449. doi:10.1073/pnas.1506943112
11. Godavarthy PS, Kumar R, Herkt SC, et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia the E-selectin - SCL/TAL1 - CD44 axis. Haematologica. 2020;105(1):136–147. doi:10.3324/haematol.2018.212365
12. Saito-Reis CA, Marjon KD, Pascetti EM, Floren M, Gillette JM, Traver D. The tetraspanin CD82 regulates bone marrow homing and engraftment of hematopoietic stem and progenitor cells. Mol Biol Cell. 2018;29(24):2946–2958. doi:10.1091/mbc.E18-05-0305
13. Huang XJ, Hu JD, Li JY, et al. Study on efficiency and safety of dasatinib in Chinese patients with chronic myelogenous leukemia who are resistant or intolerant to imatinib. Zhongguo Yi Xue Ke Xue Yuan. 2012;33(11):889–895.
14. Yu L, Qin YZ, Lai YY, Shi HX, Huang XJ, Jiang Q. Severe hematologic toxicity and its impact on treatment response in newly diagnosed patients with chronic myeloid leukemia receiving tyrosine kinase-inhibitor therapy. Zhongguo Yi Xue Ke Xue Yuan. 2019;40(4):281–287. doi:10.3760/cma.j.issn.0253-2727.2019.04.004
15. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
16. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi:10.1101/gr.107524.110
17. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
18. Lussana F, Intermesoli T, Stefanoni P, Rambaldi A. Mechanisms of resistance to targeted therapies in chronic myeloid leukemia. Handb Exp Pharmacol. 2018;249:231–250.
19. Eechoute K, Sparreboom A, Burger H, et al. Drug transporters and imatinib treatment: implications for clinical practice. Clin Cancer Res. 2011;17(3):406–415. doi:10.1158/1078-0432.CCR-10-2250
20. Iurlo A, Ubertis A, Artuso S, et al. Comorbidities and polypharmacy impact on complete cytogenetic response in chronic myeloid leukaemia elderly patients. Eur J Intern Med. 2014;25(1):63–66. doi:10.1016/j.ejim.2013.11.002
21. Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–2088. doi:10.1056/NEJMoa1205127
22. Zhang H, Li S. Molecular mechanisms for survival regulation of chronic myeloid leukemia stem cells. Protein Cell. 2013;4(3):186–196. doi:10.1007/s13238-013-2115-0
23. Loscocco F, Visani G, Galimberti S, Curti A, Isidori A. BCR-ABL independent mechanisms of resistance in chronic myeloid leukemia. Front Oncol. 2019;9:939. doi:10.3389/fonc.2019.00939
24. Bersenev A, Wu C, Balcerek J, et al. Lnk constrains myeloproliferative diseases in mice. J Clin Invest. 2010;120(6):2058–2069. doi:10.1172/JCI42032
25. Schmidt M, Rinke J, Schafer V, et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia. 2014;28(12):2292–2299. doi:10.1038/leu.2014.272
26. Bejar R, Stevenson KE, Caughey B, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol. 2014;32(25):2691–2698. doi:10.1200/JCO.2013.52.3381
27. McNerney ME, Brown CD, Wang X, et al. CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia. Blood. 2013;121(6):975–983. doi:10.1182/blood-2012-04-426965
28. Boultwood J. CUX1 in leukemia: dosage matters. Blood. 2013;121(6):869–871. doi:10.1182/blood-2012-12-473421
29. An N, Khan S, Imgruet MK, et al. Gene dosage effect of CUX1 in a murine model disrupts HSC homeostasis and controls the severity and mortality of MDS. Blood. 2018;131(24):2682–2697. doi:10.1182/blood-2017-10-810028
30. Galanis A, Levis M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica. 2015;100(3):e77–79. doi:10.3324/haematol.2014.117028
31. Heo SK, Noh EK, Kim JY, et al. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci Rep. 2017;7(1):15278. doi:10.1038/s41598-017-15492-5
32. Alfayez M, Wang SA, Bannon SA, et al. Myeloid malignancies with somatic GATA2 mutations can be associated with an immunodeficiency phenotype. Leuk Lymphoma. 2019;60(8):2025–2033. doi:10.1080/10428194.2018.1551535
33. Hsu AP, Johnson KD, Falcone EL, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(19):3830–3837. doi:10.1182/blood-2012-08-452763
34. Menendez-Gonzalez JB, Vukovic M, Abdelfattah A, et al. Gata2 as a crucial regulator of stem cells in adult hematopoiesis and acute myeloid leukemia. Stem Cell Rep. 2019;13(2):291–306. doi:10.1016/j.stemcr.2019.07.005
35. Mehta C, Johnson KD, Gao X, et al. Integrating enhancer mechanisms to establish a hierarchical blood development program. Cell Rep. 2017;20(12):2966–2979. doi:10.1016/j.celrep.2017.08.090
36. Hamey FK, Nestorowa S, Kinston SJ, Kent DG, Wilson NK, Gottgens B. Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc Natl Acad Sci U S A. 2017;114(23):5822–5829. doi:10.1073/pnas.1610609114
37. Asada S, Fujino T, Goyama S, Kitamura T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell Mol Life Sci. 2019;76(13):2511–2523. doi:10.1007/s00018-019-03084-7
38. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. doi:10.1038/nature13038
39. Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi:10.1016/j.cell.2012.06.023
40. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi:10.1056/NEJMoa1408617
41. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–121. doi:10.1056/NEJMoa1701719
42. Dorsheimer L, Assmus B, Rasper T, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA cardiol. 2019;4(1):25–33. doi:10.1001/jamacardio.2018.3965
43. Robertson NA, Hillary RF, McCartney DL, et al. Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr Biol. 2019;29(16):R786–R787. doi:10.1016/j.cub.2019.07.011
44. Sasaki K, Kanagal-Shamanna R, Montalban-Bravo G, et al. Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer. 2020;126(4):765–774.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.