Back to Journals » Journal of Inflammation Research » Volume 19
Integrated Bioinformatic Identification and Experimental Validation Reveal That Aging Exacerbates ARDS Through MAPK14/ADM/MAPK8 Axis
Authors Liu Q, Yin C, Fan L, Li D ![]() , Wang T
, Wang T ![]()
Received 17 November 2025
Accepted for publication 24 March 2026
Published 17 April 2026 Volume 2026:19 573372
DOI https://doi.org/10.2147/JIR.S573372
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Cynthia Koziol-White
Qing Liu, Cheng Yin, Long Fan, Daxian Li, Tianlong Wang
Department of Anesthesiology, Xuanwu Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Tianlong Wang, Department of Anesthesiology, Xuanwu Hospital, Capital Medical University, No. 45 Changchun Street, Xicheng District, Beijing, 100053, People’s Republic of China, Email [email protected]
Objective: To elucidate the molecular mechanisms by which aging exacerbates the severity of the Acute Respiratory Distress Syndrome (ARDS), with a specific focus on identifying and validating a novel signaling axis involving MAPK14, ADM, and MAPK8.
Methods: We performed an integrated analysis of ARDS gene expression data, focusing on aging and immune-related genes. Our approach included functional enrichment, machine learning, transcription factor prediction, and immune infiltration analysis. Computational drug repositioning identified potential therapeutics. Findings were validated in an LPS-induced murine ARDS model using young and aged mice, with assessments via qRT-PCR, H&E staining, BALF cell counts, and cytokine quantification.
Results: Our analysis revealed that MAPK8 was significantly downregulated in ARDS and exhibited a strong negative correlation with IL-10 (r = − 0.63). Conversely, MAPK14 and ADM were upregulated and demonstrated a positive association with pro-inflammatory M1 macrophages and γδ T cells. A robust positive correlation was observed between MAPK14 and the transcription factor CEBPA (r = 0.86). Co-enrichment analysis of ADM and MAPK14 at the cisbp__M0666 motif suggested a potential co-regulatory mechanism. Among the candidate therapeutics identified through drug repositioning, fostamatinib emerged as a promising agent to counteract the ARDS-associated expression signature. The in vivo experiments corroborated our computational findings, demonstrating that aging significantly exacerbated ARDS severity. Aged ARDS mice exhibited more severe lung injury, increased neutrophil infiltration, and elevated levels of IL-1β and IL-6. Furthermore, the upregulation of MAPK14 and ADM was more pronounced in aged mice compared to their younger counterparts (P < 0.05), while MAPK8 downregulation was similarly intensified with aging.
Conclusion: This integrated study reveals a novel MAPK14/ADM/MAPK8 signaling axis as a key mechanism of age-related inflammatory dysregulation in ARDS. Aging amplifies this pro-inflammatory pathway, worsening tissue damage. Targeting this axis may represent a promising therapeutic strategy for elderly ARDS patients.
Keywords: bioinformatics, ARDS, MAPK signaling pathway, inflammaging, biomarker, immune deconvolution
Introduction
Acute respiratory distress syndrome (ARDS) is a severe clinical syndrome characterized by diffuse alveolar damage, dysregulated pulmonary inflammation, and refractory hypoxemia. Although supportive care has improved, mortality remains high, particularly among older patients.1,2 Advanced age is widely recognized as an independent risk factor for ARDS incidence and poor outcomes, consistent with recent meta-regression and cohort evidence showing increasing mortality with age and reduced physiological reserve.3–5 Collectively, these observations suggest that the aging lung resides in a vulnerability-prone biological state; however, the molecular programs that render older individuals hypersensitive to inflammatory lung injury have not been fully elucidated.
ARDS pathophysiology involves early hyperinflammation driven by innate immune activation and cytokine release (eg, IL-1β, IL-6, TNF-α), along with impaired resolution that sustains tissue injury.6 Specific immune subsets, including pro-inflammatory M1 macrophages and γδ T cells, have been implicated in amplifying lung damage.7–9 In older individuals, these responses unfold on a background of “inflammaging”—chronic low-grade inflammation accompanied by immunosenescence and senescence-associated secretory phenotypes (SASP).10,11 These senescent cells constitutively release pro-inflammatory cytokines and matrix-degrading enzymes, creating a “primed” environment that amplifies cytokine storms upon secondary insults like sepsis or pneumonia.12,13 Consistent with this concept, uncontrolled systemic inflammation—often described as a cytokine storm—has been implicated as a major driver of ARDS progression toward multi-organ dysfunction and death.14 Moreover, experimental studies suggest that senescent-cell accumulation can directly impair vascular repair and promote pulmonary edema, whereas senescent-cell depletion mitigates adverse remodeling in lung-injury models.15 Together, these findings support a mechanistic link between aging biology and ARDS severity; nevertheless, the upstream coordination of age-related immune and senescence phenotypes by therapeutically actionable signaling networks remains to be fully elucidated.
A critical knowledge gap persists in linking these cellular phenotypes to specific, actionable molecular pathways. Within the landscape of inflammatory signaling, the Mitogen-Activated Protein Kinase (MAPK) pathways are central regulators. In particular, MAPK14 (p38α) is a well-established mediator of cytokine production, stress adaptation, and apoptosis, and its activation has been associated with adverse outcomes across inflammatory lung conditions and senescence-related programs.16,17 In parallel, adrenomedullin (ADM)—a vasoactive peptide with endothelial and immunomodulatory functions—is frequently elevated in sepsis and ARDS and may reflect, or contribute to, barrier stress responses.18 However, prior studies have often examined these molecules in isolation or primarily in young models, leaving unresolved whether they participate in an integrated, age-sensitive network. In addition, contemporary machine-learning approaches (eg, Random Forest and support vector machine–based feature selection) provide robust tools to prioritize biologically relevant drivers from high-dimensional transcriptomic data, although these computational outputs require experimental validation to establish their pathophysiological relevance.19,20 Consequently, an important unmet need is a coherent, experimentally supported framework that explains how MAPK14, ADM, and MAPK8 (JNK1) may interact as a signaling axis to shape the exaggerated inflammatory injury observed in aged ARDS.
To address this gap, the present study employed a rigorous “in silico to in vivo” translational strategy. We first utilized advanced machine learning algorithms, including Random Forest (RF) and Support Vector Machine-Recursive Feature Elimination (SVM-RFE), to unbiasedly screen for robust molecular drivers at the intersection of ARDS, aging, and immune regulation. This computational approach identified a novel MAPK14/ADM/MAPK8 signaling axis. Crucially, we moved beyond bioinformatic prediction to experimentally validate this axis in a murine model of LPS-induced ARDS, directly comparing young (3-month-old) versus aged (18-month-old) mice. Our findings demonstrate that aging does not merely alter the baseline but significantly amplifies the induction of this pro-inflammatory axis during lung injury. By elucidating this mechanism and identifying potential repositioned drugs such as fostamatinib, this study aims to provide a mechanistic basis for the age-related severity of ARDS and propose novel therapeutic strategies for this vulnerable population.
Materials and Methods
Data Acquisition and Preprocessing
The gene expression profile of dataset GSE76293 was retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/, accessed October 11, 2022). This dataset comprises comprehensive transcriptomic data from 12 patients with ARDS and 12 healthy controls. Raw data were processed and normalized using the limma package (version 3.58.1) in R software (version 4.3.0; R Foundation for Statistical Computing, Vienna, Austria). Differential expression analysis was performed to identify Differentially Expressed Genes (DEGs) based on the criteria of an adjusted p-value (adj.P.Val) < 0.05 and |log2 fold change (FC)| ≥ 1.0. Three curated gene sets were assembled for downstream intersection analyses: ARDS-related genes (n = 3867), sourced from the GeneCards database (https://www.genecards.org/, accessed on October 11, 2022); Aging-related genes (n = 273), retrieved from the Molecular Signatures Database (MSigDB) (https://www.gsea-msigdb.org/, accessed October 11, 2022); Immune-related genes (n = 1793), obtained from the Immunology Database and Analysis Portal (ImmPort) (https://www.immport.org/, accessed October 11, 2022).
Functional Enrichment Analysis
Functional enrichment analysis was performed using the Metascape online platform (https://metascape.org/, accessed November 2, 2022). This tool was employed for the integrated annotation of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using the list of differentially expressed genes. The analysis was conducted with the platform’s standard parameters: a minimum overlap (gene count) of 3, a p-value cutoff of 0.01, and the use of the hypergeometric test with Benjamini-Hochberg correction for multiple comparisons. Statistically significant enrichment terms were selected based on these criteria.
Machine Learning-Based Hub Gene Identification
To screen for the most robust hub genes from the DEGs, two independent machine learning algorithms were employed, and their results were intersected.
- Support Vector Machine-Recursive Feature Elimination (SVM-RFE): This algorithm was implemented using the e1071 and caret packages. The linear kernel was used. A recursive procedure embedded within a 5-fold cross-validation framework was employed to rank features. In each iteration, a linear SVM classifier (with cost parameter C=10) was trained, and features were ranked based on the squared weight coefficients (for high-dimensional data, ranked by mean (w2)/sd (w2)). The feature with the smallest ranking criterion was recursively eliminated until the optimal feature subset was identified. The final feature set was determined as the subset yielding the lowest average cross-validation error.21
- Random Forest (RF): The classification model was constructed using the randomForest package. The algorithm was configured with 500 trees (ntree = 500), and the optimal number of variables tried at each split (mtry) was determined based on the minimum out-of-bag (OOB) error rate. Feature importance was quantified using the %IncMSE (percent increase in mean squared error) metric. Genes with an importance score greater than 2.0 were retained as candidate features.
The intersection of candidate genes identified by both RF and SVM-RFE algorithms was designated as the final set of hub genes for downstream validation.
Immune Infiltration Analysis
The relative abundance of 22 distinct immune cell types in the lung tissue samples was estimated using the CIBERSORT algorithm (Cell-type Identification By Estimating Relative Subsets of RNA Transcripts, v 0.1.0, assessed November 2, 2022). This deconvolution method utilizes the LM22 leukocyte gene signature matrix. Permutations were set to 1000 to robustly estimate statistical significance. Only samples with a CIBERSORT p-value < 0.05 were included in the subsequent correlation analysis. The association between hub gene expression and immune cell infiltration levels was evaluated using Spearman’s rank correlation analysis.
Regulatory Network Construction and GSEA
To explore the transcriptional regulatory network, transcription factor (TF) enrichment analysis was performed using the RcisTarget R package (version 1.19.2). Motif enrichment was evaluated using the hg19 ranking database rcistarget.hg19.motifdb.cisbpont.500bp, which provides motif rankings for 500-bp regulatory regions as defined in the database. Enrichment was quantified by the area under the recovery curve (AUC), and the normalized enrichment score (NES) was calculated by normalizing motif AUC values to the background distribution across all motifs, following the standard RcisTarget workflow.22 Motifs with NES > 3.0 were considered significantly enriched.
Gene Set Enrichment Analysis (GSEA) was conducted to determine whether predefined gene sets were enriched at the extremes (top or bottom) of a ranked list of genes ordered by differential expression between ARDS and control samples. Signaling pathways dysregulated in high- and low-expression groups of hub genes were identified using GSEA, with 1000 permutations and a phenotype-based permutation type.
miRNA Network Construction and Drug Repositioning Analysis
Putative miRNA–mRNA interactions involving the hub genes were obtained from the Mircode database (http://mirtoolsgallery.tech/mirtoolsgallery/node/1189, assessed November 2, 2022) and visualized as a regulatory network. Potential therapeutic compounds targeting the ARDS-associated gene signature were predicted using the Connectivity Map (CMap) database (https://www.broadinstitute.org/connectivity-map-cmap, accessed on November 2, 2022). The top upregulated and downregulated genes were submitted as the query signature. Compounds with an enrichment score < −0.90 were prioritized as candidates predicted to reverse the ARDS-associated transcriptional signature.
Experimental Animals and ARDS Modeling
Ethical Approval for Animal Experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Xuanwu Hospital, Capital Medical University (approval number: XW-20231220-1). The experiments were conducted in strict accordance with a comprehensive framework that integrates Chinese national standards and internationally recognized guidelines, including the Guideline for Ethical Review of Laboratory Animal Welfare (GB/T 35892–2018), the Guideline of Assessment for Humane Endpoints in Animal Experiment (RB/T 173–2018), the NIH Guide for the Care and Use of Laboratory Animals (United States), and the ARRIVE guidelines 2.0. Humane endpoints were predefined as body weight loss exceeding 20%, severe neurological deficits, or inability to access food or water. Mice were monitored every 6 hours and any animal meeting these criteria was immediately euthanized by cervical dislocation under deep anesthesia.
Animal Housing and Conditions
Male C57BL/6 mice were divided into two age groups: Young (3-month-old, weighing 23 ± 2 g) and Aged (18-month-old, weighing 32 ± 3 g). Mice were obtained from Zhishan Institute of Healthcare Research Co., Ltd. (Beijing, China) and housed in groups of 3–5 under specific pathogen-free conditions, with environmental enrichment, at a temperature of 22 ± 2°C, 40–60% relative humidity, and a 12-hour light/dark cycle. Standard rodent diet and water were provided ad libitum. All animals were acclimatized for one week prior to experimentation.
Group Allocation and LPS-Induced ARDS
A total of 24 mice were allocated to four groups (n = 6 per group) using a computerized random number generator to ensure unbiased assignment:
- Young Control (YC): 3-month-old mice receiving normal saline
- Young ARDS (YA): 3-month-old mice receiving LPS (3 mg/kg)
- Aged Control (AC): 18-month-old mice receiving normal saline
- Aged ARDS (AA): 18-month-old mice receiving LPS (2.25 mg/kg)
After anesthesia with intraperitoneal pentobarbital sodium (90 mg/kg), mice were placed in a supine position with a 60° head-up tilt. The jaw and tongue were extended, and lipopolysaccharide (LPS, from E. coli O55:B5, Sigma-Aldrich) or normal saline (NS) was administered into the oropharynx via a 24-gauge angiocatheter (BD Biosciences). To obtain a comparable injury window and reduce excessive mortality in aged mice, the LPS dose was adjusted based on pilot titration, resulting in 3 mg/kg (young) and 2.25 mg/kg (aged). LPS was administered at a dose of 3 or 2.25 mg/kg dissolved in 50 µL of sterile normal saline. Control mice received an equal volume of NS. Aspiration of the instillate was visually confirmed. The order of intratracheal instillation was randomized across all groups. Although cage location was not systematically rotated to avoid stress, all cages were maintained in the same room under standardized environmental conditions.
Sample Size Calculation
The sample size was determined by a priori power analysis using SPSS. The primary outcome was the Bronchoalveolar Lavage Fluid (BALF) protein concentration at 24 hours post-modeling. Based on pilot data (n=3 per group) showing BALF protein concentrations of 120.59 ± 20.84 µg/mL (control) and 325.68 ± 108.1 µg/mL (LPS), an effect size (Cohen’s d) of 2.63 was estimated. For an independent t-test with 80% power and a two-tailed α of 0.05, 4 mice per group were required. The sample size was increased to 6 per group to account for potential procedural mortality.
Randomization and Blinding
Randomization was performed at the level of individual mice. An independent researcher generated the allocation sequence using the =RAND() function in Microsoft Excel and concealed it in sealed, opaque envelopes. The experimenter performing the procedures opened each envelope immediately prior to the first intervention, ensuring allocation concealment. Solutions of LPS and normal saline were prepared by an independent technician, and the operator conducting intratracheal instillation and subsequent assessments was blinded to group identity throughout the experiment. Data were analyzed by a statistician blinded to group codes, which were revealed only after the final analysis.
Sample Collection and Processing
Twenty-four hours post-instillation, mice were euthanized via anesthetic overdose. The left lung was lavaged with PBS to collect BALF, which was centrifuged, and the supernatant was stored at −80°C. The right lung was divided: one lobe was fixed in 4% paraformaldehyde for histology, and the remainder was snap-frozen for further analysis.
Histopathological Examination of Lung Tissues
For histopathological evaluation, lung tissues were fixed in 4% paraformaldehyde, processed through paraffin embedding, and sectioned. Tissue sections were then stained with hematoxylin and eosin (H&E; Vector Labs, Burlingame, CA, USA) using a standard protocol. All pathological assessments were conducted in a blinded manner by an experienced investigator using well-defined criteria, as referenced.23
BALF Total Protein and Cytokines Quantification
The total protein content in BALF supernatants was measured using a bicinchoninic acid (BCA) assay kit (Pierce Biotechnology, Rockford, IL, USA). Simultaneously, the levels of key inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-10) were evaluated using specific commercial enzyme-linked immunosorbent assay (ELISA) kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). All assays were performed in duplicate according to the manufacturers’ instructions, and sample concentrations were interpolated from standard curves.
BALF Cell Counts and Differentials
After collection, BALF samples were immediately centrifuged to pellet the cells. The total cell count was enumerated manually using a hemocytometer. For differential analysis, the cell pellets were resuspended and used to prepare cytospin slides, which were then stained with Giemsa. A blinded observer counted and classified at least 200 cells per slide based on standard morphological criteria to determine the proportions of different leukocyte types.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from the snap-frozen lung tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA concentration and purity were assessed using a NanoDrop spectrophotometer. First-strand cDNA was synthesized from 1 µg of total RNA using the PrimeScriptTM RT Reagent Kit (Takara, Shiga, Japan) as per the manufacturer’s instructions. qRT-PCR was performed on an Applied Biosystems 7500 Real-Time PCR System using the Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific, USA). The thermal cycling conditions were: initial denaturation at 95°C for 10 minutes, followed by 40 cycles of denaturation at 95°C for 15 seconds and a combined annealing/extension at 60°C for 1 minute. Each sample was run in triplicate. A melt curve analysis was performed at the end of each run to confirm amplification specificity. Relative gene expression was calculated using the 2−ΔΔCt method, with GAPDH serving as the internal normalization control. The primer sequences are designed and synthesized by Tsingke Biological Technology (Beijing, China) and the specific primer sequences are listed in Supplementary Table S1.
Statistical Analysis
All quantitative data are presented as mean ± SD unless otherwise stated. Statistical analyses were performed using R (version 4.3.0) and GraphPad Prism (version 10.1.2). For in vivo experiments involving multiple groups, differences were assessed by two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. For comparisons between two groups, an unpaired two-tailed Student’s t-test was used for normally distributed data with homogeneous variance; otherwise, the Mann–Whitney U-test was applied. Correlations were assessed using Pearson’s correlation for approximately normally distributed variables and Spearman’s rank correlation otherwise. A two-tailed P < 0.05 was considered statistically significant. No data or animals were excluded. Statistical analyses were performed in a blinded manner by two independent investigators.
Results
Identification of an ARDS-, Aging-, and Immune-Related Gene Signature
The analysis workflow is summarized in Figure 1. To systematically investigate the molecular basis of age-related ARDS severity, we first analyzed the GSE76293 dataset. Differential expression analysis identified 3428 DEGs between ARDS patients and controls (adjusted P < 0.05; Figure 2A). By intersecting these DEGs with predefined ARDS-, aging-, and immune-related gene sets, we identified 10 common genes: MAPK14, MAPK8, MAP2K1, ADM, EDNRB, RETN, PTGS2, HAMP, TFRC, and CAT (Figure 2B). Functional enrichment analysis revealed that these intersecting genes were significantly enriched in biological processes such as “response to lipopolysaccharide” and “regulation of apoptotic signaling pathway” (Figure 2C and D), suggesting that these molecules serve as critical convergence points integrating aging signals with acute inflammatory responses.
 |
Figure 1 Schematic workflow of the integrated bioinformatic and experimental study. The flowchart outlines the key steps: acquisition and processing of ARDS transcriptomic data (GSE76293), identification of differentially expressed genes (DEGs) intersecting with aging and immune gene sets, hub gene selection via machine learning (Random Forest and SVM-RFE), subsequent immune infiltration, regulatory network, and drug prediction analyses, and final experimental validation in a murine model of LPS-induced ARDS comparing young and aged mice. |
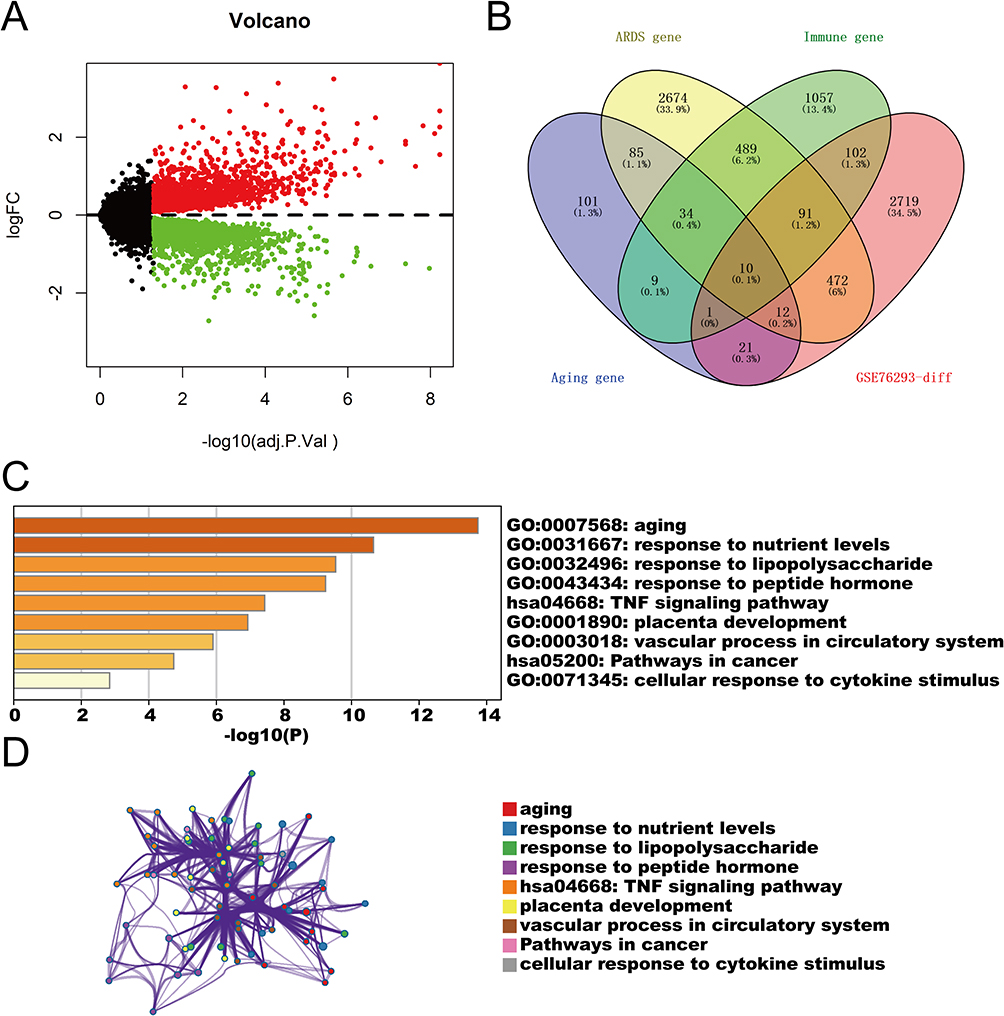 |
Figure 2 Identification of shared gene signatures linking ARDS, aging, and immune response. (A) Volcano plot displaying differentially expressed genes (DEGs) between control and ARDS samples (adjusted P < 0.05). Red and green dots represent up- and downregulated genes, respectively. (B) Venn diagram showing the intersection of 3428 DEGs with ARDS-, aging-, and immune-related gene sets, identifying 10 common candidate genes. (C) Bar graph of the top enriched functional terms (GO and KEGG) for the intersecting genes. (D) Network diagram depicting interactions among the enriched pathways. These results highlight a molecular convergence of aging and immune signaling in the pathogenesis of ARDS. |
Robust Selection of Hub Genes via Machine Learning
To pinpoint the most robust drivers among the identified candidates, we employed an integrated machine learning approach. The SVM-RFE algorithm identified a subset of genes with minimal generalization error (Figure 3A), while the Random Forest algorithm ranked genes based on classification importance (Figure 3B). The intersection of top candidates from both algorithms yielded three core hub genes: MAPK14, ADM, and MAPK8 (Figure 3C). This multi-algorithmic validation ensures that these targets are not artifacts of a single method but represent stable features of the disease state.
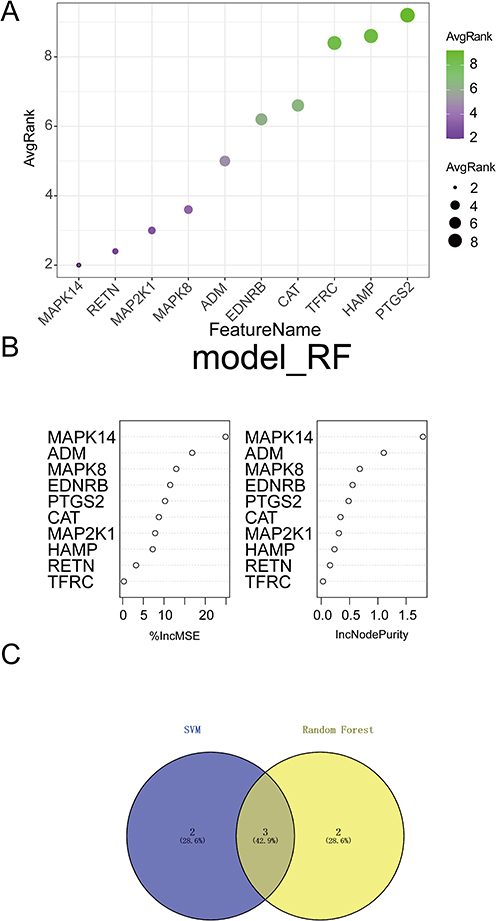 |
Figure 3 Robust identification of hub genes using machine learning algorithms. (A) Feature selection using Support Vector Machine-Recursive Feature Elimination (SVM-RFE), plotting accuracy against the number of features. (B) Ranking of candidate genes based on feature importance derived from the Random Forest (RF) algorithm. (C) Venn diagram showing the intersection of top candidates from RF and SVM-RFE. Three key hub genes—MAPK14, ADM, and MAPK8—were identified as the most robust molecular features distinguishing ARDS. |
Hub Genes Correlate with Immune Infiltration in ARDS
The immune microenvironment, composed of immune-related stromal cells, infiltrating immune cells, extracellular matrix components, and multiple cytokines and growth factors, plays an important role in disease diagnosis, prognosis, and therapeutic responsiveness. To explore potential mechanisms by which the hub genes contribute to ARDS progression, we examined the relationship between hub gene expression and immune cell infiltration in the ARDS dataset. The estimated proportions of immune cell subsets in each sample are shown in Figure 4A. Correlation analysis among immune infiltration levels revealed multiple significant relationships between immune cell subsets (Figure 4B). Compared with controls, the ARDS group exhibited significantly higher levels of naïve CD4+ T cells and γδ T cells (P < 0.05; Figure 4C). We further assessed the associations between hub genes and immune cell subsets. ADM expression was positively correlated with M1 macrophages, naïve CD4+ T cells, and γδ T cells, but negatively correlated with CD8+ T cells and follicular helper T cells (Figure 4D). MAPK8 expression was negatively correlated with M0 macrophages (Figure 4E). In addition, MAPK14 expression was positively correlated with γδ T cells and negatively correlated with CD8+ T cells and follicular helper T cells (Figure 4F). Collectively, these results suggest that the hub genes are closely associated with immune cell infiltration patterns in ARDS and may be involved in the immune microenvironment in ARDS.
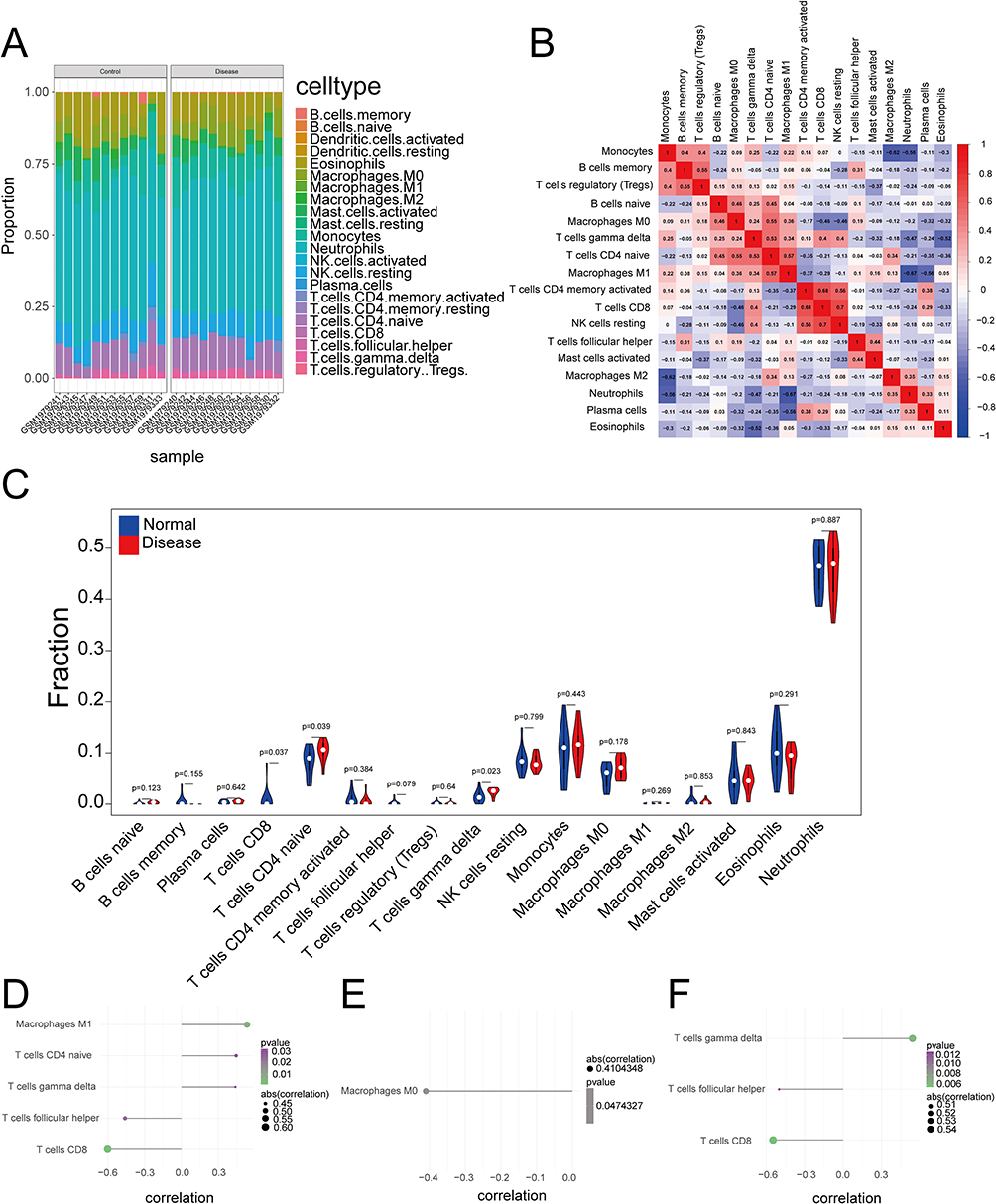 |
Figure 4 Immune landscape characterization and its correlation with hub genes. (A) Relative proportions of 22 immune cell types in each sample estimated by CIBERSORT. (B) Correlation matrix of immune cell subtypes. (C) Box plots comparing immune cell abundance between control and ARDS groups (*P < 0.05). (D–F) Scatter plots showing Spearman correlations between hub gene expression (MAPK14, ADM, MAPK8) and specific immune cell subsets. The findings demonstrate that MAPK14 and ADM expression is strongly linked to a pro-inflammatory shift involving γδ T cells and M1 macrophages. |
Functional Enrichment and Transcriptional Regulation Analysis
To elucidate the downstream mechanisms of the hub genes, we performed GSEA. High expression of MAPK14 and ADM was enriched in pathways related to “nitrogen metabolism” and “Fc gamma R-mediated phagocytosis” (Figure 5A and B), implicating them in metabolic-immune crosstalk. In contrast, MAPK8 was associated with fundamental cellular processes such as “protein export” and “autophagy regulation” (Figure 5C).
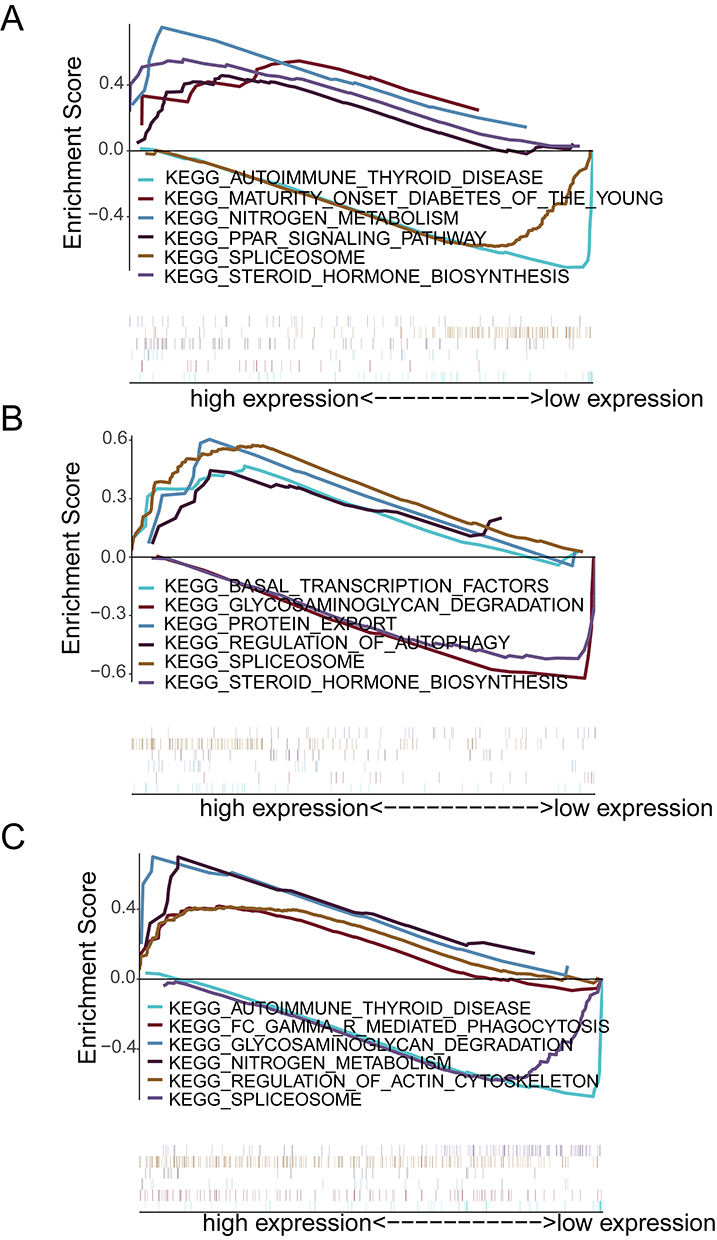 |
Figure 5 Functional enrichment analysis of individual hub genes. Gene Set Enrichment Analysis (GSEA) plots for (A) MAPK14, (B) ADM, and (C) MAPK8. Each panel displays the top signaling pathways enriched in the high-expression phenotype. These analyses suggest distinct roles for the hub genes, with MAPK14 and ADM predominantly driving metabolic and inflammatory signaling pathways. |
Motif enrichment analysis suggested candidate upstream transcriptional regulators, with cisbp_M0666 showing the highest enrichment (NES = 8.25) and identifying MAPK14 and ADM as putative motif-associated genes (Supplementary Figure S1 and Supplementary Table S2). This suggests they may be co-regulated, forming a coherent module. Furthermore, correlation analysis with established ARDS-related regulatory genes from GeneCards revealed highly significant associations: MAPK14 expression was strongly positively correlated with CEBPA (r = 0.86), a transcription factor critical in inflammation, while MAPK8 was strongly negatively correlated with IL-10 (r = −0.63), a key anti-inflammatory cytokine (Figure 6A and B). These correlations directly link the novel hub axis to established molecular players in ARDS immunopathology.
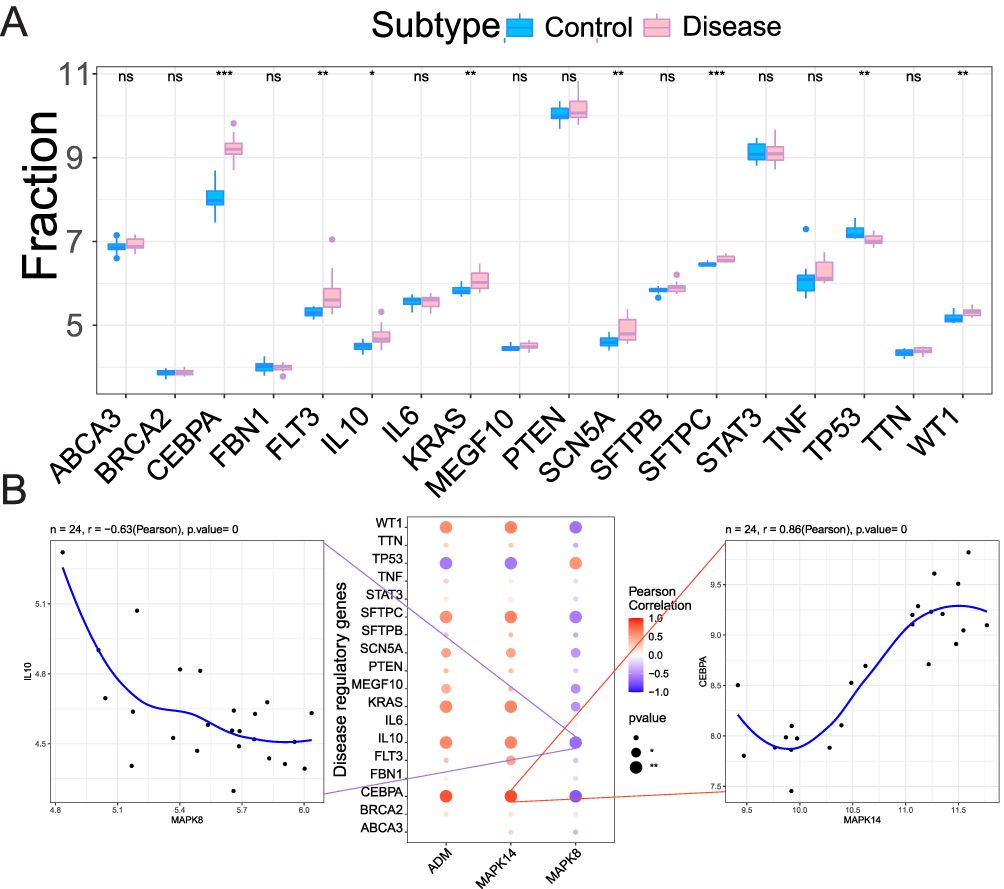 |
Figure 6 Disease gene associations and correlation analysis. (A) Box plots comparing key ARDS regulatory genes (from GeneCards) between the Control and ARDS groups. Statistical significance is indicated above each gene. * P < 0.05, ** P < 0.01, *** P < 0.001. (B) Correlation analysis between the three hub genes and the ARDS regulatory genes. Correlation coefficients (r) and P values are shown in the plots.The strong positive correlation between MAPK14 and CEBPA (r = 0.86) and the strong negative correlation between MAPK8 and IL-10 (r = −0.63) are highlighted, suggesting a potential mechanism regulating myeloid cell differentiation and function. Abbreviation: ns, not significant. |
Construction of the miRNA–mRNA Network and Drug Repositioning
To explore translational implications, we constructed a regulatory network of miRNAs targeting the three hub genes, identifying 73 miRNAs forming 122 regulatory pairs (Figure 7A). More importantly, querying the Connectivity Map database with the ARDS-specific gene signature identified several compounds whose expression signatures negatively correlated with the disease perturbation. Among the top hits were fostamatinib (a SYK inhibitor), ZG-10, mitomycin-C, and nialamide (Figure 7B–E). This in silico drug repositioning suggests these agents, particularly fostamatinib, have the potential to reverse the disease-associated gene expression pattern centered on our identified axis.
 |
Figure 7 miRNA–mRNA regulatory network and drug repositioning. (A) Regulatory network of miRNAs (pink nodes) targeting the hub gene mRNAs (purple nodes: MAPK14, ADM, MAPK8). Edges represent predicted targeting relationships. (B–E) Two-dimensional chemical structures and mechanism of action of the top four candidate therapeutic compounds identified by Connectivity Map analysis: (B) ZG-10 (JNK inhibitor), (C) Mitomycin C (DNA alkylating agent), (D) Fostamatinib (SYK inhibitor), and (E) Nialamide (monoamine oxidase inhibitor). These compounds show potential to reverse the ARDS-associated gene signature, with fostamatinib targeting upstream kinases of the identified axis. |
Aging Exacerbates ARDS Severity and Amplifies the MAPK14/ADM Axis in vivo
To experimentally validate our computational findings, we established an LPS-induced ARDS model in young (3-month) and aged (18-month) mice. Notably, despite receiving a lower dose of LPS, aged mice developed significantly more severe lung pathology, characterized by extensive alveolar septal thickening and hyaline membrane formation (Figure 8A and B). This aggravated phenotype was accompanied by greater pulmonary permeability (increased BALF protein, Figure 8C) and a more profound “cytokine storm,” with significantly higher levels of IL-1β, IL-6, and IL-10 in aged mice compared to their young counterparts (Figure 8D–G).Consistent with the immune infiltration analysis, aged ARDS mice showed increased recruitment of neutrophils and monocytes (Figure 8H–J). Most importantly, qRT-PCR analysis confirmed the specific dysregulation of the identified axis: while MAPK14 and ADM were upregulated in all ARDS mice, this induction was significantly amplified in the aged group (P < 0.05 vs. Young ARDS; Figure 8K and L). In parallel, MAPK8 downregulation was observed (Figure 8M). These in vivo data provide definitive evidence that aging does not merely alter the baseline, but specifically potentiates the MAPK14/ADM-driven inflammatory response during ARDS.
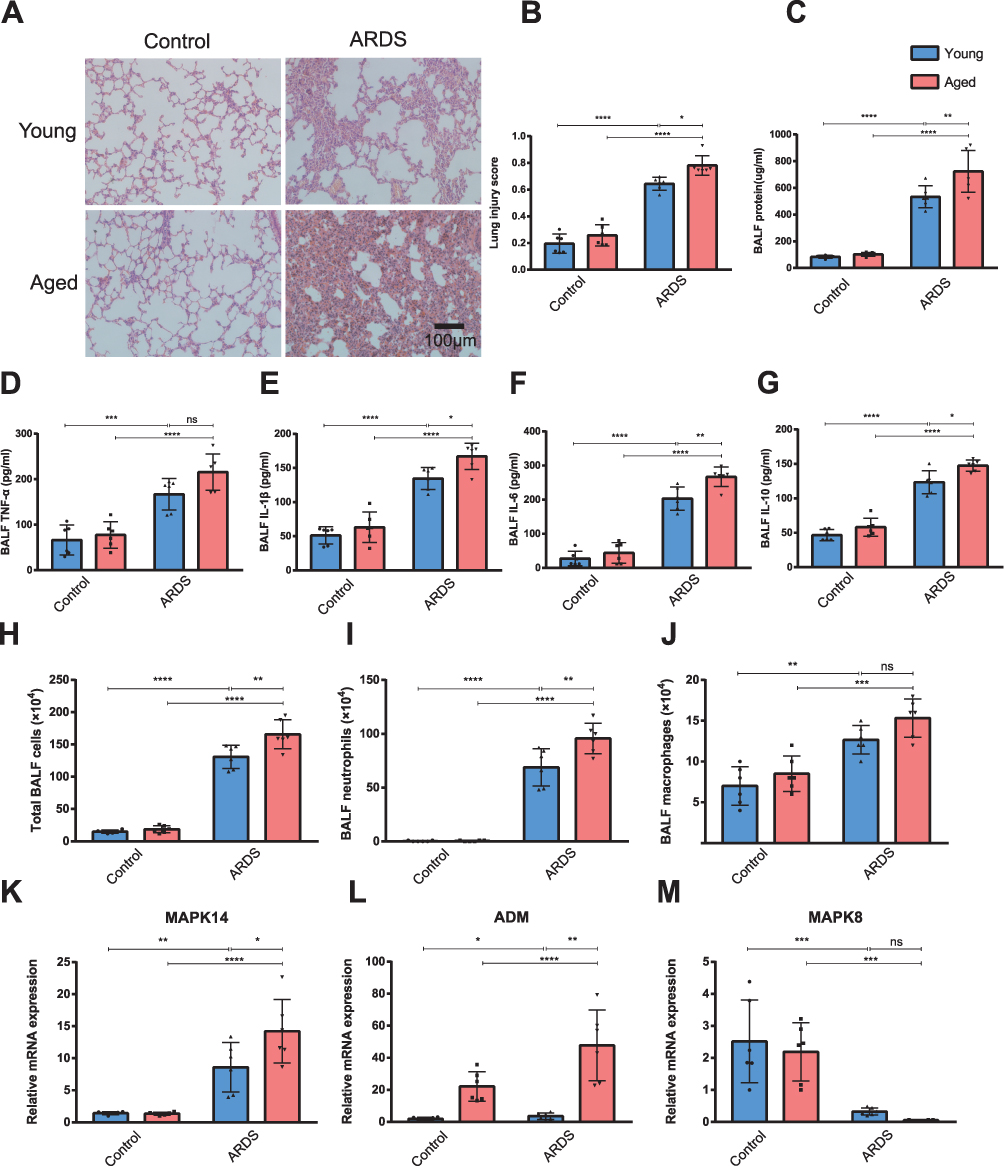 |
Figure 8 Validation of the MAPK14/ADM/MAPK8 axis in an aged murine ARDS model. (A) Representative hematoxylin and eosin (H&E)-stained lung tissue sections from young and aged mice. Scale bar: 100 μm. (B) Lung injury scoring based on histopathological features: neutrophils in alveolar and interstitial spaces, hyaline membrane formation, proteinaceous debris in airspaces, and alveolar septal thickening. (C) Total protein concentration in bronchoalveolar lavage fluid (BALF) 24 hours post-LPS exposure. (D–G) Concentrations of TNF-α, IL-1β, IL-6, and IL-10 in BALF. (H–J) Total and differential cell counts in BALF. (K–M) Relative mRNA expression of the hub genes MAPK14, ADM, and MAPK8 in lung tissues normalized to GAPDH. Data are mean ± SD (n=6). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Results demonstrate that aging significantly exacerbates lung injury and specifically amplifies the induction of MAPK14 and ADM compared to young mice. Abbreviation: ns, not significant. |
Discussion
In this study, we integrate transcriptomic bioinformatics with in vivo validation to define a focused, mechanistically informative MAPK14/ADM/MAPK8 axis associated with ARDS severity. Our central finding—that aging does not merely alter the baseline but specifically amplifies the induction of MAPK14 and ADM while suppressing MAPK8 during lung injury—supports a model of “frail” inflammatory regulation.
ARDS outcomes are disproportionately poor in older individuals, yet the molecular mechanisms linking age-associated immune dysregulation to severe inflammatory lung injury remain incompletely defined. In the present study, transcriptomic analyses integrated with in vivo validation delineated a MAPK14/ADM/MAPK8 expression pattern associated with ARDS and accentuated by aging. Aging was accompanied by greater induction of MAPK14 and ADM and deeper suppression of MAPK8 during LPS-induced lung injury, together with more severe histopathology, increased alveolar permeability, and higher inflammatory readouts. These observations were consistent with the concept of inflammaging and immunosenescence, in which chronic low-grade inflammation and impaired immune regulation predispose older hosts to exaggerated responses to acute insults.24,25
p38 MAPK14 was prioritized as an upregulated hub gene and showed stronger induction in aged ARDS mice. Prior mechanistic studies have established p38 MAPK as a central stress-activated kinase that regulates inflammatory cytokine production and has been implicated in senescence-associated secretory phenotypes (SASP).26–29 In parallel, studies in lung injury and ARDS have linked p38 MAPK signaling to inflammatory amplification, including chemokine and cytokine cascades that promote neutrophil recruitment and tissue injury.30–32 Viewed together, these lines of evidence suggest that p38 MAPK is plausibly positioned at the interface between aging-associated inflammatory priming and acute lung injury. Our results extended this literature by indicating that MAPK14-associated transcriptional activation was age-sensitive in this model and occurred alongside a broader axis shift involving ADM and MAPK8. The observed correlation between MAPK14 and CEBPA (r = 0.86) suggested that MAPK14-linked programs were associated with established inflammatory regulatory nodes; however, this relationship remained associative and did not establish directionality. Consistent with reports of prolonged neutrophilia and delayed resolution in aging-associated lung injury,31,33 neutrophil accumulation was most pronounced in aged ARDS mice, which supported the interpretation that enhanced MAPK14 induction may reflect, and potentially contribute to, an age-biased inflammatory state. Establishing causality will require targeted perturbation of MAPK14 signaling in age-stratified models.
A distinctive feature of our findings was the age-dependent amplification of ADM. Clinical studies have reported that circulating bioactive ADM or MR-proADM levels are associated with disease severity, endothelial dysfunction, and adverse outcomes in critical illness, including ARDS-related contexts, a pattern often interpreted as a marker of barrier disruption rather than unequivocal protection.34–36 Experimental studies have nevertheless suggested that ADM can reduce vascular hyperpermeability and improve outcomes in septic settings, indicating that its role may be context- and stage-dependent.37 In aging, ADM regulation appears altered, with evidence for elevated basal expression accompanied by impaired dynamic responsiveness to acute stress.38,39 In this context, the stronger ADM induction observed in aged ARDS mice suggested an intensified barrier-stress response in the aged lung. ADM also showed a positive association with M1 macrophage fractions in immune deconvolution analyses, indicating that ADM upregulation co-occurred with a pro-inflammatory immune milieu. Because computational deconvolution and correlation do not establish cellular sources or mechanistic effects, these observations should be interpreted as indicating linkage rather than proving ADM-driven macrophage polarization. Together with prior clinical and experimental evidence, our data support a testable model in which ADM reflects compensatory endothelial stress signaling that may become insufficient or dysregulated in aged ARDS.
In contrast to MAPK14 and ADM, MAPK8 was downregulated in ARDS and further suppressed in aged mice. Although JNK signaling has historically been linked to stress-induced apoptosis, more recent evidence indicates context-dependent roles in immune regulation, macrophage plasticity, and antimicrobial responses.40,41 Accordingly, reduced MAPK8 expression could indicate diminished stress-response balance or impaired immune adaptability rather than simply reduced pro-apoptotic signaling. The negative correlation between MAPK8 and IL10 (r = −0.63) suggested that MAPK8 suppression co-occurred with reduced anti-inflammatory signaling, which may indicate weakened “braking” capacity; however, this inference remained indirect and requires mechanistic validation. This interpretation is broadly consistent with the notion that aging preferentially dysregulates immune response termination rather than initiation.42 Such a shift could contribute to persistent inflammation and susceptibility to late complications, including secondary infections that are common contributors to mortality in ARDS.43
The immune remodeling patterns identified in ARDS samples were generally consistent with prior ARDS immunopathology literature implicating innate immune activation, neutrophil-driven tissue injury, and macrophage-mediated inflammatory amplification, with roles proposed for γδ T cells and pro-inflammatory macrophage states.7–9,33 In our analysis, increased γδ T-cell fractions and correlations between hub gene expression and immune-cell fractions (including MAPK14–γδ T cells and ADM–M1 macrophages) suggested that the MAPK14/ADM/MAPK8 pattern was embedded within an inflammatory cellular context rather than representing isolated transcriptional changes. Nonetheless, because immune infiltration estimates were computational, cell-type–resolved validation (eg, flow cytometry, immunostaining, or single-cell transcriptomics) will be important to confirm cellular sources and map axis activity across immune and structural cell compartments, particularly in aged lungs.
Connectivity Map analysis prioritized fostamatinib (a SYK inhibitor) as a compound predicted to reverse the ARDS-associated transcriptional signature. SYK lies upstream of multiple inflammatory cascades and may influence MAPK-related pathways, providing a plausible rationale for evaluating SYK inhibition as a strategy to dampen excessive inflammatory amplification. A randomized clinical trial reported clinical benefit of fostamatinib in hospitalized COVID-19 patients with hypoxemia,44 supporting the plausibility of SYK inhibition in severe inflammatory lung disease. However, viral hypoxemic illness is not equivalent to non-viral ARDS, and efficacy in elderly-specific ARDS populations cannot be inferred directly. Therefore, our drug-repurposing results should be viewed as hypothesis-generating and best validated through age-stratified preclinical studies that include mechanistic readouts of MAPK14/ADM/MAPK8 modulation, barrier integrity, immune resolution kinetics, and host-defense competence.
Several limitations define clear directions for future work. First, the transcriptomic findings were derived from a public dataset and computational deconvolution; replication in independent cohorts and platforms, together with protein-level validation in human samples, will strengthen generalizability and clinical relevance. Second, the present study established associations and coordinated expression changes but did not establish causality or pathway directionality within the proposed axis. This can be addressed through genetic perturbation (eg, cell-type–specific manipulation of Mapk14 or Mapk8) and pharmacologic intervention in aged models, coupled with standardized ARDS endpoints to test whether altering the axis shifts injury and resolution trajectories Third, LPS doses differed between age groups to maintain a comparable injury window while limiting excessive mortality in aged mice; additional studies incorporating severity-matched designs and complementary ARDS models will further assess robustness across injury contexts. Finally, candidate drugs were prioritized computationally; intervention experiments in aged animals will be essential to determine efficacy, optimal timing, and safety.
Conclusion
Our integrated bioinformatic and experimental study reveals that aging exacerbates ARDS severity through a novel and dysregulated MAPK14/ADM/MAPK8 signaling axis. We identified this axis as a core molecular signature linking inflammaging with acute lung injury, where aging specifically amplifies the upregulation of MAPK14 and ADM and deepens the suppression of MAPK8, correlating with a more severe pro-inflammatory immune microenvironment and tissue damage. Furthermore, computational drug repositioning nominated the SYK inhibitor fostamatinib as a promising therapeutic candidate capable of reversing this disease-associated gene signature. To advance these findings, future work must establish causality within this axis using targeted perturbations in age-stratified models, validate the therapeutic potential of candidate agents like fostamatinib in preclinical studies of aged subjects, and confirm the clinical relevance of this pathway in independent human ARDS cohorts to explore its utility for patient stratification and age-tailored therapy development.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary information files). Additional datasets supporting the findings are available from the corresponding author upon reasonable request, in alignment with efforts to promote transparency and reproducibility in scientific research.
Ethics Approval
This study utilized publicly available datasets from the Gene Expression Omnibus (GEO) database. As the research involved the secondary analysis of existing, de-identified data, it was granted an exemption from requiring ethical approval by the Review Ethics Committee (REC) of Xuanwu Hospital, Capital Medical University (reference mumber: LYS[2025]436-001). The need for informed consent was waived for the same reason. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Xuanwu Hospital, Capital Medical University (approval number: XW-20231220-1). The experiments were conducted in strict accordance with a comprehensive framework that integrates Chinese national standards and internationally recognized guidelines, including the Guideline for Ethical Review of Laboratory Animal Welfare (GB/T 35892-2018), the Guideline of Assessment for Humane Endpoints in Animal Experiment (RB/T 173-2018), the NIH Guide for the Care and Use of Laboratory Animals (United States), and the ARRIVE guidelines 2.0.
Acknowledgments
The authors sincerely thank the members of the Institute of Cerebrovascular Disease Research and the Department of Neurology at Xuanwu Hospital, Capital Medical University, for their valuable assistance throughout the animal experiments. We extend special gratitude to Prof. Yumin Luo, Dr. Feng Yan, Dr. Rongliang Wang, Dr. Yue Tian, and Dr. Yuyou Huang for their expert support and technical guidance.
Author Contributions
Qing Liu: Conceptualization, Methodology, Investigation, Funding acquisition, Writing – Original Draft.
Cheng Yin: Investigation, Validation, Writing – Review & Editing.
Long Fan: Data Curation, Visualization, Writing – Review & Editing.
Daxian Li: Data Curation, Formal analysis, Writing – Review & Editing.
Tianlong Wang: Conceptualization, Resources, Supervision, Funding acquisition, Writing – Review & Editing.
All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Post-subsidy Funds for National Clinical Research Center, Ministry of Science and Technology of China (Grant No. 303-01-001-0272-03 to T.W.), and the Xuanwu Hospital Science Program for Fostering Young Scholars (Grant No. QNPY2021024 to Q.L.).
Disclosure
Dr Qing Liu reports grants from Xuanwu Hospital Science Program for Fostering Young Scholars, during the conduct of the study; Professor Tianlong Wang reports grants from Post-subsidy Funds for National Clinical Research Center, Ministry of Science and Technology of China, during the conduct of the study. The authors declare no other competing interests.
References
1. Brown R, McKelvey MC, Ryan S, et al. The impact of aging in acute respiratory distress syndrome: a clinical and mechanistic overview. Front Med. 2020;7:589553. doi:10.3389/fmed.2020.589553
2. Smolin B, Raz-Pasteur A, Mashiach T, et al. Mechanical ventilation for older medical patients in a large tertiary medical care center. Eur Geriatr Med. 2021;13(1):253–19. doi:10.1007/s41999-021-00557-6
3. Tonetti T, Di Staso R, Bambini L, et al. Role of age as eligibility criterion for ECMO in patients with ARDS: meta-regression analysis. Crit Care. 2024;28(1):278. doi:10.1186/s13054-024-05074-z
4. Patel BM, Reilly JP, Bhalla AK, et al. Association between age and mortality in pediatric and adult acute respiratory distress syndrome. Am J Respir Crit Care Med. 2024;209(7):871–878. doi:10.1164/rccm.202310-1926OC
5. Papoutsi E, Gkirgkiris K, Tsolaki V, et al. Association between baseline driving pressure and mortality in very old patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2024;210(11):1329–1337. doi:10.1164/rccm.202401-0049OC
6. Liu X, Qiao Q, Li X, et al. Apoptotic neutrophil-mediated inflammatory microenvironment regulation for the treatment of ARDS. Nano Today. 2023;52:15. doi:10.1016/j.nantod.2023.101946
7. Xie B, Wang M, Zhang X, et al. Gut-derived memory γδ T17 cells exacerbate sepsis-induced acute lung injury in mice. Nat Commun. 2024;15(1):6737. doi:10.1038/s41467-024-51209-9
8. Arnholdt C, Kumaraswami K, Götz P, et al. Depletion of γδ T Cells Leads to Reduced Angiogenesis and Increased Infiltration of Inflammatory M1-like Macrophages in Ischemic Muscle Tissue. Cells. 2022;11(9):1490. doi:10.3390/cells11091490
9. Xiong M, Luo R, Zhang Z, et al. IL-27 regulates macrophage ferroptosis by inhibiting the Nrf2/HO1 signaling pathway in sepsis-induced ARDS. Inflamm Res. 2025;74(1):39. doi:10.1007/s00011-024-01986-2
10. Xiao J, Li HS, Satyanarayanan SK, et al. Advancements in targeting macrophage senescence for age-associated conditions. Aging Dis. 2024;16(4):2201–2236. doi:10.14336/AD.2024.0720
11. Huidobro C, Martín-Vicente P, López-Martínez C, et al. Cellular and molecular features of senescence in acute lung injury. Mech Ageing Dev. 2021;193:111410. doi:10.1016/j.mad.2020.111410
12. Hao X, Zhao B, Towers M, et al. TXNRD1 drives the innate immune response in senescent cells with implications for age-associated inflammation. Nat Aging. 2024;4(2):185–197. doi:10.1038/s43587-023-00564-1
13. Kruckow KL, Murray E, Shayhidin E, et al. Chronic TNF exposure induces glucocorticoid-like immunosuppression in the alveolar macrophages of aged mice that enhances their susceptibility to pneumonia. Aging Cell. 2024;23(6):e14133. doi:10.1111/acel.14133
14. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. 2020;93(1):250–256. doi:10.1002/jmv.26232
15. Schafer MJ, White TA, Iijima K, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi:10.1038/ncomms14532
16. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol. 2021;22(5):346–366. doi:10.1038/s41580-020-00322-w
17. Cobo I, Cheng A, Murillo-Saich J, et al. Monosodium urate crystals regulate a unique JNK-dependent macrophage metabolic and inflammatory response. Cell Rep. 2022;38(10):110489. doi:10.1016/j.celrep.2022.110489
18. Spoto S, Basili S, Cangemi R, et al. A focus on the pathophysiology of adrenomedullin expression: endothelitis and organ damage in severe viral and bacterial infections. Cells. 2024;13(11). doi:10.3390/cells13110892
19. Lin M, Xu F, Sun J, et al. Integrative multi-omics analysis unravels the host response landscape and reveals a serum protein panel for early prognosis prediction for ARDS. Crit Care. 2024;28(1):213. doi:10.1186/s13054-024-05000-3
20. Ge C, Lu Y, Shen Z, et al. Machine learning and metabolomics identify biomarkers associated with the disease extent of ulcerative colitis. J Crohns Colitis. 2025;19(2):jjaf020. doi:10.1093/ecco-jcc/jjaf020
21. Nedaie A, Najafi AA. Support vector machine with Dirichlet feature mapping. Neural Netwk. 2017;98:87–101. doi:10.1016/j.neunet.2017.11.006
22. Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi:10.1038/nmeth.4463
23. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725–738. doi:10.1165/rcmb.2009-0210ST
24. Zhou Y, Chen G, Li X, et al. Aging and lung diseases: unraveling mechanisms and therapeutic targets. Chin Med J Pulm Crit Care Med. 2025;3(4):246–272. doi:10.1016/j.pccm.2025.11.005
25. Brandenberger C, Kling KM, Vital M, et al. The role of pulmonary and systemic immunosenescence in acute lung injury. Aging Dis. 2018;9(4):553–565. doi:10.14336/AD.2017.0902
26. Zhang B, Fu D, Xu Q, et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat Commun. 2018;9(1):1723. doi:10.1038/s41467-018-04010-4
27. Haj M, Frey Y, Levon A, et al. The cGAS-STING, p38 MAPK, and p53 pathways link genome instability to accelerated cellular senescence in ATM-deficient murine lung fibroblasts. Proc Natl Acad Sci U S A. 2025;122(2):e2419196122. doi:10.1073/pnas.2419196122
28. Ozdemir SA, Faizan MI, Kaur G, et al. Heterogeneity of cellular senescence, senotyping, and targeting by senolytics and senomorphics in lung diseases. Int J Mol Sci. 2025;26(19):9687. doi:10.3390/ijms26199687
29. Easter M, Hirsch MJ, Harris E, et al. FGF receptors mediate cellular senescence in the cystic fibrosis airway epithelium. JCI Insight. 2024;9(15):e174888. doi:10.1172/jci.insight.174888
30. Lv X, Zheng L, Zhang T, et al. CLCA1 exacerbates lung inflammation via p38 MAPK pathway in acute respiratory distress syndrome. Exp Lung Res. 2024;50(1):85–95. doi:10.1080/01902148.2024.2334262
31. Gibbs KW, Chuang Key -C-C, Belfield L, et al. Aging influences the metabolic and inflammatory phenotype in an experimental mouse model of acute lung injury. J Gerontol a Biol Sci Med Sci. 2021;76(5):770–777. doi:10.1093/gerona/glaa248
32. Sayegh S, Fantecelle CH, Laphanuwat P, et al. Vitamin D3 inhibits p38 MAPK and senescence-associated inflammatory mediator secretion by senescent fibroblasts that impacts immune responses during ageing. Aging Cell. 2024;23(4):e14093. doi:10.1111/acel.14093
33. Vassallo A, Wood AJ, Subburayalu J, et al. The counter-intuitive role of the neutrophil in the acute respiratory distress syndrome. Br Med Bull. 2019;131(1):43–55. doi:10.1093/bmb/ldz024
34. Johnsson P, Fredriksson A, Tung C, et al. Plasma bioactive adrenomedullin on intensive care unit admission is associated with acute respiratory distress syndrome: an observational study. Intensive Care Med Exp. 2023;11(1):10. doi:10.1186/s40635-023-00494-7
35. Johnsson P, Sievert T, Didriksson I, et al. Plasma bioactive adrenomedullin predicts mortality and need for dialysis in critical COVID-19. Sci Rep. 2024;14(1):23787. doi:10.1038/s41598-024-74380-x
36. Spoto S, Agrò FE, Sambuco F, et al. High value of mid-regional proadrenomedullin in COVID-19: a marker of widespread endothelial damage, disease severity, and mortality. J Med Virol. 2021;93(5):2820–2827. doi:10.1002/jmv.26676
37. Hwang ISS, Fung ML, Liong EC, et al. Age-related changes in adrenomedullin expression and hypoxia-inducible factor-1 activity in the rat lung and their responses to hypoxia. J Gerontol a Biol Sci Med Sci. 2007;62(1):41–49. doi:10.1093/gerona/62.1.41
38. Temmesfeld-Wollbrück B, Hocke AC, Suttorp N, et al. Adrenomedullin and endothelial barrier function. Thromb Haemost. 2007;98(5):944–951. doi:10.1160/th07-02-0128
39. Karagiannidis C, McAuley DF, Thompson BT, et al. Safety and efficacy of inhaled PEG-ADM in ARDS patients: a randomised controlled trial. Crit Care. 2025;29(1):448. doi:10.1186/s13054-025-05617-y
40. Wang H, Wen C, Chen S, et al. ROS/JNK/C-Jun pathway is involved in chaetocin induced colorectal cancer cells apoptosis and macrophage phagocytosis enhancement. Front Pharmacol. 2021;12:729367. doi:10.3389/fphar.2021.729367
41. Eld HMS, Nielsen EM, Johnsen PR, et al. Cefoxitin treatment of MRSA leads to a shift in the IL-12/IL-23 production pattern in dendritic cells by a mechanism involving changes in the MAPK signaling. Mol Immunol. 2021;134:1–12. doi:10.1016/j.molimm.2021.02.025
42. Pattabiraman G, Palasiewicz K, Galvin JP, et al. Aging-associated dysregulation of homeostatic immune response termination (and not initiation). Aging Cell. 2017;16(3):585–593. doi:10.1111/acel.12589
43. Boumaza A, Gay L, Mezouar S, et al. Monocytes and macrophages, targets of severe acute respiratory syndrome coronavirus 2: the clue for coronavirus disease 2019 immunoparalysis. J Infect Dis. 2021;224(3):395–406. doi:10.1093/infdis/jiab044
44. Collins SP, Shotwell MS, Strich JR, et al. Fostamatinib for hospitalized adults with COVID-19 and hypoxemia: a randomized clinical trial. JAMA Network Open. 2024;7(12):e2448215. doi:10.1001/jamanetworkopen.2024.48215
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identification and Validation of Immune-Related Genes Diagnostic for Progression of Atherosclerosis and Diabetes
Fu Y, Xu L, Zhang H, Ding N, Zhang J, Ma S, Yang A, Hao Y, Gao Y, Jiang Y
Journal of Inflammation Research 2023, 16:505-521
Published Date: 10 February 2023
Preliminary Analysis of Aging-Related Genes in Intracerebral Hemorrhage by Integration of Bulk and Single-Cell RNA Sequencing Technology
Li Q, Wang B, Yang J, Wang Y, Duan F, Luo M, Zhao C, Wei W, Wang L, Liu S
International Journal of General Medicine 2024, 17:2719-2740
Published Date: 12 June 2024
The Interplay of Aging and PANoptosis in Osteoarthritis Pathogenesis: Implications for Novel Therapeutic Strategies
Liu S, Zhang G, Li N, Wang Z, Lu L
Journal of Inflammation Research 2025, 18:1951-1967
Published Date: 10 February 2025
Targeting Senescence, Oxidative Stress, and Inflammation: Quercetin-Based Strategies for Ocular Diseases in Older Adults
Medoro A, Davinelli S, Scuderi L, Scuderi G, Scapagnini G, Fragiotta S
Clinical Interventions in Aging 2025, 20:791-813
Published Date: 7 June 2025
Klotho Protein: A Multifaceted Guardian of Healthy Aging and Its Therapeutic Potential
Shen J, Bin W, Lin X, Lai Y, Lin X, Guan T, Liu H
International Journal of Nanomedicine 2025, 20:7251-7270
Published Date: 9 June 2025