Back to Journals » OncoTargets and Therapy » Volume 14
Identification of the EMT-Related Genes Signature for Predicting Occurrence and Progression in Thyroid Cancer
Authors Li Q ![]() , Jiang S
, Jiang S ![]() , Feng T, Zhu T, Qian B
, Feng T, Zhu T, Qian B
Received 8 January 2021
Accepted for publication 29 April 2021
Published 12 May 2021 Volume 2021:14 Pages 3119—3131
DOI https://doi.org/10.2147/OTT.S301127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Qiang Li,1,2,* Sheng Jiang,3,* Tienan Feng,2 Tengteng Zhu,2 Biyun Qian2
1Public Health College, Shanghai Jiao Tong University of Medicine, Shanghai, 200025, People’s Republic of China; 2Hongqiao International Institute of Medicine, Shanghai Tongren Hospital/Clinical Research Institute, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, People’s Republic of China; 3The Second Affiliated Hospital of Chengdu Medical College, China National Nuclear Corporation 416 Hospital, Chengdu, 610051, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tengteng Zhu; Biyun Qian
Hongqiao International Institute of Medicine, Shanghai Tongren Hospital/Clinical Research Institute, Shanghai Jiao Tong University School of Medicine, 227 Chongqing South Road, Shanghai, 200025, China
Email [email protected]; [email protected]
Background: The detection rate of thyroid cancer (TC) has been continuously improved due to the development of detection technology. Epithelial–mesenchymal transition (EMT) is thought to be closely related to the malignant progression of tumors. However, the relationship between EMT-related genes (ERGs) characteristics and the diagnosis and prognosis of TC patients has not been studied.
Methods: Four datasets from Gene Expression Omnibus (GEO) were used to perform transcriptomic profile analysis. The overlapping differentially expressed ERGs (DEERGs) were analyzed using the R package “limma”. Then, the hub genes, which had a higher degree, were identified by the protein–protein interaction (PPI) network. Gene expression analysis between the TC and normal data, the disease-free survival (DFS) analysis of TC patients from The Cancer Genome Atlas Thyroid Cancer (TCGA-THCA) cohort, function analysis, and immunohistochemistry (IHC) were performed to verify the importance of the hub genes. Finally, a prognostic risk scoring was constructed to predict DFS in patients with the selected genes.
Results: A total of 43 DEERGs were identified and 10 DEERGs were considered hub ERGs, which had a high degree of connectivity in the PPI network. Then, the differential expressions of FN1, ITGA2, and KIT between TC and normal tissues were verified in the TCGA-THCA cohort and their protein expressions were also verified by IHC. DFS analysis indicated upregulations of FN1 expression (P< 0.01) and ITGA2 expression (P< 0.01) and downregulation of KIT expression (P=0.01) increased risks of decreased DFS for TCGA-THCA patients. Besides, by building a prognostic risk scoring model, we found that the DFS of TCGA-THCA patients was significantly worse in high-risk groups.
Conclusion: In summary, these hub ERGs were potential biomarkers for diagnosis and prognosis of TC, which can provide a basis for further exploring the efficacy of EMT in patients with TC.
Keywords: thyroid cancer, EMT, signature, bioinformatic analysis, immunohistochemistry
Introduction
Thyroid cancer (TC) is the most common endocrine malignancy. In the past 40 years, the incidence of TC has risen steadily in many countries around the world.1 With the improvement of detection technology, the incidence of recent TC in China in 2014 was 12.40/100,000, making it one of the fastest-growing malignant tumors.2 Although the incidence of TC is increasing year by year, the mortality rate of TC is stable or decreased slightly due to early diagnosis.3,4 Ultrasonography is the main method for stratifying the risk of early cancerous thyroid nodules. Patients with suspected TC could take a fine-needle aspiration biopsy (FNAB) or surgical resection, and followed by a pathological examination to assess the nature of cancer.5 However, in some cases, due to the overlapping of cytological features of malignant and benign thyroid nodules, approximately 15%-30% of biopsies are uncertain.6 And the uncertain diagnosis may bring inappropriate treatment leading to poor quality of life, such as the surgical removal of the thyroid will seriously affect the quality of life of patients. Ultrasound and FNAB can only be diagnosed and cannot respond to the question of whether to perform surgery. Therefore, supplementing and improving traditional diagnostic methods of TC patients is still an urgent problem to be addressed.
Epithelial–mesenchymal transition (EMT) is one of the markers of carcinogenesis, which includes the re-differentiation of epithelial cells into mesenchymal cells, making the cell phenotype malignant.7–9 At present, some studies have found that EMT-related molecular markers are significantly correlated with TC occurrence and adverse outcomes.10−13 However, these studies only focused on a single genetic biomarker, which has been less effective in predicting outcomes. Studies have shown that gene signatures composed of multiple genes may be the better predictors of patients.
Some researchers have studied the relationship between EMT-related signatures and the prognosis of patients with gastric adenocarcinoma,14 endometrial cancer,15 and glioma16 by analyzing public databases. However, there has been no bioinformatics research in the field of thyroid cancer. In this study, through the analysis of existing genetic data, a transcriptomic profile of thyroid cancer of EMT-related genes (ERGs) was constructed, and further analysis was performed, as well as exploring the potential signatures for diagnosis and prognostic guidance.
Materials and Methods
Data Collection
Data screening was completed independently by two researchers, and a consistent comparison was conducted after completion. We searched in Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) for all published data related to gene expression in thyroid cancer until August 1, 2020, by using the following keywords: (thyroid cancer OR cancerous goiter OR carcinoma of thyroid OR thyroid carcinoma). Finally, a total of 4 thyroid cancer data sets from the GEO database (GSE29265, GSE33630, GSE60542, and GSE65144) were included in this study.17 Datasets inclusion criteria: (1) they contained human TC samples and normal tissue samples. (2) their sample sizes at least 20. The information of four GEO datasets was shown in Supplementary Table S1.
Differentially Expressed ERGs Analysis
A list of ERGs containing 1184 ERGs was downloaded from the EMT gene database (Supplementary Table S2).18 All data were processed using the R software (version 3.6.1). The limma package was used to identify the differentially expressed ERGs (DEERGs) between the TC samples and normal samples.19 The adjusted P-value < 0.05 and the absolute fold change (FC) ≥ 2 were considered statistically significant. Overlapping DEERGs between GEO datasets were selected using a Venn diagram (https://bioinfogp.cnb.csic.es/tools/venny/index.html).
Functional Enrichment Analysis
Gene Ontology (GO) can annotate genes, gene products and sequences with potential biological phenomena in three aspects: biological process (BP), cellular component (CC), and molecular function (MF);20 Kyoto Encyclopedia of Genes and Genomes (KEGG) is a comprehensive database for understanding advanced functions and utilities of biological systems.21 In this study, GO and KEGG analysis of the DEERGs were performed using the R package clusterProfiler.22
Hub ERGs Selection and Analyses
The online tool Search Tool for the Retrieval of Interacting Genes database (STRING) (https://string- db.org/) was used to analyze the interactive relationships of the DEERGs.23 The combined score of ≥0.4 was the cut-off value. Cytoscape software (version 3.6.0) was then used to make PPI networks.24 And top 10 DEERGs with higher node degree scores were identified hub ERGs using the Cytoscape plug-in cytoHubba.
Verification of Differential Expression of Hub ERGs
Raw read counts for hub ERGs of 497 TC samples and 56 normal ones from the TCGA database were extracted using R package TCGAbiolinks.25 They were loaded into R package DESeq2 and normalized by variance-stabilizing transformation (VST).26,27 VST counts were used for Student’s t‐test analysis by R package ggstatsplot.28 The statistical significance cut‑off level was P<0.01.
Moreover, the protein levels of hub ERGs in 5 PTC tissues and paired normal tissues from the Shanghai Tong Ren Hospital were verified using immunohistochemistry (IHC) analysis. These tissues were enrolled from October 2018 to October 2019 and all diagnoses were based on pathological and/or cytological evidence. Ethical approval was obtained from the Ethics Committee of the Shanghai Tong Ren Hospital and informed consent was given by all patients before sample collection. Five human thyroid normal and tumor tissues were fixed in 4% formaldehyde and embedded in paraffin. Paraffin-embedded samples were cut into 6 μm thick sections, and then they were placed in xylene and ethanol solution for deparaffinating and rehydration in PBS. 3% hydrogen peroxide solution and 5% donkey serum were used to block endogenous peroxidase activity and nonspecific sites after samples were placed in the citric acid solution for antigen retrieval. Sections were stained with rabbit antibodies against FIBRONECTIN (Abcam, ab268021, 1:600), ITGA2 (Abcam, ab133557, 1:200), and KIT (Abcam, ab32363, 1:200) for incubating at 4°C overnight, and then incubation in the second antibody (Abcam, ab97051, 1:1000) was carried out at room temperature for 2h. Antigen-antibody complexes were detected using ChemMateTM EnVisonTM/HRP complex (DAB) as a peroxidase substrate (GK500705, Gene Tech). Results were visualized under an optical microscope (OLYMPUS IX73, JAPAN). All pictures were taken under the same microscope camera (OLYMPUS DP80, JAPAN) with uniform parameters (OLYMPUS cellSens Standard 1.18) after sections were mounted with xylene sealant. Brown staining areas were selected with an eyedropper tool after dropping colors represented by fewer than 10 pixels, and the mean option density was calculated in the manner of integrated option density (IOD)/area using Image Pro Plus 6.0 software.
Survival Analysis of Hub ERGs, Construction of the Risk Score Model, and Correlation Analysis Between Risk Score and Clinical Characteristics
Clinical characteristics including age, gender, stage, disease-free interval (DFI) time, and DFI status for TCGA-THCA patients were downloaded from UCSC Xena datasets.29 After deleting patients with incomplete clinical characteristics and DFI time less than 0, a total of 350 patients were included in the disease-free survival (DFS) analysis. The optimal threshold was determined by the R package Survminer30 to divide patients into high- or low-expression groups and DFS was calculated using the R package survival.31 ERGs associated with DFS (P<0.05) were retained for subsequent multivariable Cox analysis to construct the ERG-related prognostic signatures. Then, a prognostic risk score model was established as follows: risk score = Σ (βn × expression of gene n). Patients were divided into low-risk group and high-risk group according to the median risk score, and the prognostic difference between the two groups was performed by the Kaplan–Meier curve. Harrell’s concordance index and calibration curves comparing predicted DFS and observed survival were used to evaluate the performance of the prognostic nomogram. Time-dependent receiver operating characteristic (ROC) curves and multivariable Cox analysis including patients’ clinical characteristics were calculated to evaluate the effectiveness of the prediction model. The relationship between risk score and age was analyzed through Pearson correlation coefficient. And the relationships between risk score and other clinical characteristics (gender, stage, and race), which are classified variables, were analyzed through t-test and ANOVA. P < 0.05 was considered statistically significant.
Results
Identification of DEERGs in TC and Data Integration
There were 116 DEERGs in GSE29265, 147 DEERGs in GSE33630, 114 DEERGs in GSE60542, 298 DEERGs in GSE65144. Among the DEERGs, 60, 86, 67, and 155 ERGs were upregulated while 56, 61, 47, and 143 ERGs were downregulated in GSE29265, GSE33630, GSE60542, and GSE65144, respectively (Figure 1A-D). After integrated analyses of the four GEO datasets, 43 ERGs (21 upregulated and 22 downregulated) were identified as the DEERGs (Figure 1E and F), and the cluster heatmap of these DEERGs is shown in Figure 1G.
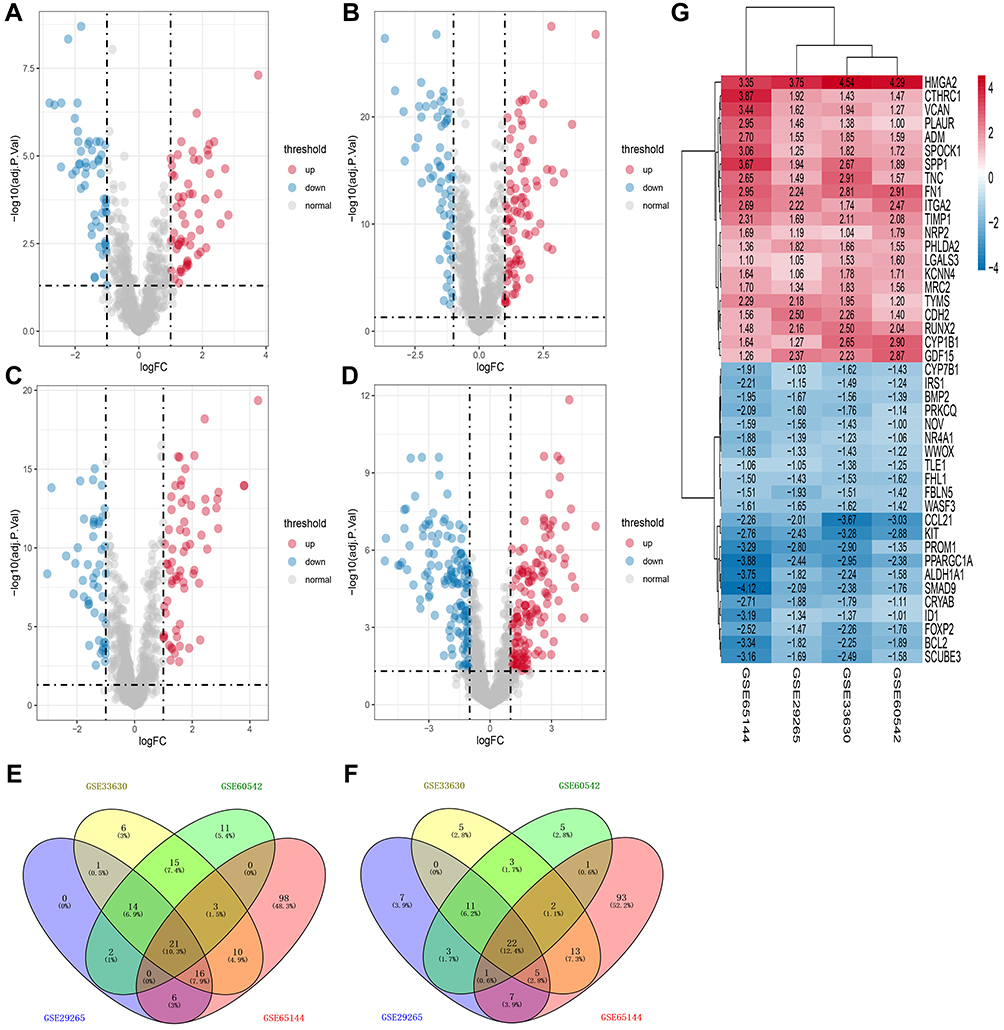 |
Figure 1 Identification of the DEERGs in TC and data integration. (A) Volcano plot of GSE29265, (B) volcano plot of GSE33630, (C) volcano plot of GSE60542, (D) volcano plot of GSE65144, (E) Venn plots of overlapping upregulated DEERGs, (F) Venn plots of overlapping downregulated DEERGs, and (G) the cluster heatmap of the overlapping DEERGs. Red, white, and blue represent higher expression levels, no expression differences, and lower expression levels, respectively. The rows and columns of the heatmap represent DEERGs and datasets, respectively, and the numbers in it represent the logFC between TC tissue and normal ones from each GEO dataset. |
Significant Functions and Pathway Enrichment Analysis
For 43 DEERGs, a pathway and process enrichment analysis were carried out using R package clusterProfiler. Terms with an adjusted P-value <0.05 were collected. GO analysis demonstrated that DEERGs were mainly related to the process of ossification, extracellular matrix (ECM), and cell growth (Figure 2A). KEGG pathway analysis showed that DEERGs were mainly related to the PI3K−Akt signaling pathway, ECM−receptor interaction, and focal adhesion (Figure 2B).
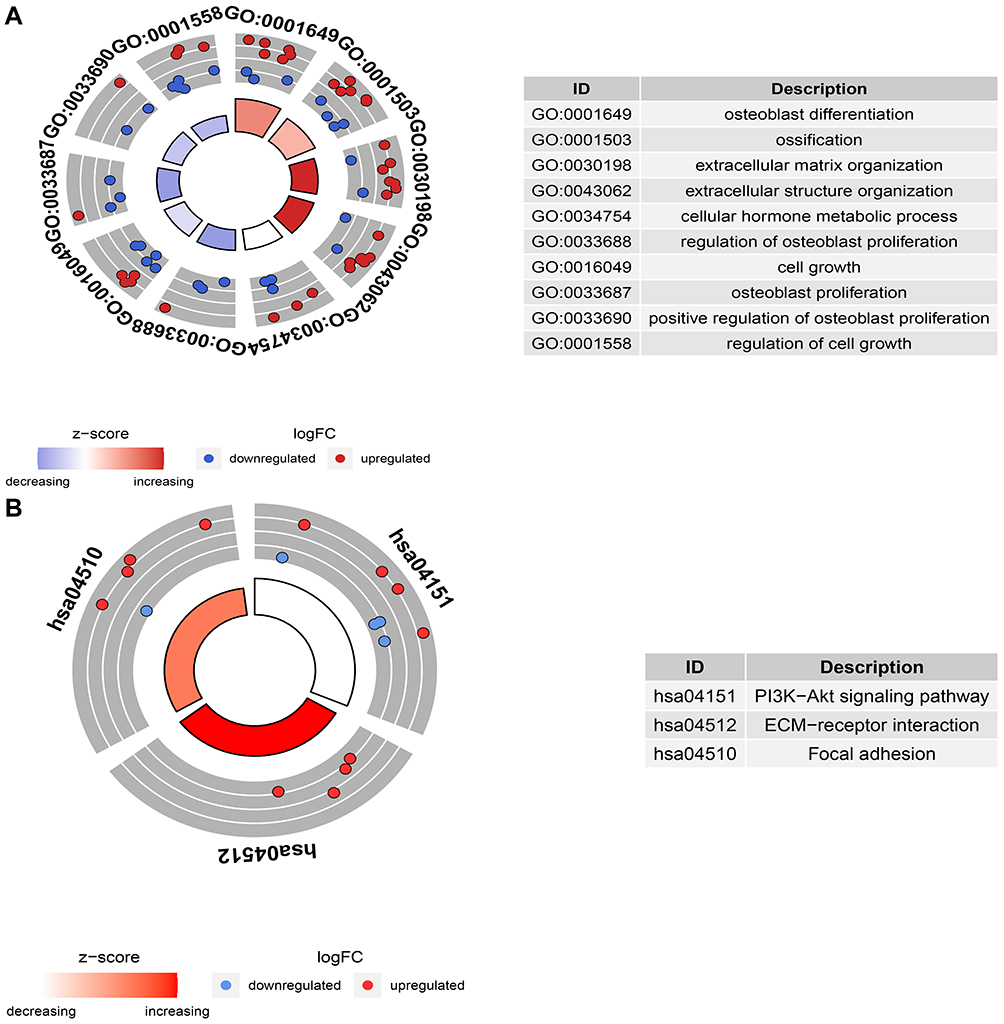 |
Figure 2 GO and KEGG pathway enrichment analysis. (A) GO circle plot of overlapping DEERGs, (B) KEGG circle plot of overlapping DEERGs. |
Hub DEERGs Identification in the PPI Network
We used the STRING database and Cytoscape to construct a PPI network to further analyze the interaction between the DEERGs (Figure 3A). There were 43 nodes and 78 edges in the network. As a result, fibronectin 1 (FN1), the tissue inhibitor of metalloproteinase 1 (TIMP1), integrin alpha 2 (ITGA2), receptor tyrosine kinase (KIT), secreted phosphoprotein 1 (SPP1), cadherin 2 (CDH2), runt-related transcription factor 2 (RUNX2), galectin 3 (LGALS3), bone morphogenetic protein 2 (BMP2), and versican (VCAN), which have a higher degree of connectivity according to the “Degree” algorithm, were identified as hub DEERGs (Figure 3B).
 |
Figure 3 The PPI network of the DEERGs. The PPI network of (A) the overlapping DEERGs, (B) top 10 hub DEERGs. Red nodes and blue nodes represent upregulated DEERGs and downregulated DEERGs, respectively in. (A). The size of the interaction score between genes was positively correlated with the depth of nodes color in (B). |
Verification of Differential Expression of Hub DEERGs
Using mRNA expression data of 497 TC samples and 56 normal ones from the TCGA-THCA dataset, upregulations of FN1 expression (P<0.01) and ITGA2 expression (P<0.01) and downregulation of KIT expression (P<0.01) was identified in TC samples compared with normal samples (Figure 4A-C). To confirm the reliability of the hub DEERGs, the IHC was used to verify the protein expression levels of the 3 hub DEERGs. Consistent with mRNA expression, the IHC results showed that FN1 and ITGA2 proteins were upregulated, while KIT protein was downregulated in the TC tissues (Figure 5A-D)
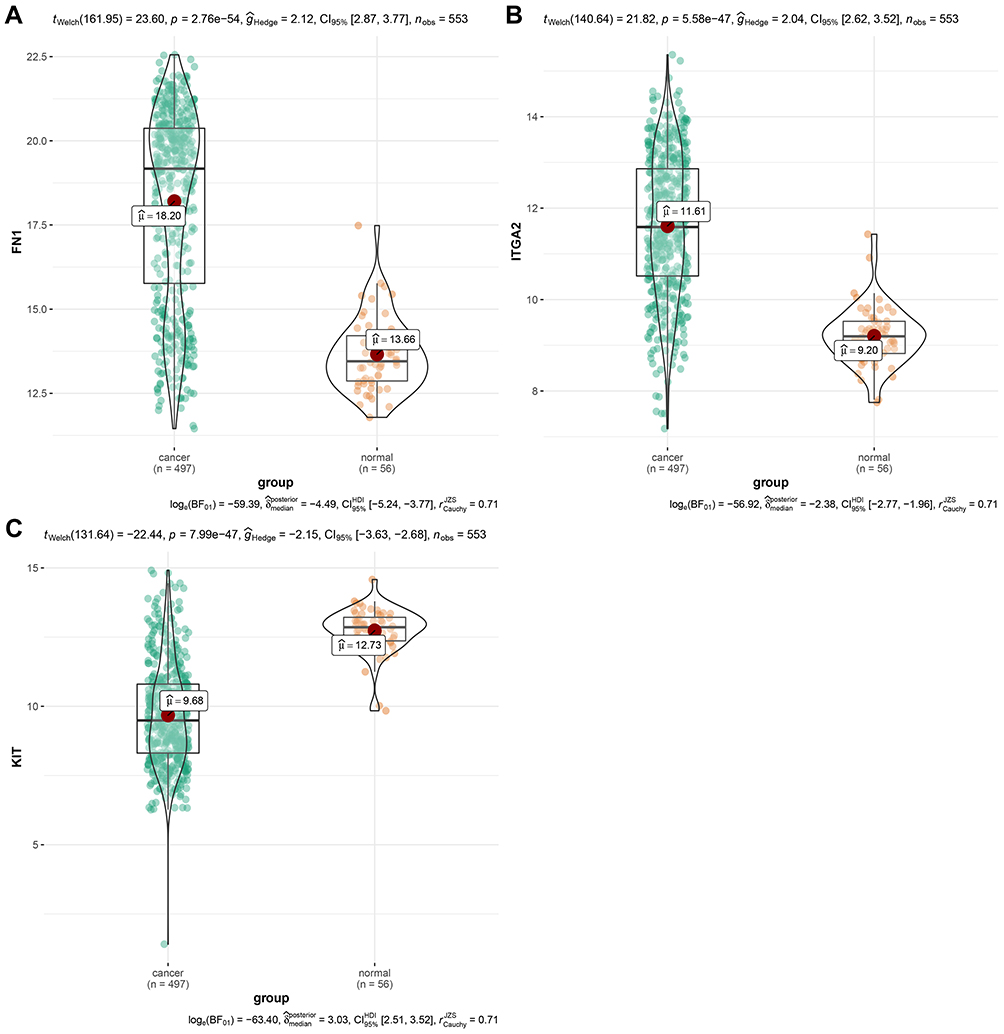 |
Figure 4 Validation of hub DEERG expressions in the TCGA-THCA dataset. (A) FN1, (B)ITGA2, and (C) KIT gene expression differences between TC tissues and normal ones. |
 |
Figure 5 Immunohistochemical staining results of the protein expressions of hub DEERGs from the clinical specimens. Brown staining represents the expression of 3 proteins and blue staining represents nucleus. Bar: 50um. A zoomed-in 40 magnification of each IHC picture is shown in better resolution at the lower left corner. (A) IHC of FN1, (B) IHC of ITGA2, (C) IHC of KIT, and (D) IHC of Blank. |
Prognostic Value of Hub DEERGs, Construction of the Risk Score Model, and Correlation between Risk Score and Clinical Characteristics
A total of 350 TCGA-THCA patients were enrolled in our survival analysis. The univariate Cox regression results showed that the high expression of FN1 (P<0.01) and ITGA2 (P<0.01) and low expression of KIT (P=0.01) were significantly associated with the worst DFS in TC patients (Figure 6A-C). Then, the prognostic signature including the above-mentioned DEERGs was developed using the multivariate Cox regression model and risk scores for every patient were determined using the coefficients (−1.32 for FN1, −0.32 for ITGA2, and 0.69 for KIT). Then, according to the median risk score, all patients were divided into high-risk (n=175) or low-risk groups (n=175) (Figure 7A). And high-risk group had worse DFS compared with low-risk group using the Kaplan–Meier curve (P<0.01) (Figure 7B). The C-index of the 3-ERG signature was 0.67, and the calibration curve further revealed that the signature had a good performance in predicting the DFS of TC patients (Figure 7C). ROC curves show that the risk score accurately predicts TC patient’s DFS (Figure 7D). In addition, the risk score calculated from the 3-ERG signature was an independent prognostic factor using multivariate Cox regression analysis (Figure 7E). The correlation analysis showed that the risk score was negatively correlated with age (P<0.01, r=−0.17) and not with other clinical characteristics (gender, stage, and race) (Supplementary Figure 1S).
 |
Figure 6 Survival analysis of hub DEERGs using TCGA-THCA dataset. Differences in DFS in patients with high and expression of (A) FN1, (B) ITGA2, and (C) KIT. |
 |
Figure 7 Survival analysis of the 3 ERGs signature using TCGA-THCA dataset. (A) Risk score based on 3 ERGs signature, survival status, and heatmap for the expressions of 3 ERGs for each patient, (B) DFS difference between high-risk and low-risk groups using the Kaplan–Meier curve, (C) calibration curves for 1-, 3-, and 5-year from the 3 ERGs signature, (D) ROC curves for 1-, 3-, and 5-year DFS predictions for the 3 ERGs signature using the ROC curves, and (E) forest plot of the risk score using multivariate Cox regression analysis. |
Discussion
Thyroid cancer (TC) is the most common endocrine malignant tumor, and its incidence increases year by year.32 Despite good prognosis, patients with recurrence or metastasis still have a high risk of death.33,34 Some studies have demonstrated that the aggressiveness of thyroid cancer is closely related to processes such as epithelial–mesenchymal transformation (EMT).34–37 At present, many studies have constructed ERG signatures to predict the survival of cancer patients. Zhong et al developed a reliable EMT-related lncRNA risk signature, which can independently predict the OS and DFS of ccRCC.14 In hepatocellular carcinoma, a total of 5 prognostic correlated ERGs were included to develop a novel prognostic risk model.38 Cao et al developed a novel EMT-related gene signature as an independent prognostic factor for bladder cancer.39 However, researches on the relationship between ERGs and the diagnosis and prognosis of TC patients are still very limited. In this study, for the first time, we used the ERG signature with higher prediction accuracy to predict the survival of TC patients and achieved good prediction results.
In this study, we comprehensively analyzed four microarray datasets from GEO, which were all published data related to gene expression in thyroid cancer and normal ones in the GEO database until August 1, 2020. A total of 43 DEERGs (21 upregulated and 22 downregulated) were identified between TC tissues and normal ones. Functional enrichment analyses demonstrated that DEERGs were mainly related to the process of ossification, extracellular matrix (ECM), and PI3K−Akt signaling pathway. In patients with thyroid cancer, there are changes in thyroid-stimulating hormone, which is closely associated with osteogenesis.40–43 The ECM plays an important role in the process of tumor shedding, adhesion, movement, degradation, and hyperplasias, such as prostate cancer,44 gastric cancer,45 breast cancer,46 and anaplastic thyroid cancer.47 The phosphoinositide 3-kinase–protein kinase B/AKT (PI3KPKB/AKT) pathway is one of the most prominent molecular signaling pathways implicated in cellular growth, proliferation, apoptosis, and metabolism.48 Activation of the PI3K-AKT signaling pathway is critical to the occurrence and development of thyroid cancer.49–51
We finally screened out that FN1, ITGA2, and KIT were hub DEERGs from the PPI network, TCGA differential expression verification, and IHC verification. Otherwise, the DFS analyses on the TCGA-THCA dataset showed that upregulations of FN1 expression (P<0.01) and ITGA2 expression (P<0.01) and downregulation of KIT expression (P=0.01) increased risks of decreased DFS for patients. Fibronectin 1 (FN1) participates in cell adhesion and migration processes and is expressed in multiple cell types.52 FN1 has also turned out to be associated with the ECM changes.53 In thyroid cancer, Sponziello M et al demonstrated that the overexpression of FN1 mediated thyroid tumor cell migration and invasion by 86 patients.54 Shaohua Zhan et al identified FN1 as a novel prognostic biomarker associated with sporadic medullary thyroid cancer pathophysiological changes.55 Furthermore, some studies found FN1 was highly expressed in TC from a series of data sets, but they did not offer a survival analysis of FN1.56–59 Integrin alpha 2 (ITGA2) is an alpha subunit, which often combines with the beta subunit to form a heterodimer α2β1, and then participates in the adhesion of platelets and other cells to the extracellular matrix.60–62 More studies suggested that ITGA2 might be closely related to tumor cell migration, invasion, and metastasis.63,64 In thyroid cancer, one study identified that ITGA2 expression was upregulated in PTC tissues but not in BRAF-positive samples,65 and another study showed that PTC patients with high ITGA2 expression had poorer relapse-free survival than PTC patients with low ITGA2 expression.66 Receptor tyrosine kinase (KIT), a form of myeloid receptor that binds the stem cell factor, plays a major role in cancer occurrence.67 Papers are showing that KIT is highly expressed in small cell lung cancer,68 leukemia cells,69 colon cancer,70 and neuroblastoma.71 But KIT expression is lower in breast cancer72 and melanoma.73 Numerous studies have investigated the expression of KIT in TC,74–76 suggesting its role in thyroid epithelial cell differentiation and growth control. These findings indicate that different ERGs jointly promote the occurrence of EMT, and these ERGs may have different functions in different tumors.
Several previous studies have constructed prognostic signatures of TC patients. Most research has focused on immune-related genes, like Gan et al77 Li et al78 Peng et al,79 and so on. They established different immune-related signatures to predict the prognosis of patients with TC. The remaining thyroid prediction models mainly focused on the study of characteristic gene sets are mainly m6A-related signature80,81 and autophagy-related signature.82 These results indicate that the characteristic signature not only has a good prediction effect on the prognosis of thyroid cancer patients but is more conducive to the comprehensive interpretation of the occurrence and development of TC and provides a theoretical basis for the use of drugs that act on this characteristic. However, no studies have been conducted to analyze the EMT characteristics of TC, which is closely related to the occurrence and development of TC, and establish relevant prediction signatures. In this study, a 3 EMT-related genes were used to construct a prognostic risk signature for TC patients. The univariate and multivariate Cox regression results showed that high-risk group had worse DFS in TC patients.
The study has several advantages. First, to our knowledge, this is the first study to analyze the EMT gene expression profile of thyroid cancer and its function. In addition, our research has constructed an excellent ERG signature as a potential predictor of TC patients, which helps to develop new strategies for diagnosing and predicting the prognosis of TC patients. Finally, calibration curves were used for internal validation of the signature, and the results showed that the signature’s prediction effect was good. However, our observations still have some limitations: 1) as the cumulation of TC samples, subtype analysis was required to perform; 2) because patients with TC have a longer survival period and fewer patients have observed death outcomes, this study only performed DFS analysis; 3) and the constructed predictive model has not been validated in a prospective cohort.
Conclusions
In conclusion, this study used existing data to construct a more comprehensive transcriptomic profile of TC and found potential biomarkers for TC diagnosis and prognosis. However, it is necessary to further study the specific role of these genes in TC.
Abbreviations
TC, thyroid cancer; EMT, epithelial–mesenchymal transition; ERGs, EMT-related genes; GEO, Gene Expression Omnibus; DEERGs, differentially expressed ERGs; PPI, protein–protein interaction; DFS, disease-free survival; TCGA-THCA, The Cancer Genome Atlas Thyroid Cancer; IHC, immunohistochemistry; FNAB, fine-needle aspiration biopsy; PTC, papillary thyroid cancer; ATC, anaplastic thyroid carcinoma; FC, fold change; GO, Gene Ontology; BP, biological process; CC, cellular component; MF, molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes; STRING, Search Tool for the Retrieval of Interacting Genes; VST, variance-stabilizing transformation; DFI, disease free interval; ROC, receiver operating characteristic; ECM, extracellular matrix; FN1, fibronectin 1; TIMP1, the tissue inhibitor of metalloproteinase 1; ITGA2, integrin alpha 2; KIT, receptor tyrosine kinase; SPP1, secreted phosphoprotein 1; CDH2, cadherin 2; RUNX2, runt-related transcription factor 2; LGALS3, galectin 3; BMP2, bone morphogenetic protein 2; VCAN, versican; PI3KPKB/AKT, phosphoinositide 3-kinase–protein kinase B/AKT.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Startup Fund for Youngman Research at SJTU (to TF, 17×100040015); Shanghai Jiao Tong University Medical and Industrial Cross Project (to TF, YG2017QN70); Young Talent Program of China National Nuclear Corportion (to SJ, CNNC201948); and The Scientific Project of Shanghai Municipal Health Commission (to BY, Grant No.2018ZHYL0202).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Chen W, Sun K, Zheng R, et al. Cancer incidence and mortality in China, 2014. Chin j Cancer Res. 2018;30(1):1. doi:10.21147/j.issn.1000-9604.2018.01.01
3. Jegerlehner S, Bulliard J-L, Aujesky D, et al. Overdiagnosis and overtreatment of thyroid cancer: a population-based temporal trend study. PLoS One. 2017;12:6. doi:10.1371/journal.pone.0179387
4. Maniakas A, Davies L, Zafereo ME. Thyroid disease around the World. Otolaryngol Clin North Am. 2018;51(3):631–642. doi:10.1016/j.otc.2018.01.014
5. Li X, Zhang S, Zhang Q, et al. Diagnosis of thyroid cancer using deep convolutional neural network models applied to sonographic images: a retrospective, multicohort, diagnostic study. Lancet Oncol. 2019;20(2):193–201. doi:10.1016/S1470-2045(18)30762-9
6. Abooshahab R, Gholami M, Sanoie M, Azizi F, Hedayati M. Advances in metabolomics of thyroid cancer diagnosis and metabolic regulation. Endocrine. 2019;65(1):1–14. doi:10.1007/s12020-019-01904-1
7. Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29(3):212–226. doi:10.1016/j.tcb.2018.12.001
8. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84. doi:10.1038/s41580-018-0080-4
9. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. doi:10.1016/j.cell.2016.06.028
10. Dong SY, Chen H, Lin LZ, et al. MFAP2 is a potential diagnostic and prognostic biomarker that correlates with the progression of papillary thyroid cancer. Cancer Manag Res. 2020;12:12557–12567. doi:10.2147/cmar.S274986
11. Jiang HC, Chen XR, Sun HF, Nie YW. Tumor promoting effects of glucagon receptor: a promising biomarker of papillary thyroid carcinoma via regulating EMT and P38/ERK pathways. Hum Cell. 2020;33(1):175–184. doi:10.1007/s13577-019-00284-y
12. Zheng C, Quan RD, Wu CY, et al. Growth-associated protein 43 promotes thyroid cancer cell lines progression via epithelial-mesenchymal transition. J Cell Mol Med. 2019;23(12):7974–7984. doi:10.1111/jcmm.14460
13. Ye D, Jiang Y, Sun Y, et al. METTL7B promotes migration and invasion in thyroid cancer through epithelial-mesenchymal transition. J Mol Endocrinol. 2019;63(1):51–61. doi:10.1530/jme-18-0261
14. Zhang D, Zhou S, Liu B. Identification and validation of an individualized EMT-related prognostic risk score formula in gastric adenocarcinoma patients. Biomed Res Int. 2020;2020:7082408. doi:10.1155/2020/7082408
15. Cai L, Hu C, Yu S, et al. Identification of EMT-related gene signatures to predict the prognosis of patients with endometrial cancer. Front Genet. 2020;11:582274. doi:10.3389/fgene.2020.582274
16. Tao C, Huang K, Shi J, Hu Q, Li K, Zhu X. Genomics and prognosis analysis of epithelial-mesenchymal transition in glioma. Front Oncol. 2020;10:183. doi:10.3389/fonc.2020.00183
17. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
18. Zhao M, Liu Y, Zheng C, Qu H. dbEMT 2.0: an updated database for epithelial-mesenchymal transition genes with experimentally verified information and precalculated regulation information for cancer metastasis. J Genet Genomics. 2019;46(12):595–597. doi:10.1016/j.jgg.2019.11.010
19. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47–e47. doi:10.1093/nar/gkv007
20. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
21. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi:10.1093/nar/gkw1092
22. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
23. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452. doi:10.1093/nar/gku1003
24. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
25. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. doi:10.1093/nar/gkv1507
26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
27. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi:10.1186/gb-2010-11-10-r106
28. Patil I, Powell C ggstatsplot: ‘ggplot2ʹ based plots with statistical details. 2018.
29. Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–678. doi:10.1038/s41587-020-0546-8
30. Kassambara A, Kosinski M, Biecek P, Fabian S survminer: drawing survival curves using ‘ggplot2ʹ. R package version 0.4.2. 2018.
31. Therneau TM. A Package for Survival Analysis in S. Version 2.38. 2015.
32. Vaccarella S, Dal Maso L. Challenges in investigating risk factors for thyroid cancer. Lancet Diabetes Endocrinol. 2020. doi:10.1016/s2213-8587(20)30426-5
33. Ho AS, Luu M, Barrios L, et al. Incidence and mortality risk spectrum across aggressive variants of papillary thyroid carcinoma. JAMA Oncol. 2020;6(5):706–713. doi:10.1001/jamaoncol.2019.6851
34. Yoo SK, Song YS, Lee EK, et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat Commun. 2019;10(1):2764. doi:10.1038/s41467-019-10680-5
35. Bhandari A, Guan Y, Xia E, Huang Q, Chen Y. VASN promotes YAP/TAZ and EMT pathway in thyroid carcinogenesis in vitro. Am J Transl Res. 2019;11(6):3589–3599.
36. Gao W, Han J. Overexpression of ING5 inhibits HGF-induced proliferation, invasion and EMT in thyroid cancer cells via regulation of the c-Met/PI3K/Akt signaling pathway. Biomed Pharmacother. 2018;98:265–270. doi:10.1016/j.biopha.2017.12.045
37. Wang SC, Chai DS, Chen CB, Wang ZY, Wang L. HPIP promotes thyroid cancer cell growth, migration and EMT through activating PI3K/AKT signaling pathway. Biomed Pharmacother. 2015;75:33–39. doi:10.1016/j.biopha.2015.08.027
38. Xiong C, Wang G, Bai D. A novel prognostic models for identifying the risk of hepatocellular carcinoma based on epithelial-mesenchymal transition-associated genes. Bioengineered. 2020;11(1):1034–1046. doi:10.1080/21655979.2020.1822715
39. Cao R, Yuan L, Ma B, Wang G, Qiu W, Tian Y. An EMT-related gene signature for the prognosis of human bladder cancer. J Cell Mol Med. 2020;24(1):605–617. doi:10.1111/jcmm.14767
40. Notsu M, Yamauchi M, Morita M, Nawata K, Sugimoto T. Papillary thyroid carcinoma is a risk factor for severe osteoporosis. J Bone Miner Metab. 2020;38(2):264–270. doi:10.1007/s00774-019-01053-5
41. Brancatella A, Marcocci C. TSH suppressive therapy and bone. Endocr Connect. 2020;9(7):R158–r172. doi:10.1530/ec-20-0167
42. Cellini M, Rotondi M, Tanda ML, et al. Skeletal health in patients with differentiated thyroid carcinoma. J Endocrinol Invest. 2020. doi: 10.1007/s40618-020-01359-6
43. Hawkins Carranza F, Guadalix Iglesias S, Luisa De Mingo Domínguez M, et al. Trabecular bone deterioration in differentiated thyroid cancer: impact of long-term TSH suppressive therapy. Cancer Med. 2020;9(16):5746–5755. doi:10.1002/cam4.3200
44. Andersen MK, Rise K, Giskeødegård GF, et al. Integrative metabolic and transcriptomic profiling of prostate cancer tissue containing reactive stroma. Sci Rep. 2018;8(1):1–11. doi:10.1038/s41598-018-32549-1
45. Hu K, Chen F. Identification of significant pathways in gastric cancer based on protein-protein interaction networks and cluster analysis. Genet Mol Biol. 2012;35(3):701–708. doi:10.1590/S1415-47572012005000045
46. Bao Y, Wang L, Shi L, et al. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell Mol Biol Lett. 2019;24(1):38. doi:10.1186/s11658-019-0162-0
47. Huang Y, Tao Y, Li X, et al. Bioinformatics analysis of key genes and latent pathway interactions based on the anaplastic thyroid carcinoma gene expression profile. Oncol Lett. 2017;13(1):167–176. doi:10.3892/ol.2016.5447
48. Hemmings BA, Restuccia DF. Pi3k-pkb/akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189. doi:10.1101/cshperspect.a011189
49. Motti ML, Califano D, Troncone G, et al. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol. 2005;166(3):737–749. doi:10.1016/S0002-9440(10)62295-X
50. Larson SD, Jackson LN, Riall TS, et al. Increased incidence of well-differentiated thyroid cancer associated with Hashimoto thyroiditis and the role of the PI3k/Akt pathway. J Am Coll Surg. 2007;204(5):764–773. doi:10.1016/j.jamcollsurg.2006.12.037
51. Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13(4):1161–1170. doi:10.1158/1078-0432.CCR-06-1125
52. Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115(20):3861–3863. doi:10.1242/jcs.00059
53. Cai X, Liu C, Zhang TN, Zhu YW, Dong X, Xue P. Down‐regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J Cell Biochem. 2018;119(6):4717–4728. doi:10.1002/jcb.26651
54. Sponziello M, Rosignolo F, Celano M, et al. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol Cell Endocrinol. 2016;431:123–132. doi:10.1016/j.mce.2016.05.007
55. Zhan S, Li J, Wang T, Ge W. Quantitative proteomics analysis of sporadic medullary thyroid cancer reveals fn1 as a potential novel candidate prognostic biomarker. The Oncologist. 2018;23(12):1415–1425. doi:10.1634/theoncologist.2017-0399
56. Liang W, Sun F. Identification of key genes of papillary thyroid cancer using integrated bioinformatics analysis. J Endocrinol Invest. 2018;41(10):1237–1245. doi:10.1007/s40618-018-0859-3
57. Zhao H, Li H. Network-based meta-analysis in the identification of biomarkers for papillary thyroid cancer. Gene. 2018;661:160–168. doi:10.1016/j.gene.2018.03.096
58. Qiu J, Zhang W, Zang C, et al. Identification of key genes and miRNAs markers of papillary thyroid cancer. Biol Res. 2018;51(1):45. doi:10.1186/s40659-018-0188-1
59. Liu L, He C, Zhou Q, Wang G, Lv Z, Liu J. Identification of key genes and pathways of thyroid cancer by integrated bioinformatics analysis. J Cell Physiol. 2019;234(12):23647–23657. doi:10.1002/jcp.28932
60. Adorno-Cruz V, Liu H. Regulation and functions of integrin α2 in cell adhesion and disease. Genes Dis. 2019;6(1):16–24. doi:10.1016/j.gendis.2018.12.003
61. Plow E, Haas T, Zhang LLJ, Smith JW. Ligand Binding To. 2000.
62. Tuckwell D, Calderwood DA, Green LJ, Humphries MJ. Integrin alpha 2 I-domain is a binding site for collagens. J Cell Sci. 1995;108(4):1629–1637. doi:10.1242/jcs.108.4.1629
63. Gong J, Lu X, Xu J, Xiong W, Zhang H, Yu X. Coexpression of UCA1 and ITGA2 in pancreatic cancer cells target the expression of miR‐107 through focal adhesion pathway. J Cell Physiol. 2019;234(8):12884–12896. doi:10.1002/jcp.27953
64. Zhao X, Wu Y, Lv Z. miR-128 modulates hepatocellular carcinoma by inhibition of ITGA2 and ITGA5 expression. Am J Transl Res. 2015;7(9):1564.
65. Chernaya G, Mikhno N, Khabalova T, et al. The expression profile of integrin receptors and osteopontin in thyroid malignancies varies depending on the tumor progression rate and presence of BRAF V600E mutation. Surg Oncol. 2018;27(4):702–708. doi:10.1016/j.suronc.2018.09.007
66. Zhai T, Muhanhali D, Jia X, Wu Z, Cai Z, Ling Y. Identification of gene co-expression modules and hub genes associated with lymph node metastasis of papillary thyroid cancer. Endocrine. 2019;66(3):573–584. doi:10.1007/s12020-019-02021-9
67. Ashman LK. The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol. 1999;31(10):1037–1051. doi:10.1016/S1357-2725(99)00076-X
68. Matsumura Y, Umemura S, Ishii G, et al. Expression profiling of receptor tyrosine kinases in high-grade neuroendocrine carcinoma of the lung: a comparative analysis with adenocarcinoma and squamous cell carcinoma. J Cancer Res Clin Oncol. 2015;141(12):2159–2170. doi:10.1007/s00432-015-1989-z
69. Yu G, Yin C, Jiang L, et al. Amyloid precursor protein cooperates with c-KIT mutation/overexpression to regulate cell apoptosis in AML1-ETO-positive leukemia via the PI3K/AKT signaling pathway. Oncol Rep. 2016;36(3):1626–1632. doi:10.3892/or.2016.4963
70. Chen EC, Karl TA, Kalisky T, et al. KIT signaling promotes growth of colon xenograft tumors in mice and is up-regulated in a subset of human colon cancers. Gastroenterology. 2015;149(3):705–717. doi:10.1053/j.gastro.2015.05.042
71. Lau S, Hansford L, Chan W, et al. Prokineticin signaling is required for the maintenance of a de novo population of c-KIT+ cells to sustain neuroblastoma progression. Oncogene. 2015;34(8):1019–1034. doi:10.1038/onc.2014.24
72. Tramm T, Kim J-Y, Leibl S, Moinfar F, Tavassoli FA. Expression of C-KIT, CD24, CD44s, and COX2 in benign and non-invasive apocrine lesions of the breast. Virchows Archiv. 2016;469(3):285–295. doi:10.1007/s00428-016-1966-1
73. Dahl C, Abildgaard C, Riber-Hansen R, Steiniche T, Lade-Keller J, Guldberg P. KIT is a frequent target for epigenetic silencing in cutaneous melanoma. J Invest Dermatol. 2015;135(2):516–524. doi:10.1038/jid.2014.372
74. Broecker-Preuss M, Sheu S-Y, Worm K, et al. Expression and mutation analysis of the tyrosine kinase c-kit in poorly differentiated and anaplastic thyroid carcinoma. Hormone Metabol Res. 2008;40(10):685–691. doi:10.1055/s-2008-1080895
75. Murakawa T, Tsuda H, Tanimoto T, Tanabe T, Kitahara S, Matsubara O. Expression of KIT, EGFR, HER‐2 and tyrosine phosphorylation in undifferentiated thyroid carcinoma: implication for a new therapeutic approach. Pathol Int. 2005;55(12):757–765. doi:10.1111/j.1440-1827.2005.01902.x
76. Tanaka T, Umeki K, Yamamoto I, et al. c-Kit proto-oncogene is more likely to lose expression in differentiated thyroid carcinoma than three thyroid-specific genes: thyroid peroxidase, thyroglobulin, and thyroid stimulating hormone receptor. Endocr J. 1995;42(5):723–728. doi:10.1507/endocrj.42.723
77. Gan X, Guo M, Chen Z, et al. Development and validation of a three-immune-related gene signature prognostic risk model in papillary thyroid carcinoma. J Endocrinol Invest. 2021. doi:10.1007/s40618-021-01514-7
78. Li Z, Lin W, Zheng J, et al. Identification of immune-related lncRNAs to improve the prognosis prediction for patients with papillary thyroid cancer. Biosci Rep. 2021;41:2. doi:10.1042/bsr20204086
79. Lin P, Guo YN, Shi L, et al. Development of a prognostic index based on an immunogenomic landscape analysis of papillary thyroid cancer. Aging (Albany NY). 2019;11(2):480–500. doi:10.18632/aging.101754
80. Wang X, Fu X, Zhang J, Xiong C, Zhang S, Lv Y. Identification and validation of m(6)A RNA methylation regulators with clinical prognostic value in Papillary thyroid cancer. Cancer Cell Int. 2020;20:203. doi:10.1186/s12935-020-01283-y
81. Xu N, Chen J, He G, Gao L, Zhang D. Prognostic values of m6A RNA methylation regulators in differentiated Thyroid Carcinoma. J Cancer. 2020;11(17):5187–5197. doi:10.7150/jca.41193
82. Han B, Yang X, Hosseini DK, et al. Development and validation of a survival model for thyroid carcinoma based on autophagy-associated genes. Aging (Albany NY). 2020;12(19):19129–19146. doi:10.18632/aging.103715
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.