Back to Journals » ImmunoTargets and Therapy » Volume 9
Immunotherapeutic Targets and Therapy for Renal Cell Carcinoma
Authors Sepe P, Mennitto A, Corti F ![]() , Procopio G
, Procopio G
Received 4 September 2020
Accepted for publication 24 October 2020
Published 13 November 2020 Volume 2020:9 Pages 273—288
DOI https://doi.org/10.2147/ITT.S240889
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Pierangela Sepe, Alessia Mennitto, Francesca Corti, Giuseppe Procopio
Genitourinary Cancer Unit, Department of Medical Oncology, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy
Correspondence: Giuseppe Procopio
Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, via Giacomo Venezian 1, Milan 20133, Italy
Tel +39 0223903650
Email [email protected]
Abstract: Over the last 20 years, different therapies have been considered as the mainstay for the treatment of patients with metastatic renal cell carcinoma (mRCC). Since angiogenesis is a key mechanism in the pathogenesis of renal carcinoma, research is still focusing on the inhibition of new vessel growth through the development of novel and potent tyrosine kinase inhibitors (TKIs), such as cabozantinib. On the other hand, a new therapeutic scenario has opened up in the forefront with immunotherapy. Immune checkpoint inhibitors (ICIs), which already represent a standard treatment option in pretreated mRCC patients, are revolutionizing the frontline therapeutic armamentarium of mRCC. Upfront combination immunotherapy as well as combinations of immunotherapy with targeted agents showed to significantly improved outcomes of mRCC patients compared to single-agent TKIs. ICIs are associated with long-lasting responses. Nonetheless, several unmet needs remain, as a small proportion of patients shows primary refractoriness to immunotherapy. Multiple treatment strategies combining different mechanisms of action or targeting immune escape pathways are emerging with the aim to improve response rates and survival outcomes. This review summarizes current immunotherapeutic targets and therapies approved for mRCC, while examining mechanisms of resistance and future directions, with the aim to address novel treatment strategies and help in improving the management of this tumor.
Keywords: metastatic renal cell carcinoma, immunotherapy, anti-PD-1, anti-CTLA-4, targeted therapy, biomarkers
Introduction
Renal cell carcinoma (RCC), due to its heterogeneity, marked angiogenesis, and immunogenicity, has been for years an intriguing test-case for innovative therapies.1 Over the last 20 years, different therapies including pure angiogenesis inhibitor monoclonal antibodies, such as bevacizumab, multitarget molecules such as vascular endothelial growth factor receptor (VEGFR), and other tyrosine kinase inhibitors (TKIs), as well as inhibitors of mammalian target of rapamycin (mTOR), have been considered as the mainstay for the first or subsequent line treatment of patients with metastatic RCC (mRCC).2–7 Since angiogenesis is a key mechanism in the pathogenesis of RCC, research is still focusing on the inhibition of new vessel growth through novel and potent TKIs, such as cabozantinib. On the other hand, a new therapeutic scenario has opened up in the forefront with immunotherapy.8,9 Recently, immune checkpoint inhibitors (ICIs) have become a viable option that has been added to the therapeutic armamentarium for mRCC.10–16 Immune checkpoint inhibitors, which already represent a standard treatment option in pretreated mRCC patients, are also revolutionizing the frontline treatment, since combination immunotherapy as well as combinations of immunotherapy with targeted agents have been shown to significantly improve the outcomes of treatment-naïve mRCC patients.11–16 In RCC, immunotherapy influences adaptive immunity, allowing the immune system to recognize tumor antigens, maintain memory, and kill neoplastic cells.17 Activation of adaptive immunity against foreign antigens is consequent to a complex chain of events, involving several receptors present on both neoplastic cells and immune cells, mediating inhibitory or activator signals.17 However, the complex biological scenario underlying the role of antitumor immunity and its modulation with treatment is not yet fully elucidated, and a deepened understanding of tumor–host immune interaction is warranted. This review summarizes current immunotherapeutic targets and therapies approved for mRCC, while examining mechanisms of resistance and future directions with the aim to address novel treatment strategies and help in improving the management of this tumor.
Immunotherapeutic Target
CD8+ T-cells and CD4+ T-cells represent the two arms of the adaptive cellular response.18 CD8+T-cells are the main effectors of the anti-tumor immune response. They can recognize antigens expressed by tumor cells and, once activated, kill malignant cells by different mechanisms.18 CD4+ T-cells help in generating an immune response by stimulating CD8+ T-cells and other immune cells, such as macrophages and B-lymphocytes. The activation of effector and memory CD8+ T-cells occurs by the interaction with antigen-presenting cells (APC) via the T-cell receptor (TCR) and major histocompatibility complex (MHC)/peptide antigen.19 The TCR-MHC/peptide interaction is a complex event amplified by the interplay of multiple costimulatory molecules. Numerous inhibitory transduction pathways, known as immunological checkpoints, in fact, are present to maintain the tolerance and homeostasis of the immune system. The most studied immune-checkpoints playing a key role in the modulation of adaptive immunity are the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) pathway and the Programmed death 1 (PD-1) with its ligand (PD-L1) (Figure 1).17,20 These molecules are the targets of numerous drugs investigated in different clinical trials and recently entered in clinical practice.
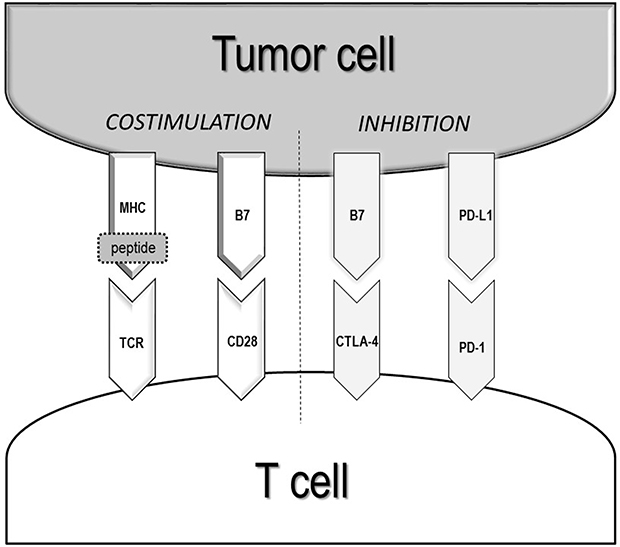 |
Figure 1 Interaction between tumor cell and T-cell. The activation of effector and memory CD8+ T-cells occurs by the interaction with antigen-presenting cells via the T-cell receptor (TCR) and major histocompatibility complex (MHC)/peptide antigen. Moreover, the complexity of the interaction TCR-MHC/peptide is amplified by the interplay of multiple costimulatory molecules. Numerous inhibitory transduction pathways, known as immunological checkpoints, maintain the tolerance and homeostasis of the immune system. The most studied immune-checkpoints playing a key role in the modulation of adaptive immunity are the Programmed Death (PD)1/Programmed Death Ligand (PD-L) 1 and the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) axes. |
Anti CTLA-4 Agents
CTLA-4 is a receptor expressed on activated CD4+ and CD8+ T-lymphocytes. Its function is to decrease the activation of T-cells by counteracting the co-stimulatory signal of CD28. In particular, CTLA-4 binds to the CD80 (also known as B7.1) and CD86 (also known as B7.2) ligands, undermining CD28 for greater affinity (Figure 1).21 This results in a reduced activation of naїve T-cells and memory. The success of ipilimumab, a CTLA-4 inhibitor, in metastatic melanoma was the proof of concept that the inhibition of this checkpoint can lead to the activation of the host immune system against tumor antigens, with consequent death of tumor cells.22 As discussed later in detail, contrary to melanoma, in mRCC ipilimumab did not demonstrate a comparable meaningful benefit.
Anti-PD-1 Agents
PD-1 is an inhibitory receptor expressed by activated T-cells, B-cells, monocytes, and natural killer (NK) cells. Two known ligands activate PD-1: PD-L1 and PD-L2. The first, also known as B7-H1 or CD274, is expressed in different cells, including APC and tumor cells. The interaction of PD-1 with PD-L1 is primarily responsible for the immunosuppressive effects of PD-1.23 Specifically, PD-1 binding PD-L1 inhibits the proliferation, survival, and function of CD8+ lymphocytes, promotes the differentiation of CD4+ T-cells into regulatory T lymphocytes (Tregs), and can induce apoptosis of infiltrating tumor cells (Figure 1). PD-L2 (also known as B7-DC or CD273), the second ligand for PD-1, is also responsible for the inhibition of T-cell activation.23
Anti-PD1 agents, such as nivolumab and pembrolizumab, and anti-PD-L1 drugs, such as atezolizumab, avelumab, and durvalumab, led to a radical change in the therapeutic algorithm of many neoplasms, including mRCC, melanoma, non-small cell lung cancer, urothelial carcinoma, Merkel cell carcinoma, and Hodgkin lymphoma.10–15,24
Tumor Microenvironment: Rationale of Combining Antiangiogenetics and Immunotherapy
Angiogenesis and immunosuppression play an important role in mRCC carcinogenesis. Neoangiogenic processes, mostly linked to the von-Hippel Lindau (VHL) gene, are key mechanisms implicated in mRCC pathogenesis. Moreover, mRCC is also an immunogenic cancer, due to its extraordinarily rich and heterogeneous immune infiltrate, encompassing T-cells, myeloid cells, macrophages, granulocytes, NK cells among others, that represent the immune reaction of the host to contain tumor growth.25 Angiogenesis and immune systems interact with each other determining changes in tumor microenvironment (TME). The TME is, in fact, a complex structure composed of different mediators involved in the cell signaling that may deeply influence sensitivity to immunotherapy.26 In particular, the inactivation of VHL tumor suppressor gene, present in about 60% of RCC, results in an imbalance in pro- and anti-angiogenic factors.27 Among the proangiogenic factors involved, the transcription factor hypoxia-inducible factor (HIF)-1α and VEGF induce the release of immunosuppressive factors, such as transforming growth factor β (TGF-β), PD-L1, and VEGF itself.28 All these factors contribute to the alteration of blood vessels with abnormal blood flow and tumor cell extravasation, inhibition of the maturation and recruitment of dendritic cells (DCs), activation of myeloid-derived suppressor cells (MDSCs), and infiltration of tumor-associated macrophages (TAMs).29–31 DCs are APC promoting self-tolerance through the control of Tregs, immunosuppressive cells that reduce the induction and proliferation of effector T-cells, NK cells, and other leukocytes.32 MDSCs and TAMs are other mediators of the immunosuppressive microenvironment. Specifically, MDSCs are progenitors of granulocytes and monocytes. They inhibit the activation of CD4+ and CD8+ lymphocytes, stimulate Tregs function, and drive monocytes’ differentiation toward activated M2 macrophages.33–35 TAMs originate from blood monocytes and are recruited to TME by cytokines produced by tumor cells. TAMs isolated from RCC tumors have been shown to stimulate tumor cells proliferation and angiogenesis through the production of proinflammatory signals, including tumor necrosis factor (TNF)α, interleukin (IL)-1β, IL-6, and C-C motif chemokine ligand (CCL) 2.36 TAMs are considered to be type 2 macrophages (M2) according to widely accepted classification of macrophage activation.36 According to different microenvironmental signals, in fact, macrophages polarize into two different phenotypes named as classically activated macrophages (M1) and alternatively activated macrophages (M2). While M1 showed a pro-inflammatory and cytotoxic function, M2 macrophages enhance tumor cells proliferation and angiogenesis through the release of angiogenic factors, such as cytokines and matrix metalloproteinases. Figure 2 depicts interaction between TME and the immune system, while Figure 3 addresses how cell signaling influences immune response and resistance to immunotherapy.
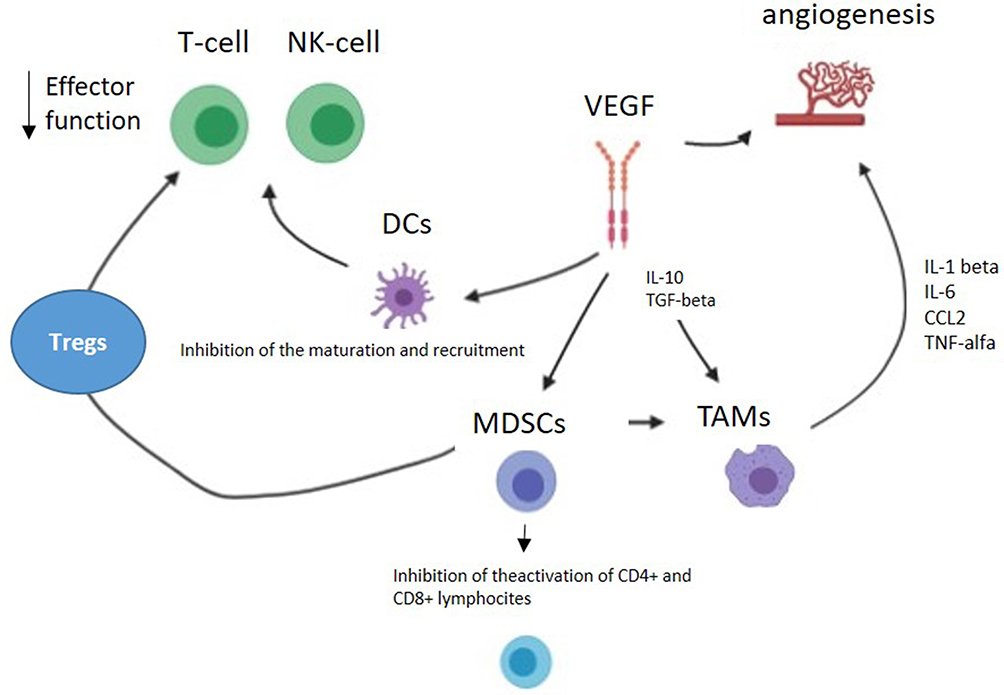 |
Figure 2 Interaction between tumor microenvironment and immune system. TME with its complexity of immune, vascular, and stromal cells could contribute to resistance to immunotherapy. |
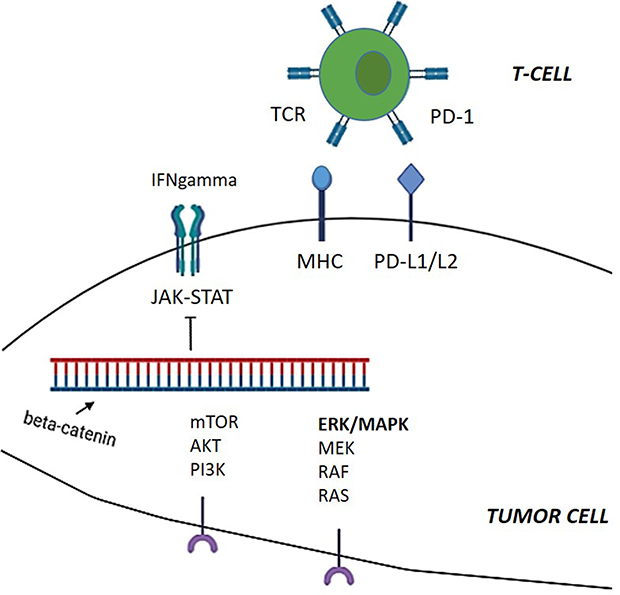 |
Figure 3 Cell signalling influences immune response and resistance to immunotherapy. Beta-catenin pathway is a canonical oncogenic pathway. Its constitutive activation might be involved in resistance to ICIs through T-cell exclusion. Abnormalities in the MAPK pathway promote oncogenesis in multiple tumors through expression of VEGF and multiple other inhibitory cytokines, resulting in immune evasion. IFNgamma signaling induces the expression of PD-L1 on tumor cells conferring adaptive resistance to tumor cells. |
The interconnected processes between the immune system and angiogenesis have implications on the therapeutic strategies to control RCC.36 In addition to their antiangiogenic activity, TKIs can exert immunomodulatory activities. The VEGFR signal blockade, in fact, leads to a modulation of TME with a recovery of the host immunity.37 This effect has been exploited to enhance the anti-tumor immune response obtained by ICIs alone.13–16 Combining ICIs with TKIs significantly improved the outcomes of mRCC patients compared to TKI monotherapy, changing the frontline treatment of these patients.13–16
The Mutational Landscape of RCC
The malignant phenotype of RCC reflects the complexity of its genomic architecture.38 Aberration in oncogenes and tumor suppressors can influence the immune response and consequently make the tumor cells resistant to ICIs.38–40 A frequently altered pathway in mRCC is the phosphoinositide 3-kinases (PI3K)/Akt/mTOR axis occurring in 16% of patients.38 This pathway plays a pivotal role in the cell cycle control, reducing apoptosis and promoting cellular proliferation when activated.41 Whereas several factors, such as epidermal growth factor (EGF), insulin-like growth factor (IGF)-1 or insulin, may constitutively activate the PI3K/AKT/mTOR signaling, PTEN is an inhibitor of the pathway.42 Loss of PTEN leads to tumorigenesis and is associated with resistance to immunotherapy due to the recruitment of immunosuppressive cells to TME through the expression of VEGF.42 Other frequently observed mutations in RCC involve chromatin remodeler genes such as PBRM1, ARID1A/B, ARID2, BRD7, or SETD2, BAP1, CDKN2A, and TP53.38–40 A genomic study performing whole-exome sequencing of 35 mRCC patients found that loss-of-function mutations in the PBRM1 gene may alter tumor-cell expression profiles and influence the response to ICIs.39 CDKN2A loss, BAP1, TP53 mutations, and an increase in mitogen-activated protein kinase (MAPK) signaling has shown to result in immune evasion through VEGF expression.38,43 Table 1 summarizes the molecular landscape of mRCC.
 |
Table 1 Summary of the Molecular Landscape in mRCC |
Moreover, three RNA signatures capturing the complex interplay between angiogenesis and immunity have been recently described. These mRNA signatures can be distinguished based on the presence or absence of the immune infiltrate.44,45 The first one identifies the T-cell-enriched signature by the expression of genes related to adaptive immunity, including the immune checkpoints CTLA4, PD-1, PDL-1, TIM3, and lymphocyte activation gene 3 protein (LAG3), or interferons (IFNs), granzyme, perforin, and Th1 cytokines. The second, the non-infiltrated signature, is identified by the expression of genes associated with angiogenic processes and immunosuppressive pathways. Finally, the third signature is characterized by an intermediate and heterogeneous milieu. These findings are relevant to further guide treatment choices and strategies.44,45
Mechanisms of Resistance and New Immunotherapeutic Target
Checkpoint inhibitors have been associated with durable long-term responses. However, a proportion of patients are primary refractory to treatment or develop a secondary resistance after a period of initial response. Mechanisms of immune escape in this context are not completely elucidated.
In different cancer types, the response rate to ICIs is associated with tumor mutational burden (TMB), even if the issue is more controversial in mRCC. Loss of neoantigen expression by cancer cells may result in resistance.46,47 The constant interaction between immune system and tumor cells can lead to immunoediting of the tumor with selection of clones lacking expression of neoantigens.48 Genetic instability with increased neoantigen load can derive from alteration of genes involved in DNA repair, such as BRCA1/2 or ATM, resulting in increased immunogenicity and contributing to high TMB as demonstrated in urothelial cancer.49 Similarly, deficiencies in DNA mismatch repair genes responsible for microsatellite instability correlate to high mutational load, with enhanced sensitivity to ICIs.50 Also loss of function in chromatin remodeler genes sensitizes to immunotherapy, increasing accessibility to IFNγ-inducible genes.39 Moreover, loss of ARID1A was associated with a failure in recruiting mismatch repair complexes, resulting in increased mutational burden.51
In addition, TME with its complexity of immune, vascular, and stromal cells could contribute to resistance to immunotherapy. Infiltrates of Tregs and MDSCs have been associated with unfavorable prognosis and poor response to ICIs.52,53 In detail, tumor cells secrete cytokines, including C-X-C Motif Chemokine Ligand (CXCL)-8/12 and CCL-2/3/4/5/17/22, recruiting immunosuppressive cells, such as MDSCs and TAMs, particularly M2 macrophages.33 All these cells promote tumor growth enhancing neoangiogenesis through the VEGF pathway, immunosuppression, and mesenchymal transition.54
Also elevated TGF-β signaling and VEGF functions were associated with poorly immunogenic tumors and resistance to ICIs, due to the recruitment of inflammatory cells with immunosuppressive functions.29–31,55,56
IFNγ plays a critical role for cancer immunoediting. First, IFNγ signaling through the JAK/STAT family of receptors upregulates MHC expression, resulting in enhanced antigen presentation. Second, the IFNγ pathway induces the expression of PD-L1 on tumor cells, conferring adaptative resistance to tumor cells and improving response to immunotherapy.57 The loss of JAK/STAT signaling has been associated to resistance to PD-1 and CTLA-4 inhibitors.58
The expression of PD-L1 on cancer cells is commonly used as a biomarker in different cancer types.58 Lack of PD-L1 expression correlates with worse outcomes with ICI treatment.59,60 The role of PD-L1 as a biomarker is controversial in mRCC. Indeed, if the negative prognostic role has been widely proven, its potential predictive value is less clear. In the CheckMate 025, a Phase III trial proving the superiority of nivolumab over everolimus in pre-treated mRCC patients, PD-L1 expression was associated with poor outcomes but not with response.10 In a different setting of patients, the CheckMate 214 phase III trial, comparing the combination of nivolumab plus ipilimumab vs sunitinib in untreated International Metastatic RCC Database Consortium (IMDC) intermediate-poor risk patients, showed a magnitude of benefit in terms of progression-free survival (PFS), but not in terms of overall survival (OS) and objective response rate (ORR), for PD-L1 positive tumors.11,12 In the JAVELIN Renal 101 trial, comparing the combination of upfront avelumab plus axitinib vs sunitinib, no difference in PFS or ORR was observed among the PD-L1 positive or negative subgroups.13 In the phase III Immotion 151 trial, comparing atezolizumab plus bevacizumab over sunitinib as first line treatment for mRCC, a trend for increased efficacy was observed in patients with higher PD-L1 expression.14,61 To date, due to the different assays and thresholds used in clinical trials, as well as different cell analyzed (tumor or immune cells) and the intratumoral heterogeneity of expression, PD-L1 cannot be used as a predictive biomarker to select mRCC patients for ICIs, and further investigation is warranted.
Another mechanism of resistance to immunotherapy is related to the indoleamine 2,3-dioxygenase (IDO1) activity, leading to an immunosuppressive microenvironment. In fact, IDO1, which is overexpressed in response to IFNγ in different cancer types, is an enzyme that catalyzes the first step in tryptophan catabolism and causes suppression of effector T-cells and resistance to ICIs.62
Lastly, upon failure of checkpoint inhibitors, a compensative overexpression of alternative receptors was described.63,64 Increased coexpression of inhibitory immune checkpoint receptors is associated with T-cell exhaustion and resistance to ICIs. Several coinhibitory receptors have been discovered so far, including LAG-3, B and T lymphocyte attenuator (BTLA), killer-cell immunoglobulin-like receptor (KIR), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), V-type immunoglobulin domain-containing suppressor of T-cell activation (VISTA), T-cell immunoglobulin, and mucin domain-containing 3 (TIM-3 or HAVCR2).65,66 All these coinhibitory receptors represent potential therapeutic targets to enhance immune response.65,66
Therapy
Cytokines
IFNγ, IFNα, and high-dose IL-2 represent the first generation immunotherapy. However, their use has been limited by significant systemic toxicities.4,67–72
Immune Checkpoint Inhibitors
Nivolumab has been the first new-generation immunotherapeutic drug to enter the therapeutic scenario of pretreated mRCC. After the results of the phase III CheckMate-025 trial, demonstrating a benefit in OS, ORR, and PFS of nivolumab compared to everolimus after failure of anti-angiogenic therapy. The median OS for nivolumab vs everolimus was 25.8 months vs 19.7 months (Hazard Ratio [HR]=0.73; 95% Confidence Interval [CI]=0.62–0.85; P<0.0001), with an ORR of 23% vs 4% (P<0.001), and a PFS of 4.2 vs 4.5 months (HR=0.84, 95% CI=0.72–0.99; P=0.0331), respectively, with a manageable toxicity (Table 2).10
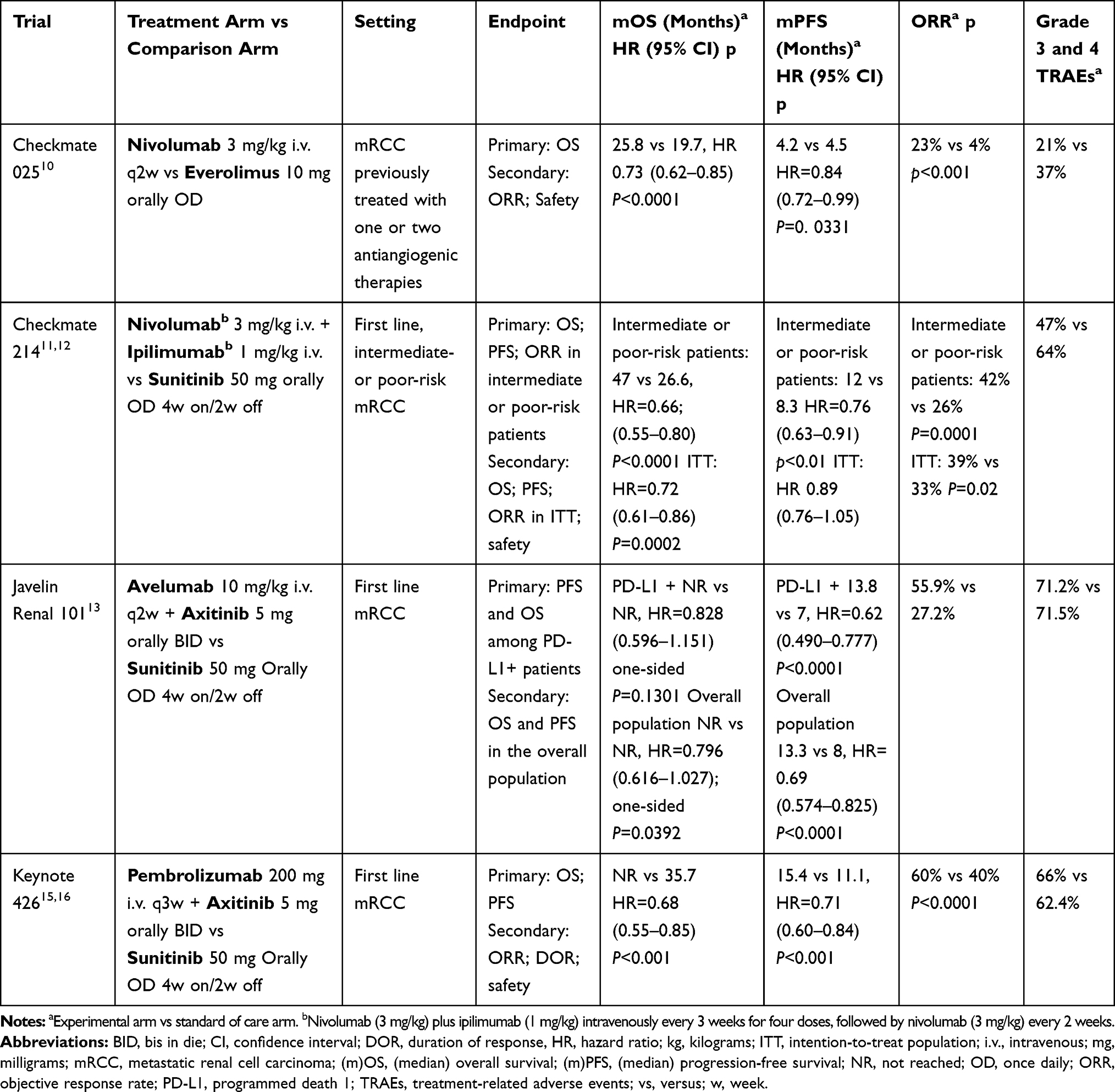 |
Table 2 Pivotal Trials of Immune Checkpoint Inhibitors in Metastatic Renal Cell Carcinoma |
Ipilimumab was also explored in mRCC. In detail, in a Phase II trial, 61 patients were treated initially with ipilimumab 3 mg/kg every 3 weeks and with ipilimumab 1 mg/kg or 3 mg/kg thereafter, obtaining 12.5% and 5% of ORR, respectively. No complete responses or long-lasting disease regressions were observed.73 Thirty-three percent of patients experienced a grade (G) 3 or 4 immune-mediated toxicity, but a strong association between toxicity and response has been documented.73 The results of trials conducted later, in which ipilimumab was combined with another checkpoint inhibitor, were encouraging.11,12
More recently, the combination of nivolumab plus ipilimumab has been approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) in treatment naïve mRCC patients at intermediate–poor risk based on the results of the CheckMate 214 trial.11,12 In this phase III trial, 847 treatment naïve patients were randomly assigned to receive nivolumab 3 mg/kg plus ipilimumab 1 mg/kg every 3 weeks, followed by nivolumab at the same dose every 2 weeks vs sunitinib 50 mg daily (schedule 4 weeks on and 2 weeks off). OS, PFS, and ORR outcomes resulted in a significantly improved combination of nivolumab plus ipilimumab compared with sunitinib among intermediate- and poor-risk patients. Long-term results at the median follow-up of 42 months confirmed the significant superiority of the ICI combination over sunitinib in terms of OS (47.0 vs 26.6 months, HR=0.66; 95% CI=0.55–0.80; P<0.0001), PFS (12.0 vs 8.3 months, HR=0.76; 95% CI=0.63–0.91; P<0.01) and ORR (42% vs 26%, respectively; P=0.0001), with a complete response rate of 9% vs 1%, respectively (Table 2). Looking at duration of response, it is widely proven that ICIs are associated with meaningful long-lasting responses. CheckMate 214 confirmed the long-term benefit for a notable proportion of patients treated with the immunotherapeutic combination. Indeed, there is an apparent flattening of the curve for nivolumab plus ipilimumab at 24-months, meaning that responses tend to be durable on the long-term compared to sunitinib, where response continues to decline.11,12
All the above studies excluded rare histologies such as collecting duct carcinoma or papillary tumors. These rare and aggressive tumors are poorly represented in phase III trials, and the therapeutic choices remain a challenge for clinicians. More knowledge about these histologies is certainly needed.
Antiangiogenic and Immune Checkpoint Inhibitors Combinations
Combining immunotherapy with antiangiogenic treatment improves outcomes in mRCC, due to a synergistic effect. As previously discussed, the underlying rationale is based on the complex interconnection between TME and immune system. VEGF inhibition could synergistically enhance the responses obtained by ICIs.
Early Phase Trials
The results of multiple early phase trials were encouraging and led to phase III studies evaluating combinations of TKIs and immunotherapeutic agents in a first line setting, which have led to the approval of pembrolizumab plus axitinib or avelumab plus axitinib combinations.13,15 Other studies are still ongoing, such as those evaluating the combination of nivolumab plus cabozantinib (Table 3).
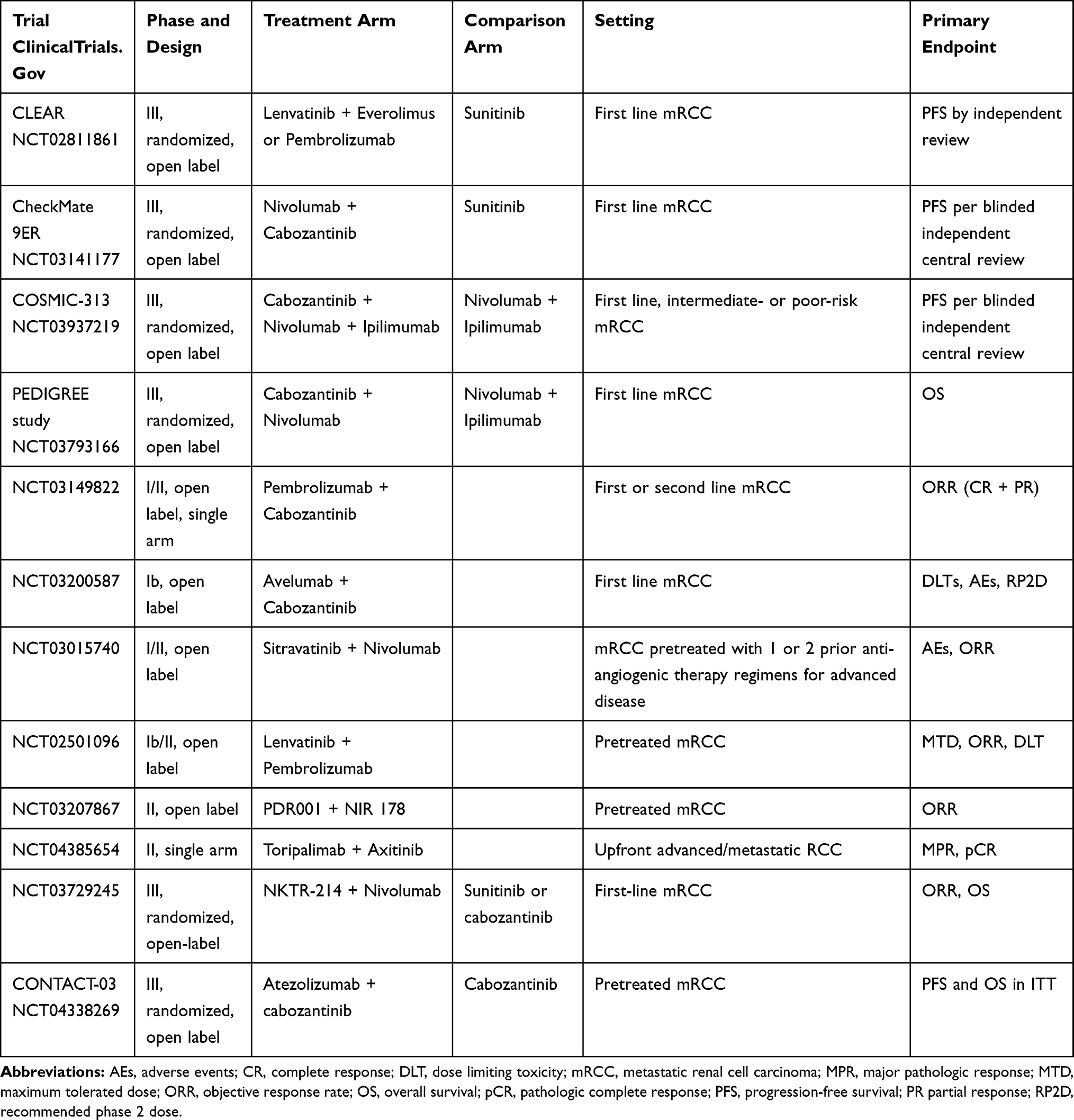 |
Table 3 Ongoing Clinical Trials Evaluating Small Molecules and Immune Checkpoint Inhibitors Combinations in Metastatic Renal Cell Carcinoma |
Several early phase studies have been limited by the high percentage of toxicity. In detail, nivolumab was evaluated in combination with sunitinib or pazopanib in the CheckMate 016, a Phase I trial dose-escalation and expansion study. However, despite the documented efficacy, the significant toxicities limited the use of these combinations, that were not considered for further evaluation.74 Pembrolizumab was also evaluated in several trials. A phase Ib/II clinical trial documented an ORR of 60.9% in 61 mRCC patients treated with pembrolizumab in combination with bevacizumab.75 The combination of pembrolizumab plus pazopanib was studied in a phase I trial (Keynote-018) but, due to significant toxicity, it was not suitable for evaluation in a larger cohort.76 A phase Ib/II study investigated nivolumab in combination with tivozanib, a TKI with minimal off-target action and presumably lower toxicity profile. Among 27 patients, 44% of whom were previously untreated, ORR was 56%, with one patient achieving complete response. Median PFS was 18.5 months for untreated patients and was not reached for previously treated patients. At the time of analysis, data were immature for OS analysis. Fifty-two percent of patients experienced G 3/4 adverse events, the most common being hypertension.77
Phase III Trials
IMmotion 151, built on the results of the phase II trial (IMmotion 150), is a phase III trial randomizing untreated mRCC patients to receive the combination of atezolizumab 1,200 mg every 3 weeks plus bevacizumab 15 mg/kg every 3 weeks vs sunitinib 50 mg daily 4 weeks on followed by 2 weeks off. After a median follow-up of 24 months, the median PFS was 11.2 months in the atezolizumab plus bevacizumab group, compared to 7.7 months for the sunitinib arm (HR=0.74; 95% CI=0.57–0.96; P=0.0217). There was no significant difference in terms of OS in the intention to treat analysis (HR=0.93; 95% CI=0.76–1.14).14 Due to the lack of benefit in OS, the combination of atezolizumab and bevacizumab was not approved by the regulatory agencies (FDA and EMA). However, the phase II IMmotion 150 trial suggests precious information on the biology of the tumor that could potentially help clinicians in future treatment choice. Exploration of the gene expression signature and correlation with treatment response distinguished two molecular subgroups based on effector T-cells, IFN-γ, and angiogenesis gene expression. A highly angiogenic signature was associated with improved ORR and PFS in the sunitinib arm. On the other hand, high expression of the T-effector gene signature was associated to improved ORR and PFS with atezolizumab plus bevacizumab compared to sunitinib.78 Exploration of biological features could be used to select patients for immunotherapy combinations over TKI and vice-versa.
Recently, the FDA and the EMA approved, in addition to nivolumab plus ipilimumab, two other combinations regardless of the risk category group: avelumab plus axitinib in 2019, and pembrolizumab plus axitinib in 2019.
The KEYNOTE-426 trial, a large phase III trial including 861 patients, showed a superiority of the combination of pembrolizumab plus axitinib over sunitinib in untreated mRCC patients. Initial data showed an improvement in OS (90% vs 78%, HR=0.53; 95% CI=0.38–0.74; P<0.0001) and ORR (59% vs 36%, P<0.001) with pembrolizumab 200 mg every 3 weeks for up to 35 cycles plus axitinib 5 mg orally twice daily compared to sunitinib 50 mg for 4 weeks in 6 week cycles. The benefit of the combination was observed across all the IMDC risk groups and regardless of PD-L1 expression.15 The updated data presented at the annual ASCO congress in 2020 confirmed a maintained benefit for the combination after a minimum follow-up of 23 months with 74% of patients alive in the combination arm at 24 months compared with 66% of patients in the sunitinib arm. Median OS in the intention-to-treat population was not yet reached for the patients assigned to receive the combination vs 35.7 months for the patients assigned to sunitinib (HR=0.68; 95% CI=0.55–0.85 P<0.001). A benefit in PFS was also documented for the combination of pembrolizumab plus axitinib, resulting in a median PFS of 15.4 months in the combination arm vs 11.1 months in the sunitinib arm (HR=0.71; 95% CI=0.60–0.84, P<0.001). In addition, the ORR was 60.2% with the combination and 40% in the sunitinib arm, with a complete response rate of 9% vs 3% (Table 2). Grouping patients by IMDC risk, significant differences in OS and PFS (HR of 0.63 for OS and 0.69 for PFS) were observed for patients with intermediate or poor risk disease, while no significant differences in OS or PFS were observed for patients with favorable-risk disease. However, the favorable-risk group had a better ORR with pembrolizumab plus axitinib vs sunitinib than did the intermediate/poor group (69.6 vs 50.4% and 55.8 vs 35.2%, respectively). Compared to CheckMate 214, the follow-up with pembrolizumab plus axitinib is not yet long enough to assess whether the curve will plateau. However, even in the absence of data about the long-term benefit, the OS curves separate since the beginning compared to the control arm along the treatment, meaning that the combination starts to work early. Discussing the safety data, G 3 or higher adverse events of any cause occurred in 66% and 62.4% of patients in the pembrolizumab-axitinib group and in the sunitinib group, respectively (Table 2).15,16
The JAVELIN Renal 101 trial evaluated the combination of a PD-L1 inhibitor, avelumab, and a multikinase inhibitor, axitinib, over sunitinib in untreated patients with mRCC. Primary endpoints were PFS and OS among patients with PD-L1–positive tumors (≥1% of immune cells staining positive within the tumor area of the tested tissue sample). PD-L1 expression was assessed at a central laboratory with the use of the Ventana PD-L1 (SP263) assay (Ventana Medical Systems). The combination was shown to be superior in terms of PFS, irrespective of IMDC risk group and PD-L1 expression. In detail, median PFS in the PD-L1 positive population was 13.8 vs 7.0 months in avelumab plus axitinib and sunitinib arms, respectively (HR=0.62, 95% CI=0.490–0.777; one-sided P<0.0001). PFS in the overall population was 13.3 vs 8.0 months in the avelumab plus axitinib and sunitinib arms, respectively (HR=0.69, 95% CI= 0.574–0.825; one-sided P<0.0001). Among the patients with PD-L1–positive tumors, the ORR was 55.9% with avelumab plus axitinib compared to 27.2% with sunitinib. The OS data are not yet mature. Adverse events during treatment occurred in 99.5% of patients in the avelumab-plus-axitinib group and in 99.3% of patients in the sunitinib group; these events were G3 or higher in 71.2% and 71.5% of the patients in the respective groups.13
Table 2 reports the design and results of this and other pivotal trials of ICIs in mRCC.
A study presented at the 2020 annual ASCO congress provided data on the use of the combination of lenvatinib 20 mg daily plus pembrolizumab 200 mg every 3 weeks for patients who had progressed on front-line immunotherapy, a setting where data are very limited. Among the evaluable cases, almost all had tumor shrinkage. The PFS was 11.7 months, with 45% of patients being progression-free at 12 months. The OS was 77% at 12 months.79
Very recently, results of the clinical phase III trial CheckMate-9ER were presented at the ESMO 2020 congress. This study met its primary endpoint showing a consistent benefit in terms of PFS for the combination of cabozantinib plus nivolumab over sunitinib in previously untreated mRCC patients (16.6 vs 8.3 months. HR=0.51; 95% CI=0.41–0.64, P=0.0001). This benefit was demonstrated in numerous subgroups including age, sex, PD-L1 expression, bone metastases, and IMDC risk group. Longer-term data for OS are certainly needed because they are still immature. These positive results support the increasing number of data showing that TKIs may create a more immune-permissive tumor microenvironment that could enhance the response to checkpoint inhibitors.80
Similarly to phase III trials investigating immunotherapeutic combinations, all the studies mentioned above combining checkpoint inhibitors with a TKI included clear cells carcinoma and excluded rare histologies such as collecting duct carcinoma or papillary tumors.
Management of Immune-Related Adverse Events
By unbalancing the immune system, ICI administration may unleash the autoreactive engagement of T-cells, leading to the development of immune-related adverse events (irAEs). Because of their peculiar pathogenesis and broad spectrum of manifestations – the most frequent involving skin, liver, lungs, gastrointestinal and endocrine system – irAEs require prompt and specific management.81
The rate and severity of irAEs vary across ICI classes: anti-CTLA-4 agents display higher rates of irAEs (̴ 90%) compared to anti-PD-1/PD-L1 (̴ 70%), and anti-PD-1 plus anti CTLA-4 combinations yield the highest safety concerns. Development of irAEs is unpredictable. A dose-dependency for an increased risk of irAEs has been described for anti-CTLA-4 agents, while there is no direct relationship with cumulative dose toxicity for anti-PD-1/anti-PD-L1 antibodies. Most irAEs occur within 3 to 6 months from ICI start. However, with the broad employment of immunotherapy in the real-world setting, the possible outbreak of late (>1 year after treatment initiation) or rare toxicities (including neurological, cardiological, renal, and hematological disorders) should be closely monitored, as these events can be irreversible or life-threatening and pose several diagnostic and therapeutic challenges.81
The mainstay for the treatment of irAEs is represented by steroids, which counteract lymphocyte activation and irAEs manifestations. According to international guidelines, low-grade G1 irAEs do not generally require to withhold ICIs, with the exception of rare neurological and cardiac toxicities of suspect autoimmune etiology. In the case of G1 irAEs, symptomatic management is recommended. For G≥2 irAEs, ICI interruption and administration of moderate-to-high doses of corticosteroids (0.5–1 mg/kg of prednisone or equivalent for G2 toxicities, 1–2 mg/kg for G3-4 toxicities) is warranted. Steroids should be tapered in 4–6 weeks. In the case of severe and potentially life-threatening irAEs, the introduction of immunosuppressants such as infliximab should be considered if symptoms or laboratory signs do not improve in the first 48–72 hours from steroid administration.82
ICI resumption may be offered after reversion of the irAEs to G1 or less, even though caution is advised for some patients who experienced G3 or early-onset disabling irAEs. The occurrence of G4 irAEs usually warrants permanent ICI discontinuation, with an exception for G4 endocrinopathies that can be adequately managed with hormone replacement.82
In the evolving therapeutic scenario of mRCC, the introduction of several combinations of ICIs or ICIs plus TKIs in the frontline setting has produced outstanding survival improvements, and the possibility of achieving long-lasting disease control. In this context, optimal management of drug-related toxicities is of utmost importance to allow long-term treatment continuation with a positive impact on tolerability and quality-of-life.
Of note, prospective data from the Italian Early Access Program for nivolumab in mRCC showed improved OS outcomes in patients reporting irAEs, thus suggesting that the occurrence of irAEs can be regarded as an hallmark of treatment activity.61,83 This observation further highlights the need for timely and effective management of irAEs in clinical practice, as a prolonged use of high-dose corticosteroids to treat misdiagnosed or lately recognized irAEs may hinder antineoplastic efficacy and yield potential detrimental effects.81
Moreover, additional challenges are posed in the management of irAEs in patients receiving the combination of ICIs plus TKIs, since most toxicities (eg, endocrine, gastrointestinal, skin, or liver toxicities) may be of difficult attribution or even cumulative. The management of this broad spectrum of toxicities warrants multidisciplinary collaboration between organ specialists, in order to improve treatment adherence and ultimately clinical outcomes.
Future Directions
Despite checkpoint inhibitors being associated with durable long-term responses, a proportion of patients are refractory to these treatments. Multiple treatment strategies combining different mechanisms of action or targeting immune escape pathways are emerging to improve response to immunotherapeutic agents.
Different small molecules with immunomodulatory effects are under evaluation. In detail, cabozantinib is an oral TKI with activity against multiple targets, such as VEGF, MET, and AXL among others. For its potent and multitarget activity on crucial kinases involved in immune escape, cabozantinib was chosen as a perfect candidate for combination therapies. A phase I trial with expansion cohorts of cabozantinib plus nivolumab and cabozantinib plus nivolumab plus ipilimumab in patients with different metastatic treatment-refractory genitourinary (GU) malignancies were presented by Nadal et al.84 Initial safety findings and promising antitumor activity were confirmed. Among the 14 patients with mRCC, the ORR was 54% and 12-month PFS and 12-month OS were 73% and 50%, respectively. Based on these positive results, several other studies investigating cabozantinib in combination with ICIs in mRCC are ongoing and are summarized in Table 3. Another ongoing trial investigating VEGF/PD-1 blockade combination is the CLEAR study (NCT02811861) evaluating lenvatinib plus everolimus or pembrolizumab vs sunitinib in a first line setting (Table 3).
Another promising therapeutic option is represented by agents targeting molecules implicated in cancer cells metabolism. To satisfy their demands of growth and proliferation, cancer cells must rewire cellular metabolism with enhancement of aerobic glycolysis, fatty acid synthesis, and glutaminolysis.85,86 In a randomized phase II trial enrolling mRCC patients after no more than two prior therapies, cabozantinib is under evaluation also in combination with CB-839 (telaglenastat), a glutaminase inhibitor (NCT03428217). CB-839 (telaglenastat) is under evaluation also in combination with everolimus vs placebo in pretreated mRCC patients (NCT03163667). Since glutaminase has emerged as a crucial enzyme in different types of cancer cells, glutaminase inhibitors (BPTES, CB-839and968) were tested showing an anti-proliferative activity in a wide range of cancers, including RCC.85,86
Contrarily to melanoma, where targeting the MAPK pathway with BRAF and MEK inhibitors has revolutionized the therapeutic scenario, this combination has not shown encouraging results in mRCC, due to the high percentage of toxicity.87 Early trials combining BRAF/MEK inhibitors with ipilimumab were prematurely stopped. However, subsequent preclinical studies combining these inhibitors with anti PD-1 or PD-L1 agents showed enhanced anti-tumor activity, giving these combinations another chance to treat mRCC.88
Another emerging treatment strategy is represented by combining immunotherapy with epigenetic modulators, such as inhibitors of histone deacetylases (HDACi) or DNA methyltransferases (DNMTi). For their demonstrated immunomodulatory effect, epi-drugs are under evaluation in multiple clinical trials in combination with immunotherapy.89
Inhibition of IDO was also used to enhance immune response. Different clinical trials evaluating combinations of ICIs and anti-IDO are ongoing.90
Other potential therapeutic targets to enhance antitumor immune response are represented by coinhibitory receptors such as LAG-3, KIR, BTLA, TIGIT, TIM-3, or costimulatory receptors including the tumor necrosis factor receptor superfamily member 4 (TNFRSF4 or OX40 or CD134), inducible T-cell co-stimulator (ICOS), tumor necrosis factor receptor superfamily member 18 (TNFRSF18 or GITR), CD137 (or 4–1BB), CD27, and CD40L.65,66 Initial evidence has been collected and appear to be promising; however, further research is needed to translate these data into clinical practice.65,66
Another upcoming frontier in the landscape of immunotherapeutic strategies for mRCC is represented by the modulation of cytokine signaling. The activity of Bempegaldesleukin (NKTR-214), a CD122-preferential IL2 pathway agonist, has been evaluated in association with nivolumab in the recent PIVOT−02 phase I clinical trial, including 22 patients with immunotherapy-naïve mRCC. In the overall study population, ORR was 59.5%, with a 18.9% rate of achieved complete responses. In the cohort of mRCC patients receiving first-line NKTR-214 plus nivolumab, ORR was 71.4%, with a median PFS of 14.2 months (1.8–not reached), whereas in the subgroup of ICI-naïve mRCC patients receiving study treatment as a second-line, ORR was 28.6%, with a median PFS of 14.3 months (0.7–not reached). The combination of NKTR-214 plus nivolumab proved safe, the most common treatment-related adverse events being flu-like symptoms (86.8%), rash (78.9%), fatigue (73.7%), and pruritus (52.6%). Overall, 21.1% of patients experienced G3/4 toxicities.91
Promising activity was observed with the HIF-2α inhibitor in treatment-naïve patients with VHL-associated tumor, with a favorable safety profile.92 Data of a randomized trial evaluating HIF-2α inhibitor vs everolimus are awaited to assess the efficacy of this emerging molecule in the ICIs pretreated setting.
A further potential treatment strategy is represented by chimeric antigen receptor (CAR) T-cell therapy, that targets cancer antigens through tumor-specific and engineered TCRs.93 Numerous expectations have been created on CAR T-cell-therapy, but it is still difficult to determine how, and when, this therapeutic strategy can be applied to all patients.
Conclusion
The therapeutic landscape of mRCC is rapidly changing, but several unmet needs remain. ICIs are associated with long-lasting responses. However, a small proportion of patients remain refractory to immunotherapy. Multiple treatment strategies combining different mechanisms of action or targeting immune escape pathways are emerging to improve responses. Certainly, the identification of biomarkers of response or resistance to immunotherapeutic agents is essential to improve outcomes. Going forward, whole genome sequencing and epigenetic analysis will probably help to understand the biology of tumor and to distinguish genomic signatures that can predict response to different treatment strategies.
Abbreviations
APC, Antigen presenting cells; BTLA, B and T lymphocyte attenuator; CAR, Chimeric antigen receptor; CCL2, C-C motif chemokine ligand 2; CD, Cluster of differentiation; CI, Confidence interval; CTLA-4, Cytotoxic T-lymphocyte-associated antigen 4; CXCL 8/12, C-X-C Motif Chemokine Ligand 8/12; DCs, Dendritic cells; DNMTi, DNA methyltransferases inhibitors; EGF, Epidermal growth factor; EMA, European Medicines Agency; FDA, Food and Drug Administration; G, Grade; HDACi, Histone deacetylases inhibitors; HIF-1α, Hypoxia-inducible factor-1α; HR, Hazard ratio; KIR, Killer-cell immunoglobulin-like receptor; ICIs, Immune checkpoint inhibitors; ICOS, Inducible T-cell co-stimulator; IDO, indoleamine 2,3-dioxygenase; IFN, Interferon; IGF-1, Insulin-like growth factor-1; IL, Interleukin; IMDC, International Metastatic RCC, Database Consortium; irAEs, immune-related adverse events; LAG3, Lymphocyte activation gene 3 protein; MAPK, Mitogen-activated protein kinase; MDSCs, Myeloid-derived suppressor cells; MHC, Major histocompatibility complex; (m)RCC, (metastatic) renal cell carcinoma; NK, Natural killer; ORR, Objective response rate; OS, Overall Survival; PD-1, Programmed-death 1; PD-L1/2, Programmed death ligand-1/2; PFS, Progression-free survival; PI3K, Phosphoinositide 3-kinase; TAMs, Tumor associated macrophages; TCR T-cell receptor; TGF-β, Transforming growth factor β; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM3, T-cell immunoglobulin and mucin domain-containing 3; TKIs, Tyrosine kinase inhibitors; TMB, tumor mutational burden; TME, Tumor microenvironment; TNF, Tumor necrosis factor; TNFRSF, Tumor necrosis factor receptor superfamily member; Tregs, Regulatory T lymphocytes; VEGF(R), vascular endothelial growth factor (receptor); VHL, Von Hippel Lindau; VISTA, V-type immunoglobulin domain-containing suppressor of T-cell activation.
Funding
The authors declare there were no funding sources.
Disclosure
Dr. Giuseppe Procopio received honoraria from Ipsen, Pfizer, BMS, AstraZeneca, and Janssen outside the submitted work, reports consultancy or advisory roles for Bayer, BMS, Janssen, Ipsen, MSD, Pfizer, and Novartis, and reports no other potential conflicts of interest for this work. The remaining authors report no conflicts of interest in this work.
References
1. Capitanio U, Montorsi F. Renal cancer. Lancet. 2016;387:894–906. doi:10.1016/S0140-6736(15)00046-X
2. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi:10.1056/NEJMoa060655
3. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi:10.1056/NEJMoa065044
4. Rini BI, Halabi S, Rosenberg JE, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28(13):2137–2143. doi:10.1200/JCO.2009.26.5561
5. Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi:10.1200/JCO.2009.23.9764
6. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1814–1823. doi:10.1056/NEJMoa1510016
7. Motzer RJ, Escudier B, Oudard S, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116:4256–4265. doi:10.1002/cncr.25219
8. Choueiri TK, Hessel C, Halabi S, Sanford B, Michaelson MD, Hahn O, et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur J Cancer. 2018 May;94:115–125. doi:10.1016/j.ejca.2018.02.012.Epub 2018 Mar 20. Erratum in: Eur J Cancer. 2018 Nov;103:287. PMID: 29550566; PMCID: PMC6057479.
9. George DJ, Hessel C, Halabi S, et al. Cabozantinib versus sunitinib for untreated patients with advanced renal cell carcinoma of intermediate or poor risk: subgroup analysis of the alliance A031203 CABOSUN trial. Oncologist. 2019;24(11):1497–1501. doi:10.1634/theoncologist.2019-0316
10. Motzer RJ, Tykodi SS, Escudier B, et al. Final analysis of the CheckMate 025 trial comparing nivolumab (NIVO) versus everolimus (EVE) with >5 years of follow-up in patients with advanced renal cell carcinoma (aRCC). J Clin Oncol. 2020;38(suppl6):617. doi:10.1200/JCO.2020.38.6_suppl.617
11. Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomized, controlled phase 3 trial. Lancet Oncol. 2019;20(10):1370–1385. doi:10.1016/S1470-2045(19)30413-9
12. Tannir NM, McDermott DF, Escudier B, et al. Overall survival and independent review of response in CheckMate 214 with 42-month follow-up: first-line nivolumab + ipilimumab (N+I) versus sunitinib (S) in patients (pts) with advanced renal cell carcinoma (aRCC). J Clin Oncol. 2020;38(6_suppl):609. doi:10.1200/JCO.2020.38.6_suppl.609
13. Choueiri TK, Motzer RJ, Rini BI, et al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol. 2020;31(8):1030–1039. doi:10.1016/j.annonc.2020.04.010
14. Rini BI, Powles T, Atkins MB, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019;393(10189):2404–2415. doi:10.1016/S0140-6736(19)30723-8
15. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1116–1127. doi:10.1056/NEJMoa1816714
16. Plimack ER, Rini BI, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib as first-line therapy for advanced renal cell carcinoma (RCC): updated analysis of KEYNOTE-426. J Clin Oncol. 2020;38(15_suppl):5001. doi:10.1200/JCO.2020.38.15_suppl.5001
17. Zhang S, Zhang E, Long J, et al. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019;110(5):1564–1572. doi:10.1111/cas.13996
18. Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015;27(4):439–449. doi:10.1016/j.ccell.2015.03.007
19. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
20. Kwon B. Is CD137 ligand (CD137L) signaling a fine tuner of immune responses? Immune Netw. 2015;15(3):121–124. doi:10.4110/in.2015.15.3.121
21. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. doi:10.3389/fonc.2018.00086
22. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma [published correction appears in N Engl J Med. 2010 Sep 23;363(13):1290]. N Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
23. Marinelli O, Annibali D, Aguzzi C, et al. The controversial role of PD-1 and its ligands in gynecological malignancies. Front Oncol. 2019;9:1073. doi:10.3389/fonc.2019.01073
24. Chevrier S, Levine JH, Zanotelli VRT, et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169:736–749. doi:10.1016/j.cell.2017.04.016
25. Drake CG, Stein MN. The immunobiology of kidney cancer. J Clin Oncol. 2018;36:3547–3552. doi:10.1200/JCO.2018.79.2648
26. Mennitto A, Huber V, Ratta R, et al. Angiogenesis and immunity in renal carcinoma: can we turn an unhappy relationship into a happy marriage? J Clin Med. 2020;9(4):930. doi:10.3390/jcm9040930
27. Rini BI, Small EJ. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol. 2005;23:1028–1043. doi:10.1200/JCO.2005.01.186
28. Na X, Wu G, Ryan CK, Schoen SR, di’Santagnese PA, Messing EM. Overproduction of vascular endothelial growth factor related to von Hippel-Lindau tumor suppressor gene mutations and hypoxia-inducible factor-1 alpha expression in renal cell carcinomas. J Urol. 2003;170(2 Pt 1):588–592. doi:10.1097/01.ju.0000074870.54671.98
29. Oyama T, Ran S, Ishida T, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-κB activation in hemopoietic progenitor cells. J Immunol. 1998;160:1224–1232.
30. Noman MZ, Desantis G, Janji B, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi:10.1084/jem.20131916
31. Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005;15:417–425. doi:10.1097/00008390-200510000-00010
32. Belkaid Y, Oldenhove G. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity. 2008;29:362–371. doi:10.1016/j.immuni.2008.08.005
33. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi:10.1038/nri2506
34. Sevko A, Umansky V. Myeloid-derived suppressor cells interact with tumors in terms of myelopoiesis, tumorigenesis and immunosuppression: thick as thieves. J Cancer. 2013;4:3–11. doi:10.7150/jca.5047
35. Fujimura T, Mahnke K, Enk AH. Myeloid derived suppressor cells and their role in tolerance induction in cancer. J Dermatol Sci. 2010;59:1–6. doi:10.1016/j.jdermsci.2010.05.001
36. Kovaleva OV, Samoilova DV, Shitova MS, Gratchev A. Tumor associated macrophages in kidney cancer. Anal Cell Pathol (Amst). 2016;2016:9307549.
37. Kwilas AR, Donahue RN, Tsang KY, Hodge JW. Immune consequences of tyrosine kinase inhibitors that synergize with cancer immunotherapy. Cancer Cell Microenviron. 2015;2:e677.
38. Linehan WM, Ricketts CJ. The cancer genome atlas of renal cell carcinoma: findings and clinical implications. Nat Rev Urol. 2019;16(9):539–552. doi:10.1038/s41585-019-0211-5
39. Miao D, Margolis CA, Gao W, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359(6377):801–806. doi:10.1126/science.aan5951
40. Shen J, Ju Z, Zhao W, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556–562. doi:10.1038/s41591-018-0012-z
41. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi:10.3389/fonc.2014.00064
42. George S, Miao D, Demetri GD, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity. 2017;46(2):197–204. doi:10.1016/j.immuni.2017.02.001
43. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203(7):1651–1656. doi:10.1084/jem.20051848
44. Şenbabaoğlu Y, Gejman RS, Winer AG, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeuticimmunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. doi:10.1186/s13059-016-1092-z
45. van den Heuvel CNAM, van Ewijk A, Zeelen C, et al. Molecular profiling of druggable targets in clear cell renal cell carcinoma through targeted RNA sequencing. Front Oncol. 2019;9:117. doi:10.3389/fonc.2019.00117
46. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi:10.1126/science.aaa4971
47. Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi:10.1126/science.aaa1348
48. Dunn GP, Old LJ, Schreiber RD. The three Es of Cancer Immunoediting. Annu Rev Immunol. 2004;22:329–360. doi:10.1146/annurev.immunol.22.012703.104803
49. Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. Clin Oncol. 2018;36(17):1685–1694. doi:10.1200/JCO.2017.75.7740
50. Le DT, Uram JN, Wang H, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733
51. Shen J, Peng Y, Wei L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5(7):752–767. doi:10.1158/2159-8290.CD-14-0849
52. Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125(Suppl. S2):S272–S283. doi:10.1016/j.jaci.2009.09.045
53. Weber R, Fleming V, Hu X, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. 2018;9:1310. doi:10.3389/fimmu.2018.01310
54. Parihar JS, Tunuguntla HS. Role of chemokines in renal cell carcinoma. Rev Urol. 2014;16:118–121.
55. Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/nature25492
56. Ohm J, Gabrilovich DI, Sempowski GD, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–4886. doi:10.1182/blood-2002-07-1956
57. Takeda K, Nakayama M, Hayakawa Y, et al. IFN-γ is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat Commun. 2017;8:14607. doi:10.1038/ncomms14607
58. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829. doi:10.1056/NEJMoa1604958
59. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015;75(11):2139–2145. doi:10.1158/0008-5472.CAN-15-0255
60. Martin AM, Nirschl TR, Nirschl CJ, et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015;18(4):325–332. doi:10.1038/pcan.2015.39
61. Raimondi A, Sepe P, Claps M, Verzoni E, Procopio G. Do biomarkers play a predictive role for response to novel immunotherapeutic agents in metastatic renal cell carcinoma? Expert Opin Biol Ther. 2019;19(11):1107–1110. doi:10.1080/14712598.2019.1651288
62. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–143. doi:10.1016/j.it.2012.10.001
63. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. 2016;6(1):e1261779.
64. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi:10.1038/ncomms10501
65. Carlo MI, Voss MH, Motzer RJ. Checkpoint inhibitors and other novel immunotherapies for advanced renal cell carcinoma. Nat Rev Urol. 2016;13:420–443. doi:10.1038/nrurol.2016.103
66. Raimondi A, Randon G, Sepe P, et al. The evaluation of response to immunotherapy in metastatic renal cell carcinoma: open challenges in the clinical practice. Int J Mol Sci. 2019;20(17):4263. doi:10.3390/ijms20174263
67. Krown SE. Interferons and interferon inducers in cancer treatment. Semin Oncol. 1986;13:207–217.
68. Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol. 2015;36:763–777. doi:10.1016/j.it.2015.10.003
69. Gleave ME, Elhilali M, Fradet Y, et al. Interferon gamma-1b compared with placebo in metastatic renal-cell carcinoma. N Engl J Med. 1998;338:1265–1271. doi:10.1056/NEJM199804303381804
70. Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–2111. doi:10.1016/S0140-6736(07)61904-7
71. Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi:10.1200/JCO.2009.26.7849
72. Fyfe G, Fisher RI, Rosenberg SA, et al. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13:688–696. doi:10.1200/JCO.1995.13.3.688
73. Yang JC, Hughes M, Kammula U, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825–830. doi:10.1097/CJI.0b013e318156e47e
74. Hammers HJ, Plimack ER, Infante JR, et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 study. J Clin Oncol. 2017;35(34):3851–3858. doi:10.1200/JCO.2016.72.1985
75. Dudek AZ, Sica RA, Sidani A, et al. Phase Ib study of pembrolizumab in combination with bevacizumab for the treatment of metastatic renal cell carcinoma: big ten cancer research consortium BTCRC-GU14-003. J Clin Oncol. 2016;34:559. doi:10.1200/jco.2016.34.2_suppl.559
76. Chowdhury S, McDermott DF, Voss MH, et al. A phase I/II study to assess the safety and efficacy of pazopanib (PAZ) and pembrolizumab (PEM) in patients (pts) with advanced renal cell carcinoma (aRCC). J Clin Oncol. 2017;35:4506. doi:10.1200/JCO.2017.35.15_suppl.4506
77. Escudier B, Barthelemy P, Ravaud A, Negrier S, Needle MN, Albiges L. Tivozanib combined with nivolumab: phase Ib/II study in meta- static renal cell carcinoma (mRCC). J Clin Oncol. 2018;36:618. doi:10.1200/JCO.2018.36.6_suppl.618
78. McDermott DF, Huseni MA, Atkins MB, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma [published correction appears in Nat Med. 2018 Dec;24(12):1941]. Nat Med. 2018;24(6):749–757. doi:10.1038/s41591-018-0053-3
79. Lee CH, Shah AY, Hsieh JJ, et al. Phase II trial of lenvatinib (LEN) plus pembrolizumab (PEMBRO) for disease progression after PD-1/PD-L1 immune checkpoint inhibitor (ICI) in metastatic clear cell renal cell carcinoma (mccRCC). J Clin Oncol. 2020;38(15_suppl):5008. doi:10.1200/JCO.2020.38.15_suppl.5008
80. Choueiri TK, Powles T, Burotto M, et al. Nivolumab + cabozantinib vs sunitinib in first-line treatment for advanced renal cell carcinoma: first results from the randomized phase 3 CheckMate 9ER trial. Ann Oncol. 2020;31(suppl_4):S1142–S1215. doi:10.1016/j.annonc.2020.08.2257
81. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. doi:10.1016/j.ejca.2015.11.016
82. Brahmer JR, Lacchetti C, Thompson JA. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: american society of clinical oncology clinical practice guideline summary. J Oncol Pract. 2018;14(4):247–249. doi:10.1200/JOP.18.00005
83. Verzoni E, Carteni G, Cortesi E, et al. Real-world efficacy and safety of nivolumab in previously-treated metastatic renal cell carcinoma, and association between immune-related adverse events and survival: the Italian expanded access program. J Immunother Cancer. 2019;7(1):
84. Nadal RM, Mortazavi A, Stein M, et al. Results of phase I plus expansion cohorts of cabozantinib (Cabo) plus nivolumab (Nivo) nd CaboNivo plus ipilimumab (Ipi) in patients (pts) with meta- static urothelial carcinoma (mUC) and other genitourinary (GU) malignancies. J Clin Oncol. 2018;36:515. doi:10.1200/JCO.2018.36.6_suppl.515
85. Cantor JR, David M, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2(10):881–898. doi:10.1158/2159-8290.CD-12-0345
86. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:890–901. doi:10.1158/1535-7163.MCT-13-0870
87. Hu-Lieskovan S, Mok S, Homet Moreno B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7(279):279ra41. doi:10.1126/scitranslmed.aaa4691
88. Liu L, Mayes PA, Eastman S, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21(7):1639–1651. doi:10.1158/1078-0432.CCR-14-2339
89. Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics. 2017;9:59.
90. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210(7):1389–1402. doi:10.1084/jem.20130066
91. Diab A, Tannir NM, Bentebibel SE, et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020;10(8):1158–1173. doi:10.1158/2159-8290.CD-19-1510
92. Choueiri TK, Plimack ER, Bauer TM, et al. Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC). J Clin Oncol. 2020;38(6_suppl):611. doi:10.1200/JCO.2020.38.6_suppl.611
93. Suarez ER, de Chang K, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–34355. doi:10.18632/oncotarget.9114
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.