Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Identification of Th17 Cell-Associated Biomarkers and Their Potential Regulatory Mechanisms in Chronic Obstructive Pulmonary Disease Through Integrated Bioinformatics Analysis and Machine Learning
Authors Li Q ![]() , Xue D
, Xue D ![]() , Yu J, Yang M
, Yu J, Yang M ![]() , Zhang Y
, Zhang Y ![]() , Obuli R
, Obuli R ![]()
Received 16 January 2026
Accepted for publication 5 June 2026
Published 11 June 2026 Volume 2026:21 596649
DOI https://doi.org/10.2147/COPD.S596649
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Qiannan Li,1,* Danhong Xue,2,* Jialin Yu,2 Man Yang,2 Yating Zhang,2 Rukiyem Obuli2
1Institute of Etiology and Prevention of Metabolic Diseases in Pamir Plateau Area, Kashi University, Kashi, Xinjiang, 844000, People’s Republic of China; 2Department of Respiratory Medicine, The 947th Hospital of the Chinese People’s Liberation Army, Kashi, Xinjiang, 844000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yating Zhang, Department of Respiratory Medicine, The 947th Hospital of the Chinese People’s Liberation Army, Kashi, Xinjiang, 844000, People’s Republic of China, Tel +86 18307031870, Email [email protected] Rukiyem Obuli, Department of Respiratory Medicine, The 947th Hospital of the Chinese People’s Liberation Army, Kashi, Xinjiang, 844000, People’s Republic of China, Tel +86 18307031870, Email [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation and chronic airway inflammation. Th17 cell-related immune pathways may contribute to COPD-associated immune dysregulation, but their molecular associations remain incompletely understood. This study aimed to preliminarily explore Th17 cell-associated biomarkers and their potential regulatory networks in COPD.
Methods: Transcriptomic profiles from COPD and control samples were analyzed to identify differentially expressed genes (DEGs). Th17 cell-related DEGs were obtained by intersecting DEGs with Th17-related genes. Random forest and Boruta algorithms were used for biomarker screening, followed by expression validation in training and validation cohorts and RT-qPCR verification. A nomogram model was constructed, and chromosome localization, immune infiltration, gene set enrichment analysis (GSEA), regulatory network analysis, and drug prediction were performed.
Results: A total of 3811 DEGs were identified, including 1408 upregulated and 2403 downregulated genes; 16 overlapped with Th17-related genes. AUC was 1.000, with a 95% confidence interval of 1.000– 1.000. Machine learning and validation analyses indicated that TGFBR2 and IKBKB may serve as COPD-associated biomarkers, with both genes showing significant downregulation in COPD samples and RT-qPCR validation (p < 0.05). Immune infiltration analysis indicated significant differences in 11 immune cell types between COPD and control groups. TGFBR2 and IKBKB were negatively correlated with Th17 cell infiltration (TGFBR2: cor = − 0.502, p < 0.05; IKBKB: cor = − 0.466, p < 0.05). IKBKB was also negatively correlated with macrophages (cor = − 0.725, p < 0.001), while TGFBR2 was negatively correlated with natural killer T cells (cor = − 0.635, p < 0.001). GSEA suggested 78 enriched pathways, including 42 related to TGFBR2 and 36 related to IKBKB. Drug prediction suggested 92 potential agents.
Conclusion: This preliminary, hypothesis-generating study suggests that TGFBR2 and IKBKB may be associated with Th17-related immune alterations in COPD and may provide candidate markers for further mechanistic and therapeutic validation. Diagram of COPD research process involving Th17, biomarkers and candidate agents.The diagram illustrates a research process related to COPD. At the center is COPD, with arrows connecting to various components. The top left shows ’Th17’ with ’immune dysregulation.’ The bottom left features ’Transcriptomics’ and ’Machine learning.’ The bottom center displays ’Biomarkers’ with ’TGFBR2’ and ’IKBKB’ both showing downward arrows, linked to ’RT-qPCR.’ The bottom right depicts ’Th17-related alteration’ with connections to ’JAK-STAT,’ ’TLR,’ and ’NOD.’ The top right shows ’Candidate agents’ with ’future validation.’ Arrows indicate the flow between these components, suggesting a cycle of research and validation.
Keywords: chronic obstructive pulmonary disease, T helper 17 cells, biomarkers, gene set enrichment analysis, immune infiltration analysis
Introduction
Chronic obstructive pulmonary disease (chronic obstructive pulmonary disease, COPD) is the third leading cause of global mortality and a major chronic respiratory disorder characterized by persistent airflow limitation, progressive airway inflammation, and parenchymal lung destruction. It results in long-term respiratory impairment and imposes a substantial socioeconomic burden. In 2019, an estimated 212.3 million individuals were living with COPD worldwide, and the disease accounted for approximately 3.3 million deaths. The burden of COPD is disproportionately concentrated among older adults and individuals with chronic exposure to tobacco smoke, ambient air pollution, or occupational hazardous particulates.1 The core pathological features of chronic obstructive pulmonary disease (COPD) include persistent airflow limitation, chronic airway inflammation, and parenchymal lung destruction—collectively contributing to substantial reductions in health-related quality of life and decreased life expectancy.2 The pathogenesis of COPD involves multiple interrelated mechanisms, including oxidative stress, protease–antiprotease imbalance, airway epithelial injury, mucin hypersecretion, structural airway remodeling, and dysregulated immune responses.3,4 Owing to the marked heterogeneity of COPD, substantial interindividual variability is observed across key clinical domains, including inflammatory endotypes, the rate of forced expiratory volume in one second (FEV1) decline, susceptibility to acute exacerbations, and therapeutic responsiveness.5,6 Current clinical management of COPD primarily relies on bronchodilators, inhaled corticosteroids, and anti-inflammatory agents—interventions aimed at symptom alleviation and reduction of acute exacerbation frequency. Nevertheless, no disease-modifying therapies are yet available that can halt or reverse the progressive decline in forced expiratory volume in one second (FEV1) or meaningfully alter the underlying disease trajectory.6 Therefore, elucidating the immune-inflammatory molecular mechanisms underlying COPD pathogenesis and identifying biomarkers with diagnostic or patient-stratification utility are critical priorities for advancing our understanding of disease heterogeneity, improving risk prediction, and uncovering novel therapeutic targets.
Within the complex pathobiology of COPD, immune dysregulation—particularly aberrations in adaptive immunity—has garnered growing scientific attention in recent years. T helper 17 (Th17) cells constitute a distinct CD4⁺ T lymphocyte subset characterized by the production of signature cytokines, including interleukin-17A (IL-17A), IL-21, and IL-22, and are critically implicated in the orchestration of chronic airway inflammation, mucosal barrier integrity, and maladaptive immune responses.7 Unlike nonspecific inflammatory pathways that reflect generalized immune activation, Th17 cells are mechanistically linked to multiple core pathological processes in COPD. First, Th17-derived cytokines, including IL-17A, IL-21, and IL-22—drive neutrophil recruitment and activation, thereby amplifying airway inflammation. Second, Th17 signaling disrupts epithelial barrier homeostasis, promotes fibroblast activation and collagen deposition, induces mucin hypersecretion, and contributes to structural airway remodeling—collectively fostering the development of persistent airflow limitation and parenchymal lung destruction.8,9 Clinical evidence consistently indicates elevated frequencies of Th17 cells and increased concentrations of interleukin-17A (IL-17A) in peripheral blood or lung tissue specimens from individuals with COPD. These immunological alterations correlate significantly with disease severity, neutrophilic airway inflammation, lung function impairment and the frequency of acute exacerbation.8–11 Collectively, these findings indicate that Th17 cells are not merely epiphenomenal in COPD-associated inflammation but represent a central immunoregulatory node linking adaptive immune dysregulation, chronic neutrophilic airway inflammation, and structural airway remodeling.
From a mechanistic standpoint, Th17-associated inflammation contributes to COPD progression via multiple interrelated pathways. IL-17 stimulates airway epithelial cells, fibroblasts, and innate and adaptive immune cells to secrete chemokines and inflammatory mediators, thereby driving neutrophilic infiltration and sustaining chronic airway inflammation.8,9 Moreover, the IL-17-related pathways may also be involved in the processes of increased mucus secretion, airway smooth muscle alteration and tissue remodeling, thereby exacerbating airflow limitation and lung structural damage.9,11 Critically, oxidative stress, bacterial or viral infections, and cigarette smoke exposure in COPD activate pro-inflammatory signaling pathways, including NF-κB, Toll-like receptor (TLR), and JAK/STAT cascades, leading to the upregulation of key cytokines such as IL-6, IL-1β, and IL-23. These cytokines precisely the important upstream signals that induce Th17 differentiation and maintain Th17 effector function. Therefore, conducting a systematic screening around Th17-related genes not only helps to explain the molecular basis of abnormal Th17 responses in COPD, but also may provide a more focused research entry point for identifying immune inflammatory-related diagnostic markers and potential intervention targets.
Although the Th17/IL-17 axis has been postulated to be potentially involved in the chronic inflammation and acute exacerbation of COPD, translational research targeting this pathway still exhibits notable limitations at present. On one hand, existing studies have primarily focused on the proportion of Th17 cells, the expression level of IL-17, or the Th17/Treg imbalance itself, while systematic analyses are still lacking regarding the differential expression profile of Th17-related genes, key regulatory nodes, and their association with the immune microenvironment in the lung tissue of COPD patients.8–11 On the other hand, the clinical translation of therapeutic strategies targeting the IL-17 pathway alone in COPD remains immature, suggesting that focusing on a single cytokine may not be sufficient to explain the complex Th17-related immune network. Therefore, it is necessary to identify the core Th17-related genes at the transcriptome level and further integrate machine learning, immune infiltration, functional enrichment, regulatory networks, and drug prediction analysis to systematically evaluate their potential biological significance and translational value in COPD.
However, at least two key knowledge gaps still exist. First, which Th17-related genes have stable differential expression characteristics in COPD and can be used as potential diagnostic or disease stratification markers have not been fully screened and verified. Second, how these Th17-related candidate genes are associated with the immune cell infiltration patterns, inflammatory signaling pathways, post-transcriptional regulatory networks, and potential drug action networks in COPD have not been comprehensively evaluated. These deficiencies limit our understanding of the molecular basis of Th17-related immune disorders in COPD and also restrict the possibility of exploring precision treatment based on the Th17 pathway.
Based on the above background, this study utilized publicly available transcriptome data to first screen for differentially expressed genes between COPD and normal samples, and then intersected them with the Th17 cell-related gene set to obtain differentially expressed Th17-related candidate genes. Subsequently, random forest and Boruta machine learning algorithms were employed to screen for key biomarkers, and their expression was verified in the training set, validation set, and RT-qPCR experiments. On this basis, this study further constructed a COPD risk prediction model and systematically evaluated the biological significance of key genes from multiple aspects, including chromosomal localization, functional enrichment, immune cell infiltration, gene set enrichment analysis, TF-miRNA-mRNA regulatory networks, and potential drug prediction. Through this integrated analysis strategy, this study aims to initially clarify the potential role of Th17-related genes in the abnormal immune microenvironment of COPD and provide candidate basis for the screening of COPD molecular markers and subsequent mechanism verification.
Materials and Methods
Data Sources
All datasets analyzed were retrieved from the publicly accessible Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) repository. For model development, the GSE38974 dataset served as the training cohort, encompassing pulmonary tissue specimens from 23 COPD cases alongside 9 healthy controls.12 Independent validation was performed using the GSE94916 dataset, comprising 6 COPD patients and 6 controls. Additionally, 87 T helper 17 (Th17) cell-related genes (Th17-RGs) were compiled following established protocols,13 specifically derived from the Th17 cell differentiation pathway catalogued within the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The GSE38974 and GSE94916 datasets were analyzed separately and were not merged for differential expression analysis or model construction. Therefore, batch-effect correction across datasets was not performed. GSE38974 was used as the training cohort for DEG identification and biomarker screening, whereas GSE94916 was used only for independent expression validation of candidate biomarkers.
Identification of DEGs and Functional Enrichment Analysis
Differential expression analysis between COPD and control samples in the GSE38974 training cohort was performed using the “limma” package in R. Genes with |log2FC| > 0.5 and adjusted p-value < 0.05 were considered differentially expressed. The Benjamini–Hochberg false discovery rate method was used for multiple-testing correction. Visualization of DEG distribution patterns was accomplished via volcano plots and heatmaps, generated through “ggplot2” and “pheatmap” packages respectively. Expression profiles of the 10 genes exhibiting the most pronounced upregulation and downregulation were illustrated using the “ComplexHeatmap” package. The identified DEGs underwent intersection with Th17-RGs, yielding differentially expressed Th17-RGs (DETh17RGs) that constituted the candidate gene pool. Functional annotation of DETh17RGs was performed through Gene Ontology (GO) and KEGG pathway enrichment analyses, implemented via “clusterProfiler” alongside the human annotation database “org.Hs.egdb”, applying a statistical cutoff of p < 0.05. Protein-protein interaction (PPI) network construction for candidate genes was executed through the STRING platform, specifying “Homo sapiens” as target species with confidence score threshold at 0.4. Network architecture was subsequently visualized in Cytoscape following importation of TSV-formatted output files.
Screening of Biomarkers Using Machine Learning Approaches and Gene Expression Validation
To reduce the risk of relying on a single feature-selection method, RF and Boruta algorithms were both applied to the 16 DETh17RGs in the GSE38974 training cohort. For the RF model, ntree was tuned from 10 to 100 in increments of 2, and ntree = 12 was selected because it yielded the lowest out-of-bag error. Gene importance was ranked according to the Gini coefficient, and the top 10 genes were retained. The Boruta algorithm was then used for complementary feature selection. Genes overlapping between RF and Boruta were considered candidate biomarkers. Given the limited sample size, these machine learning results were interpreted as exploratory and required external validation.
Exploratory Construction and Performance Assessment of a Biomarker-Based Nomogram
To preliminarily assess the combined discriminatory pattern of the candidate biomarkers, an exploratory nomogram model was constructed using the R package “rms”, based on TGFBR2 and IKBKB in the GSE38974 training cohort. Model calibration was evaluated using calibration curves to examine the apparent agreement between nomogram-estimated probabilities and observed outcomes. In addition, receiver operating characteristic (ROC) analysis was performed using the “pROC” package to calculate the area under the curve (AUC) and its 95% confidence interval. Sensitivity, specificity, and accuracy were calculated using the “caret” package. Bootstrap resampling was performed 1000 times to estimate the stability of model performance metrics. Given the limited sample size, these model performance results were interpreted as exploratory rather than confirmatory.
Functional Annotation and Genomic Localization of Biomarkers
To elucidate the biological roles and regulatory architecture of identified biomarkers, the Gene Multiple Association Network Integration Algorithm database (http://www.genemania.org) was employed. This approach enabled identification of interacting gene partners and shared biological pathways, facilitating construction of comprehensive functional association networks that contextualize biomarker mechanisms. In addition, we also use RPackage “rcircos” to do chromosome location analysis for biomarkers, determine their actual position in the genome, and lay a good genetic background for the later mechanism research.
Immune Infiltration Analysis in COPD
To find out what is the difference between COPD samples and normal samples in terms of immune cell infiltration patterns, we use ssGSEA algorithm to calculate the infiltration levels of 28 kinds of immune cells in the samples. Then Wilcoxon rank-sum test is used to compare the difference of immune cell ratio between COPD group and control group, and the screening standard is set as p < 0.05, and the results can be displayed by heat map and box chart. In addition, we did Spearman correlation analysis with R package “psychology” to see if there is any correlation between those infiltrating immune cells with differences and between these cells and candidate biomarkers. The screening criteria are |correlation coefficient (cor)| > 0.3 and p < 0.05.
GSEA in COPD
To further explore the biological pathways potentially involved by the biomarkers in COPD, the expression data from all samples in the training set were utilized. The R package “psych” was employed to calculate the Spearman cor between each biomarker and genome-wide expression values. Following gene arrangement in descending sequence according to correlation coefficients, Gene Set Enrichment Analysis (GSEA) was executed through the “clusterProfiler” package in R, employing the KEGG gene set (c2.cp.kegg.v2023.1.Hs.symbols.gmt) as reference database. Pathway enrichment significance was determined by applying statistical thresholds: p-value below 0.05, absolute Normalized Enrichment Score (|NES|) exceeding 1, and False Discovery Rate (FDR) under 0.25. Visualization of the 10 pathways demonstrating strongest enrichment for each biomarker was accomplished using the “enrichplot” package.
Regulatory Network Construction and Therapeutic Drug Prediction
To elucidate the transcriptional and post-transcriptional regulatory mechanisms of COPD biomarkers, the TRRUST (https://www.grnpedia.org/trrust/) and miRWalk (http://mirwalk.umm.uni-heidelberg.de)databases were employed to predict transcription factors (TFs) and miRNAs targeting the candidate biomarkers. The overlapping predictions from both databases were selected for subsequent analysis. High-confidence TF-miRNA regulatory relationships were retrieved from the TransmiR database (http://www.cuilab.cn/transmir). A multilayer regulatory network encompassing TF-miRNA-mRNA interactions was assembled via Cytoscape platform by integrating transcription factors, microRNAs, and messenger RNA molecules. Additionally, therapeutic compound discovery targeting candidate biomarkers was pursued through the Drug Signature Database (DSigDB, https://www.dsigdb.org), enabling prediction of pharmacological agents demonstrating significant associations with identified biomarkers. A drug-gene interaction network was visualized using Cytoscape.
Real-Time Quantitative PCR (RT-qPCR) Analysis
Total RNA was isolated from ten frozen human whole-blood samples (five Normal and five COPD) using the TRIzol-chloroform method. Briefly, 1300μL of each sample was lysed on ice, followed by phase separation, isopropanol precipitation, and washes with 75% ethanol. RNA precipitate underwent desiccation before resuspension in nuclease-free water, with concentration determined via NanoPhotometer N50 spectrophotometry. Reverse transcription was conducted on 2μg total RNA utilizing the HP All-in-one qRT Master Mix II system (incubation at 50°C for 10 minutes, followed by 85°C for 5 seconds). Complementary DNA underwent dilution and subsequent amplification on the CFX96 real-time PCR platform, employing GAPDH as internal reference standard. The amplification mixture comprised 2× Universal Blue SYBR Green qPCR Master Mix, target-specific primers for IKBKB and TGFBR2 (detailed in Table 1), and cDNA template. Thermal cycling parameters consisted of initial denaturation (95°C, 1 minute) succeeded by 40 amplification cycles: denaturation at 95°C (20 seconds), annealing at 55°C (20 seconds), and extension at 72°C (30 seconds). All reactions were performed in technical replicates to ensure reproducibility.
 |
Table 1 Relevant Primer Sequences |
Statistical Analysis
Bioinformatics computations were performed using R software version 4.3.3. Comparisons between COPD and control groups were performed using the Wilcoxon rank-sum test where appropriate. For differential expression and enrichment analyses, the Benjamini–Hochberg false discovery rate method was used for multiple-testing correction, and adjusted p-value < 0.05 was considered statistically significant. For exploratory immune infiltration and correlation analyses, nominal p-values were interpreted cautiously together with biological plausibility and validation evidence. Spearman correlation analysis was used to assess associations between biomarkers and immune cell infiltration levels. Given the limited sample size, all model-based results were regarded as exploratory. Differential expression, enrichment, immune infiltration, and correlation analyses involved multiple comparisons; therefore, the Benjamini–Hochberg false discovery rate method was applied where appropriate to reduce the risk of false-positive findings. For exploratory analyses, results were interpreted with caution, particularly when nominal p values were used together with biological plausibility and validation evidence. For model performance assessment, AUC and 95% confidence intervals were calculated by ROC analysis, and sensitivity, specificity, and accuracy were estimated. Bootstrap resampling with 1000 iterations was used to evaluate the stability of these metrics. Because of the small sample size, model performance was interpreted cautiously.
Results
Identification of Significantly Differentially Expressed Th17-RGs Between COPD and Normal Groups
Transcriptomic profiling comparing COPD patients with healthy controls in the GSE38974 training cohort identified 3811 DEGs using the criteria of |log2FC| > 0.5 and adjusted p-value < 0.05. Among these DEGs, 1408 genes were upregulated and 2,403 genes were downregulated in COPD samples (Figure 1A and B). Intersecting these DEGs with Th17-related genes yielded 16 differentially expressed Th17-related genes (DETh17RGs), which were selected as candidate genes for further analysis (Figure 1C).
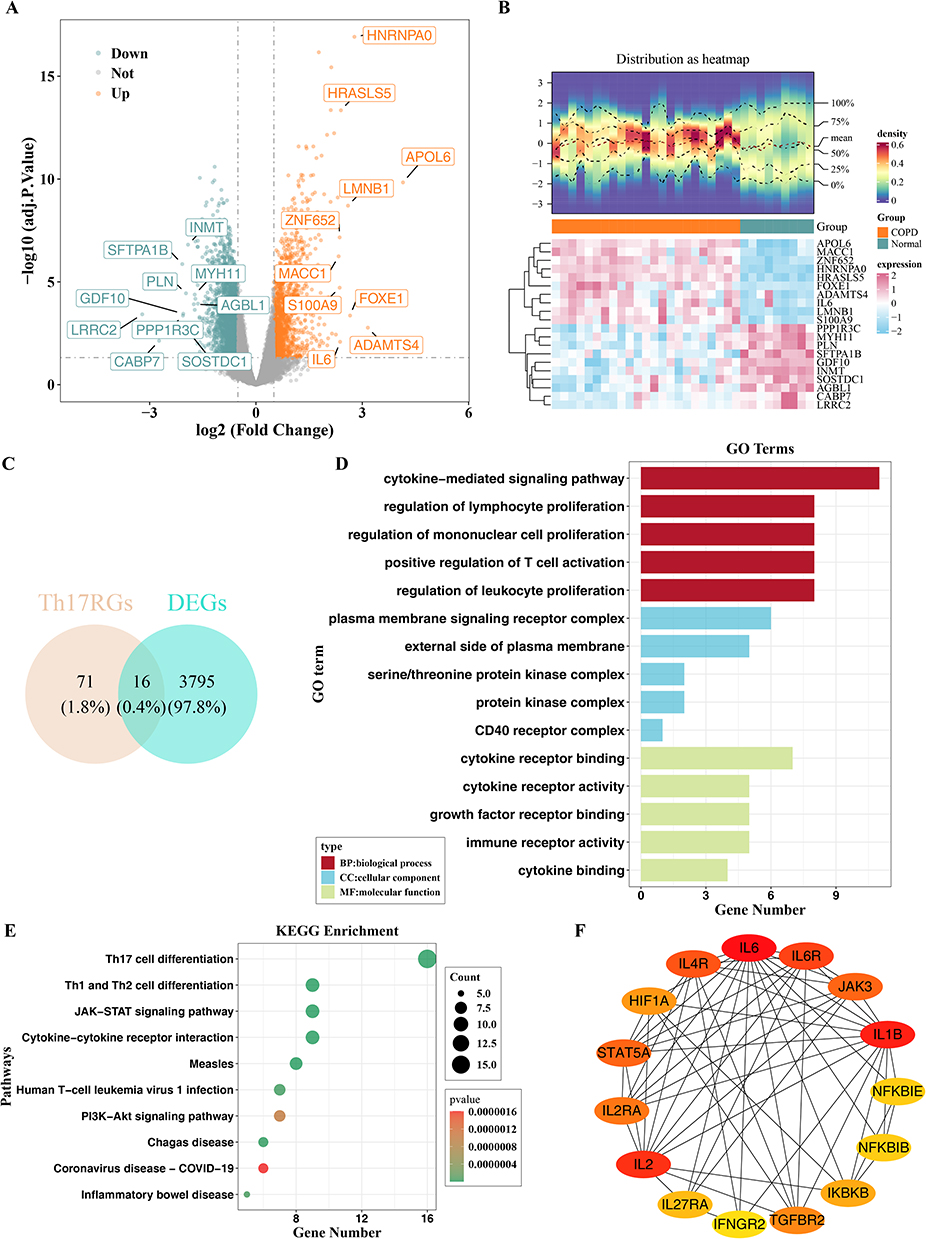 |
Figure 1 Screening of candidate genes and functional enrichment and PPI analysis of candidate genes. (A) Volcano plot of DEGs; (B) Heatmap showing the expression patterns of the top 10 DEGs (up- and down-regulated genes); (C) Venn diagram illustrating the overlap between DEGs and Th17-RGs. (D) GO enrichment terms of the 16 candidate genes. (E) KEGG pathways enriched for the 16 candidate genes. (F) PPI network of candidate genes. |
GO functional annotation of these 16 candidate genes using adjusted p-value < 0.05 revealed 746 significantly enriched terms, including 708 biological process (BP) terms, 5 cellular component (CC) terms, and 33 molecular function (MF) terms. The top five terms in each category are shown in Figure 1D. KEGG pathway enrichment analysis (p < 0.05) indicated that the 17 candidate genes were significantly enriched in 68 pathways, with the top 10 pathways—including Th17 cell differentiation, Th1 and Th2 cell differentiation—displayed in Figure 1E. Functional enrichment analysis shows that these genes are mainly involved in immune-related biological processes and pathways such as T cell differentiation and cytokine signaling, which shows that they may be may be involved in the pathogenesis of COPD. A PPI network was constructed for the 16 candidate genes using the STRING database and visualized in Cytoscape (Figure 1F). This analysis was used to display potential protein-protein interactions rather than to perform additional gene filtering. One isolated protein, AHR, was not displayed in the network.
Exploratory Machine Learning Screening Identified TGFBR2 and IKBKB as Candidate Biomarkers
During RF parameter optimization, ntree = 12 yielded the lowest OOB error and was therefore used for subsequent analysis (Figure 2A). According to the Gini coefficient, the top 10 genes were NFKBIB, IKBKB, IL2, IL4R, IFNGR2, HIF1A, TGFBR2, STAT5A, IL6R, and AHR (Figure 2B). The Boruta algorithm further confirmed 13 important genes: IL6, IL27RA, IL2RA, NFKBIE, AHR, IL2, IFNGR2, IL4R, HIF1A, NFKBIB, TGFBR2, STAT5A and IKBKB (Figure 2C). The overlap between the RF and Boruta algorithms yielded 9 candidate genes, including NFKBIB, IKBKB, IL2, IL4R, IFNGR2, HIF1A, TGFBR2, STAT5A, and AHR (Figure 2D).Among these candidates, IKBKB and TGFBR2 showed consistent expression trends and statistically significant differences between COPD and control samples in both the training and validation cohorts; therefore, they were retained as candidate biomarkers for subsequent exploratory analyses (Figure 2E and F).
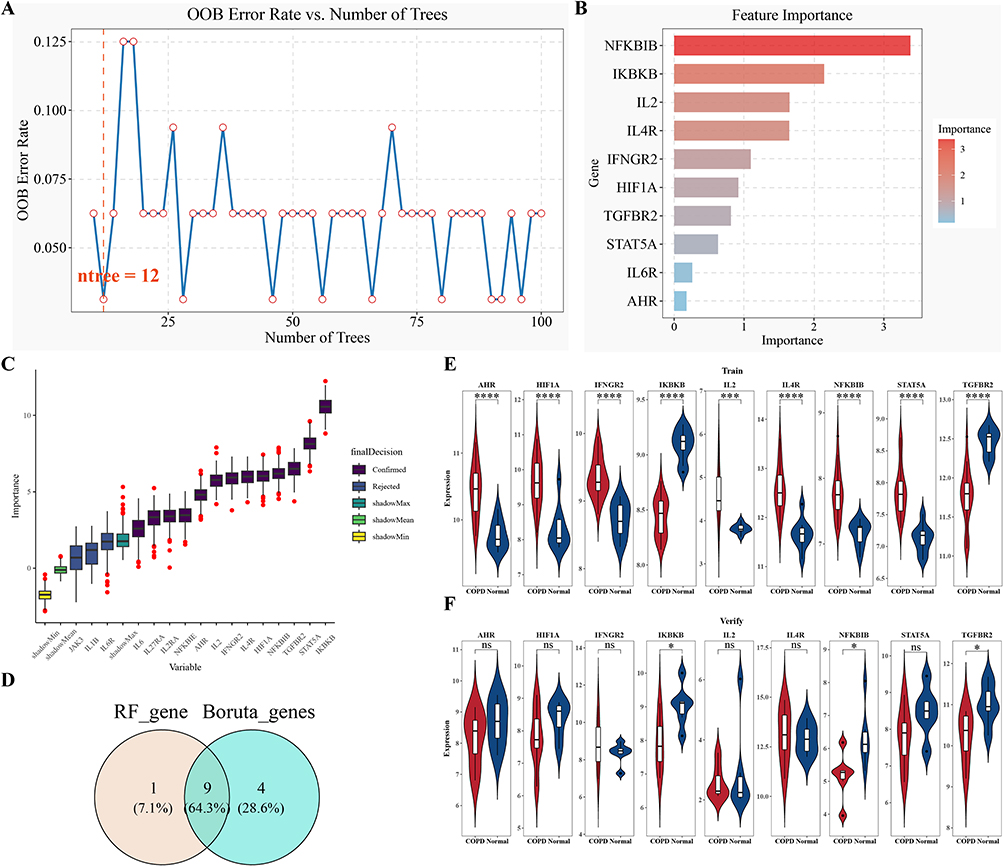 |
Figure 2 Exploratory machine learning-based screening of candidate biomarkers. (A) Relationship between OOB error rate and ntree in the RF model; (B) Importance ranking of the top 10 candidate genes in the RF model; (C) Variable selection plot generated by the Boruta algorithm; (D) Venn diagram showing the overlap between candidate genes identified by the RF and Boruta algorithms; (E and F) Expression levels of candidate biomarkers in the training set (E) and validation set (F). Statistical significance: *p < 0.05, ****p < 0.0001. |
Exploratory Construction of a Biomarker-Based Nomogram
An exploratory nomogram incorporating TGFBR2 and IKBKB was constructed in the GSE38974 training cohort to preliminarily estimate COPD-related risk (Figure 3A). The calibration curve showed an apparent agreement between predicted and observed probabilities in the training cohort (Figure 3B). To further describe the apparent model performance, ROC and bootstrap analyses were performed. After 1000 bootstrap resampling iterations, the mean AUC was 1.000, with a 95% confidence interval of 1.000–1.000. The mean sensitivity, specificity, and accuracy were 0.9998, 0.9993, and 0.9997, respectively. However, given the small sample size of the training cohort and the near-perfect performance estimates, these results should be interpreted cautiously because they may reflect model overfitting rather than stable clinical predictive performance. Therefore, this nomogram should be regarded as an exploratory model requiring validation in larger independent cohorts.
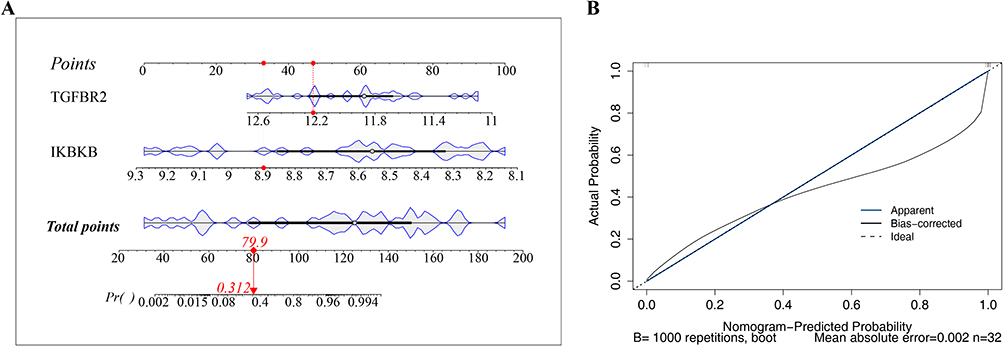 |
Figure 3 Exploratory nomogram and apparent model performance assessment. (A) Nomogram constructed using TGFBR2 and IKBKB to preliminarily estimate COPD-related classification probability in the training cohort. “Points” represents the score assigned to each candidate biomarker, “Total Points” represents the summed score, and “Risk” represents the estimated probability. The red dot and red arrow indicate an example of using the nomogram: a sample with total points of 79.9 corresponds to an estimated probability of 31.2%. (B) Calibration curve of the exploratory nomogram in the training cohort. The model requires further validation in larger independent cohorts. |
Mult-Omics Function Analysis and Genome Localization of TGF-Br2 and IKBKB in Cell Regulation
GeneMANIA analysis showed that TGFBR2 and IKBKB were involved in many biological processes, and their cell functions were showed broad functional associations (Figure 4A). Specifically, TGF-Br2 will physically interact with protein, such as CDC37 and STRAP, and may form a complex to regulate cell processes. It can be seen from the co-expression pattern that TGFBR2 and TGFBR3 will be expressed together under similar conditions, and they are likely to participate in the same biological function. The predicted interaction suggests that TGFBR2 may be related to protein such as NEDD8 and OSR1 in certain situations. The co-location relationship reflects the spatial correlation between TGFBR2 and TGFBR3 in cells. The genetic interaction shows that TGF-Br 2 and RHOA interact at the genetic level and may play a role in the same genetic pathway. Pathway correlation showed that TGFBR2 and TGFBRAP1 participated in overlapping signal pathways, which affected cell signal transduction. The sharing of protein domain indicates that TGFBR2 and TSSK1B are similar in structure, and may cooperate to function. IKBKB is similar, and it is related to many protein. These multi-level interactions can explain that TGFBR2 and IKBKB play a central role in regulating cell function. In addition, chromosome mapping analysis showed that TGFBR2 gene was on chromosome 3 and IKBKB gene was on chromosome 8 (Figure 4B). These two genes are not on the same chromosome, which also provides a structural basis for understanding their genome regulation mechanism and possible synergy in disease-related biological networks.
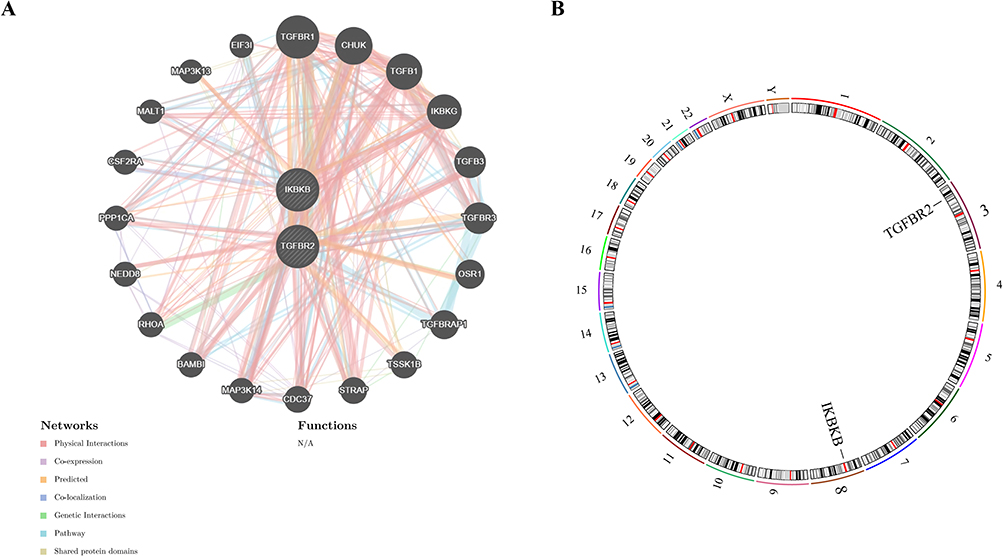 |
Figure 4 Gene MANIA and chromosomal localization analysis of biomarkers. (A) Gene MANIA interaction network of the biomarkers; (B) Chromosomal locations of the biomarkers. |
Altered Immune Cell Infiltration and Biomarker-Immune Correlations in the COPD Microenvironment
The results demonstrated significant differences in the infiltration levels of 11 immune cell types between COPD patients and the control group (p < 0.05), including activated dendritic cells, effector memory CD4⁺ T cells, gamma delta T cells, immature B cells, macrophages, myeloid-derived suppressor cells (MDSCs), memory B cells, natural killer cells, natural killer T cells, plasmacytoid dendritic cells, and Th17 cells (Figure 5A and B). These differentially infiltrated immune cells exhibited strong correlations with each other; for instance, MDSCs showed a significant positive correlation with activated dendritic cells (cor = 0.85, p < 0.05), and activated dendritic cells were significantly positively correlated with macrophages (cor = 0.86, p < 0.05) (Figure 5C). Furthermore, correlation analysis between biomarkers and immune cells (Figure 5D) revealed that TGFBR2 had the strongest positive correlation with natural killer cells (cor = 0.351, p < 0.05) and the strongest negative correlation with natural killer T cells (cor = −0.635, p < 0.001), while IKBKB showed a significant negative correlation with macrophages (cor = −0.725, p < 0.001). Notably, the expression levels of both TGFBR2 and IKBKB showed significant negative correlations with Th17 cell infiltration (TGFBR2: cor = −0.502, p < 0.05; IKBKB: cor = −0.466, p < 0.05). In summary, the immune microenvironment of COPD patients exhibits notable abnormalities in cellular infiltration and molecular-immune interactions, providing critical insights for elucidating the immune mechanisms underlying COPD.
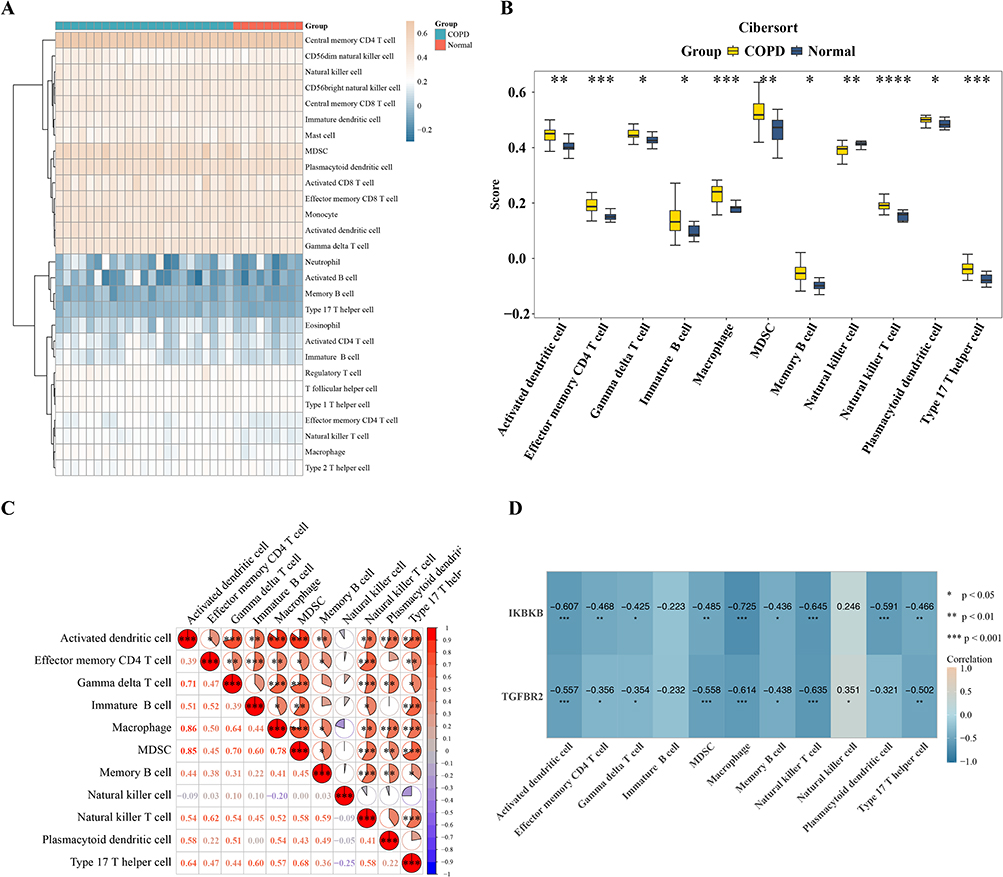 |
Figure 5 Immune infiltration analysis in COPD. (A) Heatmap showing the infiltration levels of immune cells in COPD and control samples.; (B) Differentially infiltrated immune cells between COPD and control groups; (C) Correlation analysis among differentially infiltrated immune cells. (D) Correlation analysis between biomarkers and immune cells. Cor represents Spearman correlation coefficient. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
GSEA Reveals Pathway Enrichment Profiles of TGFBR2 and IKBKB
GSEA of the two biomarkers identified a total of 78 significantly enriched pathways (p < 0.05, |NES|>1, FDR<0.25). Specifically, TGFBR2 was enriched in 42 pathways, including Cytokine-Cytokine Receptor Interaction, Toll-Like Receptor Signaling Pathway, and Jak-Stat Signaling Pathway. IKBKB was enriched in 36 pathways, such as Jak-Stat Signaling Pathway, Nod-Like Receptor Signaling Pathway, and Drug Metabolism-Cytochrome P450 (Figure 6A and B). These pathways suggest potential regulatory associations of TGFBR2 and IKBKB in COPD-related immune and inflammatory processes.
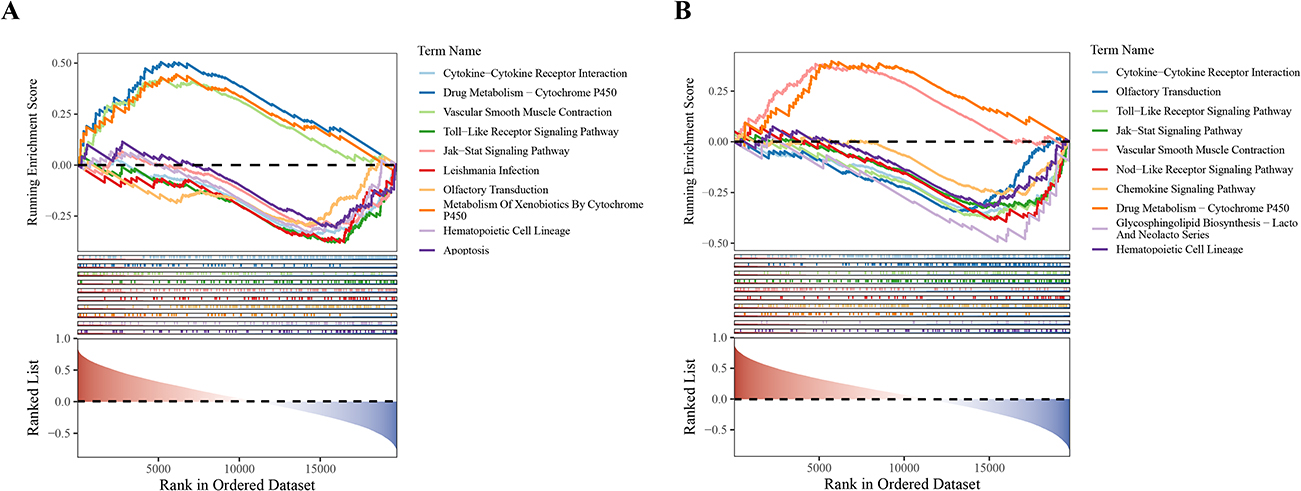 |
Figure 6 Results of GSEA. (A) Top 10 significantly enriched pathways of TGFBR2; (B) Top 10 significantly enriched pathways of IKBKB. |
Integrated Multi-Omics Analysis Reveals Regulatory Networks and Potential Therapeutic Agents for COPD
The constructed TF-miRNA-mRNA regulatory network consisted of 22 nodes and 21 edges (Figure 7A). Drug prediction identified 92 potential targeted agents, among which 50 target IKBKB, 28 target TGFBR2, and 14 agents can simultaneously act on both biomarkers. A further constructed drug-biomarker interaction network comprised 94 nodes and 106 edges (Figure 7B). In summary, this exploratory analysis constructed a multilayer regulatory network and identified predicted candidate compounds associated with TGFBR2 and IKBKB, providing preliminary resources for future mechanistic and pharmacological validation.
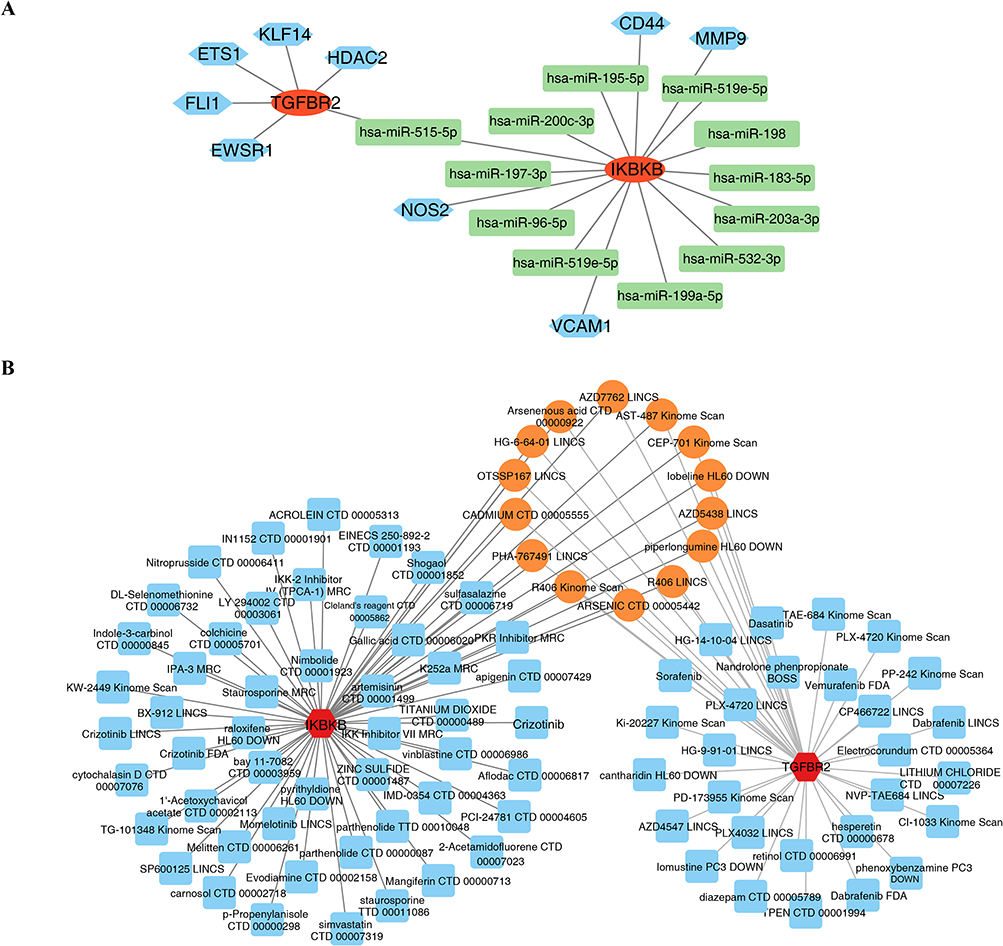 |
Figure 7 TF-miRNA-mRNA regulatory network and drug prediction analysis. (A) TF-miRNA-mRNA regulatory network; (B) Drug-biomarker interaction network. |
RT-qPCR
Quantitative RT-PCR examination demonstrated pronounced transcriptional suppression of both TGFBR2 and IKBKB within the COPD cohort relative to healthy controls, exhibiting statistical significance (p < 0.05) (Figure 8A and B). The concordance between these experimental findings and preceding computational predictions preliminarily supports the probable critical involvement of these genes in COPD pathophysiology.
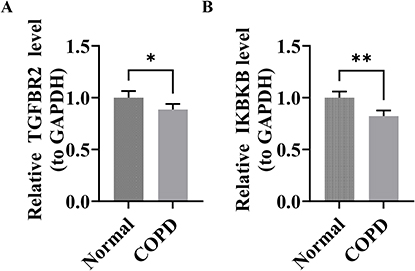 |
Figure 8 Results of the RT-qPCR analysis. (A and B) The mRNA expression levels of TGFBR2 and IKBKB. Statistical significance: *p < 0.05, **p < 0.01. |
Discussion
COPD is a chronic respiratory disease characterized by persistent airflow limitation, chronic airway inflammation and immune imbalance. Th17 cells and their related inflammatory pathways are believed to be involved in the chronic inflammatory response and immune microenvironment changes in COPD, but the related molecular network remains unclear. In this study, based on GEO transcriptome data, combined with differential expression analysis, machine learning screening, external dataset validation, immune infiltration analysis, GSEA, regulatory network prediction, drug prediction and preliminary RT-qPCR verification, two Th17 cell-associated candidate biomarkers, TGFBR2 and IKBKB, related to COPD were screened out. The results showed that both TGFBR2 and IKBKB were down-regulated in COPD samples and were significantly negatively correlated with Th17 cell infiltration; the correlation coefficient between TGFBR2 and Th17 cell infiltration was −0.502, and that between IKBKB and Th17 cell infiltration was −0.466. Additionally, immune infiltration analysis revealed 11 differences in immune cell infiltration between COPD and control samples, suggesting that COPD may be accompanied by complex immune microenvironment remodeling. The GSEA results further suggest that the genes related to TGFBR2 and IKBKB may be enriched in immune-inflammation-related pathways such as cytokine-cytokine receptor interaction, Toll-like receptor signaling pathway, Jak-Stat signaling pathway, and Nod-like receptor signaling pathway. It is important to note that the results of this study mainly indicate a potential association between TGFBR2 and IKBKB and immune abnormalities in COPD, but cannot directly prove their causal role in the occurrence and development of COPD.
TGFBR2 is an important receptor molecule in the TGF-β signaling axis. Previous studies have shown that TGF-β signaling is involved in various biological processes such as cell proliferation, differentiation, migration, apoptosis, and tissue fibrosis, and is closely related to airway remodeling and fibrotic changes in COPD.14–17 TGF-β signal transduction depends on the cooperative action of receptor complexes such as TGFBR1 and TGFBR2, so changes in TGFBR2 expression may reflect alterations in the TGF-β-related immune regulatory network. This study found that TGFBR2 was downregulated in COPD samples and negatively correlated with Th17 cell infiltration. This result suggests that the decreased expression of TGFBR2 may have a potential connection with the Th17-related immune imbalance in COPD. However, this correlation cannot be directly interpreted as the downregulation of TGFBR2 leading to an increase in Th17 cells. The downregulation of TGFBR2 may reflect the alteration of TGF-β-related immune regulatory capacity in the chronic inflammatory microenvironment of COPD, and this change may be associated with the disorder of Th17/Treg balance, inflammatory amplification, and airway remodeling.10,18,19
The GSEA results indicated that the TGFBR2-related genes were enriched in immune-related pathways such as cytokine-cytokine receptor interaction, Jak-Stat signaling pathway, and Toll-like receptor signaling pathway. Previous studies have suggested that TGF-β signaling plays a significant role in T cell differentiation and the regulation of Th17/Treg balance.18,19 Meanwhile, inflammatory factors such as IL-6 and IL-1β can affect the differentiation and function of Th17 cells through JAK/STAT-related signaling.20,21 In the chronic inflammatory environment of COPD, stimuli such as smoking, oxidative stress and infection can activate Toll-like receptor-related pathways, promoting the release of inflammatory factors such as TNF-α, IL-6 and IL-1β.22–24 In this context, the down-regulation of TGFBR2 may be related to the weakened TGF-β-mediated immune regulatory signal and may be involved in Th17-related immune abnormalities in conjunction with TLR/JAK-STAT-related inflammatory signals. However, as this study did not conduct gain-of-function or loss-of-function experiments on TGFBR2, nor did it directly detect the activity of TGF-β downstream signals, the explanation of this mechanism remains a hypothesis based on bioinformatics results and previous literature, and further experimental verification is needed.
IKBKB is an important kinase in the NF-κB canonical pathway, also known as IKKβ/IKK2, and plays a significant role in inflammatory responses, immune cell activation, and regulation of cell survival.25 Previous studies have shown that abnormal function of IKBKB can lead to immune deficiency or autoinflammatory diseases.26,27 In COPD and chronic inflammatory lung diseases, the NF-κB signaling pathway is generally considered to be involved in macrophage activation, release of inflammatory mediators, oxidative stress and airway structural change.28–30 This study found that IKBKB showed a down-regulated trend in COPD samples and was negatively correlated with Th17 cell infiltration. It should be particularly noted that the down-regulation of IKBKB expression and the activation of NF-κB pathway in COPD should not be simply understood as a direct consistency or direct causal relationship. As a key kinase in the classical NF-κB pathway, the reduced expression of IKBKB theoretically may weaken the NF-κB signaling capacity dependent on IKBKB to some extent; however, COPD is a long-term, complex and multi-cellular chronic inflammatory state. The activity of the NF-κB pathway is not only determined by the expression level of IKBKB mRNA, but also may be regulated at multiple levels, including upstream Toll-like receptor and Nod-like receptor signaling, stimulation by inflammatory factors, oxidative stress, protein phosphorylation, ubiquitination and p65 nuclear translocation.31–33
Therefore, the observed down-regulation of IKBKB in this study is more appropriately interpreted as a transcriptional alteration in the chronic inflammatory microenvironment of COPD, rather than being directly equivalent to the overall suppression of the NF-κB pathway. The decreased expression of IKBKB may reflect negative feedback regulation, adaptive inhibition, or transcriptional level changes related to alterations in cell composition following long-term inflammatory stimulation. Meanwhile, the persistent smoking stimulation, oxidative stress, TLR/NLR activation, and inflammatory factors such as IL-6 and IL-1β in COPD may still maintain NF-κB-related inflammatory responses through IKBKB-independent or partially compensatory mechanisms and promote Th17-related inflammatory responses through pathways such as JAK/STAT3.34,35 Based on this, the results of this study more cautiously suggest that the down-regulation of IKBKB may be associated with the imbalance of the NF-κB/JAK-STAT/Th17-related immune network in COPD, but it cannot be proved that the down-regulation of IKBKB directly drives NF-κB activation or directly leads to the expansion of Th17 cells. In the future, it is necessary to further clarify the relationship between the expression changes of IKBKB and the activity of NF-κB signaling by detecting the protein level, phosphorylation level, IκBα degradation, p65 nuclear translocation and the activity of the IL-6/STAT3 axis of IKBKB.
This study also conducted drug prediction analysis based on TGFBR2, IKBKB and their related pathways, and predicted 92 potential related drugs or compounds. Some of the predicted compounds include N-acetylcysteine, curcumin and quercetin, which are candidate molecules with antioxidant or anti-inflammatory effects. Oxidative stress is an important link in the pathological process of COPD, which can promote the release of inflammatory factors, airway damage and impairment of host defense function.36 Previous studies have suggested that ROS can promote the production of cytokines such as IL-6 and IL-1β through inflammatory signals like NF-κB and STAT3, and further affect the differentiation of Th17 cells and the balance of Th17/Treg.37,38 N-acetylcysteine, as an antioxidant and a precursor of glutathione, has been used in studies related to COPD or chronic bronchitis,39,40 curcumin and quercetin have also been reported to potentially influence COPD-related inflammatory responses through anti-inflammatory, antioxidant and multi-pathway regulatory effects.41,42 However, the drug results in this study are only derived from database predictions and cannot prove that these compounds can directly regulate TGFBR2 or IKBKB, nor can they prove that they can improve the clinical outcomes of COPD patients. Therefore, these drugs should be regarded as candidate molecules for future experimental validation rather than confirmed therapeutic drugs or directly translatable therapeutic strategies.
This study holds certain exploratory value. Firstly, starting from Th17 cell-related genes, this study combined machine learning and multi-dataset validation to screen out two COPD-related candidate molecules, TGFBR2 and IKBKB. Secondly, the immune infiltration analysis indicated that both TGFBR2 and IKBKB were negatively correlated with Th17 cell infiltration, providing new research clues for understanding Th17-related immune dysregulation in COPD. Thirdly, the results of GSEA, regulatory network, and drug prediction offer reference directions for subsequent mechanism experiments and candidate molecule verification. However, these results should be understood as hypothesis-generating evidence rather than mechanism conclusions confirmed by functional experiments.
This study also has several limitations. First, the research is mainly based on public transcriptome data and bioinformatics analysis. Although external datasets were verified and preliminary RT-qPCR validation was conducted, the sample size is still limited, especially as RT-qPCR only included 5 COPD and 5 control samples. The results still need to be verified in larger-scale, independent, and multi-center clinical cohorts. Second, the immune infiltration results are based on computational inference methods such as ssGSEA and cannot completely replace direct detection methods such as flow cytometry, immunohistochemistry, or single-cell sequencing. Third, this study only detected the mRNA expression levels of TGFBR2 and IKBKB, without detecting protein expression, protein activity, phosphorylation modifications, or the activation status of downstream signaling pathways. Therefore, it cannot directly infer the true functional status of the TGF-β or NF-κB pathways. Fourth, GSEA, TF-miRNA-mRNA networks, and drug predictions are all predictive analyses based on databases and algorithms and cannot prove direct regulatory relationships between molecules or the true therapeutic effects of drugs. Fifth, the relationship between the downregulation of IKBKB and the activation of the NF-κB pathway in COPD is complex and may be influenced by factors such as cell type, inflammatory stage, post-translational modifications, upstream receptor stimulation, and compensatory pathway activation. Further functional experiments are needed to clarify this. Therefore, the TGFBR2/IKBKB-Th17-related mechanism proposed in this study should be interpreted with caution, and its diagnostic value, mechanistic significance, and potential therapeutic value still need to be confirmed by further experimental and clinical studies.
In summary, this study suggests that TGFBR2 and IKBKB may be candidate molecules related to Th17-associated immune abnormalities in COPD. Both show a down-regulated trend in COPD samples and are negatively correlated with Th17 cell infiltration, indicating their possible involvement in the alteration of the COPD immune microenvironment. GSEA and regulatory network analysis further suggest that TGFBR2 and IKBKB may be associated with inflammatory and immune pathways such as TGF-β, Toll-like receptor, NF-κB, JAK-STAT, and cytokine-related pathways. However, the results of this study are mainly exploratory and hypothesis-generating findings and cannot directly prove causal mechanisms or directly support their use as confirmed diagnostic markers or therapeutic targets. Future studies still need to combine larger clinical samples, protein-level verification, cell and animal functional experiments, as well as prospective clinical research, to further confirm the specific roles of TGFBR2 and IKBKB in the immune regulation of COPD and their potential research value.
Novelty and Interpretation of Findings
The novelty of this study should be interpreted with caution. Previous research has established the association between Th17-related inflammation, dysregulation of the TGF-β signaling pathway and the NF-κB pathway, and chronic obstructive pulmonary disease (COPD). Therefore, the current findings do not reveal a completely new disease mechanism. The main contribution of this study lies in the integrated analysis of Th17-related genes, machine learning-based candidate gene screening, immune cell infiltration, pathway enrichment analysis, regulatory network construction, drug prediction, and preliminary RT-qPCR validation results. These comprehensive analyses suggest that TGFBR2 and IKBKB may be candidate molecules related to Th17-related immune alterations in COPD. However, due to the small size of the validation dataset and RT-qPCR cohort, and the potential impact of overfitting on model performance, the above findings should be regarded as preliminary conclusions and are only of a hypothetical nature.
Limitations
This study has several limitations. Firstly, the sample size of this study is relatively small: the training cohort GSE38974 contains 23 COPD samples and 9 control samples, the validation cohort GSE94916 contains 6 COPD samples and 6 control samples, and the RT-qPCR validation only used 5 COPD samples and 5 control samples. Therefore, the statistical power and generalizability of the research results may be limited. Secondly, GSE38974 and GSE94916 were analyzed as independent datasets rather than combined for integrated analysis, so no cross-dataset batch effect correction was performed. Future studies integrating multiple cohorts should adopt appropriate batch effect correction methods such as ComBat or Harmony. Thirdly, although the revised differential expression analysis used an adjusted p-value < 0.05 to reduce false positive results, some downstream exploratory analyses still relied on nominal statistical thresholds, which should be interpreted with caution. Fourthly, RF, Boruta, and nomogram analyses were all conducted in a relatively small training cohort, which may increase the risk of overfitting. Although the candidate biomarkers have been further evaluated in an independent validation cohort and preliminary RT-qPCR, larger-scale multicenter cohorts are still needed to verify their robustness. Fifthly, detailed clinical variables including age, gender, smoking history, lung function parameters, GOLD stage, history of acute exacerbation, and medication information were insufficient for systematic adjustment or subgroup analysis. Therefore, the relationship between TGFBR2/IKBKB expression and the clinical phenotype of chronic obstructive pulmonary disease (COPD) remains unclear. These findings should be regarded as preliminary conclusions and can only provide a basis for hypotheses for subsequent studies.
Although the performance evaluation based on the bootstrap method showed high apparent AUC, sensitivity, specificity and accuracy, these nearly perfect values should not be regarded as evidence of the model’s clinical applicability, as they may result from a small sample size and overfitting. A larger independent cohort is needed to assess the model’s generalization ability and clinical utility.
Conclusion
This exploratory study suggests that TGFBR2 and IKBKB may be Th17 cell-associated candidate biomarkers in COPD. Integrated bioinformatics analysis, machine learning, immune infiltration assessment, GSEA, regulatory network construction, drug prediction, and preliminary RT-qPCR verification indicated that these genes might be associated with immune microenvironment alterations, Th17-related inflammatory regulation, and COPD-related molecular pathways. The negative correlations between TGFBR2/IKBKB expression and Th17 cell infiltration may provide hypothesis-generating evidence for further investigation of immune dysregulation in COPD. In addition, the predicted compounds related to these biomarkers may represent potential candidates requiring further experimental and clinical validation rather than established therapeutic targets. Given the exploratory nature of this study, these findings should be interpreted cautiously and require confirmation in larger clinical cohorts, mechanistic experiments, and prospective validation studies before their diagnostic or therapeutic relevance can be determined.
Data Sharing Statement
The datasets analyzed in this study, including GSE38974 and GSE94916, are publicly available in the Gene Expression Omnibus (GEO) repository43 (https://www.ncbi.nlm.nih.gov/geo/). The GSE38974 dataset was used as the training cohort for DEG identification, and the GSE94916 dataset was used as the validation cohort for gene expression verification.
Ethics Approval and Consent to Participate
This study was performed in line with the principles of the Declaration of Helsinki and approved by the Ethics Committee of the Ethics Committee of Army 947th Hospital. The approval number and date of approval are as follows: [2025RR0804] and [2025-08-22]. All patients provided written informed consent at the time of clinical sample collection for experiments, ensuring that the research process complied with ethical norms and that patients’ rights and wishes were fully respected.
Acknowledgments
We would like to express our sincere gratitude to all individuals and organizations who supported and assisted us throughout this research. We extend our sincere gratitude to Ms. Zhang Suxin for her valuable contributions to the machine learning model validation and advanced bioinformatics analyses.In conclusion, we extend our thanks to everyone who has supported and assisted us along the way. Without your support, this research would not have been possible.The research reported in this project was generously supported by Open research grant from the institute of etiology and prevention of metabolic diseases in Pamir Plateau Area, Kashi University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Open research grant from the institute of etiology and prevention of metabolic diseases in Pamir Plateau Area, Kashi University (project number: YXY2025YB04).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Safiri S, Carson-Chahhoud K, Noori M, et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: results from the global burden of disease study 2019. BMJ. 2022;378:e069679. doi:10.1136/bmj-2021-069679
2. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–19. doi:10.1016/S0140-6736(22)00470-6
3. Xu J, Zeng Q, Li S, Su Q, Fan H. Inflammation mechanism and research progress of COPD. Front Immunol. 2024;15:1404615. doi:10.3389/fimmu.2024.1404615
4. Wang S, Zhong M, Deng X, et al. Based exploration of the diagnostic value of oxidative stress-related key genes in chronic obstructive pulmonary disease. Cell Biol Toxicol. 2025;41(1):69. doi:10.1007/s10565-025-10019-5
5. Corlateanu A, Mendez Y, Wang Y, Garnica RJA, Botnaru V, Siafakas N. Chronic obstructive pulmonary disease and phenotypes: a state-of-the-art. Pulmonology. 2020;26(2):95–100. doi:10.1016/j.pulmoe.2019.10.006
6. Rabin AS, Keyes CM, Oberg CL, Folch EE. Emerging interventional pulmonary therapies for chronic obstructive pulmonary disease. J Thorac Imaging. 2019;34(4):248–257. doi:10.1097/RTI.0000000000000424
7. Agalioti T, Cortesi F, Gagliani N. T(H)17 cell immune adaptation. Curr Opin Immunol. 2023;83:102333. doi:10.1016/j.coi.2023.102333
8. Ma R, Su H, Jiao K, Liu J. Role of Th17 cells, Treg cells, and Th17/Treg imbalance in immune homeostasis disorders in patients with chronic obstructive pulmonary disease. Immun Inflamm Dis. 2023;11(2):e784. doi:10.1002/iid3.784
9. Yu Y, Zhao L, Xie Y, et al. Th1/Th17 cytokine profiles are associated with disease severity and exacerbation frequency in COPD patients. Int J Chron Obstruct Pulmon Dis. 2020;15:1287–1299. doi:10.2147/COPD.S252097
10. Zhang JC, Chen G, Chen L, et al. TGF-β/BAMBI pathway dysfunction contributes to peripheral Th17/Treg imbalance in chronic obstructive pulmonary disease. Sci Rep. 2016;6:31911. doi:10.1038/srep31911
11. Mardi A, Abdolmohammadi-Vahid S, Sadeghi SA, et al. Nanocurcumin modulates Th17 cell responses in moderate and severe COPD patients. Heliyon. 2024;10(9):e30025. doi:10.1016/j.heliyon.2024.e30025
12. Ezzie ME, Crawford M, Cho JH, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67(2):122–131. doi:10.1136/thoraxjnl-2011-200089
13. Chen S, Wei P, Wang G, Wu F, Zou J. Construction of a prognostic signature based on T-helper 17 cells differentiation-related genes for predicting survival and tumor microenvironment in head and neck squamous cell carcinoma. Medicine. 2025;104(4):e41273. doi:10.1097/MD.0000000000041273
14. Stewart AG, Thomas B, Koff J. TGF-β: master regulator of inflammation and fibrosis. Respirology. 2018;23(12):1096–1097. doi:10.1111/resp.13415
15. Kraik K, Tota M, Laska J, et al. The Role of transforming growth factor-β (TGF-β) in asthma and Chronic Obstructive Pulmonary Disease (COPD). Cells. 2024;13(15):1271. doi:10.3390/cells13151271
16. Lv MY, Jin LL, Sang XQ, et al. Abhd2, a candidate gene regulating airway remodeling in COPD via TGF-β. Int J Chron Obstruct Pulmon Dis. 2024;19:33–50. doi:10.2147/COPD.S440200
17. Kilinc M, Demir I, Aydemir S, Gul R, Dokuyucu R. Elevated urotensin-II and TGF-β levels in COPD: biomarkers of fibrosis and airway remodeling in smokers. Medicina. 2024;60(11):1750. doi:10.3390/medicina60111750
18. Chen W. TGF-β regulation of T cells. Annu Rev Immunol. 2023;41:483–512. doi:10.1146/annurev-immunol-101921-045939
19. Wang J, Zhao X, Wan YY. Intricacies of TGF-β signaling in Treg and Th17 cell biology. Cell Mol Immunol. 2023;20(9):1002–1022. doi:10.1038/s41423-023-01036-7
20. Cerboni S, Gehrmann U, Preite S, Mitra S. Cytokine-regulated Th17 plasticity in human health and diseases. Immunology. 2021;163(1):3–18. doi:10.1111/imm.13280
21. Kung SP, Umbreen H, Wang JH, Tsia CM, Lin TC, Chen YT. Cucurbitacin B inhibits Th17 cell differentiation via the suppression of the JAK/STAT pathway and alleviates collagen-induced arthritis in mice. Int J Immunopathol Pharmacol. 2025;39:3946320251348715. doi:10.1177/03946320251348715
22. Dhamodharan P, Arumugam M. Multiple gene expression dataset analysis reveals toll-like receptor signaling pathway is strongly associated with chronic obstructive pulmonary disease pathogenesis. Copd. 2020;17(6):684–698. doi:10.1080/15412555.2020.1793314
23. Sidletskaya K, Vitkina T, Denisenko Y. The role of toll-like receptors 2 and 4 in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2020;15:1481–1493. doi:10.2147/COPD.S249131
24. Haw TJ, Starkey MR, Pavlidis S, et al. Toll-like receptor 2 and 4 have opposing roles in the pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2018;314(2):L298–l317. doi:10.1152/ajplung.00154.2017
25. Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)--a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008;19(2):157–165. doi:10.1016/j.cytogfr.2008.01.006
26. Cardinez C, Miraghazadeh B, Tanita K, et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J Exp Med. 2018;215(11):2715–2724. doi:10.1084/jem.20180639
27. Yu D, Liu X, Zhang G, Ming Z, Wang T. Isoliquiritigenin inhibits cigarette smoke-induced COPD by attenuating inflammation and oxidative stress via the regulation of the Nrf2 and NF-κB signaling pathways. Front Pharmacol. 2018;9:1001. doi:10.3389/fphar.2018.01001
28. Sacco K, Kuehn HS, Kawai T, et al. A heterozygous gain-of-function variant in IKBKB associated with autoimmunity and autoinflammation. J Clin Immunol. 2023;43(2):512–520. doi:10.1007/s10875-022-01395-2
29. Alanazi FJ, Alruwaili AN, Aldhafeeri NA, et al. Pathological interplay of NF-κB and M1 macrophages in chronic inflammatory lung diseases. Pathol Res Pract. 2025;269:155903. doi:10.1016/j.prp.2025.155903
30. Dang X, He B, Ning Q, et al. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-κB pathways. Respir Res. 2020;21(1):95. doi:10.1186/s12931-020-01358-4
31. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi:10.1016/j.molmed.2007.09.002
32. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: a review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. 2019;670:4–14. doi:10.1016/j.abb.2019.02.008
33. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
34. Wang H, Yu L, Cheng L, Guo Z. The roles of lncRNAs in Th17-associated diseases, with special focus on JAK/STAT signaling pathway. Clin Exp Med. 2023;23(7):3349–3359. doi:10.1007/s10238-023-01181-3
35. Ye M, Deng G, Liu Q, et al. SO(2) activates Th17 cells through the JAK1,2/STAT3 signaling pathway. Int Immunopharmacol. 2024;143(Pt 1):113263. doi:10.1016/j.intimp.2024.113263
36. Papi A, Alfano F, Bigoni T, et al. N-acetylcysteine treatment in Chronic Obstructive Pulmonary Disease (COPD) and chronic bronchitis/pre-COPD: distinct meta-analyses. Arch Bronconeumol. 2024;60(5):269–278. doi:10.1016/j.arbres.2024.03.010
37. Whitley SK, Balasubramani A, Zindl CL, et al. IL-1R signaling promotes STAT3 and NF-κB factor recruitment to distal cis-regulatory elements that regulate Il17a/f transcription. J Biol Chem. 2018;293(41):15790–15800. doi:10.1074/jbc.RA118.002721
38. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50(4):1602434. doi:10.1183/13993003.02434-2016
39. Cazzola M, Calzetta L, Page C, et al. Influence of N-acetylcysteine on chronic bronchitis or COPD exacerbations: a meta-analysis. Eur Respir Rev. 2015;24(137):451–461. doi:10.1183/16000617.00002215
40. Chen J, Cheng Y, Cui H, Li S, Duan L, Jiao Z. N‑acetyl‑L‑cysteine protects rat lungs and RLE‑6TN cells from cigarette smoke‑induced oxidative stress. Mol Med Rep. 2025;31(4):1–10. doi:10.3892/mmr.2025.13462
41. Safari S, Davoodi P, Soltani A, et al. Curcumin effects on chronic obstructive pulmonary disease: a systematic review. Health Sci Rep. 2023;6(3):e1145. doi:10.1002/hsr2.1145
42. Wu D, Dong Y, Zhang D, Wang T, Ye H, Zhang W. Efficacy and safety of dietary polyphenol supplements for COPD: a systematic review and meta-analysis. Front Immunol. 2025;16:1617694. doi:10.3389/fimmu.2025.1617694
43. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Neutrophil-Related Gene Expression Signatures in Idiopathic Pulmonary Fibrosis: Implications for Disease Characteristic and Identification of Diagnostic Hub Genes
Lin Y, Lai X, Lei T, Qiu Y, Deng Q, Liu Q, Wang Z, Huang W
Journal of Inflammation Research 2023, 16:2503-2519
Published Date: 14 June 2023
Pyroptosis-Related Genes as Diagnostic Markers in Chronic Obstructive Pulmonary Disease and Its Correlation with Immune Infiltration
Shu HM, Lin CQ, He B, Wang W, Wang L, Wu T, He HJ, Wang HJ, Zhou HP, Ding GZ
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:1491-1513
Published Date: 27 June 2024
Identification of Oxidative Stress-Associated Biomarkers in Chronic Obstructive Pulmonary Disease: An Integrated Bioinformatics Analysis
Jiang X, Wang M, Li H, Liu Y, Dong X
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:841-855
Published Date: 26 March 2025
Integrating Bioinformatics Analysis with RT-qPCR Experimental Validation to Investigate Immune Cell and Telomere-Related Biomarkers in Chronic Obstructive Pulmonary Disease
Wang S, Tang W, Yang H
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:3839-3854
Published Date: 28 November 2025
A Maltese Study in Determining the Presence of Chronic Obstructive Pulmonary Disease in Metabolic Syndrome
Gauci J, Gauci Pullicino S, Caruana E, Petroni Magri V, Formosa MM, Fenech AG, Fava S, Montefort S, Fsadni P
International Journal of Chronic Obstructive Pulmonary Disease 2026, 21:608737
Published Date: 11 June 2026