Back to Journals » Journal of Inflammation Research » Volume 16
Indole-3-Aldehyde Reduces Inflammatory Responses and Restores Intestinal Epithelial Barrier Function Partially via Aryl Hydrocarbon Receptor (AhR) in Experimental Colitis Models
Authors Wang M ![]() , Guo J, Hart AL, Li JV
, Guo J, Hart AL, Li JV
Received 28 July 2023
Accepted for publication 28 November 2023
Published 6 December 2023 Volume 2023:16 Pages 5845—5864
DOI https://doi.org/10.2147/JIR.S432747
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Mu Wang,1,2,* Jian Guo,3,* Ailsa L Hart,4 Jia V Li1
1Section of Nutrition, Division of Digestive Diseases, Department of Metabolism, Digestion and Reproduction, Faculty of Medicine, Imperial College London, London, W12 0NN, UK; 2Department of Neurology, Shanxi Provincial People’s Hospital, the Affiliated People’s Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, 030012, People’s Republic of China; 3Department of General Surgery, Shanxi Provincial People’s Hospital, the Affiliated People’s Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, 030012, People’s Republic of China; 4IBD Unit, St. Mark’s Hospital, London, HA1 3UJ, UK
*These authors contributed equally to this work
Correspondence: Jia V Li, Section of Nutrition, Division of Digestive Diseases, Department of Metabolism, Digestion and Reproduction, Faculty of Medicine, Imperial College London, London, W12 0NN, UK, Tel +44 20 7594 3230, Email [email protected]
Purpose: Indole-3-aldehyde (IAld) has been shown to improve intestinal epithelial barrier (IEB) function through the aryl hydrocarbon receptor (AhR) in murine colitis models. However, the impact of IAld on intestinal tissue inflammation remains unexplored. This study aimed to investigate the effects of IAld on the inflammatory responses of the gut both in vivo and in vitro and probe the mechanisms by which IAld attenuates colitis.
Methods: The effects of IAld on phenotypic changes, pro-inflammatory cytokines, IEB functions and the faecal bacterial composition in mice with dextran sulfate sodium salt (DSS)-induced colitis were assessed. Macrophage cells and intestinal epithelial cells were stimulated with lipopolysaccharide (LPS), and the effects of IAld on the inflammatory responses and IBE functions were measured.
Results: IAld reduced IL-6, IL-1β and TNF-α protein levels in both colonic tissues from the mice with colitis and LPS-stimulated macrophage cells. The IAld-mediated reduction of IL-6 but not IL-1β and TNF-α was through AhR activation. Furthermore, nuclear factor-κB pathway was found to be inhibited by IAld treatment via AhR activation both in vivo and in vitro. Gut permeability was significantly improved by IAld in both DSS-treated mice and LPS-stimulated Caco-2 cells. This observation is consistent with downregulation of phosphorylated myosin light chain through AhR activation. IAld did not appear to have an effect on the bacterial composition in mice with colitis despite the reduced colonic inflammatory responses.
Conclusion: IAld improved DSS-induced colitis by inhibiting the inflammatory responses and restoring IEB function, partially via AhR activation. This work provided insight into the function of IAld in modulating gut inflammation.
Keywords: indole-3-aldehyde aryl hydrocarbon receptor intestinal inflammation
Graphical Abstract:

Introduction
Ulcerative colitis (UC), a type of inflammatory bowel disease (IBD), is chronic immune-mediated inflammation occurring in the colon and rectum.1 The main contributing factors to the pathogenesis of UC include an abnormal immune response of the mucosa and impaired intestinal epithelial barrier (IEB) functions.1 These two pathogenic features result in devastating clinical consequences, such as rectal bleeding and diarrhoea. Although immunomodulators have frequently been used in treating patients with UC, therapeutic strategies remain a challenge owing to limited treatment options.2 Recent advances in characterising gut microbial metabolism and metabolite functions have highlighted several microbial metabolites that improve intestinal homeostasis, including short-chain fatty acids (SCFAs), bile acid derivatives such as isoallolithocholic acid (isoalloLCA)3 and tryptophan (Trp) metabolites.4
Trp is an essential amino acid that is exclusively supplied by diet. Trp is metabolised in the mammalian gut in three main pathways, including the kynurenine pathway, serotonin pathway and through the bacterial pathway to produce indole and indole derivatives,5 eg, indole-3-pyruvate (IPyA), indole-3-aldehyde (IAld), and indole propionic acid (IPA).6 A lower level of serum Trp was observed in patients with UC compared to healthy subjects.7 In addition, faecal levels of Trp, kynurenine and indole-3-acetic acid (IAA) in patients with UC were significantly different from those in healthy individuals.8 Trp supplementation to mice with colitis alleviated the intestinal inflammation.9,10 The anti-inflammatory effects of Trp and its metabolites such as IPA on experimental colitis have been proposed to occur through different mechanisms, one of which is through binding to receptors, such as aryl hydrocarbon receptor (AhR), to exert many immune effects.11 The beneficial effects of AhR on reducing the inflammatory response in the gut of mice with colitis were blocked by AhR antagonist, CH223191(CH-22).8,12
AhR is a nuclear receptor that is widely expressed in vertebrate cells, residing in the cytosol in an inactive form that binds to several cochaperones.13 In the canonical pathway, AhR dissociates from chaperones upon ligand binding and translocates into the nucleus, where it dimerizes with its partner gene AHR nuclear translocator (ARNT) to have functional activity by initiating downstream gene transcription, such as cytochrome P450 1A1 (CYP1A1) and CYP1B1.14 In the noncanonical pathway, AhR activation by different ligands appears to interact with several signalling pathways to regulate pathophysiological processes, such as inflammation.15 Reduced AhR expression was observed in mucosal biopsies of patients with UC16 and AhR deficiency increased the mucosal inflammatory score in a colitis mouse model.17 Indole metabolites, such as indole, IPyA and IPA, derived from bacterial metabolism of Trp are an essential source of AhR ligands.18 These indole metabolites were found to regulate the intestinal immune responses and protect the IEB function through AhR signalling, such as affecting T-cell differentiation,19 regenerating epithelial cells20 and regulating the formation of the apical junctional complex.21 IAld, a Trp metabolite produced by Lactobacillus acidophilus, L. murinus, L. reuteri, and L. johnsonii from the phylum Firmicutes, was shown to attenuate the severity of colitis through the AhR-IL-22 axis in innate lymphocytes in a dextran sulphate sodium salt (DSS)-induced colitis mouse model.22,23 In addition, IAld was reported to maintain IEB function through AhR by regulating tight junction (TJ) and cytoskeleton proteins.21 These evidence indicate that AhR activation by IAld plays a role in modulating inflammation and maintaining intestinal homeostasis. However, the underlying mechanisms remain largely uncharacterized.
The nuclear factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK) pathways are two main pathways involved in inflammation, and their activation was observed in immune cells and intestinal epitheliums from the colonic tissue of patients with UC.24–28 Targeting these pathways in macrophages has been proposed as a therapeutic strategy for patients with IBD.29 Therefore, it is valuable to investigate the impact of IAld on NF-κB and JNK pathways and understand how IAld-mediated AhR activation affects downstream cytokine expression. Despite that IAld was reported to maintain IEB function, the mechanisms through which IAld modulates IEB function and the downstream inflammatory pathways remain unknown. These mechanistic insights help further develop IAld as a therapeutic option for patients with UC.
Materials and Methods
Chemicals and Antibodies
IAld, CH-22, lipopolysaccharide (LPS, Escherichia coli O111:B4), phorbol 12-myristate 13-acetate (PMA), and FITC-dextran sulphate sodium salt (FITC-dextran, molecular weight 4 kDa) were obtained from Sigma‒Aldrich (MO, USA). DSS (molecular weight 36–50 kDa) was obtained from MP Biomedicals (CA, USA). Hank’s balanced salt solution (HBSS), phosphate buffered saline (PBS), Dulbecco’s modified Eagle’s medium (DMEM), dimethyl sulfoxide (DMSO), protease and phosphatase inhibitor, Triton X-100, and ProLong Diamond Antifade Mountant with DAPI (P36966) were obtained from Thermo Fisher Scientific. DMEM (high glucose), RPMI 1640, trypsin-EDTA (0.25%), penicillin‒streptomycin and premium foetal bovine serum (FBS) were purchased from Gibco. RNAiso Plus, PrimeScript RT reagent Kit with gDNA Eraser and Tli RNaseH Plus kit were purchased from Takara Bio.
Anti-COX-2 (ab179800) and anti-iNOS (ab179845) antibodies were purchased from Abcam. Anti-NF-κB P65 (8242), anti-p-NF-κB P65 (3033), anti-JNK (9252), anti-pJNK (9255), anti-p-MLC (3675) and MLC (8505) antibodies were purchased from Cell Signaling Technology. Anti-MLCK antibody was purchased from Sigma. Anti-ZO-1 (21773-1-AP), anti-occludin (66378-1-Ig), anti-claudin-1 (28674-1-AP) and anti-lamin B1 (12987-1-AP) antibodies were purchased from Proteintech, whereas goat anti-mouse secondary antibody-HRP, goat anti-rabbit secondary antibody-HRP, Alexa Fluor 488-conjugated anti-ZO-1 (339188), goat anti-rabbit secondary antibody-Alexa Fluor Plus 555 (A32732), goat anti-mouse secondary antibody-Alexa Fluor Plus 594 (A32742) and goat anti-rabbit secondary antibody-Alexa Fluor Plus 647 (A32733) were purchased from Thermo Fisher Scientific.
Animal Experiment and Induction of Colitis
All animals were provided by the Shanxi Provincial People’s Hospital Institutional Animal Care and Use Committee (Taiyuan, China). The study (2022–124) was approved by the animal ethics committee of Shanxi Provincial People’s Hospital, the Fifth Affiliated Hospital of Shanxi Medical University. The guidelines followed for the welfare of the laboratory is the Chinese national guidelines, People’s Republic of China National Standard GB/T 35892–2018 [Issued 6 February 2018 Effective from 1 September 2018].
Male wildtype C57BL/6 mice (6 to 8 weeks old) were housed 2/cage at 22–23°C with a 12 h/12 h light/dark cycle under specific pathogen-free conditions and ad libitum access to food and water. Mice were randomly divided into five groups (n = 8 per group), including group 1: control; group 2: IAld; group 3: DSS; group 4: DSS mice treated with IAld (DSS+IAld) and group 5: DSS mice treated with IAld and CH-22 (DSS+IAld+CH-22) (Figure 1A). After 1-week acclimatisation, IAld or CH-22 was given to mice orally at a dose of 20 and 10 mg/kg body weight in olive oil, respectively, for 7 days. Mice assigned to the control and DSS groups were orally gavaged with olive oil (10 mL/kg/day) as a vehicle. Colitis was subsequently induced by supplementation of 2.5% DSS in drinking water for 7 days followed by 2 days of regular drinking water before culling. During acute colitis induced by DSS, the disease activity index (DAI) was evaluated daily as previously described.30 The faecal samples were collected 2 days after DSS withdrawal for bacterial compositional analysis. At the end of the experiment, the mice were sacrificed by cervical dislocation. A portion of colon tissue was fixed in a 10% paraformaldehyde solution at 4°C for histological examination, and the others were snap frozen in liquid nitrogen and stored at −80°C.
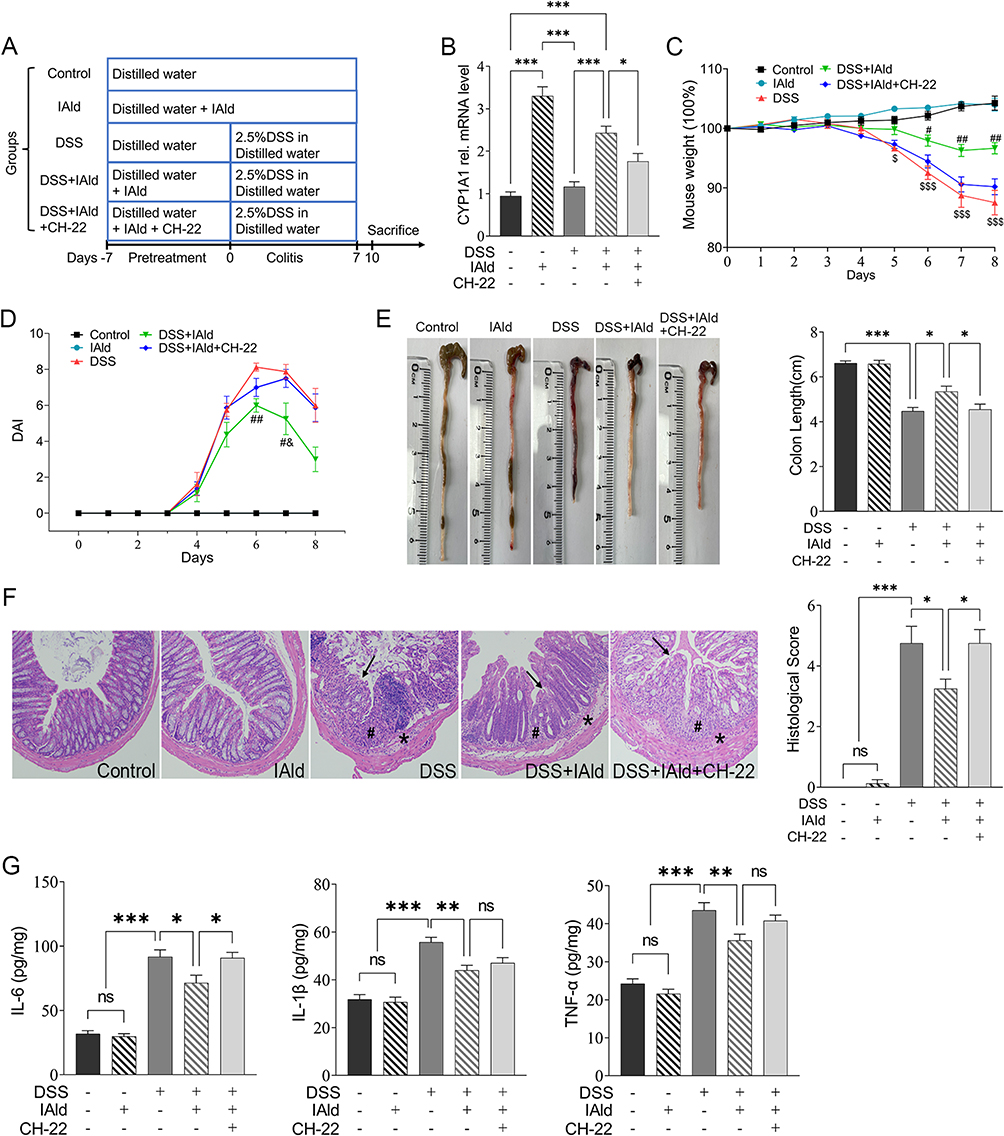 |
Figure 1 Experimental schedule and effects of IAld supplementation on basic indicators in a DSS-induced colitis mouse model. Notes: (A) Animal treatments schedule. (B) The mRNA expression of CYP1A1 in each group. (C) The measurements of bodyweight in each group. (D) The measurements of DAI scores in each group. (E) Representative images of colon length and quantitation of colon length of each group. (F) Representative images of haematoxylin and eosin staining of the colonic tissue and quantitation of histological score in each group. Arrows, ulceration; #, transmural inflammation; *, mucosal immune cell infiltration, magnification is X20 (G) The expression of IL-6, IL-1β and TNF-α in the colonic tissue were measured by enzyme-linked immunosorbent assay. The data are shown as the mean ± SEM (n = 8/group). * p < 0.05, ** p < 0.01, *** p < 0.001; # p < 0.05, ## p < 0.01 versus DSS; &p < 0.05 versus CH-22; $p< 0.05, $$$ p < 0.001 versus Control; ns, statistically non-significant. |
Haematoxylin and Eosin (H&E) Staining and Histological Examination
Colonic tissue samples were embedded in paraffin and sectioned (4 μm) before staining with H&E. The histological changes were observed with optical microscopy (DM2500, Leica, Solms, Germany). All histologic quantitation was performed by the same individual, who was blinded to the sample group information, using a scoring system previously described.31
Intestinal Permeability Test in Mice
To assess IEB function in vivo, the FITC-dextran intestinal permeability assay was performed in an independent experiment. The mouse experimental design was the same as the programme described in Section 2.2. Mice (n = 5 per group) were fasted for 4 h prior to gavage with FITC-dextran (600 mg/kg body weight) dissolved in deionised water. Blood was collected by cardiac puncture 4 h after FITC-dextran gavage, and the blood sample in EDTA-coated tubes was centrifuged at 2000 × g for 10 min at 4°C to collect plasma. The fluorescence intensity of each plasma sample was measured using a microplate reader (excitation: 490 nm; emission: 525 nm). A series of concentrations of FITC-dextran (0, 0.25, 0.5, 1, 2, 4, and 8 μg/mL) in the plasma from PBS-treated mice were used to generate a standard curve.
DNA Extraction of Faeces and Illumina MiSeq Sequencing
Microbial genomic DNA was extracted from faecal samples using the OMEGA Soil DNA Kit (M5635-02) (Omega Bio-Tek, GA, USA) following the manufacturer’s protocol. The extracted DNA was quantified using a NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific, MA, USA) and diluted to 5 ng/mL. The bacterial 16S rRNA gene V3–V4 region was amplified by the forward primer 338F (5’-ACTCCTACGGGAGGCAGCA-3’) and the reverse primer 806R (5’-GGACTACHVGGGTWTCTAAT-3’). Sample libraries were prepared following Illumina’s protocols.32 Sequencing was analysed on the Illumina MiSeq platform with MiSeq Reagent Kit v3 at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China) using paired end 2 × 250 bp chemistry.
Data Analysis of 16S rRNA Gene-Based Sequences
The sequences were analysed using R (v.4.2.2) and RStudio (v.2022.07.2+576) with the DADA2 (v.1.16.0) package. The forward and reverse reads were trimmed using a quality score of 30 as the cut-off. Taxonomic labelling of amplicon sequence variants (ASVs) was conducted with the SILVA v.138.1 database. In total, 1088 ASVs were detected. The taxon filter and prevalence control cut-off were set at ASVs ≥4 counts in 10% of the total number of samples. After the filter was applied, the resulting data set consisted of 630 ASVs. The number of reads per sample pre- and post-rarefaction is listed in Supplementary Table 1. The phyloseq package (v.1.40.0) in R was used to run further analysis. For alpha diversity, rarefaction was performed with a minimum sequence depth of 19217 counts, and the observed reads and Shannon and Simpson indices were calculated. The comparisons of alpha-diversity indices among groups were performed using ANOVA. Principal coordinate analysis (PCoA) with the Bray-Curtis distance was used to visualise the beta-diversity. Linear discriminant analysis (LDA) effect size (LEfSe) (log10 LDA scores > 2; adjusted p < 0.05) was used to determine significant changes in the abundance of the bacterial taxa between experimental groups.33 Spearman’s rank correlation was performed using R (v.4.2.2) to assess the correlation between the abundance of bacterial taxa and the inflammatory responses and the barrier function-associated parameters measured in the colonic tissues, with p < 0.05 being considered significantly different.
Cell Culture and Treatment
Murine macrophage RAW264.7 cells and human intestinal epithelium Caco-2 cells (Chinese Academy of Sciences, Shanghai, China) were cultured in DMEM with 10% FBS at 37°C under 95% air/5% CO2 and sub-cultured successively when they reached approximately 80% confluence. Human monocyte THP-1 cells (Chinese Academy of Sciences, Shanghai, China) were cultured in RPMI 1640 medium with 10% FBS at 37°C under 95% air/5% CO2 and were sub-cultured successively when the cell density reached approximately 2 × 106 cells/mL. All experiments were carried out in cell passage numbers between 20 and 30.
RAW264.7 and THP-1 cells were seeded in six-well plates at densities of 1 × 106 per well and 5 × 105 per well, respectively. THP-1 monocytes were directly differentiated into macrophages after 48h incubation with 100 ng/mL PMA and a 24 h rest period in complete RPMI medium without PMA. At the end of 72 h, THP-1 macrophages were used as M0 macrophages. LPS, IAld and CH-22 were dissolved in DMSO. Aliquots of the stock solutions of LPS, IAld and CH-22 at concentrations of 1 mg/mL, 400 mM and 20 mM, respectively, were stored at −20°C and diluted freshly into media prior to the cell treatment. The final concentration of DMSO in all wells was kept at 0.3%.
Cytotoxicity Assay
To evaluate the cytotoxicity of IAld on different cell lines, RAW264.7, THP-1 macrophages and Caco-2 cells were plated in a 96-well plate at a density of 1 × 105 cells, 5×10,4 and 1.5 × 104 per well, respectively, and treated with IAld at the concentrations of 10, 50, 100, 200 or 400 µM for 48 h. In addition, the cytotoxic effects of LPS on Caco-2 cells at different treatment times were evaluated. The treatments were added when the cells reached 60% confluency. Cell viability was then assessed using a Cell Counting Kit-834 (Proteintech, Wuhan, China) following the manufacturer’s protocol. The optical density (OD) at 450 nm was measured using a microplate reader (Epoch 2, Biotek, VT, USA). Relative cell viability (%) was calculated as (ODtreated well - ODblank well) / (ODcontrol well - ODblank well) × 100%.
Transepithelial Electric Resistance Measurement of Caco2 Monolayer
Caco-2 cells were seeded on a 12-well transwell insert (polyester membrane, diameter, 6.5 mm; pore size, 0.4 μm; Corning, MA, USA) at the density of 5 × 104 per well to form a monolayer. The permeability of the Caco-2 monolayer was monitored by measuring the electrical resistance using an EVOM2 Epithelial Voltohmmeter (EVOM2, World Precision Instruments, FL, USA) as previously described.35 After the transepithelial electric resistance (TEER) values reached a plateau (550 to 650 Ω•cm2) from 17 to 19 days, the cells were subjected to different treatments, CH-22, IAld and LPS. The Caco-2 monolayer was treated with LPS for 3 days, and the TEER was measured daily after the start of LPS treatment (Day 1 to Day 3). The blank resistance was determined by measuring the resistance across a filter without cells in HBSS. TEER was calculated according to Ohm’s law: TEER (Ω * cm2) = (Measured Resistance-Blank resistance) (Ω) × Area (cm2). The changes in TEER at Day 1–3, presented in percentage, were normalised to Day 0 (one day prior to LPS treatment) per transwell insert.
FITC-Dextran Permeability Assay for Caco-2 Cell Monolayer
To quantify the epithelial barrier function of the Caco-2 cell monolayer, the flux of FITC-dextran from the apical chambers to basolateral chambers was measured as described previously.36 At the end of LPS treatments, the monolayer was washed, and the medium was replaced with HBSS. FITC-dextran was added to the apical chamber at 1 mg/mL in HBSS and incubated for 4 h at 37°C. A total of 200 μL of HBSS were removed from the basal chamber and transferred into 96-well black opaque plates. The fluorescence intensity in the basolateral compartment was measured using a microplate reader (excitation, 490 nm: emission, 525 nm). A series of concentrations of FITC-dextran in HBSS (0, 0.25, 0.5, 1, 2, 4, and 8 μg/mL) were used to generate a standard curve.
Immunofluorescence and Confocal Analysis
RAW264.7 (2 × 105) and THP-1 (1 × 106) cells on glass coverslips or a Caco-2 monolayer on the membrane of the insert were fixed with 4% paraformaldehyde in PBS for 15 min, permeabilised with 0.1% Triton X-100 in PBS for 15 min and blocked with PBS containing 5% BSA for 1 h at room temperature. For preparations of the colonic tissue, paraffin-embedded tissue samples were boiled in antigen retrieval buffer (100 mM Tris, 5% (w/v) urea, pH 9.5) at 95°C for 10 min, cooled to room temperature, rinsed in PBS twice and fixed in 4% paraformaldehyde prior to staining. Cells or tissue samples were then incubated with primary antibodies (anti-NF-κB p65 and Alexa Fluor 488-conjugated anti-ZO1 antibodies were used at a dilution of 1:500; anti-occludin, anti-ZO-1 and anti-p-MLC antibodies were used at a dilution of 1:200) in PBS containing 5% BSA at 4°C overnight before being incubated with species-specific secondary antibodies (Alexa Fluor Plus 555, Alexa Fluor Plus 594 and Alexa Fluor Plus 647 were used at a dilution of 1:500) in the dark for 1 h in PBS containing 5% BSA. After each incubation, the cells were washed with 1 mL of PBS, and the tissue slides were washed with 5 mL of PBS 3 times. Coverslips were mounted with DAPI Prolong Diamond at 4°C overnight and imaged by a laser-scanning confocal microscope (SP8, Leica, Wetzlar, Germany). The obtained photos were analysed using ImageJ software v.1.53q. The total intensity of p65 in the nuclear was divided by that in the cytoplasm to obtain the nuclear-to-cytoplasmic ratio of NF-κB.
Reverse Transcription and Real-Time PCR
Total cellular or tissue RNA was extracted using RNAiso Plus according to the manufacturer’s instructions. Briefly, total RNA was extracted by using RNAiso Plus (700 μL), chloroform (140 μL) and isopropanol. The collected RNA was washed with 75% ethanol at 7500 x g for 5 min at 4°C. The supernatant was then discarded, and the precipitate was left at room temperature to dry. After the precipitate was dry, RNase-free water was added to dissolve the RNA. RNA concentration was measured by a microplate reader (Epoch 2, Biotek, VT, USA). For reverse transcription-PCR, 1 μg of total RNA was converted to cDNA using the PrimeScript RT Reagent Kit with gDNA Eraser in a 20 μL volume. Real-time PCR was performed using a Tli RNaseH Plus kit in a 20 μL volume containing 8 μL cDNA, 2 μL primer pairs (0.5 μM/primer) and 10 μL TB Green Premix Ex Taq II. The primer sequences for PCR are shown in Supplementary Table 2. Amplification conditions were as follows: one hold at 95°C for 60s; 40 cycles at 95°C for 5s, 55°C for 30s and 55°C for 30s; 1 cycle at 95°C for 15s, 60°C for 30s and 95°C for 15s. All values were normalised to the expression of β-actin.
Cytokine Measurement
The levels of cytokines present in colonic tissue or cell culture medium were determined by enzyme-linked immunosorbent assay (ELISA) kits against mouse interleukin (IL)-6, IL-1β and tumour necrosis factor alpha (TNF-α) (Proteintech, Wuhan, China) according to the manufacturer’s instructions.
Nuclear Extraction and p65 DNA Binding Activity
Nuclear extracts were prepared using an NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Pierce, IL, USA). The extracted nuclear fractions were used to assess p65 DNA binding activity using an NF-κB p65 Transcription Factor Assay Kit (Abcam, MA, USA) following the manufacturer’s protocol. Another aliquot of nuclear fraction was used to measure the p65 level using immunoblot.
Immunoblot Analysis
Protein expression in colonic tissue or cells was assessed by immunoblot. Tissue or cells were lysed with RIPA buffer containing phosphatase and protease inhibitors. The lysates were sonicated and centrifuged at 10,000 × g for 10 min at 4°C in an Eppendorf centrifuge to obtain a clear lysate. The supernatant was collected, and the protein concentration was determined by a BCA assay (Thermo Fisher Scientific, MA, USA). The obtained lysate containing 10 to 20 mg of protein was added to SDS loading buffer protein and boiled at 95°C for 10 min. The proteins were then separated on an SDS‒PAGE gel (8%–10%) and transferred to a PVDF membrane. The membrane was subsequently blocked with 5% dry milk in TBS-1% Tween 20 buffer for 2 h, after which the membrane was incubated with primary antibodies overnight at 4°C. Following a wash in TBS-1% Tween buffer, the membrane was then incubated in the secondary antibody for 1 h at room temperature. To measure the expression of loading control proteins, the membranes were stripped and reprobed. Proteins in the membrane were normalised to total protein concentration (TPN) using No-Stain Protein Labelling Reagent (Invitrogen, MA, USA) and analysed using a Bio-Rad Chemidoc™ XRS+ Imaging system. The results were quantified using Bio-Rad Image Lab Software to account for variation in loading among lanes.
Statistical Analysis
All experiments in the cellar study were performed with three independent biological replicates. Data are expressed as the mean ± SEM. Differences between groups were analysed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. For comparisons between only 2 groups, an unpaired, 2-tailed standard Student’s t test was used. GraphPad Prism 9 software (CA, USA) was used for statistical analysis. p <0.05 was considered statistically significant.
Results
IAld Reduces Colonic Protein Levels of Interleukin IL-6, IL-1β and TNF-α in Mice with DSS-Induced Colitis and IAld-Induced Reduction of IL-6 is Through AhR Activation
To study the effects of IAld on gut inflammation and the mechanisms associated with AhR in vivo, wild-type mice were treated with IAld or IAld and AhR inhibitor (CH-22) and DSS was used to induce colitis (Figure 1A). We showed that mRNA expression levels of AhR downstream gene CYP1A1 were upregulated in the colonic tissue by IAld (p < 0.001) and downregulated in the IAld+AhR inhibitor CH-22 group (p < 0.05) (Figure 1B). These observations confirm that IAld activated AhR signalling, and CH-22 suppressed this signalling pathway. IAld improved the phenotypic characteristics of mice with DSS-induced colitis, including body weight, DAI score and colon length, compared with DSS or DSS+IAld+CH-22 groups (Figure 1C-E). H&E staining of colon sections from mice in either the DSS or DSS+IAld+CH-22 groups showed notable ulceration, transmural inflammation and immune cell infiltration in the mucosa and submucosa, which were improved by IAld treatment, as evidenced by a significant reduction in histological scores (Figure 1F). Furthermore, we observed that IAld significantly reduced the protein levels of pro-inflammatory cytokines, such as IL-6, IL-1β and TNF-α, in the colonic tissue (Figure 1G). The IAld-induced decrease in the IL-6 level but not IL-1β and TNF-α was inhibited by CH-22 (p < 0.05).
Taken together, our results suggested that IAld suppresses the colonic proinflammatory responses induced by DSS, partially through AhR activation.
IAld Reduces the Level of NF-κB Through AhR Activation in Colonic Tissues from Mice with DSS-Induced Colitis
We next investigated if IAld impacts the NF-κB pathway, which plays a key role in regulating inflammation. As shown in Figure 2A, the protein levels of p65 from extracted nuclear fractions and the p65 DNA binding activity were significantly lower in the DSS+IAld group in contrast to the DSS group or the DSS+IAld+CH-22 group, suggesting that IAld-mediated AhR activation inhibits the NF-κB pathway. The IEB is critical in protecting the host from persistent activation of lamina propria immune cells caused by translocating microorganisms or endotoxins (eg, LPS).37 Therefore, we evaluated the gut permeability and gut integrity-related proteins in the current mouse model. The damaged IEB is related to increased myosin light-chain kinase (MLCK) expression and subsequent myosin II regulatory light chain (MLC) phosphorylation.38 Phosphorylated MLC (p-MLC) by MLCK contracts the perijunctional actomyosin ring, exerting physical tension on tight junction (TJ) proteins and increasing paracellular permeability.39 Consistent with previously reported findings,21 we also observed that IAld significantly reduced the gut permeability (p < 0.01) and the phosphorylation of MLC in the colon from mice with colitis (p < 0.001) (Figure 2B and C). The latter was reversed by inhibiting AhR activation (p < 0.05) (Figure 2C), but not the reduction in the gut permeability. As shown in Figure 2D and E, IAld significantly increased levels of TJ proteins, zonula occludens-1 (ZO-1) and occludin (p < 0.01) in the colon, but not claudin-1. Inhibiting AhR signalling resulted in a significant reduction in ZO1 levels (p < 0.05) but had no effect on IAld-induced increases in occludin (Figure 2D and E).
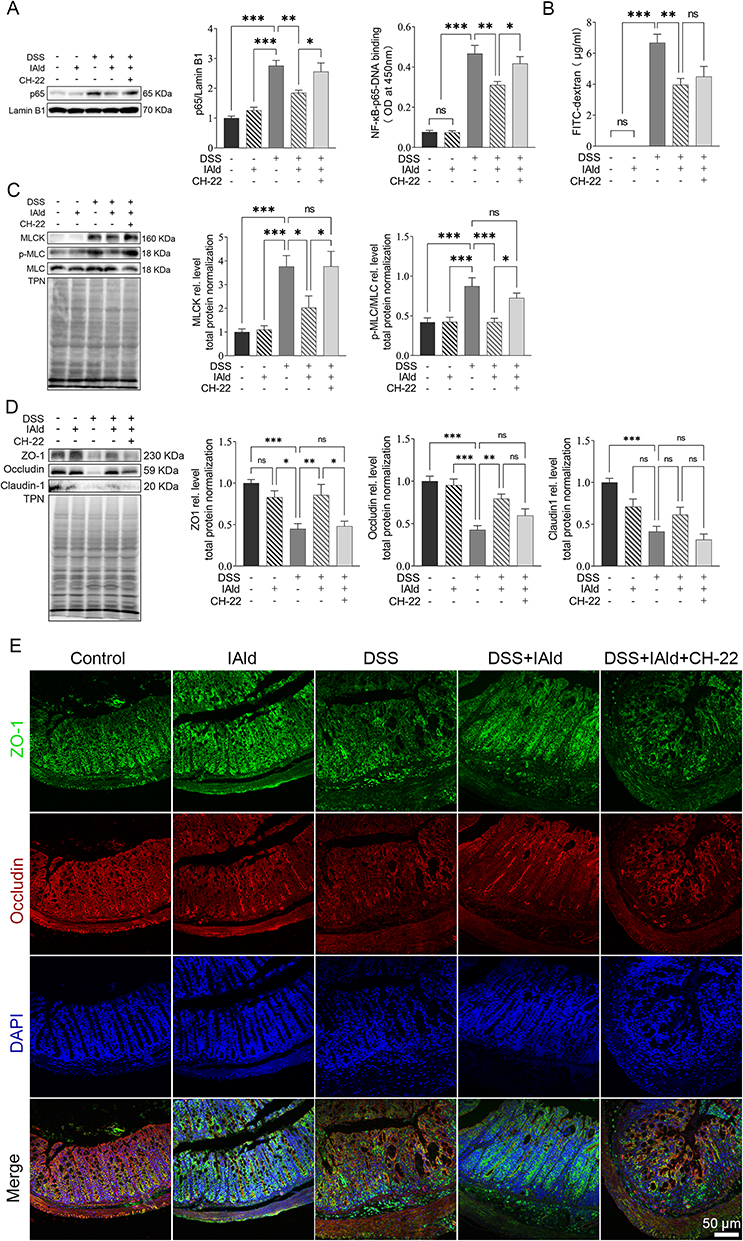 |
Figure 2 Effects of IAld supplementation on gut barrier integrity in a DSS-induced mouse model. Abbreviation: ns, statistically non-significant. Notes: (A) p65 expression (nuclear fractions) from colonic tissue was measured using immunoblot. Lamin B1 was a loading control. Data are presented as the ratio of p65/Lamin B1. p65 DNA binding activity (nuclear fractions) was determined using p65 Transcription Factor Assay. (B) The intestinal permeability was measured as the FITC-dextran levels in the serum of the mice. (C and D) The expression of and MLCK and p-MLC were measured using immunoblot. Total protein was a loading control. (E) Representative images of immunofluorescent staining of TJ proteins in colonic tissue. The data are shown as the mean ± SEM (n = 5–8/group). *p < 0.05, **p < 0.01, ***p < 0.01. |
Taken together, our findings suggested that IAld reduces NF-κB through AhR activation, which could play a role in IAld-mediated reduction of proinflammatory responses in the colon and improving the gut epithelial function.
IAld Alleviates Proinflammatory Responses in LPS-Stimulated Macrophages Partially Through AhR
To further probe the effects of IAld on the immune and epithelial cells separately, we carried out studies in vitro using RAW264.7, THP-1 macrophages, and Caco-2 colonic epithelial cells. The viability of the cells was evaluated using CCK-8 assay after 48 h exposure of IAld at a range of concentrations from 10 to 400 μM and no cytotoxic effects were observed (Supplementary Figure 1A-C). To determine an optimal treatment concentration of IAld, we treated the RAW264.7 cells with IAld at concentrations of 50, 100, 200 or 400 μM for 1 h, followed by 12 h treatment of LPS at a final concentration of 100 ng/mL. The protein levels of two common inflammatory mediators (ie, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2)) were measured, and IAld at 200 μM was chosen for subsequent investigations since it showed the most effect on iNOS and COX-2 levels (Figure 3). To examine if IAld is an agonist of AhR in RAW264.7, THP-1 and Caco-2 cell lines and how effectively CH-22 inhibits IAld-mediated AhR activation, cells were treated with IAld with or without CH-22. The mRNA levels of CYP1A1 and CYP1B1, which are AhR downstream genes,14 were measured. IAld significantly upregulated CYP1A1 and CYP1B1 mRNA expression levels in all three cell types, and the upregulated expression levels were significantly suppressed by CH-22 (Figure 4).
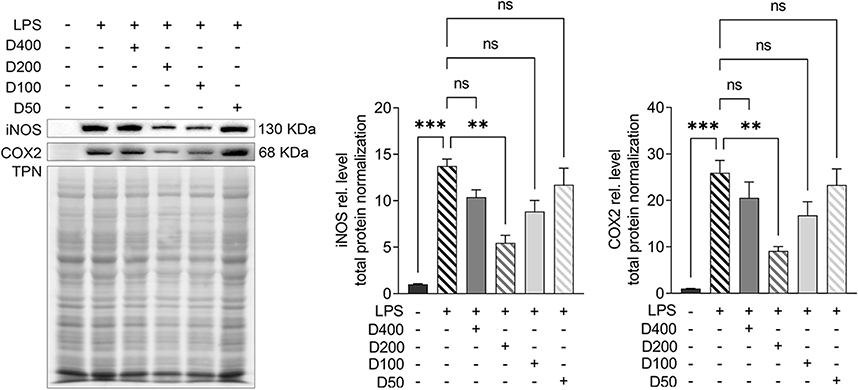 |
Figure 3 The effects of different concentrations of IAld on decreasing the level of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) in LPS stimulated RAW264.7 cells. Abbreviation: ns, statistically non-significant. Notes: Cells were treated with IAld at concentrations of 400, 200, 100, 10 μm for 1h before LPS (100ng/mL) stimulation for 12 h. Total protein was a loading control. The data are shown as the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001. |
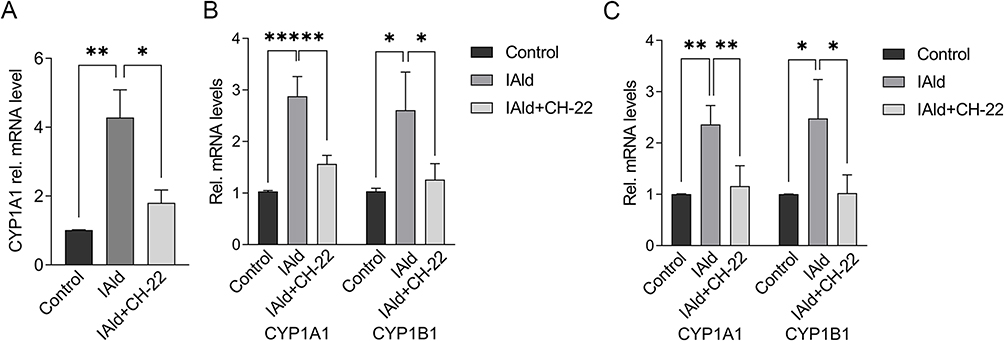 |
Figure 4 IAld activated AhR signalling in the different cell types. Notes: (A) RAW264.7 (B) THP-1 (C) Caco-2 cells were treated with IAld (200 μM) for 2 h with or without 1 h prior treatment of CH-22 (10 μM). mRNA expression of CYP1A1 and CYP1B1 was measured using qPCR. The mRNA expression of CYP1B1 in RAW264.7 cells was not detected. The data are shown as the mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. |
To examine the effects of IAld-mediated AhR activation on the LPS-induced inflammatory response in macrophages from both mice and humans, RAW264.7 and THP-1 cells were treated with IAld, CH-22 or both. We showed that the secretion of proinflammatory cytokines, IL-6, IL-1β and TNF-α was significantly increased by LPS (Figure 5). IAld treatment decreased IL-6 and IL-1β secretion from both cell lines but only decreased TNF-α secretion in mouse macrophages (Figure 5). Furthermore, as shown in Figure 5, when LPS-stimulated macrophage cells were co-treated with IAld and CH-22, the suppressive effects of IAld on IL-6 secretion, but not IL-1β and TNF-α, were abrogated, which is consistent with our findings from the mouse model.
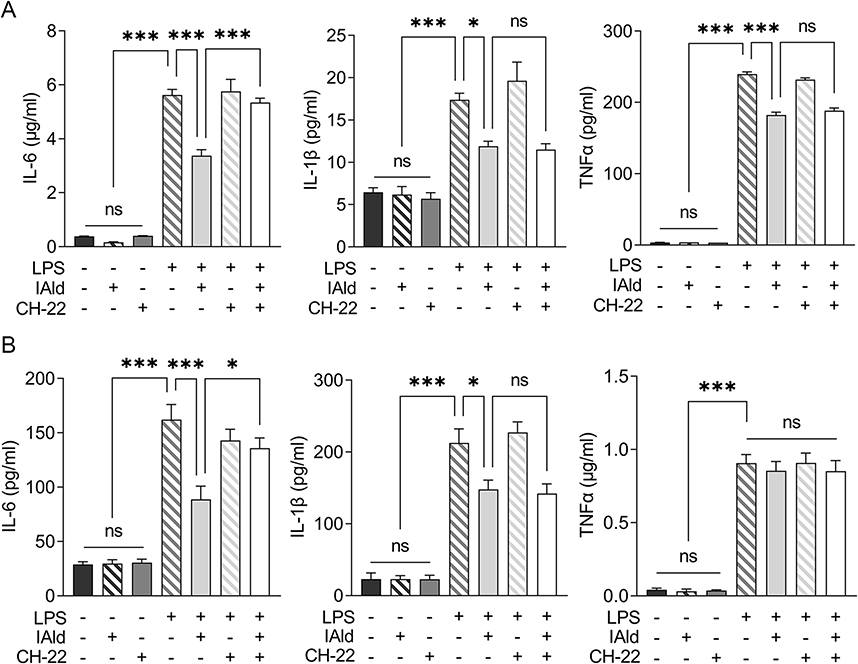 |
Figure 5 IAld decreased proinflammatory cytokine expression in LPS-stimulated macrophages partially through AhR. Abbreviation: ns, statistically non-significant. Notes: (A) RAW264.7 (B) THP-1 cells were treated with IAld (200 μM) for 1 h with or without 1 prior treatment of CH-22 (10 μM), followed by LPS (100 ng/mL) stimulation for another 12 h. The protein levels of proinflammatory cytokine IL-6, IL-1β and TNF-α in the supernatant of cell culture were measured using enzyme-linked immunosorbent assay. The data are shown as the mean ± SEM (n = 3). *p < 0.05, ***p < 0.001. |
Taken as a whole, our results reveal that IAld decreases IL-6 secretion through AhR and the IAld-induced reduction in IL-1β is independent of AhR activation in both mouse and human macrophages. IAld reduces TNF-α secretion of murine macrophages in an AhR-independent manner but poses no effects on TNF-α levels in human macrophages, suggesting that IAld suppresses the pro-inflammatory response through multiple mechanisms, which remain to be unveiled.
IAld Suppresses NF-κB and JNK Pathways in LPS-Stimulated Macrophages Through AhR
To further explore the impact of IAld-mediated AhR activation on the NF-κB and JNK inflammatory pathways in the immune cells, LPS-stimulated macrophage cells were treated with IAld, CH-22 or both. The protein levels of phosphorylated NF-κB p65 (p-p65) and JNK (p-JNK) in cells were significantly reduced by IAld (p < 0.01, p < 0.05) and CH-22 significantly inhibited this reduction (p < 0.05) (Figure 6A and B). Furthermore, IAld significantly reduced the translocation of p65 from the cytoplasm to the nucleus, which is a key step in NF-κB activation in macrophages, and this reduction was notably inhibited by CH-22 (Figure 6C and D).
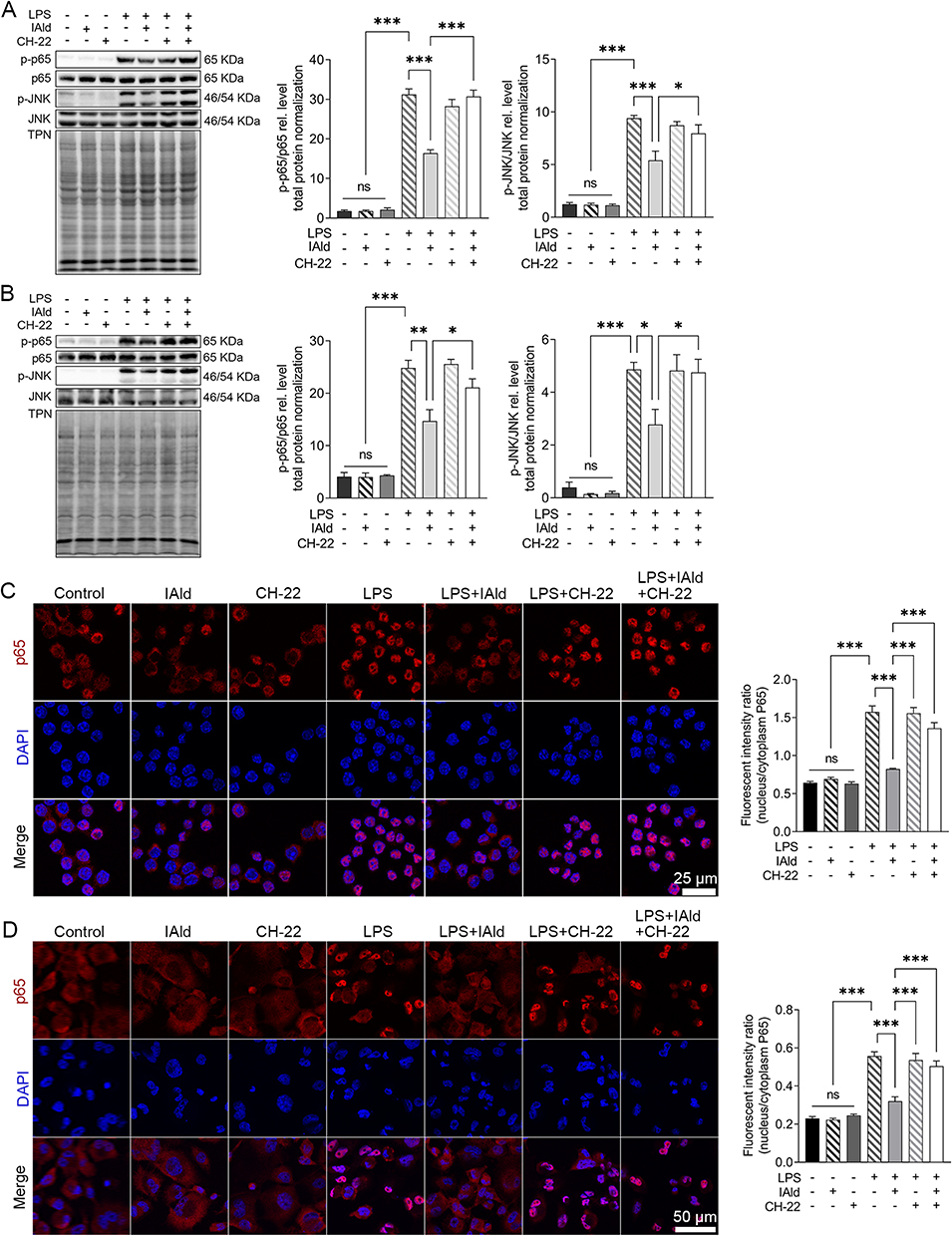 |
Figure 6 IAld suppressed NF-κB and JNK pathway through AhR in macrophage cells. Abbreviation: ns, statistically non-significant. Notes: (A) RAW264.7 (B) THP-1 cells were treated with IAld (200 μM) for 1 h with or without 1 h prior treatment of CH-22 (10 μM), followed by LPS (100 ng/mL) stimulation for 30 min, the levels of phosphorylated NF-κB p65 (p-p65) and JNK (pJNK) were determined by immunoblot. The levels of phosphorylated NF-κB p65 (p-p65) were determined by immunoblot. Total protein was a loading control. Data are presented as the ratio of p-p65/total p65 and pJNK/total JNK. Representative images of immunofluorescent staining of p65 translocation from cytoplasmic to nucleus and quantitation of p65 translocated ratio in (C) RAW264.7 (D) THP-1. All data obtained from immunoblot or IF were quantified using Image J. The data are shown as the mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. |
Taken together, our observations lead us to conclude that IAld-mediated AhR activation is necessary for reduction of p65 translocation from cytoplasm to nucleus and downregulation of NF-κB and JNK pathways in both human and murine macrophages.
IAld Attenuates IEB Dysfunction and Suppresses the NF-κB Pathway Through AhR in the LPS-Stimulated Caco-2 Monolayer
To investigate the effect of IAld on LPS-induced disruption in IEB, TEER was measured as an index of epithelial barrier integrity in Caco-2 monolayers following the treated with IAld, CH-22 or both. Figure 7A shows that the LPS reduced TEER levels (p < 0.001) and this reduction was restored by IAld (p < 0.001) but not IAld with the presence of CH-22. Consistent with the observations in the permeability of LPS-stimulated Caco-2 monolayer, CH-22 abolished the protective effect of IAld (p < 0.01) (Figure 7B). LPS at the concentration of 100 ng/mL was not cytotoxic to Caco-2 cells at 24 and 48 hours (Figure 7C), indicating that the increased permeability was due to paracellular leakage rather than LPS-induced cell death. We subsequently investigated if LPS affects the TJ proteins and if IAld can restore these protein levels. We did not observe changes in the levels of TJ proteins (ZO-1, occludine, claudine-1) induced by LPS nor IAld (Figure 7D). In contrast, LPS significantly increased MLCK and p-MLC levels compared with untreated controls and increases in these protein levels were normalised by IAld treatment (Figure 7E). Furthermore, we observed that CH-22 abolished these protective effects of IAld on MLCK and p-MLC (Figure 7E), which agrees with our observations in vivo. Immunofluorescence analysis was used to visualise the phenotype of the Caco-2 monolayer under different treatments. LPS treatment for 48 h disturbed the Caco-2 monolayer phenotype, resulting in a zigzag shape of TJ proteins, and IAld mitigated the deformation of the monolayer, and this alleviation was partly blocked by CH-22 (Figure 7F). In addition, compared with the controls, LPS-treated monolayers showed a higher proportion of merged TJs with p-MLC (Figure 7F), indicating that LPS resulted in redistribution and co-localisation of TJs and p-MLC. These effects were inhibited by IAld, as shown by the increased portion of separate staining of TJ and p-MLC in the cells, and CH-22 partly eliminated these effects (Figure 7F).
 |
Figure 7 IAld attenuated Caco-2 epithelial barrier dysfunction through AhR activation. Abbreviation: ns, statistically non-significant. Notes: Polarized Caco-2 monolayers were pre-treated with or without CH-22 (10 μM) for 1 h before IAld (200 μM) was added to the apical chamber for another 1 h, followed by adding LPS (100 ng/mL) into the basolateral chamber for next 48 h. Epithelial permeability was measured by (A) TEER or (B) FITC-dextran paracellular transport from the apical to basolateral compartment. (C) The cytotoxic effects of LPS on Caco-2 monolayer at different time points were measured. (D and E) The expression of tight junction (TJ) proteins (eg, ZO-1, occludin, and claudin-1), and MLCK and p-MLC were determined by immunoblot. Total protein was a loading control. (F) Representative images of immunofluorescent (IF) staining of TJ and p-MLC in Caco-2 monolayer. Arrows, the disrupted phenotypes of TJ proteins in LPS-stimulated Caco-2 monolayer. All data obtained from IF was quantified using Image J. The data are shown as the mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001; #p < 0.001 versus LPS; &p < 0.01 versus CH-22; $p < 0.001 versus Control. |
The NF-kB pathway has been suggested to be involved in upregulating MLCK and p-MLC to cause an increase in Caco-2 monolayer permeability;40 therefore, we next explored the effects of IAld-mediated AhR activation on NF-kB in Caco-2 cells. We observed that LPS upregulated the NF-kB pathway (p<0.01), and this upregulation was prevented by IAld-mediated AhR activation (p<0.05) (Figure 8A). IAld-mediated AhR activation prevented the translocation of p65 from the cytoplasm to the nucleus in LPS-stimulated Caco-2 cells (Figure 8B and C). In contrast, the protein levels of JNK were not affected by LPS at a concentration of 100 ng/mL nor IAld (Supplementary Figure 2).
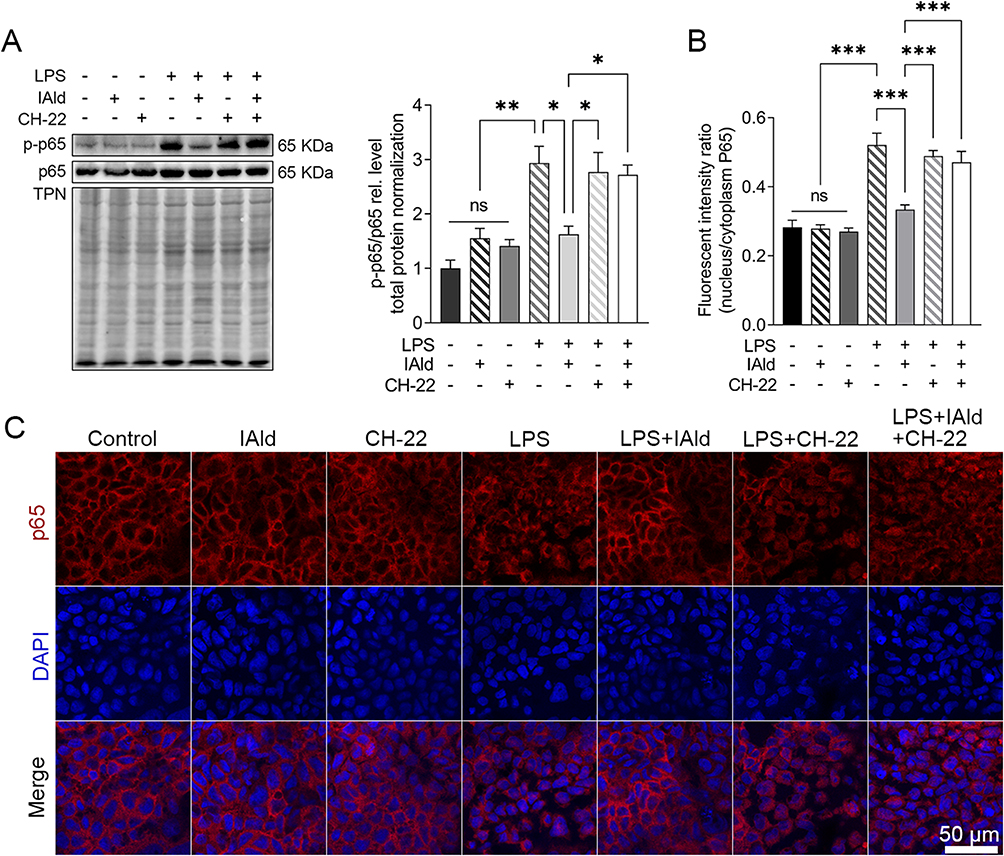 |
Figure 8 IAld supressed NF-κB through AhR in Caco-2 cells. Abbreviation: ns, statistically non-significant. Notes: Polarized Caco-2 cells were treated with IAld (200 uM) for 1 h with or without 1 h prior treatment of CH-22 (10 μM), followed by LPS (100 ng/mL) stimulation for 4 h. (A) The levels of phosphorylated NF-κB p65 (p-p65) were determined by immunoblot. Total protein was a loading control. Data are presented as the ratio of p-p65/total p65. (B and C) Quantitation of p65 translocated ratio in Caco-2 cells and representative images of immunofluorescent staining of p65 translocation from cytoplasmic to nucleus. All data obtained from immunoblot or IF were quantified using Image J. The data are shown as the mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. |
Taken together, our data suggest that IAld ameliorates LPS-induced disruptions in epithelial barrier integrity by downregulating MLCK and p-MLC/MLC and NF-κB through AhR activation.
Faecal Relative Abundances of the Certain Bacterial Families Including Akkermansiaceae Were Changed by IAld in Mice with DSS-Induced Colitis and are Correlated with Tight Junction Proteins and Inflammation
As the intestinal inflammation and increased gut permeability are frequently accompanied by compositional changes in the gut microbiota,41 the regulatory effect of IAld on the gut bacterial composition in DSS-induced colitis mice was studied using 16S rRNA gene sequencing technique. Alpha-diversity (Shannon and Simpson indices) was not significantly different among the groups (Figure 9A). Non DSS-treated groups were separated from the DSS-treated groups along the 2nd component (Axis 2) in the PCoA score plot (p = 0.001) (Figure 9B). We observed that the relative abundance of bacterial family Lactobacillaceae was higher in the control mice compared to the other groups (Figure 9C). LefSe analysis revealed that the relative abundances of 16 bacterial families were significantly changed owing to DSS treatment and gram-negative bacteria, including Prevotellaceae, Acholeplasmataceae, Oscillospirales UCG_010, Bacteroidaceae and Christensenellaceae were enriched (Figure 9D). We further observed significant changes in the abundances of 14 bacterial families induced by IAld-treated mice with colitis, for example, increased relative abundances of Akkermansiaceae and Enterobacteriaceae and decreases in Bifidobacteriaceae and Fusobacteriaceae (Figure 9E). Moreover, treatment with IAld and CH-22 in mice with colitis increased the relative abundances of these bacterial families that showed a reduction in IAld-treated mice, such as, Bifidobacteriaceae, Clostridiacaea, Peptostreptococcaceae and Christensenellaceae (Figure 9F).
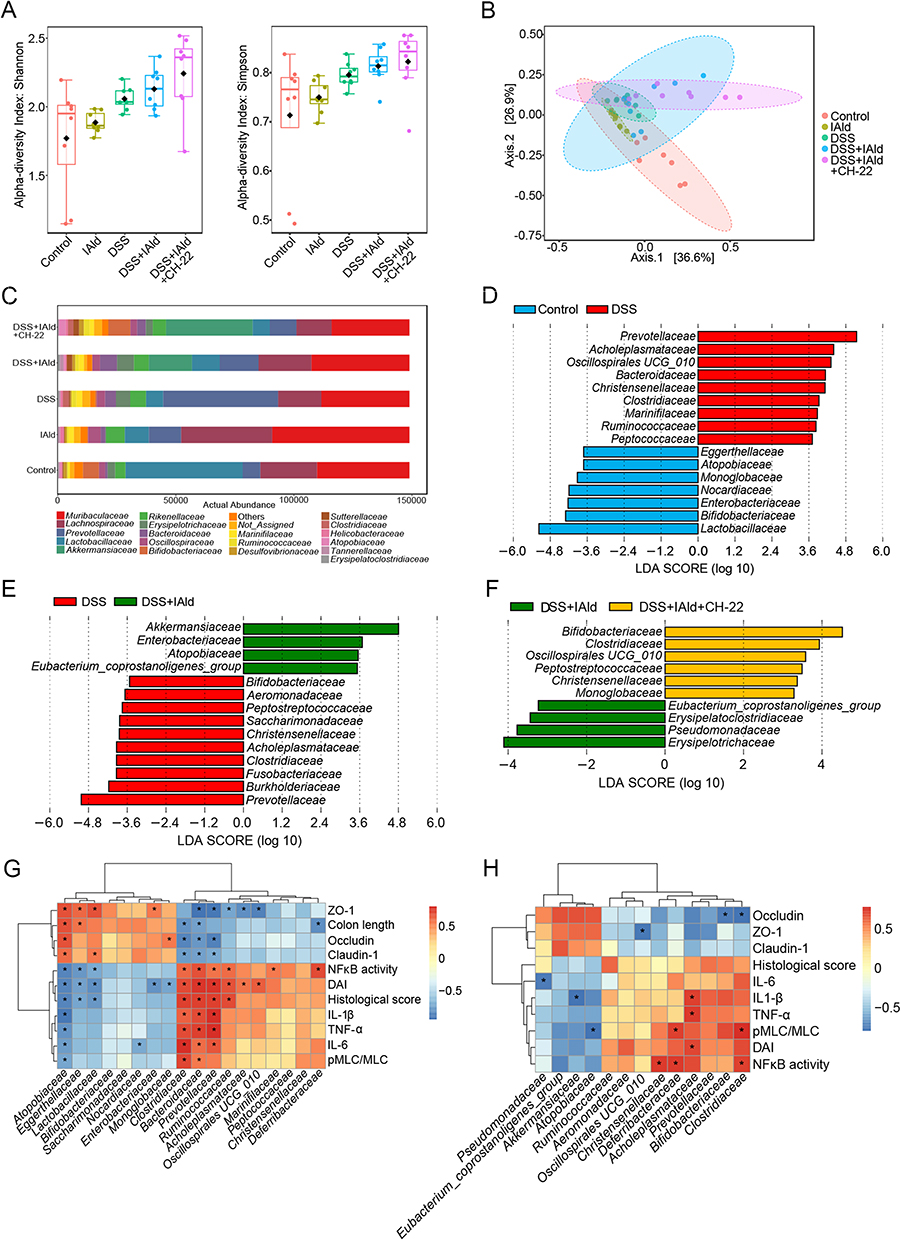 |
Figure 9 Effects of IAld and AhR activation on the gut microbiome composition in a DSS-induced mouse model. Notes: (A) α-diversity was determined using Simpson index and Shannon index. (B) β-diversity was determined using weighted UniFrac distance-based Principal Coordinates Analysis (PCoA) (R2 = 0.49613 P = 0.001). (C) The relative abundance of bacteria of different groups at family level. (D-F) LDA scores from LefSe analysis were performed on relative ASV between groups of Control vs DSS; DSS vs DSS+IAld; DSS+IAld vs DSS+IAld+CH-22. (G and H) Correlation heatmap was calculated by using Spearman correlation coefficient. The data are shown as the mean ± SEM (n = 5–8/group). *p < 0.05. |
In the control and DSS groups, the bacterial families, eg, Atopobiaceae, Eggerthellaceae and Lactobacillaceae showed a strongly negative correlation with inflammatory cytokines and displayed a highly positive correlation with TJ proteins, whereas Clostridiaceae, Bacteroidaceae, Prevotellaceae and Ruminococcaceae showed the opposite correlation (Figure 9G). In the DSS and DSS+IAld groups, Christensenellaceae, Deferribacteraceae, Acholeplasmataceae and Clostridiaceae were positively correlated with inflammatory parameters and pMLC/MLC, while Bifidobacteriaceae and Clostridiaceae were negatively correlated with TJ protein levels (Figure 9H). In particular, Akkermansiaceae was significantly and negatively correlated with IL-1β (Figure 9H) No statistically significant correlations were found in the DSS+IAld and CH-22 groups.
Taken as a whole, these observations prompt us to conclude that IAld-mediated improvement in gut epithelial integrity and reductions in gut tissue inflammation are associated with gut bacteria changes. Investigating their causal relationship warrants further studies.
Discussion
The bacterial metabolites of Trp have been increasingly studied owing to their biofunctional properties in regulating host physiology.5 IAld, one of the Trp metabolites by the gut bacteria, has been shown to have anti-inflammatory properties in multiple disease models, such as endometritis, dermatitis, and experimental autoimmune encephalomyelitis.12,22,42,43 In this study, we report that IAld reduces pro-inflammatory cytokines, namely, IL-6, IL-1β and TNF-α in both colon tissues from the mice with DSS-induced colitis and LPS-stimulated macrophages. Pro-inflammatory cytokines are commonly increased in mucosal biopsies from patients with UC or mice with colitis.44–46 Therapies of suppressing these pro-inflammatory cytokines such as anti-TNF-α and anti-IL-6 have been shown to be effective in certain cohorts of patients.47,48 In line with our findings, IPA and IAld have been reported to upregulate epithelial IL-10R1 activity to reduce colitis severity and promote epithelial wound healing.9 These evidence as a whole suggest that IAld not only enhances anti-inflammatory cytokines but also reduces pro-inflammatory cytokines to exert anti-inflammatory effects. Furthermore, we report that IAld-induced reduction of IL-6 is AhR activation-dependent, but the reductions of IL-1β and TNF-α are likely through AhR-independent mechanisms, which remain to be explored. Our observations agree with a previous study, where AhR deficiency was found to increase IL-6 expression at the protein level in LPS-stimulated macrophages,49 indicating that AhR activation is involved in the downregulation of IL-6 levels. IAld was found to reduce mRNA expression levels of IL-1β and CH-22 partially inhibited this reduction in RAW264.7 cells, of which heat-killed Mycobacterium tuberculosis was used to induce the inflammatory responses; however, the functional protein level of IL-1β was not measured.50
Another profound finding was that IAld reduces NF-κB and JNK pathways through AhR activation in LPS-stimulated macrophages. AhR suppresses p65 activation induced by TNF-α and Poly I:C;51 on the other hand, NF-κB activates the transcription of AhR and enhances the activities of AhR-regulated genes.52 We provide experimental evidence that IAld-mediated AhR activation suppresses the NF-κB pathway by both reducing the phosphorylated p65 expression and inhibiting p65 translocation. In contrast, little is known about the effect of AhR on the JNK pathway.53 The AhR ligands of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) was observed to activate JNK pathway to regulate cell plasticity. Previous studies have shown that these two pathways regulate the expression of a number of pro-inflammatory cytokines, such as IL-6 in macrophages.54–57 Therefore, these evidence together suggest that IAld-mediated AhR activation reduces LPS-induced secretion of IL-6, which is likely through inhibiting NF-κB and JNK pathways in both murine and human macrophages in the current study.
In addition to reduction in pro-inflammatory cytokine secretion, our investigation revealed that through AhR activation IAld resulted in improved IEB function and reduced protein levels of MLCK and p-MLC in LPS-stimulated Caco-2 monolayers which are in agreement with several published studies where AhR activation was shown to ameliorate IEB dysfunction under different intestinal stresses.21,58–60 A previous study using TNF-a as a stimulant for Caco-2 cells found that IAld reduced the permeability of the monolayer21 which is consistent with our observations. Furthermore, we observed a down-regulatory effect of IAld on NF-κB pathway through AhR activation, evidenced by reduced levels of p-p65 and inhibition of p65 translocation to from cytoplasm to nucleus in the LPS-stimulated Caco-2 cells. Inhibition of NF-κB by siRNA knockdown of p50/p65 was reported to prevent MLCK expression and maintain the permeability of LPS-stimulated Caco-2 monolayers.40 As the previous study showed that MLCK inhibitor prevented the TNF-α induced increase in the permeability of the Caco-2 monolayer,61 indicating that gut permeability is regulated by MLCK. These evidence together suggest that IAld-mediated AhR activation inhibits NF-κB pathway, which downregulates both IL-6 secretion in macrophages and MLCK/p-MLC expression in the intestinal epithelial cells to improve the gut permeability and IEB function.
In contrast to the findings from human Caco-2 cells, we noted a significant reduction in the gut permeability induced by IAld in mice with DSS-induced colitis; however, this improvement was not fully abolished by the AhR inhibitor. This observation suggests that IAld-induced protective effects on the gut integrity could be through some AhR-independent mechanisms. This finding differs from a previous study using Ahr-/- mice, where IAld at a dose of 1000 mg per kg of body weight,21 a much higher than the physiological concentration found in human stool,62 was shown to improve the gut permeability in an AhR-dependent manner.21 This inconsistency could be due to the differences in the mouse models and IAld doses.
Our further investigation on the faecal bacterial composition showed that neither alpha- nor beta-diversity was significantly affected by IAld treatment. However, the relative abundance of Akkermansiaceae family was higher in IAld-treated mice with colitis compared to the non-treated mice, and this increase was inversely correlated with colonic IL-1β expression. Akkermansia muciniphila is a well-studied species of Akkermansiaceae family and has been found to reduce the risk of metabolic disorders.63,64 A previous study reported that IAld at above 5 µM promoted the growth of A. muciniphila BAA-835T in vitro,65 which is in line with our observations in vivo. A. muciniphila BAA-835T was also shown to metabolise Trp to indole, indole-3-acetic acid, indole-3-lactic acid and IAld.65 These data together led us to speculate that IAld promotes the growth of Akkermansiaceae bacteria in the gut, which in turn increases the bioavailability of Trp metabolites to improve IEB function, forming a positive feedback loop. This hypothesis warrants further investigation by assessing bacterial function and Trp metabolite concentrations in the lumen following IAld treatment.
Conclusion
In summary, our data showed that IAld-mediated AhR activation suppressed the NF-κB pathway, which is key in regulating inflammation (down-regulation of pro-inflammatory cytokine IL-6 secretion) and IEB function (reduced expression of MLCK/p-MLC and improved gut permeability). We also demonstrated that IAld could exert its beneficial effects such as reduction of IL-1β and TNF-α via AhR-independent mechanisms, as well as through potential gut microbiota-mediated mechanisms (eg, growth of Akkermansiaceae). The latter will require further studies to probe the complex crosstalk amongst Trp metabolites, the gut microbiota and the host. These anti-inflammatory effects of IAld on intestinal inflammation provide us with new perspectives as a potential adjuvant therapeutic approach to alleviate colitis symptoms in patients with UC.
Funding
Mu Wang received the grant from Natural Science Foundation of Shanxi Province [202203021212076]. Jian Guo received the grant from Natural Science Foundation of Shanxi Province [20210302124560]. Jia V Li thanks the salary support from the ERC Starting Grant [715662].
Disclosure
The authors report no conflicts of interest in this work. Jia Li declared consultancy work for the University of Cardiff, the work is not relevant to the submitted manuscript.
References
1. Chang JT. Pathophysiology of Inflammatory Bowel Diseases. N Engl J Med. 2021;384(14):1378. doi:10.1056/NEJMc2101562
2. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14(5):269–278. doi:10.1038/nrgastro.2016.208
3. Li W, Hang S, Fang Y, et al. A bacterial bile acid metabolite modulates T(reg) activity through the nuclear hormone receptor NR4A1. Cell Host Microbe. 2021;29(9):1366–1377 e9. doi:10.1016/j.chom.2021.07.013
4. Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(4):223–237. doi:10.1038/s41575-019-0258-z
5. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9(1):3294. doi:10.1038/s41467-018-05470-4
6. Gutierrez-Vazquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity. 2018;48(1):19–33. doi:10.1016/j.immuni.2017.12.012
7. Nikolaus S, Schulte B, Al-Massad N, et al. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology. 2017;153(6):1504–1516 e2. doi:10.1053/j.gastro.2017.08.028
8. Lamas B, Richard ML, Leducq V, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22(6):598–605. doi:10.1038/nm.4102
9. Alexeev EE, Lanis JM, Kao DJ, et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am J Pathol. 2018;188(5):1183–1194. doi:10.1016/j.ajpath.2018.01.011
10. Islam J, Sato S, Watanabe K, et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J Nutr Biochem. 2017;42:43–50. doi:10.1016/j.jnutbio.2016.12.019
11. Ala M. Tryptophan metabolites modulate inflammatory bowel disease and colorectal cancer by affecting immune system. Int Rev Immunol. 2022;41(3):326–345. doi:10.1080/08830185.2021.1954638
12. Zhao C, Bao L, Qiu M, et al. Dietary Tryptophan-Mediated Aryl Hydrocarbon Receptor Activation by the Gut Microbiota Alleviates Escherichia coli-Induced Endometritis in Mice. Microbiol Spectr. 2022;10(4):e0081122. doi:10.1128/spectrum.00811-22
13. Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36(2):189–204. doi:10.1016/s1357-2725(03)00211-5
14. Larigot L, Juricek L, Dairou J, Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open. 2018;7:1–9. doi:10.1016/j.biopen.2018.05.001
15. Ishihara Y, Kado SY, Bein KJ, et al. Aryl Hydrocarbon Receptor Signaling Synergizes with TLR/NF-kappaB-Signaling for Induction of IL-22 Through Canonical and Non-Canonical AhR Pathways. Front Toxicol. 2021;3:787360. doi:10.3389/ftox.2021.787360
16. Monteleone I, Rizzo A, Sarra M, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141(1):237–48, 248 e1. doi:10.1053/j.gastro.2011.04.007
17. Alvik K, Shao P, Hutin D, Baglole C, Grant DM, Matthews J. Increased sensitivity to chemically induced colitis in mice harboring a DNA-binding deficient aryl hydrocarbon receptor. Toxicol Sci. 2023;191(2):321–331. doi:10.1093/toxsci/kfac132
18. Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018;11(4):1024–1038. doi:10.1038/s41385-018-0019-2
19. Cervantes-Barragan L, Chai JN, Tianero MD, et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science. 2017;357(6353):806–810. doi:10.1126/science.aah5825
20. Hou Q, Ye L, Liu H, et al. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018;25(9):1657–1670. doi:10.1038/s41418-018-0070-2
21. Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2020;117(32):19376–19387. doi:10.1073/pnas.2000047117
22. Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39(2):372–385. doi:10.1016/j.immuni.2013.08.003
23. Wilck N, Matus MG, Kearney SM, et al. Salt-responsive gut commensal modulates T(H)17 axis and disease. Nature. 2017;551(7682):585–589. doi:10.1038/nature24628
24. Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168(10):5342–5351. doi:10.4049/jimmunol.168.10.5342
25. Dahan S, Roda G, Pinn D, et al. Epithelial: lamina propria lymphocyte interactions promote epithelial cell differentiation. Gastroenterology. 2008;134(1):192–203. doi:10.1053/j.gastro.2007.10.022
26. Mandic AD, Bennek E, Verdier J, et al. c-Jun N-terminal kinase 2 promotes enterocyte survival and goblet cell differentiation in the inflamed intestine. Mucosal Immunol. 2017;10(5):1211–1223. doi:10.1038/mi.2016.125
27. Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med. 2008;263(6):591–596. doi:10.1111/j.1365-2796.2008.01953.x
28. Andresen L, Jorgensen VL, Perner A, Hansen A, Eugen-Olsen J, Rask-Madsen J. Activation of nuclear factor kappaB in colonic mucosa from patients with collagenous and ulcerative colitis. Gut. 2005;54(4):503–509. doi:10.1136/gut.2003.034165
29. Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. 2019;16(9):531–543. doi:10.1038/s41575-019-0172-4
30. Murano M, Maemura K, Hirata I, et al. Therapeutic effect of intracolonically administered nuclear factor kappa B (p65) antisense oligonucleotide on mouse dextran sulphate sodium (DSS)-induced colitis. Clin Exp Immunol. 2000;120(1):51–58. doi:10.1046/j.1365-2249.2000.01183.x
31. Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2(3):541–546. doi:10.1038/nprot.2007.41
32. Mullish BH, Pechlivanis A, Barker GF, Thursz MR, Marchesi JR, McDonald JAK. Functional microbiomics: evaluation of gut microbiota-bile acid metabolism interactions in health and disease. Methods. 2018;149:49–58. doi:10.1016/j.ymeth.2018.04.028
33. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
34. Cai L, Qin X, Xu Z, et al. Comparison of Cytotoxicity Evaluation of Anticancer Drugs between Real-Time Cell Analysis and CCK-8 Method. ACS Omega. 2019;4(7):12036–12042. doi:10.1021/acsomega.9b01142
35. Gupta V, Doshi N, Mitragotri S. Permeation of insulin, calcitonin and exenatide across Caco-2 monolayers: measurement using a rapid, 3-day system. PLoS One. 2013;8(2):e57136. doi:10.1371/journal.pone.0057136
36. Sanders SE, Madara JL, McGuirk DK, Gelman DS, Colgan SP. Assessment of inflammatory events in epithelial permeability: a rapid screening method using fluorescein dextrans. Epithelial Cell Biol. 1995;4(1):25–34.
37. Ghosh SS, Wang J, Yannie PJ, Ghosh S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J Endocr Soc. 2020;4(2):bvz039. doi:10.1210/jendso/bvz039
38. Su L, Nalle SC, Shen L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145(2):407–415. doi:10.1053/j.gastro.2013.04.011
39. Turner JR, Rill BK, Carlson SL, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273(4):C1378–85. doi:10.1152/ajpcell.1997.273.4.C1378
40. Nighot M, Rawat M, Al-Sadi R, Castillo EF, Nighot P, Ma TY. Lipopolysaccharide-Induced Increase in Intestinal Permeability Is Mediated by TAK-1 Activation of IKK and MLCK/MYLK Gene. Am J Pathol. 2019;189(4):797–812. doi:10.1016/j.ajpath.2018.12.016
41. Selvakumar D, Evans D, Coyte KZ, et al. Understanding the development and function of the gut microbiota in health and inflammation. Frontline Gastroenterol. 2022;13(e1):e13–e21. doi:10.1136/flgastro-2022-102119
42. Rothhammer V, Mascanfroni ID, Bunse L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22(6):586–597. doi:10.1038/nm.4106
43. Yu J, Luo Y, Zhu Z, et al. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J Allergy Clin Immunol. 2019;143(6):2108–2119 e12. doi:10.1016/j.jaci.2018.11.036
44. Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94(1):174–181. doi:10.1111/j.1365-2249.1993.tb05997.x
45. Ishiguro Y. Mucosal proinflammatory cytokine production correlates with endoscopic activity of ulcerative colitis. J Gastroenterol. 1999;34(1):66–74. doi:10.1007/s005350050218
46. Kai Y, Takahashi I, Ishikawa H, et al. Colitis in mice lacking the common cytokine receptor gamma chain is mediated by IL-6-producing CD4+ T cells. Gastroenterology. 2005;128(4):922–934. doi:10.1053/j.gastro.2005.01.013
47. Schreiber S, Aden K, Bernardes JP, et al. Therapeutic Interleukin-6 Trans-signaling Inhibition by Olamkicept (sgp130Fc) in Patients With Active Inflammatory Bowel Disease. Gastroenterology. 2021;160(7):2354–2366 e11. doi:10.1053/j.gastro.2021.02.062
48. Friedrich M, Pohin M, Powrie F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity. 2019;50(4):992–1006. doi:10.1016/j.immuni.2019.03.017
49. Kimura A, Naka T, Nakahama T, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009;206(9):2027–2035. doi:10.1084/jem.20090560
50. Langan D, Perkins DJ, Vogel SN, Moudgil KD. Microbiota-Derived Metabolites, Indole-3-aldehyde and Indole-3-acetic Acid, Differentially Modulate Innate Cytokines and Stromal Remodeling Processes Associated with Autoimmune Arthritis. Int J Mol Sci. 2021;22(4). doi:10.3390/ijms22042017
51. Ovrevik J, Lag M, Lecureur V, et al. AhR and Arnt differentially regulate NF-kappaB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun Signal. 2014;12:48. doi:10.1186/s12964-014-0048-8
52. Vogel CF, Khan EM, Leung PS, et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-kappaB. J Biol Chem. 2014;289(3):1866–1875. doi:10.1074/jbc.M113.505578
53. Diry M, Tomkiewicz C, Koehle C, et al. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene. 2006;25(40):5570–5574. doi:10.1038/sj.onc.1209553
54. Wang Y, Sun D, Song F, Hu Y, Smith DE, Jiang H. Expression and regulation of the proton-coupled oligopeptide transporter PhT2 by LPS in macrophages and mouse spleen. Mol Pharm. 2014;11(6):1880–1888. doi:10.1021/mp500014r
55. Shin GT, Lee HJ, Kim H. GADD45gamma regulates TNF-alpha and IL-6 synthesis in THP-1 cells. Inflamm Res. 2012;61(11):1195–1202. doi:10.1007/s00011-012-0515-x
56. Zhang W, Shen X, Xie L, Chu M, Ma Y. MicroRNA-181b regulates endotoxin tolerance by targeting IL-6 in macrophage RAW264.7 cells. J Inflamm. 2015;12:18. doi:10.1186/s12950-015-0061-8
57. Ma P, Liu H-T, Wei P, et al. Chitosan oligosaccharides inhibit LPS-induced over-expression of IL-6 and TNF-α in RAW264. 7 macrophage cells through blockade of mitogen-activated protein kinase (MAPK) and PI3K/Akt signaling pathways. Carbohydr. Polym. 2011;84(4):1391–1398. doi:10.1016/j.carbpol.2011.01.045
58. Cho H, Kim E, Lee H, et al. BJ-1108, a 6-Amino-2, 4, 5-trimethylpyridin-3-ol analogue, ameliorates DSS-induced colitis in mice. J Crohns Colitis. 2018;12. doi:10.1093/ecco-jcc/jjx180.224
59. Yu M, Wang Q, Ma Y, et al. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int J Biol Sci. 2018;14(1):69–77. doi:10.7150/ijbs.22259
60. Han B, Sheng B, Zhang Z, et al. Aryl Hydrocarbon Receptor Activation in Intestinal Obstruction Ameliorates Intestinal Barrier Dysfunction Via Suppression of MLCK-MLC Phosphorylation Pathway. Shock. 2016;46(3):319–328. doi:10.1097/SHK.0000000000000594
61. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-alpha modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G422–30. doi:10.1152/ajpgi.00412.2004
62. Dong F, Hao F, Murray IA, et al. Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microbes. 2020;12(1):1–24. doi:10.1080/19490976.2020.1788899
63. Shin NR, Lee JC, Lee HY, et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63(5):727–735. doi:10.1136/gutjnl-2012-303839
64. Roopchand DE, Carmody RN, Kuhn P, et al. Dietary Polyphenols Promote Growth of the Gut Bacterium Akkermansia muciniphila and Attenuate High-Fat Diet-Induced Metabolic Syndrome. Diabetes. 2015;64(8):2847–2858. doi:10.2337/db14-1916
65. Yin J, Song Y, Hu Y, et al. Dose-Dependent Beneficial Effects of Tryptophan and Its Derived Metabolites on Akkermansia In Vitro: a Preliminary Prospective Study. Microorganisms. 2021;9(7). doi:10.3390/microorganisms9071511
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.