Back to Journals » ImmunoTargets and Therapy » Volume 9
Immunotherapy for Medulloblastoma: Current Perspectives
Authors Kabir TF, Kunos CA, Villano JL, Chauhan A
Received 28 January 2020
Accepted for publication 29 March 2020
Published 20 April 2020 Volume 2020:9 Pages 57—77
DOI https://doi.org/10.2147/ITT.S198162
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Tanvir F Kabir,1 Charles A Kunos,2 John L Villano,3 Aman Chauhan3
1Department of Internal Medicine, University of Louisville, Louisville, KY, USA; 2Cancer Therapy Evaluation Program, National Cancer Institute, Bethesda, MD, USA; 3Department of Internal Medicine-Medical Oncology, Markey Cancer Center, University of Kentucky, Lexington, KY, USA
Correspondence: Aman Chauhan Email [email protected]
Background: Immune-mediated therapies have transformed the treatment of metastatic melanoma and renal, bladder, and both small and non-small cell lung carcinomas. However, immunotherapy is yet to demonstrate dramatic results in brain tumors like medulloblastoma for a variety of reasons. Recent pre-clinical and early phase human trials provide encouraging results that may overcome the challenges of central nervous system (CNS) tumors, which include the intrinsic immunosuppressive properties of these cancers, a lack of antigen targets, antigenic variability, and the immune-restrictive site of the CNS. These studies highlight the growing potential of immunotherapy to treat patients with medulloblastoma, a disease that is a frequent cause of morbidity and mortality to children and young adults.
Methods: We conducted an inclusive review of the PubMed-indexed literature and studies listed in clinicaltrials.gov using combinations of the keywords medulloblastoma, immunotherapy, CNS tumors, brain tumors, vaccines, oncolytic virus, natural killer, and CAR T to identify trials evaluating immunotherapy in preclinical experiments or in patients with medulloblastoma. Given a limited number of investigations using immunotherapy to treat patients with medulloblastoma, 24 studies were selected for final analysis and manuscript citation.
Results: This review presents results from pre-clinical studies in medulloblastoma cell lines, animal models, and the limited trials involving human patients.
Conclusion: From our review, we suggest that cancer vaccines, oncolytic viral therapy, natural killer cells, and CAR T therapy hold promise against the innate immunosuppressive properties of medulloblastoma in order to prolong survival. There is an unmet need for immunotherapy regimens that target overexpressed antigens in medulloblastoma tumors. We advocate for more combination treatment clinical trials using conventional surgical and radiochemotherapy approaches in the near-term clinical development.
Keywords: medulloblastoma, immunotherapy, vaccines, oncolytic virus, natural killer, CAR T, review
Introduction
Primary brain and CNS malignancies are among the most common solid tumors in the pediatric population, and medulloblastoma is the most prevalent brain tumor in children.1 Medulloblastomas originate from the cerebellar vermis and usually in proximity to the fourth ventricle, commonly metastasizing through cerebrospinal fluid pathways.2,3 Medulloblastoma accounts for 8–10% of pediatric brain tumors and the 5-year survival rate in children is 75–85% with conventional treatments.4–6 However, the current standard treatment, which includes surgery with subsequent chemotherapy and radiation, often results in severe neurological and endocrine deficits.2,7-9 New therapies are vital to improve treatment outcomes but require penetration of the blood–brain barrier. Although the blood–brain barrier remains a significant challenge, activated T cells and other elements of the immune system can traverse the capillary tight junctions formed in the blood-brain barrier, unlike many chemotherapy agents.10 Immunotherapy is an attractive targeted approach to eliminate cancer cells while simultaneously sparing adjacent brain tissue.2 Tumor targeting T cells can be activated in vivo via cancer vaccines and oncolytic viruses while other ex vivo engineered therapies can be transfused into patients to stimulate the host immune system. Immunotherapy has shown clinical benefit in a variety of cancers like melanoma, lung cancer, and leukemias. Yet several challenges exist in targeting central nervous system (CNS) tumors such as medulloblastoma including the lack of known immunogenic antigens.10 Encouraging results were demonstrated with immunotherapy for brain tumors including glioblastoma, and recent studies documented the overexpression of certain antigens on medulloblastoma that can potentially serve as targets for vaccines, CAR T, and other forms of immunotherapy.
We conducted a review of PubMed-indexed literature and clinicaltrials.gov using combinations of the keywords medulloblastoma, immunotherapy, CNS tumors, brain tumors, vaccines, oncolytic virus, natural killer, and CAR T to find as many completed and ongoing trials as possible that evaluated immunotherapy as treatment for patients with medulloblastoma. This paper presents a review of these findings with a discussion of the investigations categorized by therapeutic modalities: cancer vaccines, oncolytic viruses, checkpoint inhibitors, natural killer cells, radiotherapy, and CAR-T cell therapy. Current FDA-approved studies evaluating immunotherapy in medulloblastoma will also be discussed and outlined in Table 1.
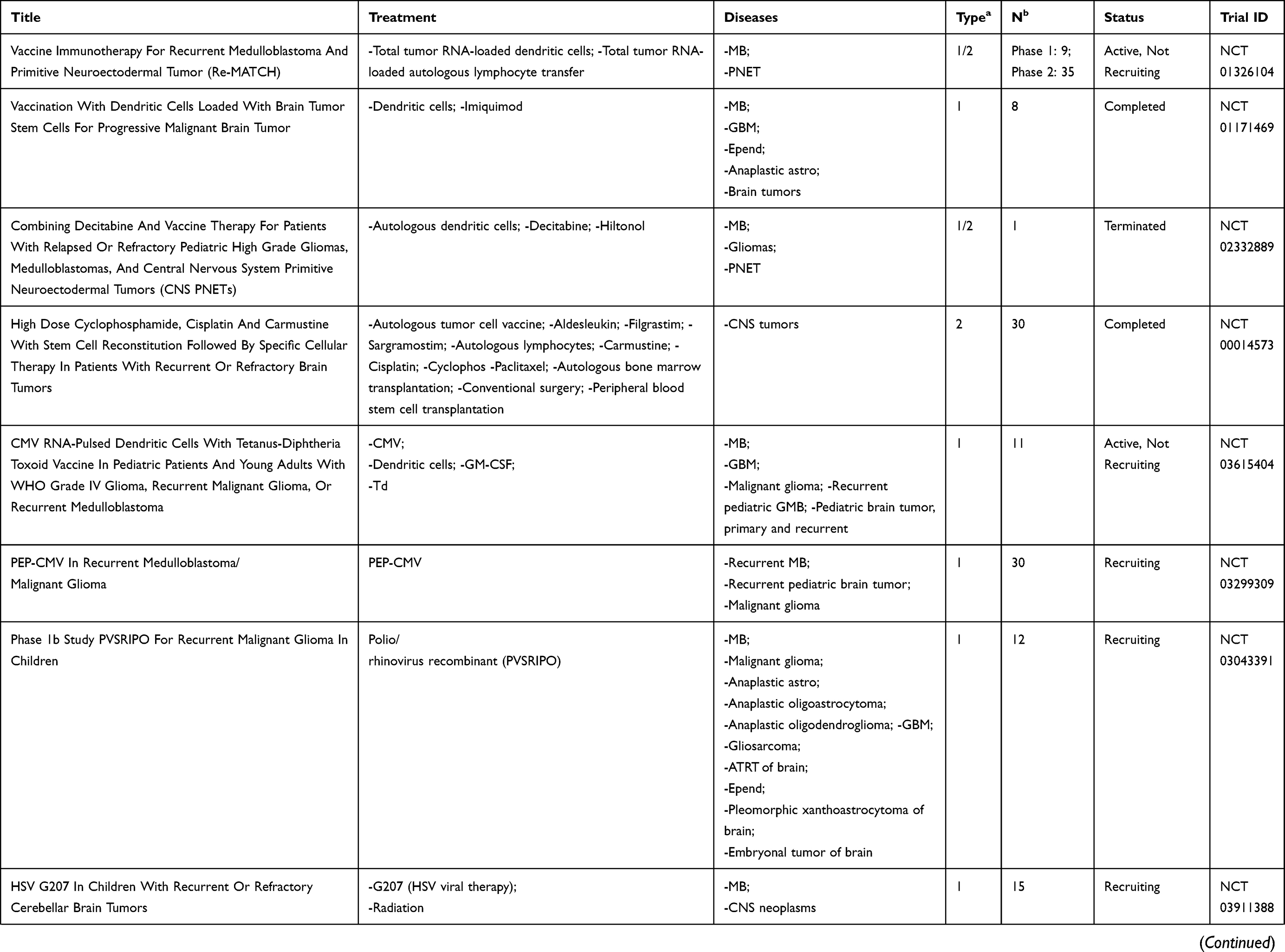 | 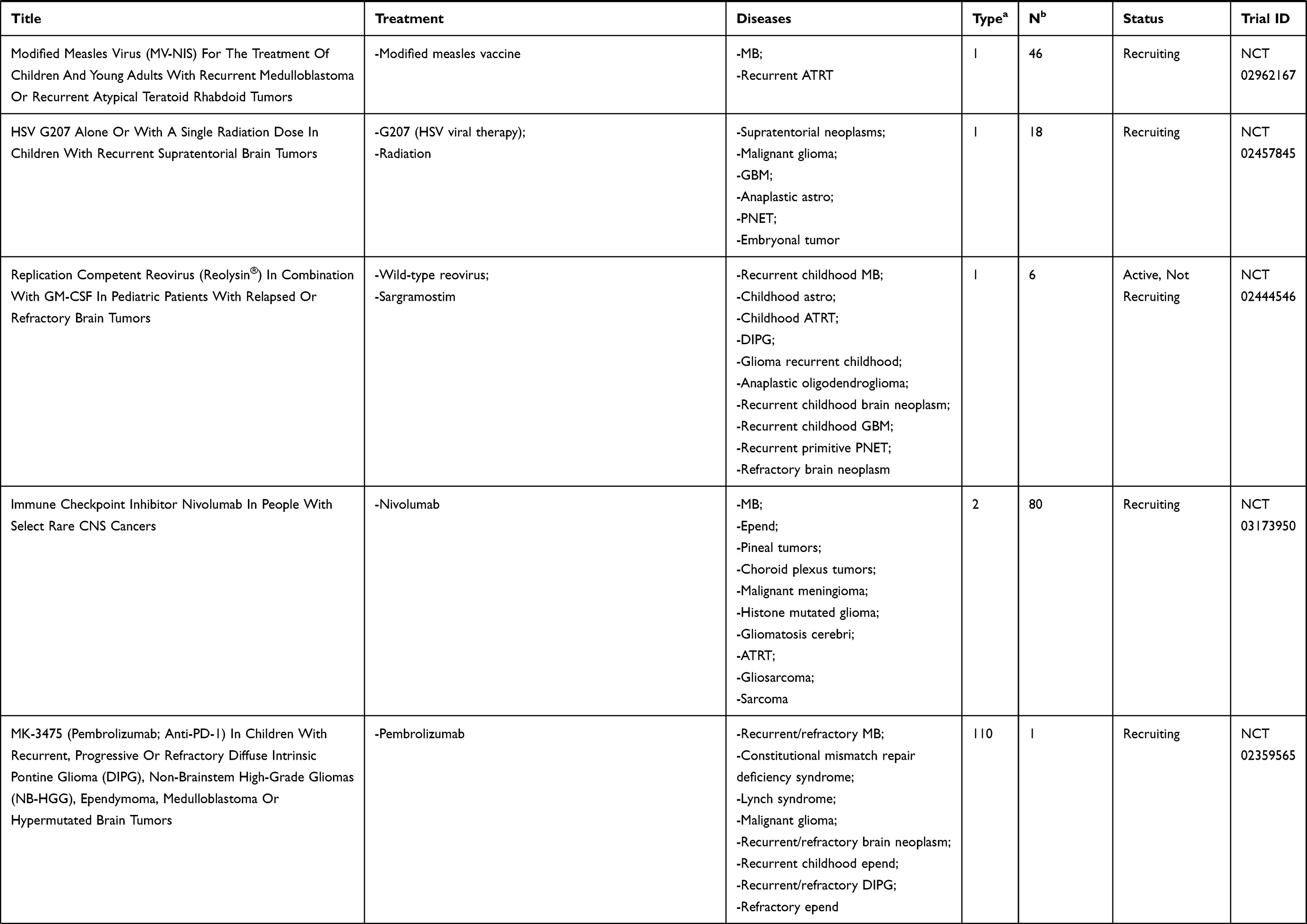 | 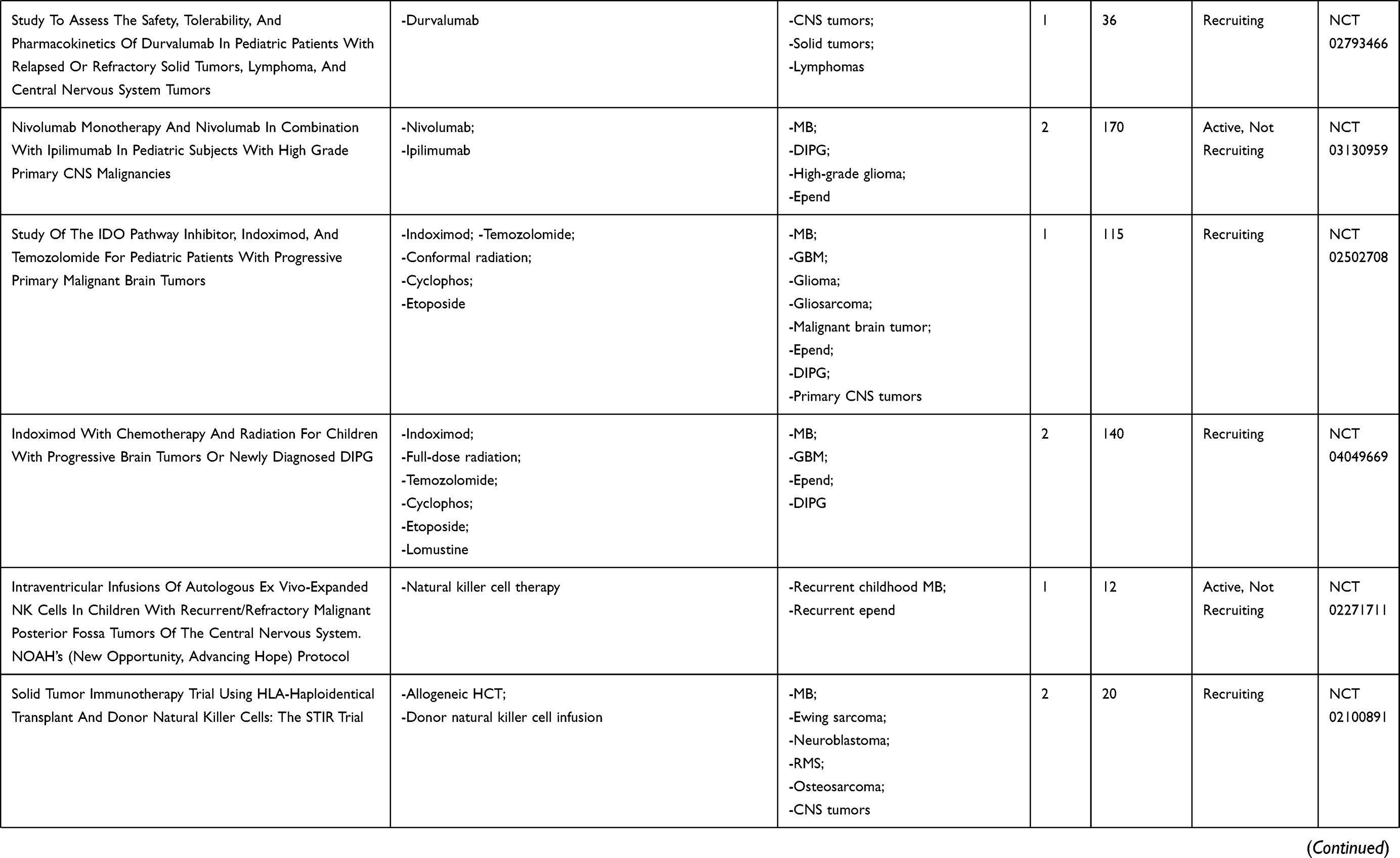 | 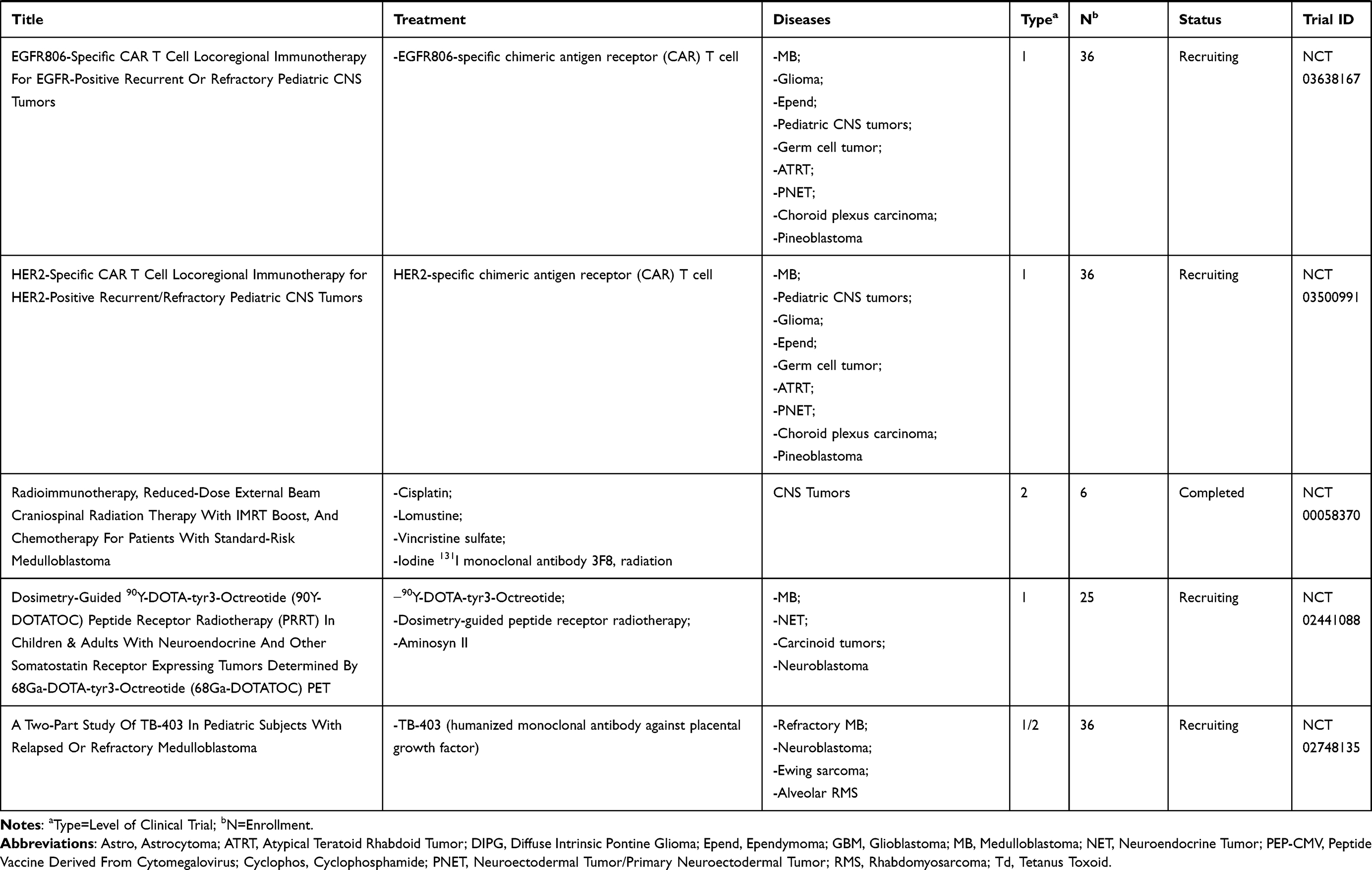 |
Table 1 Current Immunotherapy Clinical Trials in Patients with Medulloblastoma |
Background
The World Health Organization classifies medulloblastomas into four histological groups: large cell, anaplastic, nodular desmoplastic, and extensive nodularity.11 In addition, a newer, revised classification system is based on the molecular profile of medulloblastoma and divided into four subgroups: sonic hedgehog activated medulloblastoma (SHH, further subdivided into TP53-mutant and Tp53-wildtype), WNT-activated, group 3, and group 4.11 The first two subgroups are caused by alterations in the SHH and WNT pathways, respectively, while less is known about the etiology of groups 3 and 4. Furthermore, advancements in gene sequencing have shown that the subgroups can also be divided into 12 subtypes with distinct activated pathways, somatic copy-number aberrations, and clinical outcomes: SHH α, SHH β, SHH γ, SHH δ, WNT α, WNT β, group 3α, group 3β, group 3γ, group 4α, group 4β, and group 4γ.12 For example, SHH α tumors demonstrate the most MYCN and GLI2 gene amplification and have worse prognosis compared to SHH β tumors, which exhibit increased PTEN gene deletion.12
Embryologically, SHH medulloblastomas originate from cerebellar granule neuron precursor cells (GNPCs), while WNT tumors originate from precursor cells within the dorsal brain stem.13,14 Group 3 medulloblastomas also appear to originate from GNPCs but through a different pathway than SHH; interestingly, these tumors express photoreceptor-encoding genes resembling rod precursor cells at week 15 of retinal development.15 Group 4 cells arise from GNPCs as well, and their gene expression is similar to cerebellar glutamatergic granule neurons at late fetal stages of development.15 WNT medulloblastomas have the best prognosis of 95% 5-year survival but are the least common subgroup.16,17 Group 4 medulloblastoma is the most common subgroup.
Most of our current knowledge of CNS immunology is based on glioma research, which showed a deficient immune response in patients and animal models with gliomas.2,18–20 Similarly, immunosuppression was first reported over 30 years ago in medulloblastoma patients who had reduced peripheral T cell activity, shown by skin hypersensitivity reactions.21,22 However, given the less deleterious side effects of immunotherapy compared to conventional therapies, researchers nonetheless strive to circumvent the immunosuppressive properties of these tumors and elicit a host response. Furthermore, immunotherapy can be used in conjunction with chemotherapy, as various preclinical and clinical studies have shown that vaccination concurrent with chemotherapy improves antigen-specific T cell activity.10,23–25 For an immune response to be elicited against brain tumors, appropriate tumor-specific antigens must be targeted. Antigens not expressed on normal tissues are the ideal target; however, these antigens are likely personalized to the tumor in each individual patient. CNS tumors, including medulloblastomas, exhibit a low mutational burden.26 Alternative targets may include normal tissue antigens that are overexpressed on tumor cells.27 As brain tumors, including medulloblastomas, have limited gene mutations, there are a number of epigenetic deregulatory pathways that may increase the re-expression of fetal antigens in medulloblastomas.28–30 These fetal, developmental antigens may be tumor-specific and targeted as they are not expressed in normal tissue.10
Until recently, accepted dogma was that the CNS was resistant to immunotherapy based on the limited access to antibodies.26,31 However, the occurrence of autoimmune syndromes including multiple sclerosis that is regulated by the immune system suggests that T cell immunosurveillance does occur.26 Furthermore, the incidence of John Cunningham virus progressive multifocal leukoencephalopathy in patients treated with rituximab shows that immunosuppression can lead to a re-activation of viruses in the CNS.26 While it was previously thought that the brain did not contain lymphatics, studies have shown that lymphatics do indeed exist in the dura and meninges and that lymphocytes exit the CNS via the deep cervical lymph nodes.32 T cells do not enter through the blood–brain barrier but rather access the parenchyma through the choroid plexus or pia mater.33 Thus, theoretically, antigens created through tumor mutations should be detected by the immune system within the deep cervical lymph nodes, and T cells administered through systemic infusion should be able to target tumors via these routes.26 One recent study showed that the disruption of dorsal meningeal lymphatic vessels in mice impaired tumor fluid drainage and dissemination of tumor cells to deep cervical lymph nodes; however, dendritic cell trafficking to these lymph nodes was also diminished.34 Consequently, the disrupted dorsal meningeal lymphatics reduced the efficacy of combined anti-CTLA-4/PD-1 checkpoint immunotherapy in striatal tumor models.
However, many challenges remain in treating brain tumors with immunotherapy. The highly specialized blood-brain barrier, which protects the brain from circulating toxins and viruses, can prevent chemotherapy and large-sized molecules from entering the CNS. The brain also appears to have a different set of immune cells than the periphery and does not generate an immune response in the same manner. Furthermore, components of the immune system, including tumor-infiltrating T lymphocytes, appear depleted surrounding certain tumors including glioblastoma.35 As glioblastoma has been the most widely investigated brain tumor using immunotherapy, much of our knowledge on immunotherapeutic mechanism of action and toxicity within the CNS is based on such research. It is encouraging that clinical trials for glioblastoma have progressed to Phase III; however, results have so far been less than promising. The first ever Phase III clinical trial evaluating the PD-1 checkpoint inhibitor nivolumab in glioblastoma (Checkmate 143) failed to increase survival compared to bevacizumab. Another phase III clinical trial evaluating a vaccine known as rindopepimut that targets EGFRvIII, overexpressed in many glioblastomas, also failed to improve survival.36
Current treatment regimens for medulloblastoma have been relatively unchanged for the past two decades. Medulloblastomas have typically been more radiosensitive than other pediatric brain tumors, including glioblastomas.4 For children older than 3 years, external beam radiation to the spine and brain combined with multidrug chemotherapy and surgery has been standard.37,38 Treatment is stratified based on risk: average risk and high risk.39 Average risk includes residual tumor post-surgery less than 1.5 cm2 with no metastasis, while high risk includes metastases and greater than 1.5 cm2 residual disease.40 While both are treated with radiation, higher-risk patients are given larger boosts of radiation. Both groups typically receive four 28-day cycles of cisplatin, vincristine, and cyclophosphamide.39,41,42 Children younger than 3 years are given high dose chemotherapy but no radiation due to the adverse effects on the developing brain. However, surgery can result in multiple complications including cerebellar mutism syndrome.43 Radiation in older children has been linked to reduced IQ and induction of secondary cancers, vasculopathy, hearing loss, and future strokes.44–48
The main cause of death from medulloblastoma is due to metastasis. While metastatic tumors have the same molecular subtype as the primary medulloblastoma, the genomics of the secondary tumor often differ.49,50 Additionally, pediatric brain tumors differ from adult brain tumors in clinical presentation in addition to gene mutations, embryological origins, and microenvironment of the tumor.51 Even more problematic, major subpopulations of medulloblastoma patients do not respond to current immunotherapy due to lack of antigenic mutations and/or immuno-resistant properties of medulloblastoma cells.52
However, there have been many advances in our understanding of the mechanisms of immunosuppression in patients with medulloblastoma. Such knowledge will help researchers exploit these pathways to develop immunotherapies that can circumvent the tumor’s immunosuppressive properties. Already documented is the finding that immune response correlates with prognosis in patients with medulloblastoma. For example, there is evidence that inflammation and immunological markers correlate with survival of patients with medulloblastoma. In one study, 144 patients with medulloblastoma were divided into subgroups (SHH, WNT, group 3, group 4) and evaluated for preoperative hematological markers and overall survival.53 The study found that an increased preoperative neutrophil-to-lymphocyte ratio (NLR) as well as platelet-to-lymphocyte ratio (PLR) predicted worse prognosis in childhood medulloblastoma patients but not in adult patients. Preoperative monocyte-to-lymphocyte ratio, mean platelet volume, platelet distribution width, and albumin-to-globin ratio did not correlate with prognosis in either age group. Furthermore, the levels of NLR and PLR in group 3 were higher than in WNT.
The remainder of this paper will discuss the most recent findings on various forms of immunotherapy for the treatment of medulloblastoma.
Vaccine Therapy in Medulloblastoma
Cancer vaccines are a form of immunotherapy that can be categorized into several groups: whole tumor cell, peptide, DNA, RNA, etc. Cancer vaccines must activate an immune system that has become tolerant of the host cancer.54 Whole tumor cell and peptide cancer vaccines deliver tumor antigens directly to antigen-presenting cells (APCs) for processing. These APCs then present the antigens as epitopes on the surface of their own MHC class I and II molecules, which are then recognized by T cells.10,55 One example is the FDA-approved sipuleucel-T vaccine for prostate cancer, which consists of a fusion protein antigen containing a tumor-specific antigen, prostate acid phosphatase, and expanded with granulocyte-macrophage colony stimulating factor (GM-CSF). The fusion protein is incubated for many days with antigen-presenting cells taken from the patient, then re-infused into the patient.56 Sipuleucel-T has increased overall survival by 4 months in hormone-resistant prostate cancer.57
In contrast, DNA- and RNA-based cancer vaccines bypass the human leukocyte antigen (HLA) haplotype restriction; this system allows a person’s own cellular processes to generate proteins via transcription/translation for more personalized processing.10,58–61 One research group generated RNA-loaded autologous dendritic cells from patients with medulloblastoma; these cells were matured ex vivo and activated with inflammatory cytokines.62 The activated dendritic cells were then incubated with a personalized group of tumor messenger RNA amplified from that individual’s cDNA library including tumor-specific transcriptome. The dendritic cells were cultured with T cells and then re-administered with the activated T cells in the patient. This research is currently in Phase I and II studies involving patients with medulloblastoma (NCT01326104). The challenge of using dendritic cells is that it requires tumor collection from each patient, which results in extensive processing.63 In another study, autologous monocyte-derived dendritic cells were loaded with tumor lysate, which served as a source of tumor antigens.64 Patients with high-grade glioma and atypical teratoid-rhabdoid tumors responded more favorably to the vaccine treatment than the medulloblastoma patients.
Pre-clinical trials evaluating vaccines showed promise in medulloblastoma but have yet to be translated into human trials. More favorable responses have been observed in other brain tumors such as glioma thus far. Two of the most successful vaccines used in gliomas include epidermal growth factor receptor variant III (EGFRvIII) junctional epitope combined with GM-CSF and whole tumor cell antigen pools presented by autologous dendritic cells.2,65–68 Rindopepimut, the vaccine consisting of EGFRvIII-specific peptide, stimulates the immune system to target the EGFRvIII protein on intracerebral melanoma tumors. In one study, C3H mice with intracerebral melanoma expressing EGFRvIII and treated with the vaccine had a 600% increase in median survival compared to controls.56,69 However, a phase III trial of 165 glioblastoma patients with EGFRvIII was stopped due to lack of efficacy.36 EGFRvIII, while specific in a variety of tumors, can downregulate and escape immune system surveillance.70,71
Unfortunately, recent clinical trials to evaluate cancer vaccines in medulloblastoma patients have been relatively unsuccessful. A phase I/II trial assessed a cancer vaccine consisting of autologous dendritic cells targeting tumor proteins NY-ESO-1 melanoma antigen gene-A1 (MAGE-A1) and melanoma antigen gene-A3 (MAGE-A). The vaccine, administered in combination with decitabine and hiltonol to patients with refractory medulloblastoma was terminated after enrolling one patient due to a serious adverse event and/or disease progression (NCT02332889). Another phase I study evaluated the safety and tolerability of a cancer vaccine consisting of autologous dendritic cells loaded with allogeneic brain tumor stem cells and imiquimod (NCT01171469). Eight patients enrolled in this study, and the trial was terminated with no results posted. Similarly, an earlier Phase II trial that evaluated an autologous tumor cell vaccine concurrent with standard chemotherapy (cyclophosphamide, cisplatin, carmustine), stem cell transplantation, and interleukin-2 was terminated without any results published (NCT00014573). To some degree, the failure of these trials may be attributed to limited viable tumor tissue available for processing and inconsistent expression of tumor antigens.
Yet a number of cancer vaccines with more sophisticated processing combined with adjuvant therapy to enhance the immune response are currently in clinical trial for the treatment of medulloblastoma. In the previously mentioned phase I/II trial using total tumor RNA-loaded dendritic cells, researchers showed that sufficient RNA can be amplified with as few as 500 tumor cells, which allows vaccine preparation from surgical biopsies (NCT01326104). Furthermore, the immunotherapy is infused during recovery from chemotherapy, an advantage as lymphodepletive conditioning is the most effective therapeutic strategy for the treatment of metastatic melanoma. Finally, ex-vivo expanded RNA-loaded autologous lymphocytes will also be infused into the patient to mount an increased immune response, a novel approach distinct from earlier studies. Results are eagerly awaited.
Oncolytic Viral Therapy in Medulloblastoma
Similar to vaccines, oncolytic viral therapy attempts to stimulate the host’s innate immune response. Oncolytic viruses are intended to propagate within tumor cells until they are recognized by the immune system and targeted for rejection.10,72–77 When a virus-infected tumor cell lyses, a systemic immune response is stimulated that targets tumor antigens that have shed; this process is known as epitope spreading. Viral therapy has been studied in a variety of cancers with encouraging results, and one study showed that oncolytic herpes simplex virus expressing murine IL-12 (interleukin-12) cured most mice in two different glioma models.78 In a phase I trial consisting of human patients with recurrent glioma, the oncolytic adenovirus DNX-2401 was injected directly into the primary tumor and reportedly shrunk the tumor by at least 95% in 3/25 of the patients.79 Five of the treated patients lived for at least 3 years post-treatment. The researchers theorized that the dramatic tumor response was a direct effect of the virus eliciting a long-lasting immune-mediated response.
In medulloblastoma, oncolytic viral therapy using poliovirus has demonstrated efficacy in vitro. One study evaluated the expression of the poliovirus receptor CD155 in medulloblastoma and the ability of that virus to infect as well as inhibit tumor proliferation.80 A rhinovirus recombinant form of polio (PVSRIPO) was used, in which part of the virus in the live polio vaccine was removed and replaced with a reciprocal part of a common cold virus. The viral replacement with the reciprocal part of the common cold virus ensures that the modified virus does not cause polio in the host. Poliovirus receptor mRNA expression was studied in 763 medulloblastoma samples from two group 3 cell lines: D283 and D341. CD155 was expressed in the medulloblastoma cell lines, and PVSRIPO infection resulted in decreased tumor proliferation in the D341 cell line at 48 hours resulting in cellular death. The study noted that poliovirus receptor is highest in medulloblastoma subtypes WNT α, WNT β, and group 3γ. This study concluded that PVSRIPO is capable of infecting and killing group 3 medulloblastoma.
Though controversial, there is evidence that human cytomegalovirus (CMV) infection is linked to medulloblastoma, possibly by promoting an inflammatory environment.81–83 This pro-inflammatory state occurs, in part, by an up-regulation of cyclooxygenase-2 (COX-2), and up-regulation of COX-2 has been demonstrated in medulloblastomas. Even more, COX-2 induces synthesis of inflammatory stimulants including prostaglandin E2 (PGE2), which stimulates the proliferation of medulloblastomas.84 Human CMV has been found in glioblastomas and up to 99% of brain metastases of colorectal and breast cancers, but it is still controversial whether CMV infection is linked to the development of medulloblastomas.85 Several research groups reported CMV infection in human medulloblastomas including cell lines and xenografts.86 One study showed that the antiviral drug ganciclovir results in reduced clonogenic capability of medulloblastomas in vitro.84,86 The COX-2 inhibitor celecoxib also diminishes clonogenic ability by decreasing PGE2 levels, which reduced stem cell-like ability and increased sensitivity to radiation in medulloblastoma transplanted into mice.86–88 Synergistically, valganciclovir and celecoxib reduce tumor growth of CMV-infected tumor cells both in vitro and in mice engrafted with human medulloblastoma cells.86
Currently, a host of modified viruses are under evaluation for the treatment of medulloblastoma. A combination of CMV RNA-pulsed dendritic cells, tetanus-diphtheria toxoid vaccine, and GM-CSF is currently in a phase I trial (NCT03615404). PEP-CMV, a vaccine comprised of a synthetic long peptide from human CMV matrix protein pp65, is another investigational viral agent in phase I trial and given after the patient receives a pre-conditioning tetanus-diphtheria toxoid vaccine (NCT03299309). The previously mentioned poliovirus with polio/rhinovirus recombinant (PVSRIPO) is also under phase I testing in patients with a variety of brain tumors including medulloblastoma (NCT03043391). In this ongoing study, the recombinant virus is injected directly into the primary tumor in the brain. Even a modified measles vaccine is under investigation in a phase I trial. An altered virus is injected directly into the primary tumor or via lumbar puncture if the disease is disseminated (NCT02962167). Similarly, a re-engineered reovirus known as reolysin, in combination with GM-CSF, is under investigation in patients with high-grade brain tumors (NCT02444546). Finally, a phase I trial is currently investigating an oncolytic herpes simplex virus-1 engineered to introduce viral mutations that enable it to selectively replicate within tumor cells. Lysed tumor cells then disseminate to subsequently target other tumor cells while also mounting a host immune response by exposing shed cancer antigens (NCT03911388). In addition, low dose radiation is administered to enhance viral replication and promote tumor lysis. The safety of this regimen, in addition to radiographic and pathologic evidence of anti-tumor response, has been successfully demonstrated in three phase I trials with adult glioma patients and an ongoing trial in children with recurrent supratentorial brain tumors (NCT02457845).
Checkpoint Inhibitors in Medulloblastoma
Checkpoint inhibitors have shown promise in a variety of cancers including melanoma, small cell lung cancer, and renal cell carcinoma. Activity of checkpoint inhibitors such as nivolumab depends on expression of PD-L1 on tumor cells. However, there are limited data on PD-L1 expression in brain tumors including medulloblastomas. One study with 89 patients in Korea showed that 0% of medulloblastoma patients demonstrated expression of PD-L1 (0 out of 28 patients).89 This was in direct contrast to other pediatric brain tumors including 40% of atypical teratoid/rhabdoid tumor (8 out of 20), 20% of ependymoma (4 out of 20), and 19% of high-grade glioma (4 out of 21) that demonstrate expression of PD-L1. This study concluded that medulloblastoma may not be a suitable candidate for PD-1 checkpoint blockade. These results are consistent with another study, which found a complete absence of PD-L1 on medulloblastomas, further suggesting limited value for PD-1 blockade to treat medulloblastoma.90
However, PD-1 blockade may depend on the subtype of medulloblastoma. Given that human studies evaluating PD-1 blockade are limited in human medulloblastoma patients, one study using immunocompetent murine models of human SHH-driven and group 3 medulloblastoma showed that PD-1 blockade demonstrated greater anti-tumor efficacy in group 3 medulloblastoma tumors compared to SHH tumors.31 This study showed that response to immune checkpoint blockade differs across medulloblastoma subtypes, at least in murine models. Of note, anti-PD-1 antibody binding was evident in peripheral T cells and not tumor-infiltrating lymphocytes within the brain’s tumor microenvironment. But this peripheral PD-1 blockade did result in increased CD3 T cells within the tumor microenvironment. This study concluded that effective PD-1 blockade does not require peripheral antibodies to penetrate the tumor microenvironment within the brain.
While certain studies documented limited levels of PD-L1 in medulloblastoma, one study showed that the SHH variant has higher PD-L1 expression than the other groups (and lowest MYC expression).91 Groups 3 and 4 had the highest levels of MYC expression (and lowest PD-L1 expression). This study subsequently showed in vitro IFN-y-induced expression of PD-L1 in all cell lines while radiation stimulated variable expression. Altering the expression of MYC did not change the expression of PD-L1. The study concluded that TH1 cytokine induction is the most potent stimulator of PD-L1 expression in vitro, suggesting that an inflamed microenvironment is required for PD-1 activation in medulloblastoma.
Another study showed that in brain tumors resistant to PD-1 blockage, lineage negative hematopoietic stem and progenitor cells (HSCs), derived from bone marrow that expresses C-C chemokine receptor type 2 (CCR2+), reversed resistance by sensitizing mice to PD-1 blockage.92 PD-1 blockage with HSC transfer increased T cell activation within preclinical models of medulloblastoma. The HSC transfer also diminished resistance to adoptive cellular therapy against medulloblastoma.
In human trials so far, immune checkpoint inhibitors have failed as monotherapy in recurrent glioblastoma.63,93 Lack of efficacy is believed due to minimal T cell infiltration into glioblastoma tumors, with similar findings reported in medulloblastomas.63,90 One study showed that cytotoxic T cells infiltrate medulloblastomas with variable activation and do not correlate with increased survival.90 Medulloblastoma cells have decreased antigen presentation via downregulated MHC-1 expression of PD-L1. Thus, it is possible that PD-1/PD-L1 contributes to immune escape in medulloblastoma.63
Nevertheless, the checkpoint inhibitors against PD-1, including nivolumab, pembrolizumab, and durvalumab, are each under investigation in clinical trials recruiting medulloblastoma patients and other CNS tumors (NCT03173950, NCT02359565, NCT02793466). Furthermore, nivolumab with and without ipilimumab, another checkpoint inhibitor that targets a different pathway known as CTLA-4, is also under investigation in a phase II trial of patients with high-grade CNS malignancies including medulloblastoma (NCT03130959). In addition, an inhibitor of the immune checkpoint pathway indoleamine 2,3-dioxygenase (IDO), in combination with radiochemotherapy, is currently in a phase I trial involving pediatric brain tumors. This study was recently expanded into a new phase II trial in August 2019. Although results from the first trial have not been released, the acceleration into phase II investigation is encouraging.
Natural Killer Cells in Medulloblastoma
Adoptive cell therapy, also known as cellular immunotherapy, involves isolation of immune cells from the host for modification and subsequent transplantation back into the host for direct targeting of tumor cells. Natural killer (NK) cells, a subset of the innate immune system often involved in controlling viral infections, have been studied to potentially serve as a type of adoptive cell therapy. Natural killer cells target medulloblastoma cells, primarily through activation of receptors such as natural killer group 2 member D activator receptor (NKG2D) and others (DNAM-1, NKp30, and NKp46), via ligands on medulloblastoma cells.94 Resistance to natural killer cell activity may be one mechanism by which medulloblastoma tumors can evade the natural immune response. One study analyzed tumor samples from 54 medulloblastoma patients for ligands of NKG2D, which included UL16 binding protein (ULPB-2) and major histocompatibility complex class I-related chains A (MICA). The study found that a certain medulloblastoma cell line, HTB-186, was relatively resistant to in vitro NK cell toxicity.95 By blocking the NKG2D receptor on the natural killer cells and blocking MICA/ULBP-2 on the HTB-186 tumor cells, the resistance to natural killer cell lysis increased. On the other hand, blocking the HLA class I on these cells and incubation with NK cells treated with IL-15 resulted in increased killing of tumor cells in vitro. The study found that at least in vitro, interactions of NKG2D/MICA-ULBP-2 play a role in natural killer cytotoxicity of medulloblastoma, and increased expression of HLA class I may increase medulloblastoma resistance to natural killer cell toxicity.
Cord blood natural killer cells may potentially be an effective immunotherapy due to their recognition of tumor cells and ability to be expanded exponentially in the laboratory.96 Natural killer cells lyse tumor cells without immunization and secrete granules containing granzymes and perforin.63,97 However, their efficacy is limited by immunosuppressive cytokines released in the medulloblastoma microenvironment such as transforming growth factor B (TGF-B). One study modified cord blood natural killer cells to express a dominant negative TGF-B receptor.96 When unmodified natural killer cells were added to TGF-B rich medulloblastoma media, their cytotoxic ability was reduced. However, the cytotoxic effect with modified natural killer cells was stronger and unaffected by the environment. The study concluded that medulloblastoma may be rendered susceptible to natural killer cytotoxicity with neutralization of TGF-B.
Other studies showed that CD1d, an antigen-presenting molecule for natural killer T cells, is highly expressed on medulloblastoma cells. This molecule is particularly over-expressed in the SHH subtype.98 One study showed that 9 of 20 primary medulloblastoma tumors express CD1d, which means it may be a target for immunotherapy. Another study showed that 13 of 38 medulloblastomas (34%) expressed CD1d. The majority (82%) of groups 3 and 4 medulloblastomas in that study were CD1d-negative.
One study evaluated lymphokine-activated killer cells (LAK) gathered by harvesting non-antigen primed monocytes in the peripheral blood of HLA-matched donors or the patients themselves, then expanding the monocytes by stimulation with anti-CD3 antibodies.99,100 In vitro studies showed that LAK cells target medulloblastomas but spare healthy brain tissue.99,101 This therapy was expanded clinically and published in a case series involving 8 patients with medulloblastomas that involved CSF dissemination.2 Intrathecal injection of the LAK cells provided a complete response within 3 months and a durable response up to 20 months in three patients.100,102 However, only two other reports of successful treatment with LAK have been published in the past 20 years.103,104
Currently, an ongoing phase I clinical trial is evaluating the safety and efficacy of ventricular infusion of autologous ex-vivo expanded NK cells in patients with recurrent CNS tumors of the posterior fossa (NCT02271711). In another phase II trial, NK cells from a donor are administered to patients after allogenic hematopoietic cell transplantation and reduced intensity radiochemotherapy in order to stimulate graft versus tumor response (NCT02100891).
CAR T Cell Therapy in Medulloblastoma
While oncolytic viral therapy and cancer vaccines aim to stimulate a specific T cell response, a number of steps must be orchestrated including APC activation and presentation to the T cells. The T cells must then penetrate the microenvironment of the tumor. However, tumor cells can downregulate MHC class I, which disables T cell recognition. Engineered chimeric antigen receptor T cells (CAR T) can overcome MHC downregulation by cancer cells and directly target surface antigens on the malignant cells.10,105–110 As another form of adoptive cell therapy, CAR T therapy involves use of a patient’s own re-engineered T cells and was first reported in the 1980s.56,111 The technology has been translated clinically only recently. CAR T therapy consists of hybrid receptors, which fuse T lymphocytes and other cells with an antibody-binding domain. The T lymphocyte can then theoretically target an antigen of choice.112 These synthesized receptors are retrovirally integrated into the patient’s own T cells and then reinfused back into the patient. But design of CAR T is complex, requiring a short-chain variable fragment (ScFv) to bind to an antigen of choice, a transmembrane domain, costimulatory domain, hinge-linker region, and intracellular CD3ζ tail.56,112 While first-generation CAR T therapy lacked a costimulatory domain resulting in limited expansion, the incorporation of CD28 and CD137 co-stimulating domains produced second-generation CARs effective against many blood cancers including relapsed B cell acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia, and non-Hodgkin lymphoma.112,113 CAR T therapy does not require a systemic immune response and is theoretically useful for tumors that lack a high tumor mutational burden.56 One study showed that intrathecal and intraventricular administration of CAR T targeting IL-13 receptor a2, an antigen associated with glioma, led to tumor regression in an adult patient with relapsed glioblastoma.114 Of note, the patient relapsed 6 months later when a tumor did not express the antigen. But the therapy was well tolerated without cytokine release syndrome or severe neurotoxicity.
Multiple trials using CAR T targeting the CD19 antigen have proven that CAR T can cross the blood–brain barrier, as these cells were detected in the CSF via immunofluorescence post-treatment.115 Other studies involving glioblastoma and B cell ALL patients also reported CAR T cells in brain tissue following infusion as demonstrated by flow cytometry.116 However, severe grade 3 neurotoxicity was reported in several of the B cell ALL patients following infusion revealing new challenges that must be investigated. CAR T cells essentially contain the cytotoxicity of T cells with specificity of antibodies; however, side effects have been noted in the brain including neurotoxicity, inflammation, and increased intracranial pressure. The development of CAR T cells that are activated only when stimulated by multiple tumor antigens simultaneously may decrease the incidence of side effects in the future.
CAR T cells showed anti-tumor activity against glioblastoma by targeting human epidermal growth factor receptor 2 (HER2).114,117 Since HER2 is overexpressed in certain medulloblastomas and anti-HER2 CAR T therapy has been successfully used against other cancers, it is plausible that such therapy can be used against medulloblastoma. One study successfully evaluated HER2-BBz-CAR T cells in mice and non-human primates.118 The CAR T cells cleared the medulloblastoma implanted in the posterior fossa of the mice without significant toxicity. The therapy showed strong efficacy both in vitro and in the murine medulloblastoma models, leading to possible future testing in humans. Another study showed that the antigen B7-H3 (CD276) could successfully be used as a target for CAR T cell therapy in a variety of pediatric cancers.119 The B7-H3 CAR T cells induced tumor regression in xenograft models of medulloblastoma, osteosarcoma, and Ewing sarcoma.
Challenges in developing the next generation of CAR T cell therapy for medulloblastoma include accessing the specialized tumor microenvironment within the CNS and variability of target antigens in tumor cells.56,115 So far, current CAR T cell proliferation has been poor with inadequate cytokine secretion. Furthermore, there is the challenge of tumor variants losing antigen, thus theoretically rendering CAR T ineffective without a target to hone in on. A new generation of CAR T cells will need to be engineered to circumvent these issues. Gene editing technologies are currently in development to increase cytokine overexpression, target a variety of antigens simultaneously, and provide more intricate control of CAR T signaling.
Antigenic escape must also be resolved for CAR T to become an established treatment against medulloblastoma. Antigenic escape has been one of the primary causes of tumor resistance and relapse in the leukemia patients treated with CD19-targeted CAR T therapy.56,109,120 This issue will be especially challenging in CNS tumors including medulloblastoma with known antigen heterogeneity. Decreased expression of tumor antigens below the level that can be detected by CAR T cells has been reported in multiple trials including the groundbreaking CAR T trial in brain tumors where a glioblastoma patient relapsed with an IL13-Ra2-negative tumor that lost antigen expression.114 CAR T cells must be designed to target multiple tumor antigens to prevent antigenic escape and relapse. For example, trivalent CAR T cells were able to overcome antigenic variability in patients with glioblastoma.121
Toxicity of treatment must be seriously considered, particularly in children with developing nervous systems. It is still unclear what the long-term effects of immunotherapy-induced inflammation in the CNS are. Vaccine and antibody therapy have yet to report serious adverse events in the CNS. However, neurotoxicity was reported in multiple CAR T cell trials targeting CD19 and included aphasia, encephalopathy, seizures, and memory loss in leukemia patients.109,122–124 The increased neurological disease burden in patients with CNS tumors may demonstrate even greater neurotoxicity risk.115
Currently, HER2-specific CAR T therapy is under investigation in HER2-positive recurrent CNS tumors including medulloblastoma using autologous CD4+ and CD8+ T cells lentivirally transduced to express a HER2 chimeric antigen receptor and EGFRt (NCT03500991). Similarly, autologous CD4+ and CD8+ lentivirally transduced to express an EGFR806 chimeric antigen receptor and EGFRt is currently in a phase I trial recruiting patients with EGFR-positive CNS tumors including medulloblastoma (NCT03638167).
Radiation with Immunotherapy in Medulloblastoma
The future of medulloblastoma therapy may incorporate radiotherapy that synergistically enhances the efficacy of immunotherapy. Studies have demonstrated that ionizing radiation can enhance the surface expression of MHC class I molecules and alter cellular proteins, enhancing the visibility of treated tumor cells to the immune system.125,126 One study evaluated the effect of low dose radiation on the functional immunological responses of medulloblastoma cell lines (DAOY, D283, D341).52 The study found that low dose radiation upregulated the expression of HLA class I and HLA class II molecules in medulloblastoma cells by more than 20% in vitro, including subtypes MAGE C1, CD137, and ICAM-1. Even more, an increase in reactive oxygen species resulted in altered cell surface expression of monoclonal antibody target molecules HER2 and vascular endothelial growth factor (VEGF), suggesting that low dose radiation and monoclonal antibodies can create a synergistic effect. While no studies have demonstrated increased survival using combination radiation and immunotherapy in patients with medulloblastoma, one study did show improved response of immunotherapy in patients with melanoma brain metastasis when given within 1 month of radiation.127 While various other trials have similarly shown greater efficacy when combining radiation with immunotherapy in lung malignancies, this study shows that the synergistic effect can be demonstrated in malignancies within the CNS.128,129 The induction of immune-mediated medulloblastoma cell death coupled with radiation-induced tumor necrosis may 1 day be part of the future paradigm in the treatment of medulloblastoma.
One recent phase II trial completed in June 2019 evaluated the use of radiolabeled monoclonal antibodies to target tumors and deliver radioactive substances to the cells in combination with radiation and chemotherapy in patients who have undergone surgery for medulloblastoma (NCT00058370). Patients first received iodine 131I monoclonal antibody 3F8 followed by 6 weeks of radiation, vincristine, lomustine, and cisplatin. Six patients enrolled in the trial, and results are pending. In another trial, a radiopharmaceutical, 90Y-DOTA-tyr3-octreotide, and its renal protectant, aminosyn II, were infused in patients with somatostatin receptor-positive tumors including medulloblastoma, with the intent to deliver radiation directly into tumor cells (NCT02441088).
Novel Antigenic Targets in Medulloblastoma
One of the major challenges in treating medulloblastoma with immunotherapy is the low immunogenicity and mutational load. Despite the low mutation burden, one study did find immunogenic peptides specific to the medulloblastoma tumors in human patients.130 Induction of a CD8 T cell response was successfully initiated for the neoepitopes derived from neuraminidase 2 (NEU2), programmed cell death 10 (PDCD10), supervillain (SVIL), histidine ammonia-lyase (HAL), tRNA splicing endonuclease subunit 54 (TSEN54), and proprotein convertase subtilisin (PCSK9) variants. Tumor-derived neoantigens were specific to each patient and confirm that T-cell therapy can be designed for the individual patient in the future.
Another recent study in 2018 analyzed 36 medulloblastomas (18 adults and 18 pediatric samples) via next-generation sequencing to identify overexpressed proteins. Although PD-L1 expression was uncommon, there were various other proteins overexpressed that may be used as therapeutic targets.131 Testing on samples was performed at the discretion of the physician, so not every sample was evaluated for all mutations. Multidrug resistance-associated protein 1 (MRP1) was found in 89% of the medulloblastomas (8 out of 9), tubulin beta 3 class 3 (TUBB3) was found in 86% of samples (18 out of 21), and thymidylate synthase (TS) was found in 80% of the samples (24 out of 31). Phosphatase and tensin homolog (PTEN) was evident in 85% of tumors (28 out of 33), topoisomerase 2A (TOP2A) was overexpressed in 84% of tumors (26 out of 31), and ribonucleotide reductase M1 (RRM1) was found in 71% of tumors (15 out of 21). Even more, topoisomerase 1 (TOP1) was found to be overexpressed in 90% of metastatic tumors (9 out of 10) relative to posterior fossa medulloblastomas that did not metastasize (50%, 10 out of 20). Additionally, PGP expression was found solely in the pediatric medulloblastomas.
Another study found that the tumor-associated antigen PRAME was detectable in 82% of medulloblastomas, independent of subgroup.132 Since PRAME has limited expression in normal tissues, this antigen can serve as a potent target for immunotherapy. This study also evaluated PRAME as a viable target in immunotherapy using genetically altered T cells with PRAME-specific T-cell receptors. The T cells were found to control tumor growth in a murine model of HLA-A*02 medulloblastoma, concluding that PRAME-specific T-cell receptor therapy may show promise in the treatment of HLA-A*02 medulloblastoma. Another study analyzed 37 tumor samples and showed that PRAME was overexpressed in 84% of the tumor samples.133 Of note, the study did not find any association between PRAME overexpression and patient survival.
Cancer testis antigens (CTAs) are among the best-described antigens in medulloblastomas, specifically the subtypes MAGE and GAGE proteins.2,134,135 These proteins were first described in melanoma and shown to stimulate T cell anti-tumor responses. These antigens can also be found in lung, breast, esophageal, and hepatocellular cancers.134 Even more, expression of these antigens has been found to correlate with aggressiveness of tumor and resistance to chemotherapy.134 One study explored expression on eight medulloblastomas.2 MAGE-4 expression was evident in 50% of the medulloblastomas, SCP-1 was found in 50% of the medulloblastomas, and the other CTAs were not found. Another study that used Western blot analysis found GAGE, MAGE-A family, and MAGE-A1 in 84%, 62%, and 46% of medulloblastoma samples, respectively. This same study showed that the downregulation of GAGE and MAGE led to an increased response to cisplatin. MAGE has been successfully targeted by vaccine therapy in various other cancers.136–139
CD47, a protein on the surface of a variety of solid tumors including medulloblastoma, is involved in evading the host immune response and may be a viable antigenic target. Preclinical trials have shown that antagonizing CD47 with a humanized monoclonal antibody can activate macrophages to phagocytize cancerous cells in several CNS tumor models.140 Finally, a subset of the VEGF group, known as placental group factor (PIGF), was secreted by various pediatric brain tumors including medulloblastoma.141 Inhibition of PIGF via the receptor neuropilin 1 pathway delayed tumor growth in murine models of medulloblastoma.
A current, ongoing phase I/II trial is testing various doses of a humanized monoclonal antibody known as TB-403 against PIGF in patients with refractory medulloblastoma (NCT02748135). Patients with other cancers that exhibited PIGF secretion in past studies, including Ewing sarcoma and alveolar rhabdomyosarcoma, are also eligible for this trial. These studies demonstrate the vast potential of immunotherapy when tumor-specific antigens can be identified.
Challenges in Treating Medulloblastoma with Immunotherapy
Various cell lines derived from medulloblastoma have existed since the 1980s.51,142 However, tumor implantation models do not reflect the normal events of tumor initiation and growth. Furthermore, experiments are often conducted in immunodeficient mouse models to allow tumors to be transplanted without eradication by the host immune response. Genetically engineered animal models must be developed to introduce genetic mutations that drive the development of tumors and provide more accurate insight into tumor development and response to treatment. Many genetically engineered models of medulloblastoma have been developed.143,144
The blood–brain barrier also continues to be a major challenge in targeting brain tumors. IgG antibodies penetrate the blood–brain barrier poorly, due to their large size. Tight junctions of epithelial cells in the blood–brain barrier prevent passage of molecules larger than 500 Da.145 This may be one of the reasons that checkpoint inhibitors have failed to improve survival in recurrent GBM in large multicenter trials such as the Phase III trial Checkmate-143.146 On the other hand, more encouraging results were found in clinical trials evaluating checkpoint inhibitors in melanoma and NSCLC that had metastasized to the brain.147,148 While it is likely that the difference in trial results is due to differences in the biology of the tumor, it is possible that patients with metastatic brain tumors have a disrupted blood–brain barrier since tumor cells were able to initially cross the barrier.149 Current trials are evaluating checkpoint inhibitors combined with radiotherapy against glioblastoma multiforme with the hope of disrupting the blood–brain barrier (NCT02617589, NCT03576612).
Smaller peptides less than 50 residues (which can cross cell membranes) have been shown to circumvent the blood–brain barrier via receptor-mediated transcytosis.150 Anti-human epidermal growth factor (HER2) antibodies conjugated with cell-penetrating peptides were able to cross the blood–brain barrier and prolong overall survival in BT-474 mice with brain tumors.151 It has also been demonstrated that CAR T cells can bypass the blood–brain barrier, particularly when conjugated to cell-penetrating peptides less than 40 residues that can undergo transcytosis. Coupling of vaccines or antibody therapies with cell-penetrating peptides may be necessary to bypass the blood–brain barrier.
Conclusion
Future forms of immunotherapy in medulloblastoma must overcome tumor antigen heterogeneity, the blood–brain barrier, and mechanisms of tumor immunosuppression (Figure 1). Currently, vaccine and CAR T therapy are limited by lack of documented tumor antigens.115 Preclinical research must be continued to identify new antigen targets and engineer therapies that can target multiple antigens simultaneously. By counteracting tumor immunosuppression and activating immunity via vaccines, viruses, and/or adoptive T cell therapy, combination therapy should be the hallmark of future treatment of medulloblastoma to improve survival outcomes.
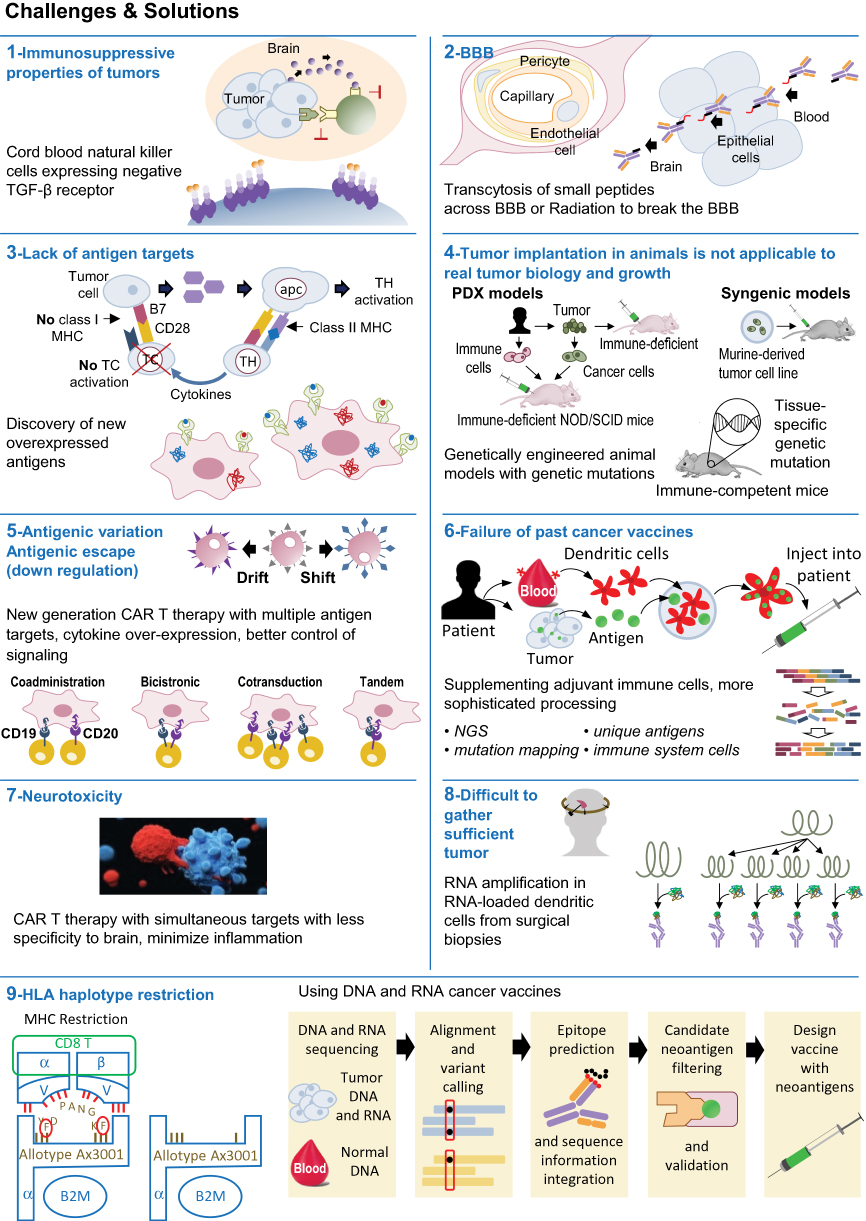 |
Figure 1 Immunotherapy in medulloblastoma – Challenges and Solutions. |
Disclosure
Dr Aman Chauhan reports grants from BMS and Clovis, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Pollack IF, Agnihotri S, Broniscer A. Childhood brain tumors: current management, biological insights, and future directions. J Neurosurg Pediatr. 2019;23(3):261–273. doi:10.3171/2018.10.PEDS18377
2. Sonabend AM, Ogden AT, Maier LM, et al. Medulloblasoma: challenges for effective immunotherapy. J Neurooncol. 2012;108(1):1–10. doi:10.1007/s11060-011-0776-1
3. Thurnher MM. 2007 World Health Organization classification of tumours of the central nervous system. Cancer Imaging. 2009;9(Spec No A):S1–S3. doi:10.1102/1470-7330.2009.9001
4. Archer TC, Mahoney EL, Pomeroy SL. Medulloblastoma: molecular classification-based personal therapeutics. Neurotherapeutics. 2017;14(2):265–273. doi:10.1007/s13311-017-0526-y
5. Packer RJ, Finlay JL. Chemotherapy for childhood medulloblastoma and primitive neuroectodermal tumors. Oncologist. 1996;1(6):381–393. doi:10.1634/theoncologist.1-6-381
6. Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21(Supplement_5):v1–v100. doi:10.1093/neuonc/noz150
7. Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the children’s cancer group 921 randomized phase III study. J Clin Oncol. 1999;17(3):832–845. doi:10.1200/JCO.1999.17.3.832
8. Ris MD, Packer R, Goldwein J, Jones-Wallace D, Boyett JM. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a children’s cancer group study. J Clin Oncol. 2001;19(15):3470–3476. doi:10.1200/JCO.2001.19.15.3470
9. Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom children’s cancer study group PNET-3 study. J Clin Oncol. 2003;21(8):1581–1591. doi:10.1200/JCO.2003.05.116
10. Sayour EJ, Mitchell DA. Immunotherapy for pediatric brain tumors. Brain Sci. 2017;7(10)137.
11. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
12. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754 e736. doi:10.1016/j.ccell.2017.05.005
13. Gibson P, Tong Y, Robinson G, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–1099. doi:10.1038/nature09587
14. Grammel D, Warmuth-Metz M, von Bueren AO, et al. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012;123(4):601–614. doi:10.1007/s00401-012-0961-0
15. Hooper CM, Hawes SM, Kees UR, Gottardo NG, Dallas PB, Alsina B. Gene expression analyses of the spatio-temporal relationships of human medulloblastoma subgroups during early human neurogenesis. PLoS One. 2014;9(11):e112909. doi:10.1371/journal.pone.0112909
16. Northcott PA, Jones DT, Kool M, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12(12):818–834. doi:10.1038/nrc3410
17. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. doi:10.1007/s00401-011-0922-z
18. Wood GW, Morantz RA. In vitro reversal of depressed T-lymphocyte function in the peripheral blood of brain tumor patients. J Natl Cancer Inst. 1982;68(1):27–33.
19. Wood GW, Morantz RA. Depressed T lymphocyte function in brain tumor patients: monocytes as suppressor cells. J Neurooncol. 1983;1(2):87–94. doi:10.1007/BF00182953
20. Yamasaki T, Handa H, Yamashita J, Namba Y, Hanaoka M. Characteristic immunological responses to an experimental mouse brain tumor. Cancer Res. 1983;43(10):4610–4617.
21. Gerosa M, Amadori G, Semenzato C, et al. Immunobiology of paediatric intracranial tumours. A preliminary report. Acta Neurochir (Wien). 1979;50(1–2):49–54. doi:10.1007/BF01813548
22. Gerosa MA, Amadori G, Semenzato P, Gasparotto G, Carteri A. Immunobiology of primary CNS tumors in infancy and childhood. B- and T-cell dependent immunity and cytotoxicity and cell kinetic evaluation. Childs Brain. 1980;6(2):92–102. doi:10.1159/000119890
23. Mitchell DA, Cui X, Schmittling RJ, et al. Monoclonal antibody blockade of IL-2 receptor α during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood. 2011;118(11):3003–3012. doi:10.1182/blood-2011-02-334565
24. Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–369. doi:10.1038/nature14320
25. Batich KA, Swartz AM, Sampson JH. Preconditioning vaccine sites for mRNA-transfected dendritic cell therapy and antitumor efficacy. Methods Mol Biol. 2016;1403:819–838.
26. Sampson JH, Maus MV, June CH. Immunotherapy for brain tumors. J Clin Oncol. 2017;35(21):2450–2456. doi:10.1200/JCO.2017.72.8089
27. Hao C, Tian J, Liu H, Li F, Niu H, Zhu B. Efficacy and safety of anti-PD-1 and anti-PD-1 combined with anti-CTLA-4 immunotherapy to advanced melanoma: a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2017;96(26):e7325. doi:10.1097/MD.0000000000007325
28. Alizadeh AA, Aranda V, Bardelli A, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(8):846–853. doi:10.1038/nm.3915
29. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
30. Fontebasso AM, Gayden T, Nikbakht H, et al. Epigenetic dysregulation: a novel pathway of oncogenesis in pediatric brain tumors. Acta Neuropathol. 2014;128(5):615–627. doi:10.1007/s00401-014-1325-8
31. Pham CD, Flores C, Yang C, et al. Differential immune microenvironments and response to immune checkpoint blockade among molecular subtypes of murine medulloblastoma. Clin Cancer Res. 2016;22(3):582–595. doi:10.1158/1078-0432.CCR-15-0713
32. Schlager C, Korner H, Krueger M, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature. 2016;530(7590):349–353. doi:10.1038/nature16939
33. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353(6301):766–771. doi:10.1126/science.aag2638
34. Hu X, Deng Q, Ma L, et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020. doi:10.1038/s41422-020-0287-8
35. Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24(17):4175–4186. doi:10.1158/1078-0432.CCR-17-1846
36. Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international Phase 3 trial. Lancet Oncol. 2017;18(10):1373–1385. doi:10.1016/S1470-2045(17)30517-X
37. Mueller S, Chang S. Pediatric brain tumors: current treatment strategies and future therapeutic approaches. Neurotherapeutics. 2009;6(3):570–586. doi:10.1016/j.nurt.2009.04.006
38. Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24(25):4202–4208. doi:10.1200/JCO.2006.06.4980
39. Merchant TE, Kun LE, Krasin MJ, et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70(3):782–787. doi:10.1016/j.ijrobp.2007.07.2342
40. Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–820. doi:10.1016/S1470-2045(06)70867-1
41. Wahba HA, Abu-Hegazy M, Wasel Y, Ismail EI, Zidan AS. Adjuvant chemotherapy after reduced craniospinal irradiation dose in children with average-risk medulloblastoma: a 5-year follow-up study. J BUON. 2013;18(2):425–429.
42. Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a children’s cancer group study. J Clin Oncol. 1999;17(7):2127–2136. doi:10.1200/JCO.1999.17.7.2127
43. Tamburrini G, Frassanito P, Chieffo D, Massimi L, Caldarelli M, Di Rocco C. Cerebellar mutism. Childs Nerv Syst. 2015;31(10):1841–1851. doi:10.1007/s00381-015-2803-6
44. Merchant TE, Kiehna EN, Li C, et al. Modeling radiation dosimetry to predict cognitive outcomes in pediatric patients with CNS embryonal tumors including medulloblastoma. Int J Radiat Oncol Biol Phys. 2006;65(1):210–221. doi:10.1016/j.ijrobp.2005.10.038
45. Bansal LR, Belair J, Cummings D, Zuccoli G. Late-onset radiation-induced vasculopathy and stroke in a child with medulloblastoma. J Child Neurol. 2015;30(6):800–802. doi:10.1177/0883073814538501
46. Benson PJ, Sung JH. Cerebral aneurysms following radiotherapy for medulloblastoma. J Neurosurg. 1989;70(4):545–550. doi:10.3171/jns.1989.70.4.0545
47. Neglia JP, Robison LL, Stovall M, et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst. 2006;98(21):1528–1537. doi:10.1093/jnci/djj411
48. Ullrich NJ, Pomeroy SL. Molecular genetics of pediatric central nervous system tumors. Curr Oncol Rep. 2006;8(6):423–429. doi:10.1007/s11912-006-0070-0
49. Wang X, Dubuc AM, Ramaswamy V, et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015;129(3):449–457. doi:10.1007/s00401-015-1389-0
50. Morrissy AS, Garzia L, Shih DJ, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016;529(7586):351–357. doi:10.1038/nature16478
51. Wang J, Garancher A, Ramaswamy V, Wechsler-Reya RJ. Medulloblastoma: from molecular subgroups to molecular targeted therapies. Annu Rev Neurosci. 2018;41(1):207–232. doi:10.1146/annurev-neuro-070815-013838
52. Das A, McDonald D, Lowe S, et al. Immunological low-dose radiation modulates the pediatric medulloblastoma antigens and enhances antibody-dependent cellular cytotoxicity. Childs Nerv Syst. 2017;33(3):429–436. doi:10.1007/s00381-016-3305-x
53. Li K, Duan WC, Zhao HB, et al. Preoperative neutrophil to lymphocyte ratio and platelet to lymphocyte ratio are associated with the prognosis of group 3 and group 4 medulloblastoma. Sci Rep. 2019;9(1):13239. doi:10.1038/s41598-019-49733-6
54. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
55. Jahnisch H, Fussel S, Kiessling A, et al. Dendritic cell-based immunotherapy for prostate cancer. Clin Dev Immunol. 2010;2010:517493. doi:10.1155/2010/517493
56. Wang SS, Bandopadhayay P, Jenkins MR. Towards immunotherapy for pediatric brain tumors. Trends Immunol. 2019;40(8):748–761. doi:10.1016/j.it.2019.05.009
57. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi:10.1056/NEJMoa1001294
58. Holtkamp S, Kreiter S, Selmi A, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. doi:10.1182/blood-2006-04-015024
59. Kranz LM, Diken M, Haas H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401. doi:10.1038/nature18300
60. Kreiter S, Selmi A, Diken M, et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70(22):9031–9040. doi:10.1158/0008-5472.CAN-10-0699
61. Coban C, Kobiyama K, Aoshi T, et al. Novel strategies to improve DNA vaccine immunogenicity. Curr Gene Ther. 2011;11(6):479–484. doi:10.2174/156652311798192815
62. Nair SK, Driscoll T, Boczkowski D, et al. Ex vivo generation of dendritic cells from cryopreserved, post-induction chemotherapy, mobilized leukapheresis from pediatric patients with medulloblastoma. J Neurooncol. 2015;125(1):65–74. doi:10.1007/s11060-015-1890-2
63. Majd N, Penas-Prado M. Updates on management of adult medulloblastoma. Curr Treat Options Oncol. 2019;20(8):64. doi:10.1007/s11864-019-0663-0
64. Ardon H, De Vleeschouwer S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54(4):519–525. doi:10.1002/pbc.22319
65. Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773–2779. doi:10.1158/1535-7163.MCT-09-0124
66. Kim W, Liau LM. Dendritic cell vaccines for brain tumors. Neurosurg Clin N Am. 2010;21(1):139–157. doi:10.1016/j.nec.2009.09.005
67. Liau LM, Black KL, Martin NA, et al. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides: case report. Neurosurg Focus. 2000;9(6):e8. doi:10.3171/foc.2000.9.6.9
68. Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. doi:10.1158/1078-0432.CCR-05-0464
69. Heimberger AB, Archer GE, Crotty LE, et al. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery. 2002;50(1):
70. Felsberg J, Hentschel B, Kaulich K, et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) positivity in EGFR-amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res. 2017;23(22):6846–6855. doi:10.1158/1078-0432.CCR-17-0890
71. van den Bent MJ, Gao Y, Kerkhof M, et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol. 2015;17(7):935–941. doi:10.1093/neuonc/nov013
72. Aurelian L. Oncolytic viruses as immunotherapy: progress and remaining challenges. Onco Targets Ther. 2016;9:2627–2637. doi:10.2147/OTT.S63049
73. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. doi:10.1038/nrd4663
74. Killock D. Skin cancer: T-VEC oncolytic viral therapy shows promise in melanoma. Nat Rev Clin Oncol. 2015;12(8):438. doi:10.1038/nrclinonc.2015.106
75. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22(5):1048–1054. doi:10.1158/1078-0432.CCR-15-2667
76. Kanerva A, Nokisalmi P, Diaconu I, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013;19(10):2734–2744. doi:10.1158/1078-0432.CCR-12-2546
77. Yu YA, Galanis C, Woo Y, et al. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1h68. Mol Cancer Ther. 2009;8(1):141–151.
78. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253–267 e255. doi:10.1016/j.ccell.2017.07.006
79. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. doi:10.1200/JCO.2017.75.8219
80. Thompson EM, Brown M, Dobrikova E, et al. Poliovirus receptor (CD155) expression in pediatric brain tumors mediates oncolysis of medulloblastoma and pleomorphic xanthoastrocytoma. J Neuropathol Exp Neurol. 2018;77(8):696–702. doi:10.1093/jnen/nly045
81. Hortal AM, Vermeulen JF, Van Hecke W, Bovenschen N. Oncogenic role of cytomegalovirus in medulloblastoma? Cancer Lett. 2017;408:55–59. doi:10.1016/j.canlet.2017.08.024
82. Cinatl J
83. Michaelis M, Baumgarten P, Mittelbronn M, Driever PH, Doerr HW, Cinatl J
84. Baryawno N, Sveinbjornsson B, Eksborg S, et al. Tumor-growth-promoting cyclooxygenase-2 prostaglandin E2 pathway provides medulloblastoma therapeutic targets. Neuro Oncol. 2008;10(5):661–674. doi:10.1215/15228517-2008-035
85. Taher C, Frisk G, Fuentes S, et al. High prevalence of human cytomegalovirus in brain metastases of patients with primary breast and colorectal cancers. Transl Oncol. 2014;7(6):732–740. doi:10.1016/j.tranon.2014.09.008
86. Baryawno N, Rahbar A, Wolmer-Solberg N, et al. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J Clin Invest. 2011;121(10):4043–4055. doi:10.1172/JCI57147
87. Yang MY, Lee HT, Chen CM, Shen CC, Ma HI. Celecoxib suppresses the phosphorylation of STAT3 protein and can enhance the radiosensitivity of medulloblastoma-derived cancer stem-like cells. Int J Mol Sci. 2014;15(6):11013–11029. doi:10.3390/ijms150611013
88. Raso A, Mascelli S, Biassoni R, et al. High levels of PROM1 (CD133) transcript are a potential predictor of poor prognosis in medulloblastoma. Neuro Oncol. 2011;13(5):500–508. doi:10.1093/neuonc/nor022
89. Hwang K, Koh EJ, Choi EJ, et al. PD-1/PD-L1 and immune-related gene expression pattern in pediatric malignant brain tumors: clinical correlation with survival data in Korean population. J Neurooncol. 2018;139(2):281–291. doi:10.1007/s11060-018-2886-5
90. Vermeulen JF, Van Hecke W, Adriaansen EJM, et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology. 2018;7(3):e1398877. doi:10.1080/2162402X.2017.1398877
91. Martin AM, Nirschl CJ, Polanczyk MJ, et al. PD-L1 expression in medulloblastoma: an evaluation by subgroup. Oncotarget. 2018;9(27):19177–19191. doi:10.18632/oncotarget.24951
92. Flores CT, Wildes TJ, Drake JA, et al. Lin(-)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade. Nat Commun. 2018;9(1):4313. doi:10.1038/s41467-018-06182-5
93. Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674–686. doi:10.1093/neuonc/nox208
94. Castriconi R, Dondero A, Negri F, et al. Both CD133+ and CD133– medulloblastoma cell lines express ligands for triggering NK receptors and are susceptible to NK-mediated cytotoxicity. Eur J Immunol. 2007;37(11):3190–3196. doi:10.1002/eji.200737546
95. Fernandez L, Portugal R, Valentin J, et al. In vitro natural killer cell immunotherapy for medulloblastoma. Front Oncol. 2013;3:94. doi:10.3389/fonc.2013.00094
96. Yvon ES, Burga R, Powell A, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19(3):408–418. doi:10.1016/j.jcyt.2016.12.005
97. Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49. doi:10.1126/science.1198687
98. Liu D, Song L, Brawley VS, et al. Medulloblastoma expresses CD1d and can be targeted for immunotherapy with NKT cells. Clin Immunol. 2013;149(1):55–64. doi:10.1016/j.clim.2013.06.005
99. George RE, Loudon WG, Moser RP, Bruner JM, Steck PA, Grimm EA. In vitro cytolysis of primitive neuroectodermal tumors of the posterior fossa (medulloblastoma) by lymphokine-activated killer cells. J Neurosurg. 1988;69(3):403–409. doi:10.3171/jns.1988.69.3.0403
100. Okamoto Y, Shimizu K, Tamura K, et al. An adoptive immunotherapy of patients with medulloblastoma by lymphokine-activated killer cells (LAK). Acta Neurochir (Wien). 1988;94(1–2):47–52. doi:10.1007/BF01406615
101. Kang SG, Ryu CH, Jeun SS, et al. Lymphokine activated killer cells from umbilical cord blood show higher antitumor effect against anaplastic astrocytoma cell line (U87) and medulloblastoma cell line (TE671) than lymphokine activated killer cells from peripheral blood. Childs Nerv Syst. 2004;20(3):154–162. doi:10.1007/s00381-003-0898-7
102. Shimizu H, Sasaki K, Takaue Y, Ota S, Fzjimoto T. Studies of children with acute lymphoblastic leukemia (ALL) who relapsed. Relationship of site of relapse, time and prognosis. Rinsho Ketsueki. 1989;30(7):999–1004.
103. Sankhla SK, Nadkarni JS, Bhagwati SN. Adoptive immunotherapy using lymphokine-activated killer (LAK) cells and interleukin-2 for recurrent malignant primary brain tumors. J Neurooncol. 1996;27(2):133–140. doi:10.1007/BF00177476
104. Salmaggi A, Dufour A, Silvani A, Ciusani E, Nespolo A, Boiardi A. Immunological fluctuations during intrathecal immunotherapy in three patients affected by CNS tumours disseminating via CSF. Int J Neurosci. 1994;77(1–2):117–125. doi:10.3109/00207459408986024
105. Garfall AL, Maus MV, Hwang WT, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373(11):1040–1047. doi:10.1056/NEJMoa1504542
106. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi:10.1056/NEJMoa1215134
107. Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi:10.1126/scitranslmed.3002842
108. Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. doi:10.1182/blood-2011-05-354449
109. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222
110. Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi:10.1126/scitranslmed.aac5415
111. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–10028. doi:10.1073/pnas.86.24.10024
112. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi:10.1158/2159-8290.CD-12-0548
113. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. doi:10.1126/science.aar6711
114. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497
115. Petersen CT, Krenciute G. Next generation CAR T cells for the Immunotherapy of high-grade glioma. Front Oncol. 2019;9:69. doi:10.3389/fonc.2019.00069
116. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–1154. doi:10.1517/14712598.2015.1046430
117. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. doi:10.1001/jamaoncol.2017.0184
118. Nellan A, Rota C, Majzner R, et al. Durable regression of medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer. 2018;6(1):30. doi:10.1186/s40425-018-0340-z
119. Majzner RG, Theruvath JL, Nellan A, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560–2574. doi:10.1158/1078-0432.CCR-18-0432
120. Tasian SK, Gardner RA. CD19-redirected chimeric antigen receptor-modified T cells: a promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther Adv Hematol. 2015;6(5):228–241. doi:10.1177/2040620715588916
121. Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20(4):506–518. doi:10.1093/neuonc/nox182
122. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. doi:10.1126/scitranslmed.3008226
123. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi:10.1016/S0140-6736(14)61403-3
124. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
125. Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–1271. doi:10.1084/jem.20052494
126. Vermeer DW, Spanos WC, Vermeer PD, Bruns AM, Lee KM, Lee JH. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int J Cancer. 2013;133(1):120–129. doi:10.1002/ijc.28015
127. Qian JM, Yu JB, Kluger HM, Chiang VL. Timing and type of immune checkpoint therapy affect the early radiographic response of melanoma brain metastases to stereotactic radiosurgery. Cancer. 2016;122(19):3051–3058. doi:10.1002/cncr.30138
128. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage iii non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. doi:10.1056/NEJMoa1709937
129. Shaverdian N, Lisberg AE, Bornazyan K, et al. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017;18(7):895–903. doi:10.1016/S1470-2045(17)30380-7
130. Blaeschke F, Paul MC, Schuhmann MU, et al. Low mutational load in pediatric medulloblastoma still translates into neoantigens as targets for specific T-cell immunotherapy. Cytotherapy. 2019;21(9):973–986. doi:10.1016/j.jcyt.2019.06.009
131. Hashimoto Y, Penas-Prado M, Zhou S, et al. Rethinking medulloblastoma from a targeted therapeutics perspective. J Neurooncol. 2018;139(3):713–720. doi:10.1007/s11060-018-2917-2
132. Orlando D, Miele E, De Angelis B, et al. Adoptive immunotherapy using PRAME-specific T cells in medulloblastoma. Cancer Res. 2018;78(12):3337–3349. doi:10.1158/0008-5472.CAN-17-3140
133. Vulcani-Freitas TM, Saba-Silva N, Cappellano A, Cavalheiro S, Toledo SR. PRAME gene expression profile in medulloblastoma. Arq Neuropsiquiatr. 2011;69(1):9–12. doi:10.1590/S0004-282X2011000100003
134. Kasuga C, Nakahara Y, Ueda S, et al. Expression of MAGE and GAGE genes in medulloblastoma and modulation of resistance to chemotherapy. Laboratory investigation. J Neurosurg Pediatr. 2008;1(4):305–313. doi:10.3171/PED/2008/1/4/305
135. Oba-Shinjo SM, Caballero OL, Jungbluth AA, et al. Cancer-testis (CT) antigen expression in medulloblastoma. Cancer Immun. 2008;8:7.
136. Atanackovic D, Altorki NK, Stockert E, et al. Vaccine-induced CD4+ T cell responses to MAGE-3 protein in lung cancer patients. J Immunol. 2004;172(5):3289–3296. doi:10.4049/jimmunol.172.5.3289
137. Kim SH, Castro F, Gonzalez D, Maciag PC, Paterson Y, Gravekamp C. Mage-b vaccine delivered by recombinant listeria monocytogenes is highly effective against breast cancer metastases. Br J Cancer. 2008;99(5):741–749. doi:10.1038/sj.bjc.6604526
138. Sypniewska RK, Hoflack L, Tarango M, et al. Prevention of metastases with a mage-b DNA vaccine in a mouse breast tumor model: potential for breast cancer therapy. Breast Cancer Res Treat. 2005;91(1):19–28. doi:10.1007/s10549-004-6454-7
139. Toh HC, Wang WW, Chia WK, et al. Clinical benefit of allogeneic melanoma cell lysate-pulsed autologous dendritic cell vaccine in MAGE-positive colorectal cancer patients. Clin Cancer Res. 2009;15(24):7726–7736. doi:10.1158/1078-0432.CCR-09-1537
140. Gholamin S, Mitra SS, Feroze AH, et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. 2017;9(381). doi:10.1126/scitranslmed.aaf2968
141. Snuderl M, Batista A, Kirkpatrick ND, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152(5):1065–1076. doi:10.1016/j.cell.2013.01.036
142. Xu J, Erdreich-Epstein A, Gonzalez-Gomez I, et al. Novel cell lines established from pediatric brain tumors. J Neurooncol. 2012;107(2):269–280. doi:10.1007/s11060-011-0756-5
143. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi:10.1158/2159-8290.CD-18-0367
144. Weiss WA, Burns MJ, Hackett C, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 2003;63(7):1589–1595.
145. Pardridge WM. Brain drug development and brain drug targeting. Pharm Res. 2007;24(9):1729–1732. doi:10.1007/s11095-007-9387-0
146. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget. 2017;8(53):91779–91794. doi:10.18632/oncotarget.21586
147. Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, Phase 2 trial. Lancet Oncol. 2012;13(5):459–465. doi:10.1016/S1470-2045(12)70090-6
148. Goldberg SB, Gettinger SN, Mahajan A, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17(7):976–983. doi:10.1016/S1470-2045(16)30053-5
149. Tiwary S, Morales JE, Kwiatkowski SC, Lang FF, Rao G, McCarty JH. Metastatic brain tumors disrupt the blood-brain barrier and alter lipid metabolism by inhibiting expression of the endothelial cell fatty acid transporter Mfsd2a. Sci Rep. 2018;8(1):8267. doi:10.1038/s41598-018-26636-6
150. Razpotnik R, Novak N, Curin Serbec V, Rajcevic U. Targeting malignant brain tumors with antibodies. Front Immunol. 2017;8:1181. doi:10.3389/fimmu.2017.01181
151. Regina A, Demeule M, Tripathy S, et al. ANG4043, a novel brain-penetrant peptide-mAb conjugate, is efficacious against HER2-positive intracranial tumors in mice. Mol Cancer Ther. 2015;14(1):129–140. doi:10.1158/1535-7163.MCT-14-0399
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.