Back to Journals » International Journal of Nanomedicine » Volume 16
Integration of Dual Targeting and Dual Therapeutic Modules Endows Self-Assembled Nanoparticles with Anti-Tumor Growth and Metastasis Functions
Authors Chen B, Dong X ![]() , Dong X, Wang Q, Wu M, Wu J, Lou X, Xia F, Wang W, Dai J, Wang S
, Dong X, Wang Q, Wu M, Wu J, Lou X, Xia F, Wang W, Dai J, Wang S ![]()
Received 11 November 2020
Accepted for publication 12 January 2021
Published 18 February 2021 Volume 2021:16 Pages 1361—1376
DOI https://doi.org/10.2147/IJN.S291285
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel Rubinstein
Biao Chen,1 Xiaoqi Dong,2 Xiyuan Dong,1 Quan Wang,2 Meng Wu,1 Jun Wu,2 Xiaoding Lou,2 Fan Xia,2 Wenwen Wang,1 Jun Dai,1 Shixuan Wang1
1Department of Obstetrics and Gynecology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 2Engineering Research Center of Nano-Geomaterials of the Ministry of Education, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan, 430074, People’s Republic of China
Correspondence: Wenwen Wang; Jun Dai 1095 Jiefang Avenue, Wuhan, Hubei, People’s Republic of China
Tel +86-0276868907
; +86-0276868907
Email [email protected]; [email protected]
Object: High targeting and efficient cytotoxicity toward tumor cells endow NPs excellent anti-tumor activity. Herein, a peptide polymer possessing dual-targeting ability and double therapeutic activity was developed and named TGMF, which can form NPs through self-assembly. It is composed of four functional modules: 1) Active targeting peptide TMTP1 (T) deliver NPs to tumors specifically; 2) Therapeutic peptide GO-203 (G), which can significantly inhibit tumor growth by disrupting the redox balance in cells; 3) A passively targeted enzyme-responsive peptide PLGLGA (M), which can be cleaved specifically by metalloproteinase-2 (MMP-2) highly expressed in the tumor microenvironment (TME); and 4) Hexadecyl (F), which has strong hydrophobicity, can promote the self-assembly of TGMF NPs.
Methods: Five modular peptide probes, namely, TGF, TMF, TGM, GMF, and TGMF were synthesized and self-assembled into NPs in solution. The characterization, enzyme reactivity, and cytotoxicity of NPs were evaluated in vitro, and the pharmacokinetics, bio-distribution, anti-tumor activity of NPs were investigated in vivo. In addition, transcriptome sequencing identified the intracellular signaling pathway-related genes involved in the anti-tumor effect of TGMF.
Results: Upon enzyme cleavage, two types of nanostructure, NPs and nanofibers (NFs), were detected under TEM. Moreover, the cytotoxicity and anti-invasion activity of TGMF against tumor cells used were strongest among the five modular probes examined in vitro. TGMF increased reactive oxygen species (ROS) levels in cytoplasm and produced numerous NFs in extracellular interval and intracellular space. Transcriptome sequencing revealed that TGMF caused 446 genes’ down-regulation and 270 genes’ up-regulation in HeLa cells. In vivo, TGMF has a good anti-tumor effect, effectively prolonging the survival time of HeLa-tumor-bearing mice without systemic side effects.
Conclusion: Integration of multiple functional modules into NPs could be a promising strategy for the future of nanomedicine design towards tumor treatment.
Keywords: modular peptide, nanofiber, anti-tumor, anti-metastasis, transcriptome sequencing
Introduction
Malignant tumors are recognized as one of the most challenging diseases to human health.1 Because of diversity, complexity, and heterogeneity, varieties of malignant tumors are not able to be thoroughly eliminated. Chemotherapy plays an important role in the initial treatment of tumors and subsequently controls the tumor growth at the following therapy stages. Inevitably, the poor selectivity and specificity of chemotherapy induce quick drug-resistance and severe side effects, finally resulting in organ dysfunction and increased mortality of the patients.2–5 To achieve an effective treatment, self-assembled biomaterials based on peptides which form the well-organized and flexible structures that enjoy multiple functions have recently attracted great interest. So far, varieties of peptide biomaterials have demonstrated a large number of functions.6–8 In particular, the peptide amphiphiles, peptide modules and peptide-conjugated vectors have been utilized as novel agents to construct supramolecular systems,6 nanomaterials7, and peptide-conjugated AIEgens,8 respectively, rendering the utilization of peptides for nanostructure development becoming a new trend in material design and morphological control. In these materials, peptide-based nanoparticles (NPs) bring about numerous strategies that integrate multiple therapeutic agents and achieve remarkable treatment effects.9–12 The ideal strategy, however, is that the NPs possess specific targeting and responsive abilities applied for improving enhanced permeability and retention (EPR) effects and leasing the biocomponents under stimulation. Otherwise, peptide NP locating tumor cells difficultly or enabling poor tumor-specific reactions, the cellular functions of individual agents could be compromised and induce therapeutic effect unfulfilled.
Tumor cells exist in a particular tumor microenvironment (TME), gradually evolve into a complicated biological microstructure, and finally develop into a group of abnormal tissues. TME displays unique characteristics via unique metabolism and usually features over-production of specific enzymes, such as matrix metalloproteinase-2 (MMP-2), which plays a significant role in proteolytically activating TGF-β for the cancer metastasis.13,14 Such characteristics of TME could be extensively utilized for the responsive strategy of suppressing the undesired leakage in the normal conditions and consequent releasing the drug under stimulation in cancer cells, resulting in an accurate temporal and spatial cargo delivery.15,16 On the other hand, peptide biomaterials owned high specific recognition of cancer cells has great significance in improving the efficiency of cancer therapy. Attaching the specific-targeting peptide sequences to the NPs are able to transform them into versatile vessels with high specificity and sensitivity to cancer cells.17–19 In view of this, NPs that combine the superiorities of responsive and targeting peptide modules and exhibit synergistic and complementary benefits are urgently needed.
Besides, peptide’s functions are based on the specific tumor cell binding and can select enzyme interaction in TME, groups of peptide drugs with highly efficient therapy have also been frequently reported in the last few years.20–22 In comparison with the traditional chemotherapy and novel gene/photodynamic therapy, the therapeutic peptide is able to address unsatisfied medical needs in clinical trials and be selected as a prominent complementary treatment, as well as a preferable alternative to biological and chemo therapies. Recently, several inhibitor peptides were integrated into peptide-based NPs and demonstrated exciting suppressive effects on tumor growth and metastasis. For example, the peptide GO-203 in tetrablock NPs was reported to target the intracellular MUC1-C oncoprotein for decreasing the survival of cancer cells in vitro and inhibiting tumor growth in vivo.23,24 Developing peptide-based drugs becomes a popular trend and provided a novel perspective in the tumor therapeutic area. Moreover, a nanofiber network was also introduced to construct compositional complexity through self-assembly and engineer heterogeneous cell microenvironments, which led to TME reconstruction or even apoptosis of tumor cells.25,26
Inspired by the peptide drugs' strategy, we developed a self-assembled nanoparticle TGMF based on peptide materials for combinational specific tumor targeting and therapeutic peptide delivery.19,27,28 The TGMF consisted of four multifunctional modules: peptide TMTP1 (T), PLGLGA (M), GO-203 (G) and hydrophobic tail Hexadecyl (F) (Scheme 1). First, TMTP1 peptide exhibited excellent targeting properties for malignant tumor cells29–31 and was then employed to construct peptide NPs for differentiating normal and tumor cells. Second, the peptide module PLGLGA was able to exclusively interact with high-expressed MMP-2 in TME, as a result of the cleavage via enzymic digestion and the subsequent release of therapeutic residue in real-time. More importantly, GO-203 could serve as a peptide drug to block intracellular Mucin 1 (MUC1) function and significantly inhibit the survival of cancer cells both in vitro and in vivo,23,24 providing an efficient treatment for tumor therapy. Finally, the Hexadecyl (F), which encapsulated in the hydrophobic core of NPs by the self-assembly, rendered the nanofibers (NFs) available under MMP-2 cleavage and hence induced cellular apoptosis in tumors.25,26 By virtue of such a rational design, this novel self-assembled peptide NPs was generated by introducing specific tumor-targeting and TME responsive peptides,32,33 and provided an advanced peptide drug for ovarian cancer treatment. In the end, transcriptome sequencing was conducted to identify involved genes in the signaling pathways of therapeutic actions.
 |
Scheme 1 Schematic of the molecular composition and anti-tumor mechanism of TGMF. (A) TGMF is composited of four modules: a TMTP1 peptide (T), a GO-203 (G), a hexapeptide (M) and a hexadecyl chain (F). (B) Proposed process and mechanism of TGMF anti-tumor effect. |
Materials and Methods
Synthesis and Characterization of Modular Probe
Five modular peptide probe (TGF, TMF, TGM, GMF, and TGMF) derived from diverse combinations of four modules including TMTP1 (T), GO-203 (G), MMP-2 cleavage peptide (M) and Hexadecyl (F) were synthesized by a biochemistry company (GL Biochem (Shanghai) Ltd., purity 95%). For self-assembly observation, TGMF was dissolved in PBS at a concentration of 200 μM and stood for 30 minutes at room temperature. The sample was stained with 0.3% phosphotungstic acid solution and observed on a transmission electron microscope (TEM, FEI Tecnai G2 12 TEM instrument) with an accelerating voltage of 100 kV.
TGMF Cleavage by MMP-2
MMP-2 enzyme was activated by 4-aminophenylmercuric acetate (APMA) before used for the enzyme-responded test. APMA (2.5 mM) and MMP-2 (5 μg) were both added into 50 mM Tris-HCl solution and then the solute was incubated at 37 °C for 2 h. After that, activated MMP-2 was used to cleave TMF, TGM, GMF, and TGMF at 37 °C. Target molecules were identified by high-resolution mass spectra (HRMS) on a Bruker micrOTOF II mass spectrometer system under MALDI-TOF mode.
Cell Culture
Five cancer cell lines including SKOV-3, A375, HeLa, PC3 and A549 were used in this study and all the cell lines were purchased from the National Infrastructure of Cell Line Resource (China). Cells were incubated with the proper medium as recommended by the provider (http://www.cellresource.cn/) in a humidified incubator with 5% CO2 at 37 °C.
ELISA Assay
After 24 hours, cell culture supernatant was collected for protein quantification without dilution. MMP-2 was quantified using a Human MMP-2-specific ELISA kit (EK1M02-96, Multi Science, China) according to the manufacturer’s recommendations.
Western Blot
Whole protein was extracted by lysis buffer and denatured through heating at 100 °C for 5 min. Protein concentration in the supernatant was quantified by a BCA Protein Assay Reagent kit (Pro.23228, Thermo Fisher Scientific, USA). Dodecyl sulfate-polyacrylamide electrophoresis was conducted with 20 μg total protein loaded in each well. After electrophoresis, separated proteins were transferred to a nitrocellulose membrane (Millipore, Italy) in buffer. Following primary antibody (MUC1, Proteintech, 19976-1-AP, China; XPNPEP2, GeneTex, GTX109995, Irvine, CA, USA) and secondary antibody (Proteintech, 10230-1-AP, China) incubation, target protein bands were detected by an enhanced chemiluminescence kit (NCI5079, Thermo Fisher).
Cytotoxicity Assay
About 1 × 105 cells were seeded per well in 96-well plates for viability assay. After overnight culture at 37°C, cells were treated with PBS, TGM, TGF, TMF, GMF or TGMF (20 μM), respectively, following a further 24 hours culture. Then the media was changed with fresh culture media and cell viability was evaluated by Cell Counting Kit-8 (CCK-8) system.
Transwell Assay
HeLa cells were digested and suspended in a serum-free medium at 106 cells/mL. Cell suspension (100 μL) was added to the upper chamber (Corning, 8 μm pore) pre-coated with Matrigel (BD, 356234) that then was put into the lower chamber loaded with 600 μL NPs-concocted medium (20 μM). The chambers were put in an incubator for 24 hours. Cells on the upper side of the polycarbonate film were removed. After PBS washing, the upper chamber was immersed in methanol for 30 min and 0.1% crystal violet for 20 min. The film was observed under the microscope when air-dried and cells adhering on its surface were counted.
Reactive Oxygen Species Measurement
After overnight culture at 37°C, HeLa cells were treated with PBS, TGM, TGF, TMF, GMF or TGMF (20 μM), respectively, following another 24 hour culture. Changes of intracellular ROS level were detected using a specified kit (S0033 Beyotime, China). Cells were incubated for 30 min with 10 μM DCFH-DA at 37 °C. After PBS washing, the fluorescence of oxidized DCF was measured at an excitation of 488 nm and an emission of 525 nm.
Anti-Tumor Study in vivo
This study was approved by the Animal Ethics Committee of Tongji Hospital, Huazhong University of Science & Technology and institutional guidelines for proper usage of research animals were followed. To establish the xenograft model, 6-week-old female BALB/c nude mice were inoculated subcutaneously into the left flank with approximately 1 × 107 HeLa cells. When the tumor grew to 50 mm3, all tumor-bearing mice were randomly divided into 6 groups: PBS, TGM, TMF, TGF, GMF, and TGMF. Mice were injected PBS, TGF, TGF, TMF, GMF and TGMF (200 µM, 100 μL) via tail vein, respectively. Tumor size was calculated through the formula: tumor volume = length × width × width/2. The mice unless died were continuously monitored until 60 days after cell inoculation and then were sacrificed. Organs and tumors were collected immediately after death or killing in 4% formaldehyde solution for a histopathology examination.
Histopathology
Harvested organs and tumors were fixed in 4% paraformaldehyde solution 24 hours. After regular dehydration with graded ethanol and vitrification with dimethylbenzene, tissue blocks were embedded in paraffin and subsequently sectioned at 5 µm thickness. Slides were stained with hematoxylin and eosin (HE) and sealed with resin. Sections were carefully observed and photographed under a white-light microscope equipped with a camera.
TUNEL Assay
Apoptosis was analyzed using a One Step TUNEL kit (C1086, Beyotime, China).Slides were in turn treated with Xylene, ethyl alcohol, and distilled water. Protease K solution (20 µg/mL) free from DNase was dropped on each slide for 20 min incubation. Then we washed slides with PBS and added 50 µL TUNEL liquid that containing Terminal Deoxynucleotidyl Transferase (TdT) and FITC-labeled dUTP to each well. All the sections were kept in dark for an additional 60 min at room temperature and then washed twice with PBS solution. Green fluorescence indicated cell apoptosis at excitation wavelength 488 nm and emission wavelength 525 nm.
Next Generation RNA Sequencing Analysis
Briefly, Total RNA was isolated from cell lysate and checked for quality, quantity, purity and integrity with appropriate methods. Then the mRNA was enriched by Oligo(dT) magnetic beads and fragmented in a proper buffer. Using the mRNA as templates, first-strand cDNA was synthesized in the presence of random hexamers and then the second-strand cDNA was produced with the addition of specified regents. After purification, the cDNA collect received ends repair, dA-tailing, adaptor ligation, and fragment size selection. The rest of each sample was amplified by PCR and loaded on an Illumina HiSeq 2500 platform for sequencing with a 2 × 150 bp paired-end (PE) configuration according to the manufacturer’s instructions. Original image signals were converted to sequenced reads by base calling procedure and saved in fastq format. Technical sequences were filtered and the quality of clean data was checked. The clean reads were aligned to the human reference genome assembly using Hisat2. The FeatureCounts Software was used to estimate the relative abundance and distribution of gene expression for each sample. Differential expressions of genes were analyzed by edgeR with adjusted P-value sated < 0.05 and |log2FoldChange| >1 and visualized as heatmap and volcanomap. Enrichment of functions and signaling pathways of the differential expression genes were also processed by edgeR based on the Gene Ontology (GO, p < 0.05) and Kyoto Encyclopedia of Genes and Genomes (KEGG, FDR < 0.05) database.
Statistical Analysis
All results were presented as Mean ± SD from at least three independent experiments. Statistical differences were examined using Student’s t-test or one-way ANOVA test. * p < 0.05, ** p < 0.01, *** p < 0.001, n.s. not significant.
Results and Discussion
Preparation and Characterization of NPs
In order to verify the function of different peptide modules, both TGMF and four control peptide backbone GMF (without TMTP1 module), TGF (without MMP-2 cleavage module), TGM (without fiber unit), and TMF (without GO-203 module) were designed and synthesized (Scheme 1A; Figure S1). All the products were confirmed by HRMS (Figures S2-S6). Except for TMF, the zeta potential of other products (TGMF, GMF, TGM and TGF) is about 30 mV (Figure S7). Through the hydrophobic interaction, TGMF formed stable NPs in aqueous solution, and the particle size was about 300 nm (Figures S8 and S9). Based on our design, the enzyme-responsive peptide PLGLAG was selectively cleaved by the cancer-associated protease MMP-2, which played an important role in regulating the morphology of NFs by MMP-2 cleavage as well as facilitating the peptide drug GO-203 release process. As shown in Figure 1A-D, except TGF, the other four peptide skeletons could be successfully separated into two fragments upon MMP-2 cleavage, and resultant peptide residues were further confirmed by HRMS. It was further confirmed by high performance liquid chromatography (HPLC) that TGMF was cleaved by MMP-2, and its cleavage efficiency reached 90.8% (Figure S10). More importantly, NPs were formed via the self-assembly of amphiphilic TGMF in solution and observed on the transmission electron microscope (TEM) (Figure 1E). After MMP-2 digestion in solutions, two different types of nanostructure, NPs (Figure 1F, Left) and NFs (Figure 1F, Right), were demonstrated by TEM. At the same time, we monitored the change of TGMF particle size before and after MMP-2 treatment by DLS (Figure S11). The average particle size of TGMF NPs is 274.2 nm and PDI is 0.195 (Figure S11A). After MMP-2 treatment, the average particle size of TGMF increased to 343.2 nm, while PDI was 0.529, which may be due to the formation of NFs, resulting in uneven particle size (Figure S11B). As can be seen from Figure 1F, MMP-2 cleavage resulted in a smaller NPs and a large number of NFs, suggesting the efficient protease-responsive strategy depends on MMP-2 digestion.
 |
Figure 1 MMP-2 cleavage. In the presence of MMP-2, molecules containing M module including TGMF (A), GMF (B), TMF (C) and TGM (D) were successfully cleaved into two pieces, which was confirmed by HRMS. In aqueous solution, TGMF self-assembled into NPs that could be verified on TEM (E). Upon MMP-2 cleavage, two types of nanostructure, NPs (F, Left) and NFs (F, Right), were detected under TEM. |
TGMF Inhibits Tumor Cell Viability and Invasion in vitro
Based on the design rationale, the therapeutic responses of TGMF was established by the release of peptide modules before and after the interaction with protease in tumor cells. As a result, the different levels of proteases in TME were examined in this section. First, XENPEP2 is an important protease distributed on the cell membrane and served as a binding receptor for TMTP1.34 The expression of XENPEP2 was associated with the different cell lines, among which two highest levels were discovered by A375 and HeLa cells (Figure 2A). Second, because of the significant role of MMP-2 in tumor metastasis, their concentrations were determined in culture medium among SKOV-3, A375, HeLa, PC3, and A549 cells. Our data showed that MMP-2 was highly expressed by SKOV-3, A375 and HeLa cell, and exhibited fairly low concentrations in PC3, and A549 cells (Figure 2B). On the other hand, the reported literature proved that GO-203 was able to compromise the redox balance in cells and hence used to induce apoptosis.23,24 Investigated as the intracellular target of GO-203, Mucin-1 (MUC1) functioned a pivotal role in preventing cellular apoptosis and promoting tumor invasion, and its expression was recognized as a key feature of the tumor therapy treated with TGMF. Therefore, we explored the expression of MUC1 in SKOV-3, A375, HeLa, PC3, and A549 cell lines by Western blotting and found that MUC1 was widely existing in all five cells (Figure 2C), suggestive of the MUC1’s occurrence. Moreover, to understand the therapeutic effects of NPs, cell viabilities were quantified by using the CCK-8 assay. As a negative control, PBS did not affect cell viability (Figure 2D). However, many viable tumor cells were presenting when treated with TGM, which indicated that therapy efficiency kept weak in the presence of peptide drug GO-203 (Figure 2E). When constructed without peptide drug GO-203, TMF exhibited a lower background cell viability than that of TGM group (Figure 2E and G), implying that fiber units played a paramount role in inducing cellular apoptosis. When without MMP-2 responsive module in TGF, cleavage was completely inhibited to impede the self-assembly of NFs and monofunctional TGF led to an increased tumor cell amount and resulted in compromised cellular therapy (Figure 2F). More importantly, employing the MMP-2-cleavable site in GMF increased tumor therapies and consequently led to a significant decrease in cell viability (Figure 2H). In a comparison of TGM, TGF and TMF groups, the GMF held an MMP-2 cleavage peptide module as well as peptide drug and fiber units, and could impose more effective suppression on tumor cell growth (Figure 2E–H). Such a difference could be ascribed to increased cell growth inhibition by releasing active peptide GO-203 and free aggregated NFs, which was critical to cell death. The integration of all peptide modules into NPs was undoubtedly a most powerful combination, which gave the weakest cell viability in all cell lines and opened up a novel avenue to the efficient theragnostic biomaterials design (Figure 2I). In addition, we also calculated the synergistic effect of GO-203 and NFs in vitro.35,36 According to the therapeutic doses of TGMF (GO-203 + NFs), TGM (GO-203) and TMF (NFs), we calculated the combination index (CI) of GO-203 and NFs to be 0.72, which means that there is a synergistic effect between GO-203 and NFs. Tumor progression is a unique phase in tumor development feature with cell invasive growth and metastasis.37 Since inhibition of invasive growth is of great importance in maintaining therapy efficacy, the NPs defense against invasion growth of tumor cells was assessed by using transwell assay. As elicited in Figure 2J and K, HeLa cells were used, and the results were much consistent with cell viability analysis. A large number of invasive cells were observed in the PBS control group and TGM group, suggestive of a very weak inhibition effect of TGM on cells invasion. The invasive cells gradually vanished under treatment of TGF, TMF, and GMF, and reached the smallest quantity by using TGMF (Figure 2J and K), implying that TGMF possessed a stronginhibiting ability of the HeLa cell invasion. When in the absence of an MMP-2 responsive module in TGF, the release of fiber units was restricted, which generated inadequate NFs and led to an inefficient inhibition of tumor cell invasion (Figure 2J and K). In this vein, TGMF, not monofunctional TGF, was able to aggregate into NFs to inhibit tumor cell growth and effectively prevent cellular invasion.
 |
Figure 2 TGMF inhibits tumor cell viability and invasion. (A) XPNPEP2 expresses in SKOV-3, A375, HeLa, PC3 and A549 cell lines. (B) Elisa was used to detect the relative expression of MMP-2 in the culture medium of SKOV-3, A375, HeLa, PC3 and A549 cell lines. (C) Western blot was used to detect the expression of MUC1 in SKOV-3, A375, HeLa, PC3 and A549 cell lines. The effect of PBS (D) or TGF (E), TMF (F), TGM (G), GMF (H) and TGMF (I) on cell viability tested by CCK-8 assay. (J) The invasion ability of HeLa cells treated with PBS, TGM, TGF, TMF, GMF and TGMF was detected by transwell assay. (K) Compared to PBS, TGF, TMF, GMF and TGMF rather than TGM significantly restrained the migration of Hela cells and TGMF suppressed cell movement most significantly. Data were presented as the mean ± SD (n =3) and analyzed by two-sided Student’s t-test. * p <0.05, ** p <0.01 *** p <0.001. |
ROS Process and NFs Release Profile of TGMF in vitro
The previous findings demonstrated that MUC1-C is one of the druggable targets, and inhibition of the MUC1-C subunit was able to block its oncogenic function with aim of inducing cell death.38 The GO-203 peptide was initially designed to selectively silence the MUC1-C function and consequently reduce the levels of glutathione (GSH), resulting in an increased amount of ROS. In this vein, cellular redox balance was disturbed, which damaged cell membrane integrity and caused cell elimination.23,24 As shown in Figure 3, bright green fluorescence (DCF) in Hela cells were generated under TGF, GMF, TGM, and TGMF treatment, indicating an overexpressed ROS level in cytoplasm, while no DCF signal detected in PBS and TMF treated cells. The NPs integrate GO-203 peptide selectively targeted MUC1-C and contributed to ROS generation which affected cell behaviors and amplified antitumor efficacy. In addition, because GO-203 peptide is rich in arginine, this structure may be able to enhance the penetration effect of NPs, thus enhancing the therapeutic effect.37,39,40 On the other hand, Bio-TEM images offered direct information for the understanding of nanofiber-involved cellular morphology change. Numerous NFs were generated in the extracellular interval and intracellular space (Figure 4A and B). Because of the highly accumulated MMP-2 in TME,41 TGMF was cut off to release fiber fragments and thus self-assemble into NFs around and within the tumor cells, which inhibited tumor cell migration as well as destroyed the intracellular structure.42–45 The combination of GO-203 module and fiber units through MMP-2 cleavable site could readily afford TGMF blocks the MUC1-C function and generates NFs. Such a strategy was able to decrease GSH concentrations and consequently disturb cellular redox balance, and for another, provide sufficient self-assembled NFs that retard tumor cell metastasis and interfere with the intracellular structure.
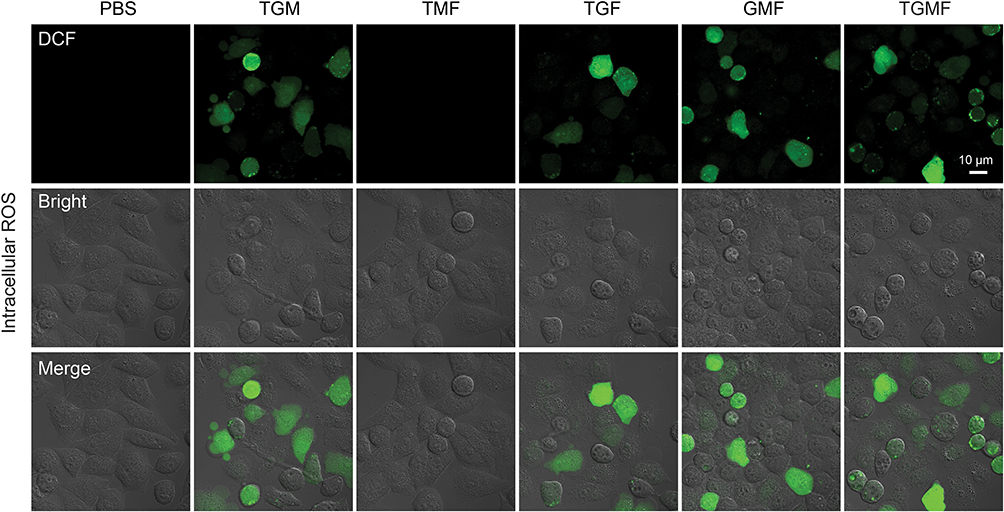 |
Figure 3 ROS measurement and derived NFs in vitro. In Hela cells, TGF, GMF, TGM and TGMF, but not PBS and TMF, treatment generated bright green fluorescence in cytoplasm indicating massive increase of intracellular ROS. |
 |
Figure 4 The NFs were detected by TEM. Numerous NFs could be detected whether in intracellular (A) or extracellular (B) in vitro. Red arrows indicate NFs. |
Transcriptome Sequencing Reveals the Gene Involved in TGMF Treatment
In order to elucidate the TGMF therapeutic pathway, the transcriptome sequencing was employed to detect the mRNA expression in HeLa cells under TGMF treatment. Six samples from the TGMF group and three samples from the PBS group were parallelly used to unveil their genetic information. First, to assess the overall difference of gene expression level between two groups and evaluate sample repeatability within a group, we performed PCA analysis within tested samples and observed two distinct subpopulations, which generally reflected the separate source of samples and satisfactory repeatability within a group (Figure 5A). The samples from the TGMF group and PBS group were analyzed by PCA analysis with 74.2 to 76.4% results located in coding sequence regions (CDS), among which 96.17 to 99.6% genetic mapping rate was achieved in the reference genome, suggestive of a high-performance probing. To authenticate genetic targets of TGMF intervention, gene expression in two groups was analyzed and compared at the transcription level, which was distributed on a volcano plot (Figure 5B). Genes with a fold change of log2 transformed FPKM > 1.0 were designated as differential expression genes (DEGs) (Figure 5B). Then, we further characterized DEGs with a thermogram. The results showed that TGMF caused 446 genes' down-regulation and 270 genes' up-regulation compared with the PBS group (Figure 5C). Although the therapeutic effects of TGMF could be ascribed to the redox balance disruption and cell migration inhibition, the important roles of TGMF in regulating gene functions remained to be explored during the treatment. Hence, the gene-involved signal pathways were analyzed by the Kyoto Encyclopedia of Genes and Genomes (KEGG), and DEGs were categorized into the different top 19 signal pathways (Figure 5D). Among them, over 20 genes were involved in the metabolic pathways, presenting the most gene counts in the up-regulated process (Figure 5D). On the contrary, the genes in down-regulated signal pathways were widely distributed, such as Tight junction, Ras/Rap1 signaling pathway, and pathways in cancer (Figure 5D), implying a comprehensive cellular response under TGMP treatment against tumor cells. Wnt family member 1 (WNT1) is a protein coding gene, and its decreased expression can promote tumor cell apoptosis and inhibit tumor invasion. Interestingly, our transcriptome sequencing showed a significant decrease in WNT1 expression in HeLa cells treated with TGMF (Figure S7). In fact, regulation of gene expression is one of the principal approaches of pharmacotherapeutics. In this respect, our data provided deep insight for understanding potential mechanisms for the detrimental effect of TGMF on tumors.
 |
Figure 5 (A) PCA of the transcriptome of RNA-seq data from HeLa cells with PBS or TGMF treatment. (B) Volcano plot of the transcriptome of RNA-seq data from HeLa cells with PBS or TGMF treatment. (C) Differential gene expression heat maps for RNA-seq data of HeLa cell treated with PBS or TGMF. (D) The top 19 pathway terms enriching DEGs in Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. |
Therapeutic Efficiency of TGMF in vivo
To investigate the therapeutic efficiency of TGMF in vivo, we established subcutaneous HeLa tumor xenografts. As shown in Figure 6A–G, in comparison with other groups, the tumor volumes in HeLa subcutaneous tumor models were significantly suppressed by treating TGMF for 38 days (p < 0.001, Figure 6G). While TGM, TGF, TMF, and GMF were slightly or slowly active in restraint of the tumor growth (Figure 6B-5G), suggestive of the effective tumor inhibition required the conjugation of four modules (T, G, M, and F) into NPs. Moreover, compared with other groups, employing TGMF was the most effective way to increase the survival rate of HeLa tumor models ((p < 0.001, Figure 6H), which was calculated to be almost 40% under 60 days' treatment, confirming the synergistic combination effect of TGMF on HeLa tumor models through controlling tumor metastasis and inhibiting tumor viability.
 |
Figure 6 Individual tumor growth curve of each Hela tumor-bearing mouse in PBS (A), TGM (B), TMF (C), TGF (D), GMF (E) and TGMF (F) groups. (G) Tumor growth curves of groups and comparisons among groups. Data were presented as the mean ± SD (n =10) and analyzed by two-sided Student’s t-test. *** p <0.001. (H) Survival curves of tumor-bearing mice treated with PBS, TGM, TMF, TGF, GMF and TGMF respectively. The data were analyzed by two-sided Log rank test. *** p< 0.001. |
The Anti-Tumor Effects of TGMF in vivo
To understand the therapeutic mechanism of TGMF, the morphological changes during HeLa cell apoptosis were investigated by using the HE staining analysis. As shown in Figure 7A, a large number of necrotic cells were presenting when treated with TGMF and GMF group, while only a few necrotic cells existing under treatment by TGM, TGF, TMF, and PBS, which indicated that a combination of targeting and dual therapeutic modules was the most effective way to induce HeLa cell death. Then the tumor tissues were stained with TUNEL and the concentration of positive TUNEL cells after culturing with TMGF were much stronger than that in the other 6 groups (Figure 7B). Obviously, TGMF finally resulted in the highest ratio of apoptosis tumor cells. Next, the antitumor effect was further evaluated by investigating the aggregated NFs in vivo, a large number of NFs were observed in TMF and TGMF groups (Figure 7C, the black arrow is the cell membrane, and the red arrow is the NFs), while in the TGM and PBS group no obvious fabrication of NFs between the treated and control groups was observed, which was ascribed to the loss of fiber module. On the other hand, inadequate NFs were provided by TGF treated groups. Because of the absence of an MMP-2-cleavable site, TGF was unable to generate sufficient free fiber molecules as well self-assembly of NFs in tumor cells. Similarly, without targeting modules, GMF provided a weak targeted recognition of tumors and consequently released few NFs among tumor cells. We believed that highly efficient therapy of tumors was achieved by conjugating four modules in the peptide skeleton to generate sufficient GO-203 and NFs. In the previous transcriptome sequencing results, TGMF may promote tumor death and inhibit its invasion and metastasis by inhibiting WNT1 expression (Figure S12). Herein, the IHC staining was employed to explore the expression of WNT-1 (Figure 7D) in tumors during treatment. The IHC staining of WNT1 in PBS, TGM, TMF, TGF, and GMF treated cells was stronger than that of TGMF, which indicated that TGMF did down-regulate WNT1 expression in HeLa cells and exert its anti-tumor function.46 The above results show that TGMF can achieve an anti-tumor effect through the WNT1 pathway.
 |
Figure 7 (A) He staining was used to detect the morphological changes of HeLa tumor after receiving PBS, TGM, TMF, TGF, GMF and TGMF treatment. (B) TUNEL staining was used to detect the proportion of apoptotic cells in HeLa tumor after receiving PBS, TGM, TMF, TGF, GMF and TGMF treatment. (C) The presence of NFS in HeLa tumor cells was detected by bio-TEM (the black arrow is cell membrane, and the red arrow is NFs). (D) The expression of WNT1 in HeLa tumor was detected by IHC staining. |
Minimal Side Effects of TGMF in vivo
The toxicity of TGMF to multiple organs was evaluated by staining with HE. As a result, the pathological changes of heart, lung, liver, spleen and kidney were not observed in all groups (Figure 8A). Aspartate (AST) levels validated that TGMF was not involved to influence the normal liver function (Figure 8B). Together with unchanged levels of glucose (GLU) (pancreas, Figure 8C) and creatinine (CRE) (kidney, Figure 8D), the results verified limited side effect of TGMF on different organs and rendered a reliable biomaterial to treat the fatal tumors in vivo. Meanwhile, different blood cells, such as blood cells (WBC) (Figure 8E), red blood cells (RBC) (Figure 8F), and platelets (PLT) (Figure 8G), were also monitored to investigate the effect of TGMF on their concentration in volume. It was found that there was no significant reduction of all kinds of blood cells in the treatment, implying the restricted side effect of TMGF on blood cells. Undoubtedly, the low toxicity and minimal side effects made TGMF promising for efficient therapeutic biomaterials and served as a valuable clue for the molecular design of tumor cell-selective theragnostic NPs.
 |
Figure 8 (A) Histological changes of organs including heart, lung, liver, spleen and kidney were observed by HE staining after PBS, TGM, TGF, TMF, GMF and TGMF interventions. Biochemical markers in blood including alanine aminotransferase (B), glucose (C) and creatinine (D) were examined. Blood cells including white cell (E), red cell (F) and platelet (G) were also tested. Data were presented as the mean ± SD (n =3). |
Conclusion
Creating a reliable strategy for delivery of the peptide drug and fiber units and reducing side effects in pursuit of improved treatment rate of tumors remains a challenge. In this vein, we constructed a peptide skeleton containing four modules and obtained TGMF NPs by the self-assembly. The TGMF was delivered into the tumor sites via TMTP1 active targeting, and the high concentration of MMP-2 in TME facilitated a cleavage of MMP-2 responsive linkin the peptide chain, which broke the bond to generate active peptide drug GO-203 and nanofiber precursors. Nevertheless, limited MMP-2 in normal tissues and organs only initiated a cleavage in a low concentration region to prevent the corresponding side effects. The therapeutic studies in HeLa cells have demonstrated the synergistic effect upon redox balance disruption and NFs-involved cellular structure interference. In HeLa tumor models, TGMF specifically enriched in the tumor sites after intravenous injection and significantly inhibited the tumor growth. The cell enzymic expression further validated that TGMF promoted cellular apoptosis through down-regulation of WTN1 levels. Moreover, different index and blood cell expression proved the limited side effect of TGMF to the key organs during therapy. These results indicated a versatile biomaterial co-loading peptide drug and fiber precursors for efficient tumor therapy, and an advanced design for the future development of therapeutic reagents based on peptide biomaterials with minimal side effects.
Acknowledgments
This work is supported by the National Key R&D Program of China (2020YFA0211200), the National Natural Science Foundation of China (21525523, 21722507, 21874121, 21974128 and 81701420), the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University (to Jun Dai and Jun Wu).
Disclosure
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–31. doi:10.3322/caac.21590
2. Zhou J, Yu G, Huang F. Supramolecular chemotherapy based on host–guest molecular recognition: a novel strategy in the battle against cancer with a bright future. Chem Soc Rev. 2017;46(22):7021–7053. doi:10.1039/C6CS00898D
3. Wang H, Yu JH, Lu XB, He XM. Nanoparticle systems reduce systemic toxicity in cancer treatment. Nanomedicine. 2016;11(2):103–106. doi:10.2217/nnm.15.166
4. Eisenhauer EA. Real-world evidence in the treatment of ovarian cancer. Ann Oncol. 2017;28:viii61–viii65. doi:10.1093/annonc/mdx443
5. Weingart SN, Zhang L, Sweeney M, Hassett M. Chemotherapy medication errors. Lancet Oncol. 2018;19(4):e191. doi:10.1016/S1470-2045(18)30094-9
6. Hendricks MP, Sato K, Palmer LC, Stupp SI. Supramolecular Assembly of Peptide Amphiphiles. Acc Chem Res. 2017;50(10):2440–2448. doi:10.1021/acs.accounts.7b00297
7. Li LL, Qiao ZY, Wang L, Wang H. Programmable Construction of Peptide-Based Materials in Living Subjects: from Modular Design and Morphological Control to Theranostics. Adv Mater. 2018;31(45):1804971. doi:10.1002/adma.201804971
8. Xia F, Wu J, Wu X, Hu QY, Dai J, Lou XD. Modular Design of Peptide- or DNA-Modified AIEgen Probes for Biosensing Applications. Acc Chem Res. 2019;52(11):3064–3074. doi:10.1021/acs.accounts.9b00348
9. Cheng Y, Dai J, Sun CL, et al. An Intracellular H2O2 -Responsive AIEgen for the Peroxidase-Mediated Selective Imaging and Inhibition of Inflammatory Cells. Angew Chem Int Ed. 2018;57(12):3123–3127. doi:10.1002/anie.201712803
10. Cheng Y, Sun CL, Liu R, et al. A Multifunctional Peptide‐Conjugated AIEgen for Efficient and Sequential Targeted Gene Delivery into the Nucleus. Angew Chem Int Ed. 2019;58(15):5049–5053. doi:10.1002/anie.201901527
11. Cheng Y, Sun CL, Ou XW, Liu BF, Lou XD, Xia F. Dual-targeted peptide-conjugated multifunctional fluorescent probe with AIEgen for efficient nucleus-specific imaging and long-term tracing of cancer cells. Chem Sci. 2017;8(6):4571–4578. doi:10.1039/C7SC00402H
12. Cheng Y, Huang FJ, Min XH, et al. Protease-Responsive Prodrug with Aggregation-Induced Emission Probe for Controlled Drug Delivery and Drug Release Tracking in Living Cells. Anal Chem. 2016;88(17):8913–8919. doi:10.1021/acs.analchem.6b02833
13. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278(1):16–27. doi:10.1111/j.1742-4658.2010.07919.x
14. Detry B, Erpicum C, Paupert J, et al. Matrix metalloproteinase-2 governs lymphatic vessel formation as an interstitial collagenase. Blood. 2012;119(21):5048–5056. doi:10.1182/blood-2011-12-400267
15. Li J, Xiao S, Xu Y, et al. Smart Asymmetric Vesicles with Triggered Availability of Inner Cell-Penetrating Shells for Specific Intracellular Drug Delivery. ACS Appl Mater Interfaces. 2017;9(21):17727–17735. doi:10.1021/acsami.7b02808
16. Zhu L, Wang T, Perche F, Taigind A, Torchilin V. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proce Nat Acad Sci. 2013;110(42):17047–17052. doi:10.1073/pnas.1304987110
17. Dai J, Cheng Y, Wu J, et al. Modular Peptide Probe for Pre/Intra/Postoperative Therapeutic to Reduce Recurrence in Ovarian Cancer. ACS Nano. 2020;14(11):14698–14714. doi:10.1021/acsnano.9b09818
18. Yuan QM, Cheng Y, Lou XD, Xia F. Rational Fabrication and Biomedical Application of Biomolecule-Conjugated AIEgens through Click Reaction. Chin J Chem. 2019;37(10):1072–1082. doi:10.1002/cjoc.201900211
19. Dai J, Li Y, Long Z, et al. Efficient Near-Infrared Photosensitizer with Aggregation-Induced Emission for Imaging-Guided Photodynamic Therapy in Multiple Xenograft Tumor Models. ACS Nano. 2020;14(1):854–866. doi:10.1021/acsnano.9b07972
20. Grossmann TN, Yeh JTH, Bowman BR, Chu Q, Moellering RE, Verdine GL. Inhibition of oncogenic Wnt signaling through direct targeting of. Proce Nat Acad Sci. 2012;109(44):17942–17947. doi:10.1073/pnas.1208396109
21. Kumar M, Gupta D, Singh G, et al. Novel Polymeric Nanoparticles for Intracellular Delivery of Peptide Cargos: antitumor Efficacy of the BCL-2 Conversion Peptide NuBCP-9. Cancer Res. 2014;74(12):3271–3281. doi:10.1158/0008-5472.CAN-13-2015
22. Yan J, He WX, Yan SQ, et al. Self-Assembled Peptide–Lanthanide Nanoclusters for Safe Tumor Therapy: overcoming and Utilizing Biological Barriers to Peptide Drug Delivery. ACS Nano. 2018;12(2):2017–2026. doi:10.1021/acsnano.8b00081
23. Hasegawa M, Sinha RK, Kumar M, et al. Intracellular Targeting of the Oncogenic MUC1-C Protein with a Novel GO-203 Nanoparticle Formulation. Clin Cancer Res. 2015;21(10):2338–2347. doi:10.1158/1078-0432.CCR-14-3000
24. Ahmad R, Alam M, Hasegawa M, et al. Targeting MUC1-C inhibits the AKT-S6K1-elF4A pathway regulating TIGAR translation in colorectal cancer. Mol Cancer. 2017;16(1):33. doi:10.1186/s12943-017-0608-9
25. Tanaka A, Fukuoka Y, Morimoto Y, et al. Cancer Cell Death Induced by the Intracellular Self-Assembly of an Enzyme-Responsive Supramolecular Gelator. J Am Chem Soc. 2015;137(2):770–775. doi:10.1021/ja510156v
26. He HJ, Wang JQ, Wang HM, et al. Enzymatic Cleavage of Branched Peptides for Targeting Mitochondria.J Am Chem Soc. 2018;140(4):1215–1218. doi:10.1021/jacs.7b11582
27. Zhang K, Yang -P-P, He -P-P, et al. Peptide-Based Nanoparticles Mimic Fibrillogenesis of Laminin in Tumor Vessels for Precise Embolization. ACS Nano. 2020;14(6):7170–7180. doi:10.1021/acsnano.0c02110
28. Dai J, Wu X, Ding SY, et al. Aggregation-Induced Emission Photosensitizers: from Molecular Design to Photodynamic Therapy. J Med Chem. 2020;63(5):1996–2012. doi:10.1021/acs.jmedchem.9b02014
29. Yang WH, Luo DF, Wang SX, et al. TMTP1, a Novel Tumor-Homing Peptide Specifically Targeting Metastasis. Clin Cancer Res. 2008;14(17):5494–5520. doi:10.1158/1078-0432.CCR-08-0233
30. Liu RH, Xi L, Luo DF, et al. Enhanced targeted anticancer effects and inhibition of tumor metastasis by the TMTP1 compound peptide TMTP1-TAT-NBD. J Controlled Release. 2012;161(3):893–902. doi:10.1016/j.jconrel.2012.05.002
31. Wei R, Jiang GY, Lv MQ, et al. TMTP1-modified Indocyanine Green-loaded Polymeric Micelles for Targeted Imaging of Cervical Cancer and Metastasis Sentinel Lymph Node in vivo. Theranostics. 2019;9(24):7325–7344. doi:10.7150/thno.35346
32. Xu Y, Zhang JN, Liu XY, et al. MMP-2-responsive gelatin nanoparticles for synergistic tumor therapy. Pharm Dev Tech. 2017;9(8):1002–1013. doi:10.1080/10837450.2019.1621899
33. Yao Q, Liu Y, Kou LF, Tu Y, Tang X, Zhu L. Tumor-targeted drug delivery and sensitization by MMP2-responsive polymeric micelles. Nanomedicine. 2019;19:71–80. doi:10.1016/j.nano.2019.03.012
34. Zhou Y, Jiang GY, Wang W, et al. A Novel Near-Infrared Fluorescent Probe TMTP1-PEG4-ICG for in Vivo Tumor Imaging. Bioconjugate Chem. 2018;29:4119–4126.
35. Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6:222ra18.
36. Li J, Li Y, Wang Y, et al. Polymer Prodrug-Based Nanoreactors Activated by Tumor Acidity for Orchestrated Oxidation/Chemotherapy. Nano Lett. 2017;17:6983–6990.
37. Riveros A, Eggeling C, Riquelme S, et al. Improving Cell Penetration of Gold Nanorods by Using an Amphipathic Arginine Rich Peptide. Int J Nanomedicine. 2020;15:1837–1851.
38. Swain S, Sahu PK, Beg S, Babu SM. Nanoparticles for Cancer Targeting: current and Future Directions. Curr Drug Deliv. 2016;13:1290–1302.
39. Cheng T, Li R, Kao YJ, et al. Synthesis and characterization of Gd-DTPA/fucoidan/peptide complex nanoparticle and in vitro magnetic resonance imaging of inflamed endothelial cells. Mater Sci Eng C Mater Biol Appl. 2020;114:111064.
40. Mehrotra N, Kharbanda S, Singh H. Peptide-based combination nanoformulations for cancer therapy. Nanomedicine. 2020;15:2201–2217.
41. Yin J, Liu DK, Bao LC, et al. Tumor targeting and microenvironment-responsive multifunctional fusion protein for pro-apoptotic peptide delivery. Cancer Lett. 2019;452:38–50.
42. Gao Y, Yang ZM, Kuang Y, et al. Enzyme‐instructed self‐assembly of peptide derivatives to form nanofibers and hydrogels. Biopolymers. 2010;94:19–31.
43. Wang HM, Feng ZQQ, Xu B. D-amino acid-containing supramolecular nanofibers for potential cancer therapeutics. Adv Drug Deliver Rev. 2017;110:102–111.
44. Zhou J, Du XW, Chen X, et al. Enzymatic Self-Assembly Confers Exceptionally Strong Synergism with NF-κB Targeting for Selective Necroptosis of Cancer Cells J. Am Chem Soc. 2018;140:2301–2308.
45. Rasouli R, Barhoum A, Bechelany M, Dufresne A. Nanofibers for Biomedical and Healthcare Applications. Macromol Biosci. 2019;19:1800256.
46. Tang Z, Cai HF, Wang R, Cui Y. Overexpression of CD300A inhibits progression of NSCLC through downregulating Wnt/β-catenin pathway. Onco Targets Ther. 2018;11:8875–8883.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.