Back to Journals » Drug Design, Development and Therapy » Volume 16
Identification of Drug Combination Therapies for SARS-CoV-2: A Molecular Dynamics Simulations Approach
Authors Abdel-Halim H ![]() , Hajar M, Hasouneh L, Abdelmalek SMA
, Hajar M, Hasouneh L, Abdelmalek SMA ![]()
Received 14 March 2022
Accepted for publication 31 August 2022
Published 9 September 2022 Volume 2022:16 Pages 2995—3013
DOI https://doi.org/10.2147/DDDT.S366423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Georgios Panos
Heba Abdel-Halim,1,*,† Malak Hajar1,†, Luma Hasouneh1,†, Suzanne MA Abdelmalek2,†
1Department of Medicinal Chemistry, Faculty of Pharmacy and Medical Sciences, University of Petra, Amman, Jordan; 2Department of Pharmacology and Biomedical Sciences, Faculty of Pharmacy and Medical Sciences, University of Petra, Amman, Jordan
†Dr Abdel-Halim passed away on July 14, 2022
*These authors contributed equally to this work
Correspondence: Suzanne MA Abdelmalek, Department of Pharmacology and Biomedical Sciences, Faculty of Pharmacy and Medical Sciences, University of Petra, P.O. Box 961343, Amman, 11196, Jordan, Tel +962-6-5799555 Ext. (8310), Fax +962-65715570, Email [email protected]
Purpose: The development of effective treatments for coronavirus infectious disease 19 (COVID-19) caused by SARS-Coronavirus-2 was hindered by the little data available about this virus at the start of the pandemic. Drug repurposing provides a good strategy to explore approved drugs’ possible SARS-CoV-2 antiviral activity. Moreover, drug synergism is essential in antiviral treatment due to improved efficacy and reduced toxicity. In this work, we studied the effect of approved and investigational drugs on one of SARS-CoV-2 essential proteins, the main protease (Mpro), in search of antiviral treatments and/or drug combinations.
Methods: Different possible druggable sites of Mpro were identified and screened against an in-house library of more than 4000 chemical compounds. Molecular dynamics simulations were carried out to explore conformational changes induced by different ligands’ binding. Subsequently, the inhibitory effect of the identified compounds and the suggested drug combinations on the Mpro were established using a 3CL protease (SARS-CoV-2) assay kit.
Results: Three potential inhibitors in three different binding sites were identified; favipiravir, cefixime, and carvedilol. Molecular dynamics simulations predicted the synergistic effect of two drug combinations: favipiravir/cefixime, and favipiravir/carvedilol. The in vitro inhibitory effect of the predicted drug combinations was established on this enzyme.
Conclusion: In this work, we could study one of the promising SARS-CoV-2 viral protein targets in searching for treatments for COVID-19. The inhibitory effect of several drugs on Mpro was established in silico and in vitro assays. Molecular dynamics simulations showed promising results in predicting the synergistic effect of drug combinations.
Keywords: molecular dynamics simulations, ligand docking, multiple binding sites, drug synergy, SARS-CoV-2
Graphical Abstract:

Plain Language Summary
The outbreak of COVID-19 was declared a pandemic by the World Health Organization in March 2020. Almost 520 million cases and more than 6 million deaths were reported. Many drugs that have been used since the outbreak’s start were proven futile in compacting the infection. Recently, a newly developed drug was approved for clinical use in severe or critical COVID-19 conditions. However, more drugs are still needed to reduce the impact of this pandemic. Due to the limited time and the urgent need for a treatment, we proposed an in silico approach to explore the effect of approved drugs on one of the important virus targets, the main protease (Mpro). In this work, we have established the inhibitory effect of three drugs and two drug combinations on this enzyme.
Introduction
COVID-19 is caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2).1 With more than 520 million people infected and around 6 million reported deaths, COVID-19 represents a serious menace to the world’s public health and economies.2 Many of the treatment approaches that have been employed since the beginning of the pandemic were proven ineffective in targeting the virus.3–5 Therefore, there is an imminent need to identify drugs that can be effectively used to treat COVID-19.
Coronaviruses are enveloped single-stranded positive-sense RNA viruses that belong to the family Coronaviridae. They are widely distributed in humans and other mammals.6 The SARS-CoV has a large genome (comprised of ≈29,700 nucleotides) that encodes structural and non-structural proteins.7 The virus replication and transcription occurs at cytoplasmic membranes and involves RNA synthesis mediated by the viral replicase gene products. The replicase gene of SARS-CoV-2 encodes two overlapping polyproteins–pp1a and pp1ab–that are required for viral replication and transcription.8 Extensive proteolytic processing of these polypeptides takes place by the activity of the 33.8-kDa Mpro protease (also known as 3C-like protease), which digests the polyproteins at a minimum of 11 conserved sites. The functional importance of Mpro in the viral life cycle, combined with the absence of closely related homologs in humans, identify Mpro as an attractive target for the design of antiviral drugs.9 Other proteins that have been implicated in the virus pathogenesis were identified, for example, the spike glycoprotein, which is responsible for viral entry, through binding to the angiotensin-converting enzyme 2 (ACE2) receptor that is broadly expressed in human tissues.10
Previous efforts aimed to develop anti-SARS-CoV-2 treatments were based on designing drugs that would interfere with the viral life cycle, and these included: 1) inhibiting viral entry to the cell by blocking either the viral spike protein or its molecular human target ACE2; 2) inhibiting viral proteases, and 3) preventing viral replication through inhibition of viral RNA dependant RNA polymerase (RdRp).11,12 Most of these studies depended on drug repurposing strategy to identify new therapeutics from already approved drugs, thus accelerating the drug discovery process and offering the advantage of immediate use of drugs that have already demonstrated safety.13 Since de novo identification of drugs is a costly and lengthy process, research groups worldwide have established the effect of approved drugs on this virus. One study showed that emodin, omipalisib, and tipifarnib had an RdRp inhibitory effect.11 Other studies demonstrated the possible binding of cytarabin, raltitrexed, tenofovir, cidofovir, lamivudine, and fludarabine to the viral spike protein.14,15 Clinically, antiviral drugs, such as ritonavir and ribavirin, were used in the treatment of COVID-19.16–19 Recently, the FDA approved a newly developed antiviral drug combination (paxlovid) for use in severe or critical cases.20 However, more drugs are still needed to reduce the impact of this pandemic.
Since the start of the pandemic, many three-dimensional (3D) crystal structures have been reported for Mpro in its dimer form,21–23 which provided a solid basis for the search of potential inhibitors for this enzyme’s action. One of the crystal structures reported for Mpro, was found to be crystalized with an inhibitor bound within its binding site,22 while the second set of crystal structures were reported in the bound and unbound forms.21 Using the available crystal structures, many research groups were able to identify potential Mpro inhibitors from known drugs to be repurposed for use as COVID-19 therapy.24–27 Although many covalent and noncovalent inhibitors have been designed to inhibit this enzyme, none have been FDA approved.28
Combination therapies are more effective than single drugs for many diseases,29,30 including COVID-19.31 Once established, synergistic combinations achieve more potent therapeutic effects, decreasing the dose used and thus a reducing observed side effects.32,33 Synergistic drug combination is particularly useful in drug repurposing as many of the identified active compounds show weak activities and do not give the required effect if used alone.34 Despite their benefits, finding drug combinations is mainly dependent on a trial and error process that requires high-throughput testing of the various drug combinations on each of the known drug targets.35
Various in silico approaches have provided promising tools to establish the anti-SARS-CoV-2 activity of different compounds, including molecular docking, molecular docking with follow-up molecular dynamics simulations, structure-guided machine learning, network-based, and hybrid methods.21,22,27 These studies mainly depended on the availability of crystal structures of potential macromolecule targets co-crystallized with the natural substrate and/or a drug.36
The computational approach followed in this project is shown in Figure 1. Herein, we report a high-throughput screening in which the authors used a systematic in silico approach to identify all possible druggable sites within the Mpro enzyme cavities. Docking studies coupled with the molecular mechanics/generalized Born surface area (MM/GBSA) were subsequently used to assess the binding of 4000 approved and investigational drugs in each identified binding site. Three sites showed good binding affinities between the screened drugs and the enzyme. Consequently, complexes of the identified drugs, bound in their respective Mpro binding sites, were analyzed using molecular dynamics (MD) simulations to determine the effect of each drug’s binding on the overall enzyme conformation and hence determine the possible impact on allosteric binding of other drugs. Subsequently, the inhibitory activity and the synergistic effect of the combinations of the drug candidates were established through in vitro testing.
 |
Figure 1 Workflow of the work implemented in this research. |
Materials and Methods
Binding Sites Identification
“SiteMap” module from Schrödinger37 was used to identify potential ligand binding sites within the single subunit and at the interface between the two subunits of Mpro. Potential druggable sites were identified by searching for regions on or near the protein surface suitable for ligand binding. Any point that overlaps with protein atoms is insufficiently enclosed by the protein, or has a small van der Waals interaction energy with the protein is removed, and each grid of points is then grouped into a site. Site contour maps that show the hydrophobic and hydrophilic regions of the binding site are then generated. All sites were scored based on their enclosure, size, and hydrophilicity and ordered according to the “SiteScore” scoring function.37 The identified sites were used for subsequent ligand docking.
Ligand Docking
Docking was carried out using “Glide” (Grid-based Ligand Docking with Energetics) module of Schrödinger.38 Ligand docking proceeded as follows: 1) ligand preparation, 2) protein preparation and refinement, 3) active site grid generation, and 4) flexible ligand docking.39
Ligands Preparation
An in-house library of more than 4000 compounds was created using the Maestro 3D interface. This library included all approved drugs, chemical compounds in different stages of clinical trials, and Mediterranean natural product compounds. “Ligand preparation” was performed to optimize the ligand structures before docking.40 The “OPLS_2005” force field was used for ligand minimization, keeping the initial stereochemistry of the active compounds, tautomeric states were generated, and the ligands were desalted. Ionization states for all compounds were generated at a pH range of 7.0±2.0 using “Epik”.41
Protein Preparation and Refinement
The crystal structures of SARS-CoV-2 main protease, Mpro, were downloaded from PDB. One Mpro was crystalized in complex with the inhibitor, N3, and was reported with a resolution of 2.16 Å (PDB code: 6LU7).22 A series of crystal structures in the bound and unbound forms of Mpro were identified. In this work, we used the dimer form of the enzyme that was reported with a resolution of 2.2 Å (PDB code: 6Y2G).21 Different molecules bound to the protein and waters were removed. The quality of the docking results is highly dependent on the input receptor structure; the two protein structures were prepared and refined for docking using the “Protein Preparation” module from Schrödinger.41
Active Site Grid Generation
The shape and properties of the binding site are represented on a “grid” that provides more accurate scoring of the ligand poses. Active site grids to be used in docking were generated using the “Receptor Grid Generation” module in Glide from Schrödinger. Glide uses a filter to locate the ligand in the receptor’s active site. Grids were generated on the refined protein to determine the shape and properties of the part of the receptor that will be involved in ligand binding.38 Grids can be generated to include the binding cavity surrounding the crystal structure-bound ligand. The co-crystalized inhibitors in the 6LU7,22 and 6Y2G21 crystal structures were not identified as ligands by the “Receptor Grid Generation” module. Therefore, for all sites, grids were generated as a centroid of the amino acid residues that were found to line the corresponding cavity (Table 1).
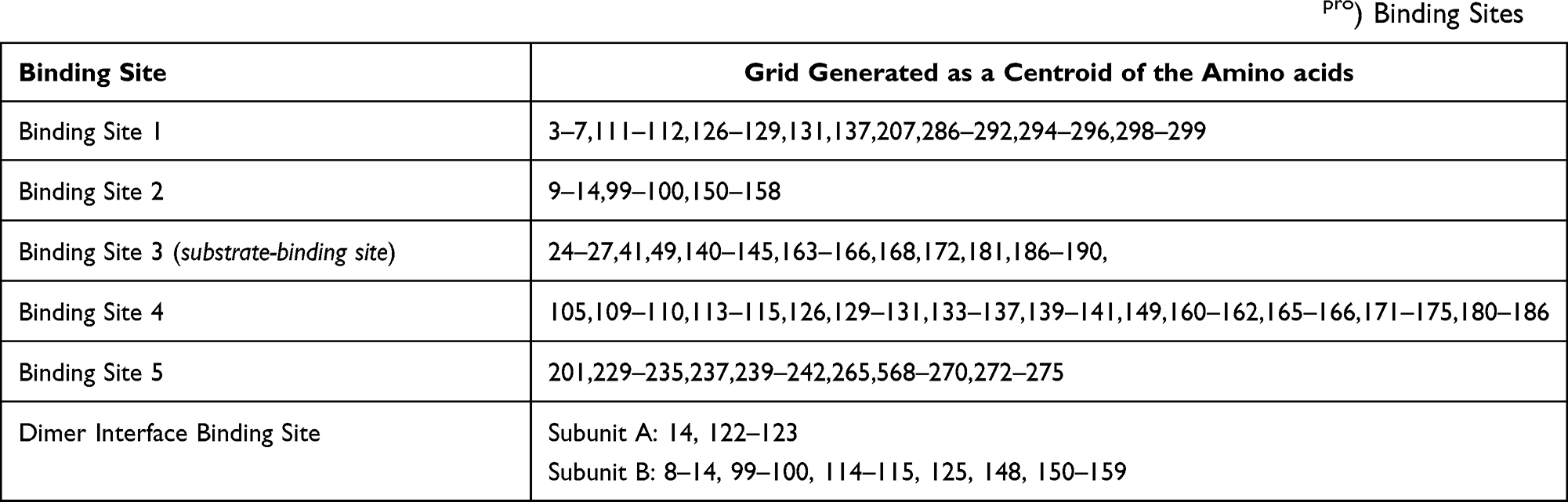 |
Table 1 Amino Acid Residues Used to Generate Grids of Different SARS-CoV-2 Main Protease Receptor (Mpro) Binding Sites |
Glide does not allow for flexible receptor docking; however, ligand-protein binding is a dynamic process that involves movements from both the ligand and the protein. Scaling down the van der Waals radii (vdW) of protein nonpolar atoms would allow easier binding of the larger ligands.39 To allow more freedom of ligand docking and to increase the number of bound compounds, all grids were generated with a scaled down van der Wall radii to 0.8 (vdW = 0.8) and without scaling down for the second grid generation (vdW = 1) using the centroid of the same amino acid residues of the respective binding site. Dock ligands with length ≤20 Å preference was maintained for all grid generation runs.
Flexible Ligand Docking
Flexible ligand docking predicts the possible ligand binding conformation and interactions within the macromolecule. Flexible docking refers to the conformational search performed on the ligand before docking.42 Extra precision (XP) flexible ligand docking was carried out using the prepared ligands’ list. The optimum ligand conformation(s) to be docked into the binding site were identified.39 The final scoring on the energy-minimized ligand poses was performed, and all interactions within the receptor binding site were calculated. Ligand poses were ranked according to their GlideScore (GScore) scoring function, the best pose of each ligand having the lowest GScore value.43 GScore was calculated using the following equation:
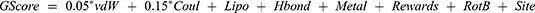
where vdW: the van der Waals energy and Coul: the Coulomb energy both associated with formal charges, Lipo: the lipophilic rewarding hydrophobic interactions, Hbond: the hydrogen-bonding, Metal: the metal-binding term involving anionic or highly polar atoms interactions, Rewards: rewards and penalties such as hydrophobic enclosure, correlated hydrogen bonds and amide twist, RotB: a penalty assigning freezing rotatable bonds, and Site: a term for polar interactions within the active site.42
Initially, to increase the number of reported compounds, in a similar manner to the grid generation step, docking was performed with the scaling down of the vdW radii of nonpolar ligand atoms (vdW = 0.8), while the second set of docking runs were performed selecting the no scaling down (vdW = 1) option. All docking runs using the different generated grids and docking preferences were visually inspected and ordered according to their GScores. The run using the scaling down of grid (vdW = 0.8) and no scaling down (vdW = 1) XP docking preferences resulted in ligands with best docked poses and highest GScores. Table 1S reports the docking results of all binding sites for future reference, more than ninety compounds have shown potential binding to Mpro different identified cavities. It is worth mentioning that cephalosporins were the only compounds to be docked within Binding Site 5 (data not shown in Table 1S). Due to the large number of drugs that passed this docking selection process, more refinement of the results was needed. Ligands that were bound in “Binding Sites 1” (BS1), “Binding Site 3” (BS3), and at the “Dimer Interface Binding Site” (DIBS) showed the highest scores of all docked ligands. The shortlisted compounds that included the top scoring ligands in each binding site were passed to the next step.
Molecular Mechanics/Generalized Born Surface Area Calculations
Due to the limitations of ligand docking and scoring functions in predicting binding energies, a more accurate computational method is required to study drug ligand interactions. Molecular mechanics/generalized Born surface area calculations (MM-GBSA) calculates binding free energies for the docked complexes. Prime-MMGBSA module incorporating OPLS3 force field and VSGB dissolvable model from Schrödinger was used to determine the binding free energies of the top docked complexes obtained from the XP docking process.44 To increase the accuracy of the calculations, amino acid residues within 8 Å and the ligand structure were relaxed during the calculations. The binding free energy for the protein-ligand complex follows:
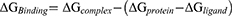
where ∆GBinding is the binding free energy; whereas ∆Gcomplex, ∆Gprotein and ∆Gligand represent the free energy of complex, protein, and ligand, respectively.45 The protein-ligand complexes were ranked based on their GScores and the calculated free energies. Compounds that showed both the highest GScore and binding free energies were selected for subsequent molecular dynamics simulations.
Molecular Dynamics Simulations
All molecular dynamics (MD) simulations were performed using the NAMD 2.13 package46,47 and the CHARMM36 force field.48 The parameters for the top docking results were generated using the CHARMM general force field (CgenFF).49 TIP3P explicit solvation model was used, and the periodic boundary conditions were set with a dimension of the dimensions x:105.17 Å, y: 108.28 Å, and z:110.37 Å. The MD protocols involved minimization, annealing, equilibration, and production. The atoms of the protein backbone were restrained in the minimization and annealing simulations, while the Cα atoms of the protein were restrained in the 1 ns equilibration simulation, and no atoms were restrained in the 100 ns MD production simulation. The isothermal–isobaric (NPT) ensemble and a 2 fs time step of integration were chosen for all MD simulations. Pressure was set at 1 atm using the Nose´–Hoover Langevin piston barostat,50,51 with a Langevin piston decay of 0.2 ps and a period of 0.4 ps. The temperature was set at 298.15 K using the Langevin thermostat.52 A distance cutoff of 10 Å was applied to short-range non-bonded interactions with a pair list distance of 12 Å, and Lennard Jones interactions were smoothly truncated at 8.0 Å. Long-range electrostatic interactions were treated using the particle-mesh Ewald (PME) method,53,54 with a grid spacing of 1.0 Å. All covalent bonds involving hydrogen atoms were constrained using the SHAKE algorithm.55 The same preferences were applied for all MD simulations. Binding free energy for the systems were calculated as mentioned above. CF analysis was performed using the contactFreq module on VMD with a cutoff of 4 Å and hydrogen bond (H-bond) interactions for each relevant residue for the binding was generated by VMD.56 The stability of the structures during and at the end of MD simulations was evaluated through the determination of the root-mean-square deviation (RMSD), and the root-mean-square fluctuation (RMSF). To analyze the conformational changes in the surface area and volume of different protease cavities after the MD simulations, CASTp 3.0 was used.57
Flexible Ligand Docking and Molecular Mechanics/Generalized Born Surface Area Calculations
To analyze the effect of MD simulations on the binding and binding affinities of different compounds, XP docking and MM-GBSA analysis were performed using the minimized structures generated from the MS simulations step. New grids were generated for the binding sites using the previously mentioned residues (Table 1). Grids and docking were performed with no scaling down (vdW = 1). MM-GBSA was repeated using the same preferences as mentioned above, and ∆GBinding was determined.
In vitro Assay
The “3CL Protease (SARS-CoV-2) Assay Kit” from “BPS Bioscience” was used to test the inhibitory activity of the identified compounds as well as to study the potential synergistic effect between the identified hits (drugs), the kit was used according to manufacturer’s instructions.58 Briefly, various concentrations of the drugs were mixed with the enzyme. Purified 3CL Protease cleaves a fluorogenic peptide substrate between glutamine and serine to generate a highly fluorescent peptide fragment. Thus, the fluorescence intensity increases proportionally to the activity of 3CL protease. Activity of the enzyme was measured using a Fluorescent microplate reader at 360 nm/460 nm. Compounds to be tested were obtained from local pharmaceutical drug manufacturers with established purity; all compounds were found to be >95% pure by HPLC analysis.
A range of five different concentrations, 0.1, 0.2, 0.5, and 1.2 µM, of each drug were tested against Mpro, and the %inhibition of the enzyme was determined. The drug concentration that resulted in the highest %inhibition was subsequently used in the synergy assay step. The possible combinations of the drugs were mixed and the %inhibition and the coefficient of drug interaction (CDI) were calculated for the different mixtures. CDI was calculated using the following equation:

Results
Prediction of Mpro Binding Sites
The 3D structure of the Main Protease Receptor Mpro (PDB:6Y2G)21 binding sites was analyzed using the “SiteMap” module from Schrödinger.37 Six druggable sites in the single protease subunit and one binding site at the interface between the two adjacent domains of the dimer were identified (Figure 2). All sites were considered as potential active sites and were screened in search for possible inhibitors from our in-house chemical library. All seven identified sites were found to have a “SiteScore” of ≥0.79.
 |
Figure 2 Crystal structure of SARS-CoV-2 main protease receptor Mpro, showing the potential binding sites identified using “SiteMap”. Site points are represented as red dots. |
Docking Studies
We used our in-house built chemical library of more than 4000 compounds in the flexible ligand docking. Grids to be used in the docking process were determined as the centroid of the amino acid residues lining the identified cavities (Table 1). Extra precision (XP) flexible ligand docking was employed to explore the druggability of the identified sites. With the exception of the ligand binding site, all sites were not reported to be bound to any ligand in the crystal structures. This lack of reference ligand has limited the validation of the docking preferences, which is usually done by comparing the docked ligand to its original crystal structure-bound counterpart.39 Visual inspection of the docked ligands and the GlideScore (GScore) scoring function were used to evaluate the docking results.43 Through docking, many currently marketed or under investigation drugs showed potential protease inhibitory activity; some were found to bind to more than one of the identified sites. A recent study for the “Assessment of Evidence for COVID-19-Related Treatments” released by the American Society of Health-System Pharmacists has reported the clinical evidence for the effect of some of the compounds identified in this work in the treatment of COVID-19 without any information about their mechanism of action.59 This list included antiviral drugs, nonsteroidal anti-inflammatory drugs, statins lipid lowering agents, angiotensin converting enzyme inhibitors, and angiotensin II receptor blockers.60 As further studies are required to establish the type of effect of these drugs, in our study we have established the in silico effect of some of these drugs as potential Mpro inhibitors. In addition to the above mentioned compounds, our study has also identified potential effect of drugs that belong to the following therapeutic classes: chemotherapeutic agents, kinase inhibitors, and nucleoside and nucleotide analogues. More than ninety drugs have shown potential to be used, alone or in combination, as inhibitors of Mpro and thus in the treatment of COVID-19 infections (Table 1S). Moreover, Binding Site 5” docking results were very interesting as only cephalosporins were found to bind within this binding site (data not shown in Table 1S).
The highest GScores were reported from the different docking runs in three of the studied sites, BS1, BS3, and DIBS. Upon studying the amino acid residues involved in the binding of different drugs, some overlap was observed. Interestingly, some ligands were bound to common amino acid residues of BS4 and DIBS, suggesting the ability of these compounds to bind within the protein cavity or to prevent the formation of the dimer.
Due to the large number of drugs that passed the docking selection process, more refinement of the results was needed. The GScore cut-off value was set for the selection of hits to be further refined by molecular mechanics/generalized Born surface area (MM-GBSA) calculations. According to their GScores, the top ten ligand-Mpro complexes for BS1 and BS3, and the DIBS were chosen for the next step.
Molecular Mechanics/Generalized Born Surface Area Calculations
Docking studies resulted in identifying a large number of potential inhibitors, a further more accurate computational tool was used for lead hit identification, MM-GBSA, that calculates free energies of binding for the docked complexes. The best binding affinity of all MM-GBSA calculations were found to be for cefixime in BS1, carvedilol in BS3, and favipiravir in DIBS (Table 2). In accordance with the crystal structure and mutation data, carvedilol was found to form interactions with the binding site residues: Thr25, His41, Leu141, Gly143, Cys145, Glu166, and Gln189 (Figure 3A).21,61 Cefixime, on the other hand, was found to bind deeper into BS1 forming interactions with amino acid residues from the facing subunit (Table 2 and Figure 3B). Favipiravir interactions were as expected, and interactions were formed between the drug and amino acid residues from both dimer subunits (Table 2 and Figure 3C). Compounds that showed both the highest GScore and ∆G binding were selected for MD simulations.
 |
Table 2 Molecular Interactions of Potential Mpro Inhibitors Within Their Respective SARS-CoV-2 Main Protease Receptor (Mpro) Binding Sites |
 |
Figure 3 Interaction of different SARS-CoV-2 main protease receptor Mpro, inhibitors within their respective binding sites. (A) carvedilol in binding site 3, (B) cefixime in binding site 1, and (C) favipiravir in the Dimer Interface Binding Site. |
Molecular Dynamics Simulations
MD simulations have the advantage of assessing the ligand binding in aqueous environment mimicking physiological conditions which would result in a more accurate prediction of binding interactions than those obtained from docking. In addition, an optimization of ligand/protein contacts is achieved through the MD simulations’ conformational changes in the protein, including binding site residues, that occur upon ligand binding. These optimizations are critical in our work as we try to explore the effect of any ligand’s binding on the different enzyme cavities, thus allowing the study of the possible changes that may affect other drug’s binding.
The resulting systems of the different MD simulations were studied for their stability through the determination of root-mean-square deviation (RMSD), and root-mean-square fluctuation (RMSF) of backbone atoms between simulated and initial structures. RMSF was also used to evaluate the impact of ligand interaction on different amino acid residues. In addition, an analysis of contact frequency (CF) (Figure 1S) and hydrogen bond (H-bond) interactions was performed.
Carvedilol
Carvedilol, a beta-adrenergic receptor blocker, was found to bind to the original substrate-binding site with the highest scores; therefore, this complex was first used to explore the changes in the Mpro dimer conformation through MD simulations. MD simulation confirmed the conclusion from initial docking and MM-GBSA results, and the compound remained bound to this conserved binding site. An increase in RMSD relative to the crystal Mpro structure has occurred after 10–20 ns, after which no significant development of RMSD could be observed. The ligand-protein exhibited successful conversion following 60 ns of MD simulation start, and the simulation converged between 2 and 3 Å. The high RMSD fluctuation of the ligand is expected, as carvedilol is a highly flexible small molecule that is bound to a relatively large binding site (Figure 4A). RMSF showed relative stabilization for the amino acid residues reported to be involved in ligand binding throughout the simulation (Figure 5A). MD binding showed similar interactions and H-bonds formation between the ligand and the binding site residues, we believe that these are more to represent the actual ligand binding due to the new conformation of the binding site after the MD run (Table 2S). To evaluate the binding between carvedilol and Mpro, CF analysis was performed, Figure 1SA shows the residues with higher CF during the simulation, residues that showed more than 80% CF were His41, Met49, and Thr25, and a CF of more than 70% was reported for Cys44, Ser46, and Glu166. MD simulation results confirmed the previously reported residues to be involved in ligand binding in the Mpro main binding site that included Thr25, His41, Ser46, Met49, Asn142, Cys145, Met165, Glu166, Pro168, and Gln189 (Figure 1SA and Table 2S).62 The highest H-bond occupancy reported was for Glu166, and Thr25 with a value of 29.33% and 4.60%, respectively. Since we are not performing MD simulations for different ligands within this same binding site, we did not do any further analysis of the H-bond occupancy results.
 |
Figure 4 Root mean square deviation (RMSD) plots for 100 ns long MD simulations with respect to initial structure for: (A) carvedilol in binding site 3, (B) cefixime in binding site 1, and (C) favipiravir in the Dimer Interface Binding Site of SARS-CoV-2 main protease receptor Mpro. Lines represent; black: ligand-dimer complex, red: ligand, and blue: protein. |
 |
Figure 5 Root-mean-square fluctuation (RMSF) plots for of the Cα atoms for SARS-CoV-2 main protease receptor Mpro during 100ns long MD simulations for: (A) carvedilol in binding site 3, (B) cefixime in binding site 1, and (C) favipiravir in the Dimer Interface Binding Site. |
The ∆G binding of carvedilol in the MD simulation was calculated to be −19.1 Kcal/mol (∆G MM-GBSA = −66.0 Kcal/mol). To study the effect of carvedilol binding on the overall conformation of the protein and hence the different cavities, docking and MM-GBSA were performed in BS1, and DIBS (refer to materials and methods). An increase in the binding affinity of cefixime in BS1, and favipiravir in DIBS was observed. It is interesting that both compounds showed the highest affinity among the docked ligands which confirms their binding to these sites as mentioned earlier. The ∆G binding of cefixime, and favipiravir were −71.2 and −33.7 Kcal/mol, respectively (Table 2). Interestingly, an increase in ∆G binding was calculated for cefixime and favipiravir in BS3 after the MD simulation. Cefixime showed an increase in the binding affinity from −57.6 to −69.7 Kcal/mol, while the increase was from −26.8 to −38.1 Kcal/mol for favipiravir. An increase in the binding site volume of BS1 from 670 to 802 Å3 and in the surface area from 797 to 884 Å2 was observed, while the dimer has opened up and an increase in the solvent accessible area was seen for the DIBS.
Cefixime
MD simulations of cefixime, a third-generation cephalosporin, in BS1 showed intermediate system stability among the studied systems (Figure 4B). The highest increase in RMSD was observed during the first 30 ns of the simulation indicating significant changes in the conformation of Mpro structure upon ligand binding. The whole system converged at the end of the simulation. This stability was also seen in the RMSF analysis for the amino acid residues involved in ligand binding throughout the simulation (Figure 5B). The simulation confirmed the position and interactions that were reported in the docking and MM-GBSA calculations, Arg4, Lys5, Val125, Tyr-126, Gln-127, Phe291, and Ser284 showed CF of more than 90% (Figure 1SB). On the other hand, Lys5 and Trp207 showed the highest H-bond occupancy during the simulation (Table 3S). ∆G binding of cefixime from the MD simulation was calculated to be −28.7 Kcal/mol (∆G MM-GBSA=−50.3 Kcal/mol). Similar to the first MD simulation, a study of the effect of cefixime binding on the overall conformation of the protein was performed, and a decrease in the binding site volume of BS1 from 410 to 336 Å3 was observed, while an increase in the surface area from 315 to 455 Å2 was seen. Similar to the effect of carvedilol binding, the dimer has opened up the solvent accessible area of the DIBS and an increase in the volume has been measured.
Docking and MM-GBSA were performed in BS3, and DIBS to measure any changes of binding affinity of the different molecules as a result of the conformational changes from cefixime binding. A decrease in ∆G binding for carvedilol and favipiravir was observed as compared to initial data reported in the unminimized enzyme (Table 2). Interestingly, the binding affinity of cefixime and favipiravir was found to increase in BS3 in the simulated enzyme over the original one. A ∆G binding of −74.0 was reported for cefixime, and −38.0 Kcal/mol for favipiravir in BS3, an increase from their original of −57.6 and −26.8 Kcal/mol, respectively.
Favipiravir
Favipiravir, an RNA-dependent RNA polymerase inhibitor, was previously reported to have an effect on Mpro.63,64 The third MD simulation run was performed with favipiravir bound within DIBS. Unfortunately, the system showed great instability as was seen in RMSD and RMSF analysis (Figures 4C and 5C). The increase within RMSD trajectories (from 2.7 to 5.2 Å) following the 70 ns is more to suggest a dramatic escape of the ligand out of the binding site; in fact, after studying the different trajectories, favipiravir molecule was found to escape the binding site also early in the simulation. Unlike previous simulations, the instability of the complex was reflected on RMSF analysis (Figure 5C), higher fluctuations were observed for all amino acids during the simulation timeframe. Some intervals of the 100 ns simulations showed binding of favipiravir to its binding site. Favipiravir interactions to different binding site residues were studied, and CF and H-bond interactions were analyzed. The CF performed, confirmed previous docking and MM-GBSA results in which favipiravir was mainly bound at the interface between the two subunits of Mpro binding site. Contacts were reported with the following residues; subunit A: Ser121 and Pro122, and subunit B: Phe8, Pro9, Lys12, Ile152, Tyr154, and Val303 (Figure 1S and Table 4S).
In vitro Assay
The inhibitory activity of the identified hits was established after testing five different concentrations, 0.1, 0.2, 0.5, and 1.2 µM, of each drug (carvedilol, cefixime, and favipiravir) against Mpro and the %inhibition of the enzyme was determined. The following concentrations: 2 µM carvedilol, 2 µM cefixime and 1 µM favipiravir were shown to exhibit the highest inhibitory effect on the enzyme. The different possible combinations of the three drugs were mixed at the specified concentration, and the %inhibition and the coefficient of drug interaction (CDI) were calculated.
Table 3 summarizes the %inhibition of the drug combinations: favipiravir/cefixime, and favipiravir/carvedilol. The %inhibition and the CDI for favipiravir/cefixime were 95.9 and 0.61 and for favipiravir/carvedilol were 98 and 0.89, respectively. A significant increase in the %inhibition of the drug combinations was observed (P < 0.001, Figure 6). The CDI value for the two combinations was <1, indicating the synergistic effect of both drug combinations.
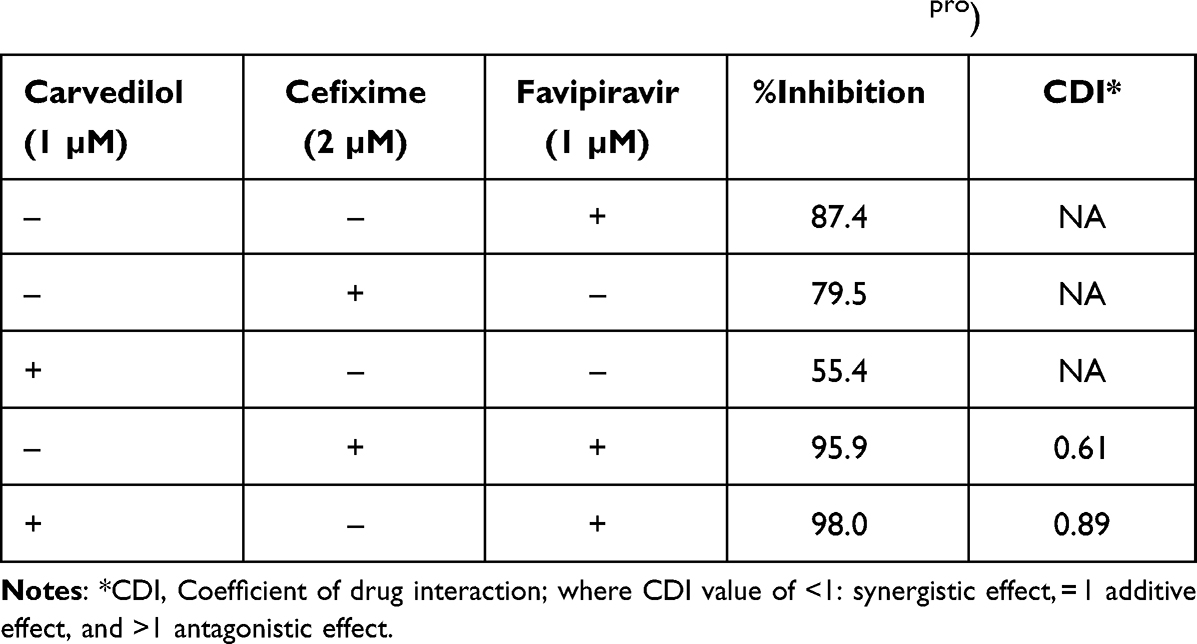 |
Table 3 %Inhibition of the Individual Drugs and the Drug Combinations: Favipiravir/Cefixime, and Favipiravir/Carvedilol Using the Same Concentrations on SARS-CoV-2 Main Protease Receptor (Mpro) |
 |
Figure 6 %inhibition of the individual drugs: carvedilol (1 µM), cefixime (2 µM), and favipiravir (1 µM), and the %inhibition of the drug combinations: favipiravir/cefixime, and favipiravir/carvedilol using the same concentrations on SARS-CoV-2 main protease receptor (Mpro). P < 0.001. |
Discussion
The urgent need to find a safe and effective remedy for COVID-19 makes drug repurposing one of the most appropriate options. Using an approved drug to treat a new disease holds huge promise to have a fast clinical effect with a much lower cost than de novo drug development.65 Despite the attractive opportunities presented by drug repurposing, limitations and factors must be considered for a drug to be used for a new ailment.66 In general, known compounds show weak activities and may not give the required effect if used alone.34 Different examples of drug combinations have been used to treat many infectious diseases with increased therapeutic efficacy and reduced toxicity.67,68 However, the process of establishing new drug combinations is hard and depends on a lengthy trial and error process of testing the different combinations of available drugs on each of the known drug targets.35
In this work, we studied the essential SARS-CoV-2 main protease (Mpro) and identified all druggable binding sites in search for potential drugs from a list of approved or under investigation drugs. Initially, Mpro reported structures were optimized, and complexes bound to the protein molecule, and water molecules were removed. Although water molecules may play a critical role in protein–ligand and protein–protein interaction,69 we decided to remove all water molecules to free the cavities to be able to identify all possible binding sites in Mpro. This can be acceptable as for most ligands, upon binding to a protein surface, they tend to displace bound water molecules in the binding site to interact directly with the protein.70
The search for potential druggable sites has identified seven binding sites, including the reported inhibitor–bound crystal structure site. The identified sites were evaluated according to “SiteScore”, where a score between 0.8 and 1 suggests a druggable site.37 The top seven identified binding sites were found to have a “SiteScore of ≥0.79. Despite that some of these sites may not provide the optimum characteristics of a druggable binding site, we decided to perform docking in all top-ranked sites to exploit all possibilities. Typically, the search for novel inhibitors would involve virtual high-throughput screening (vHTS) to screen large libraries of compounds against the drug target.71 In general, only a small percentage of known compounds can be classified as drug candidates, and compounds identified in the vHTS need to pass through a long and costly process to be approved and safely used.72 Due to the limitations of time and the urgency to find new COVID-19 treatments, we limited our search for potential drug targets and used our in-house library of compounds to streamline the inhibitor discovery process. The first docking runs were performed using grids generated with a scaling down of the van der Waals radii (vdW) of protein nonpolar atoms to 0.8 (vdW = 0.8), and docking was performed with the scaling down of the vdW radii of nonpolar ligand atoms to 0.8 (vdW = 0.8). These preferences would allow greater freedom for ligands to be docked as it eases protein rigidity restrains. For more refined results, we generated new grids and repeated the docking without scaling down (vdW = 1). It is believed that this would allow for a more accurate fit of the ligand pose within the binding site and hence would limit the number of ligands to be docked.38 The refinement of the docking preference and the analysis of docking results have led to the identification of a list of marketed or under investigation drugs as potential protease inhibitors (Table 1S). Many of the initially identified drugs were proposed to have an effect on COVID-19,60 however, their exact mechanism for combating the virus was not established.
Many limitations are associated with ligand docking; the most important one is that docking is performed in a rigid receptor and does not consider the different conformational changes that a protein can adopt upon ligand binding.73 Another limitation is the low confidence in the results of the scoring functions in giving accurate binding energies as some intermolecular interactions cannot be predicted accurately, such as the solvation effect and entropy change.74 Therefore, the more accurate MM-GBSA computational method was used in the lead hit identification. By calculating binding free energies for the best docked complexes, we were able to identify hits based on a more realistic binding affinities prediction. The flexibility of the receptor was accounted for as amino acid residues within 8 Å and the ligand structure were relaxed during calculations and more precise binding poses were generated.
A wide range of conformational changes are expected to occur in the protein structure upon ligand binding, the interaction between the inhibitor and Mpro may result in alterations in the protein structure that may affect the size and shape of other cavities, and hence the binding affinity of different drugs. These structural changes in the inhibitor-bound, versus the free, receptor may provide increased possibility of synergistic or combined effect.75 Therefore, the top ranked complexes from the MM-GBSA calculations, based on their GScore and ∆G, were selected for MD simulations. With the advantages of this technique in mind, it would be appropriate to study the results of MD simulations both at the ligand docked site and the other identified sites. Both carvedilol and cefixime-bound receptors showed excellent stability throughout the 100 ns run, unlike what was observed for favipiravir, which was seen in the RMSD analysis of all MD runs (Figure 4). Both compounds were found to have a perfect fit and remained bound within their respective binding sites. Figure 7 shows a comparison of the MD simulations of the three drugs. A higher stability was observed for carvedilol in its binding site; this may be expected as this is the orthostatic site of the enzyme, which is usually characterized by high structural stability and may not be subject to great variation of the conformation upon substrate and/or ligand binding.75 Similarly, cefixime showed consistent binding within its binding site, albeit the dimer as a whole showed more conformational changes than in carvedilol bound complex. This may be attributed to the effect of cefixime binding to an allosteric less stable site. The small size of the favipiravir molecule and the large size and water accessibility of the DIBS may have contributed to the instability of the bound structure (Figure 7).
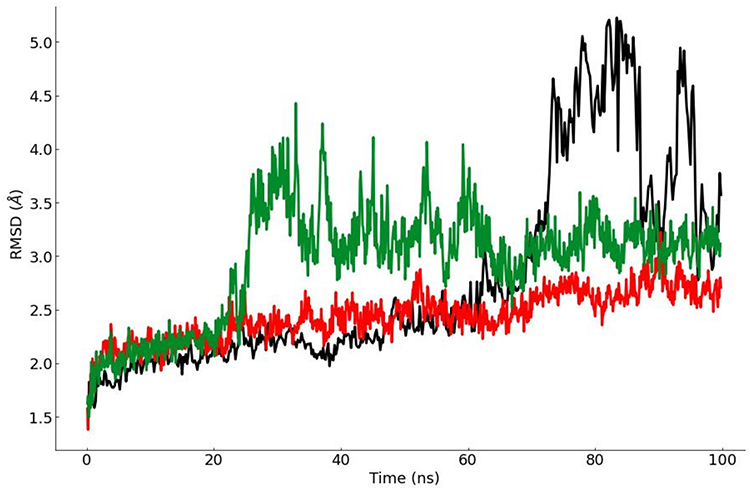 |
Figure 7 Comparison of the root mean square deviation (RMSD) plots for the three 100 ns long MD simulations with respect to initial structure of SARS-CoV-2 main protease receptor Mpro. Lines represent; red: carvedilol in binding site 3, green: cefixime in binding site 1, and black: favipiravir in the Dimer Interface Binding Site. |
With the accuracy of MD predictions, we were able to confirm the various residues involved in the different ligands binding (Tables 2S–4S). RMSF analysis for carvedilol and cefixime established the stability of the residues involved in ligand binding over other residues (Figure 5). This result of the correct conformation of binding sites, not previously reported, would provide a tool to design and optimize new Mpro inhibitors targeting a cavity different than the one identified in the crystal structures.
In general, an increase in the binding site volumes and/or surface areas was observed after the convergence of all simulations. This was reflected by the better binding affinities calculated for the different compounds in their respective binding sites (Table 2), and may indicate an additive or synergistic effect of the different drugs. The MD simulation is expected to reflect the conformational changes of the protein upon ligand binding which will lead to a change in the size and the orientation of the amino acid residues lining the different sites. In addition, the increase observed in ∆G binding of cefixime and favipiravir in BS3 after the MD simulation suggests that the synergistic effect may not only be attributed to binding of cefixime in BS1 or favipiravir in DIBS, but it can also be due to the enhancement of the binding of these two compounds in the second main binding site in the second subunit of the dimer. Results of the in vitro assay confirmed the in silico suggested synergistic effect; however, due to the lack of crystal structure information about the specific site(s) that each of the drugs was able to bind to, more studies are required to confirm the mechanisms of inhibition caused by each compound.
Conclusions
In this work, we studied one of SARS-CoV essential proteins, the main protease (Mpro), in search for treatments for COVID-19. Favipiravir, cefixime, and carvedilol were established as Mpro inhibitors in both in silico and in vitro assays. MD simulations were used to study the different conformational changes in the enzyme that can be induced by ligands’ binding. The analysis of these changes suggested potential drug combinations that gave a synergistic effect once tested on Mpro. Two drug combinations: favipiravir/cefixime, and favipiravir/carvedilol, were found to be active against this enzyme. Due to lack of facilities and the restrictions imposed under the pandemic circumstances, we did not establish the effect of these drug combinations using cell and/or animal models, which will be considered in our future work. Therefore, these findings are not for clinical application at this moment.
Abbreviations
SARS-CoV-2, acute respiratory syndrome coronavirus-2; RdRp, RNA-dependent RNA polymerase; Mpro, SARS-CoV-2 main protease; ACE2, angiotensin converting enzyme 2; GScore, GlideScore; BS1, binding sites 1; BS3, binding site 3; BS4, binding site 4; IBDS, in-between dimer binding site; MM-GBSA, molecular mechanics/generalized Born surface area; MD, molecular dynamics; RMSD, root-mean-square deviation; RMSF, root-mean-square fluctuation; vHTS, virtual high-throughput screening.
Acknowledgment
The authors of this manuscript deeply regret the loss of Dr. Heba Abdelhalim, a sincere, hardworking colleague who passed suddenly during the final stages of this manuscript preparation. She was the corresponding author till that date. May her soul rest in peace.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gorbalenya AE, Baker SC, Baric RS, et al. Severe acute respiratory syndrome-related coronavirus: the species and its viruses – a statement of the Coronavirus Study Group. bioRxiv. 2020;4:1–15.
2. Kola L, Kohrt BA, Hanlon C, et al. COVID-19 mental health impact and responses in low-income and middle-income countries: reimagining global mental health. Lancet Psychiatry. 2021;535–550. doi:10.1016/S2215-0366(21)00025-0
3. Alattar R, Ibrahim TBH, Shaar SH, et al. Tocilizumab for the treatment of severe COVID-19. J Med Virol. 2020;19:1–12. doi:10.1002/jmv.25964
4. Deng L, Li C, Zeng Q, et al. Arbidol combined with LPV/r versus LPV/r alone against Corona Virus Disease 2019: a retrospective cohort study. J Infect. 2020;1–5. doi:10.1016/j.jinf.2020.03.002
5. Singh AK, Singh A, Shaikh A, Singh R, Misra A. Chloroquine and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: a systematic search and a narrative review with a special reference to India and other developing countries. Diabetes Metab Syndr. 2020;14:241–246. doi:10.1016/j.dsx.2020.03.011
6. Richman DD, Whitley RJ, Hayden FG. Clinical Virology.
7. Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100(23):13190–13195. doi:10.1208/s12248-012-9390-1
8. Ziebuhr J. The coronavirus replicase. Curr Top Microbiol Immunol. 2005;287:57–94. doi:10.1007/3-540-26765-4_3
9. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. In: Maier HJ, Bickerton E, Britton P, editors. Coronaviruses: Methods and Protocols. New York: Springer New York; 2015:1–23.
10. Mittal A, Manjunath K, Ranjan RK, Kaushik S, Kumar S, Verma V. COVID-19 pandemic: insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020;16(8):e1008762. doi:10.1371/journal.ppat.1008762
11. Jang WD, Jeon S, Kim S, Lee SY. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc Nat Acad Sci. 2021;118:e2024302118. doi:10.1073/pnas.2024302118
12. Aronskyy I, Masoudi-Sobhanzadeh Y, Cappuccio A, Zaslavsky E. Advances in the computational landscape for repurposed drugs against COVID-19. Drug Discov Today. 2021;26:2800–2815. doi:10.1016/j.drudis.2021.07.026
13. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. doi:10.1038/nrd.2018.168
14. Abhithaj J, Sadasivan C. Drug repurposing to identify therapeutics against COVID 19 with SARS-Cov-2 spike glycoprotein and main protease as targets: an in silico study; 2020.
15. Sharanya C, Abhithaj J, Sadasivan C. Drug repurposing to identify therapeutics against COVID 19 with SARS-Cov-2 spike glycoprotein and main protease as targets: an in silico study; 2020.
16. Cao B, Wang Y, Wen D, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. New Engl J Med. 2020;382:1787–1799. doi:10.1056/NEJMoa2001282
17. Tong S, Su Y, Yu Y, et al. Ribavirin therapy for severe COVID-19: a retrospective cohort study. Int J Antimicrob Agents. 2020;56:106114. doi:10.1016/j.ijantimicag.2020.106114
18. Farne H, Kumar K, Ritchie AI, Finney LJ, Johnston SL, Singanayagam A. Repurposing existing drugs for the treatment of COVID-19. Ann Am Thorac Soc. 2020;17:1186–1194. doi:10.1513/AnnalsATS.202005-566FR
19. Edwards A. What are the odds of finding a COVID-19 drug from a lab repurposing screen? J Chem Inf Model. 2020;60:5727–5729.
20. Mahase E. Covid-19: pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ. 2021;n2713. doi:10.1136/bmj.n2713
21. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi:10.1126/science.abb3405
22. Jin Z, Du X, Xu Y, et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020;582:289–293. doi:10.1038/s41586-020-2223-y
23. Redhead MA, Owen CD, Brewitz L, et al. Bispecific repurposed medicines targeting the viral and immunological arms of COVID-19. Sci Rep. 2021;11:13208–13222. doi:10.1038/s41598-021-92416-4
24. Mótyán JA, Mahdi M, Hoffka G, Tőzsér J. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: lessons learned from HIV-1 protease. Int J Mol Sci. 2022;23:3507. doi:10.3390/ijms23073507
25. Sharma P, Joshi T, Mathpal S, et al. Identification of natural inhibitors against Mpro of SARS-CoV-2 by molecular docking, molecular dynamics simulation, and MM/PBSA methods. J Biomol Struct Dyn. 2022;40:2757–2768. doi:10.1080/07391102.2020.1842806
26. Papaj K, Spychalska P, Kapica P, et al. Evaluation of Xa inhibitors as potential inhibitors of the SARS-CoV-2 Mpro protease. PLoS One. 2022;17:e0262482. doi:10.1371/journal.pone.0262482
27. Bello M, Martínez-Muñoz A, Balbuena-Rebolledo I. Identification of saquinavir as a potent inhibitor of dimeric SARS-CoV2 main protease through MM/GBSA. J Mol Model. 2020;26:1–11. doi:10.1007/s00894-020-04600-4
28. Noroozi Tiyoula F, Aryapour H. Reconstruction of the unbinding pathways of noncovalent SARS-CoV and SARS-CoV-2 3CLpro inhibitors using unbiased molecular dynamics simulations. PLoS One. 2022;17:e0263251. doi:10.1371/journal.pone.0263251
29. Yilancioglu K, Cokol M. Design of high-order antibiotic combinations against M. tuberculosis by ranking and exclusion. Sci Rep. 2019;9:1–11. doi:10.1038/s41598-019-48410-y
30. Tan X, Hu L, Luquette LJ 3rd, et al. Systematic identification of synergistic drug pairs targeting HIV. Nat Biotechnol. 2012;30:1125–1130. doi:10.1038/nbt.2391
31. Riva L, Yuan S, Yin X, et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020;586:113–119. doi:10.1038/s41586-020-2577-1
32. Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–466. doi:10.1038/nchembio817
33. Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3:1–11. doi:10.1002/prp2.149
34. Sun W, Sanderson PE, Zheng W. Drug combination therapy increases successful drug repositioning. Drug Discov Today. 2016;21:1189–1195. doi:10.1016/j.drudis.2016.05.015
35. Cheng F, Kovacs IA, Barabasi A-L. Publisher Correction: network-based prediction of drug combinations. Nat Commun. 2019;10(1):1–11. doi:10.1038/s41467-019-09692-y
36. Berman HM. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
37. Halgren T. New method for fast and accurate binding-site identification and analysis. Chem Biol Drug Des. 2007;69:146–148. doi:10.1111/j.1747-0285.2007.00483.x
38. Halgren T, Murphy R, Friesner R, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi:10.1021/jm030644s
39. Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi:10.1021/jm0306430
40. Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi:10.1007/s10822-013-9644-8
41. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi:10.1007/s10822-013-9644-8
42. Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J Med Chem. 2006;49:6177–6196. doi:10.1021/jm051256o
43. Hou T, Wang J, Chen L, Xu X. Automated docking of peptides and proteins by using a genetic algorithm combined with a tabu search. Protein Eng, Design Selection. 1999;12:639–648. doi:10.1093/protein/12.8.639
44. Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins: Struct, Funct, Bioinfo. 2011;79:2794–2812. doi:10.1002/prot.23106
45. Misini Ignjatović M, Caldararu O, Dong G, Muñoz-Gutierrez C, Adasme-Carreño F, Ryde U. Binding-affinity predictions of HSP90 in the D3R Grand Challenge 2015 with docking, MM/ GBSA, QM/MM, and free-energy simulations. J Comput Aided Mol Des. 2016;30:707–730. doi:10.1007/s10822-016-9942-z
46. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi:10.1002/jcc.20289
47. Ribeiro JV, Bernardi RC, Rudack T, et al. QwikMD - Integrative molecular dynamics toolkit for novices and experts. Sci Rep. 2016;6:26536. doi:10.1038/srep26536
48. Best RB, Zhu X, Shim J, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi:10.1021/ct300400x
49. Yu W, He X, Vanommeslaeghe K, MacKerell AD
50. Nosé S, Klein ML. Constant pressure molecular dynamics for molecular systems. Mol Phys. 1983;50:1055–1076. doi:10.1080/00268978300102851
51. Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 1984;52:255–268. doi:10.1080/00268978400101201
52. Grest GS, Kremer K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys Rev a Gen Phys. 1986;33:3628–3631. doi:10.1103/physreva.33.3628
53. Darden T, York D, Pedersen L. Particle mesh Ewald: anN⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi:10.1063/1.464397
54. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. doi:10.1063/1.470117
55. Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. doi:10.1016/0021-9991(77)90098-5
56. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):27–38. doi:10.1016/0263-7855(96)00018-5
57. Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46(W1):W363–W367. doi:10.1093/nar/gky473
58. Morse JS, Lalonde T, Xu S, Liu WR. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. ChemBioChem. 2020;21:730–738. doi:10.1002/cbic.202000047
59. Snow EK, Miller JL, Kester L, et al. Creation and maintenance of a table for assessment of evolving evidence for COVID-19–related treatments. Am J Health Syst Pharm. 2020;78:154–157. doi:10.1093/ajhp/zxaa334
60. Assessment of evidence for COVID-19-related treatments; 2020. Available from: https://www.ashp.org/-/media/8CA43C674C6D4335B6A19852843C4052.ashx.
61. Joshi RS, Jagdale SS, Bansode SB, et al. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J Biomol Struct Dyn. 2021;39:3099–3114. doi:10.1080/07391102.2020.1760137
62. Farag A, Wang P, Ahmed M, Sadek H. Identification of FDA approved drugs targeting COVID-19 virus by structure-based drug repositioning; 2020.
63. McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res. 2020;157:104859. doi:10.1016/j.phrs.2020.104859
64. Rafi MO, Bhattacharje G, Al-Khafaji K, et al. Combination of QSAR, molecular docking, molecular dynamic simulation and MM-PBSA: analogues of lopinavir and favipiravir as potential drug candidates against COVID-19. J Biomol Struct Dyn. 2020;1–20. doi:10.1080/07391102.2020.1850355
65. Corsello SM, Bittker JA, Liu Z, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23:405–408. doi:10.1038/nm.4306
66. Oprea TI, Mestres J. Drug repurposing: far beyond new targets for old drugs. AAPS J. 2012;14:759–763. doi:10.1208/s12248-012-9390-1
67. Zheng W, Sun W, Simeonov A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br J Pharmacol. 2018;175:181–191. doi:10.1111/bph.13895
68. Abdel-Halim H, Al Dajani A, Abdelhalim A, Abdelmalek S. The search of potential inhibitors of the AcrAB–TolC system of multidrug-resistant Escherichia coli: an in silico approach. Appl Microbiol Biotechnol. 2019;103:6309–6318. doi:10.1007/s00253-019-09954-1
69. Guo Z, Li B, Cheng L-T, Zhou S, McCammon JA, Che J. Identification of protein-ligand binding sites by the level-set variational implicit-solvent approach. J Chem Theory Comput. 2015;11:753–765. doi:10.1021/ct500867u
70. Mancera RL. Molecular modeling of hydration in drug design. Curr Opin Drug Discov Devel. 2007;10:275–280.
71. Chen JJF, Visco DP Jr. Developing an in silico pipeline for faster drug candidate discovery: virtual high throughput screening with the Signature molecular descriptor using support vector machine models. Chem Eng Sci. 2017;159:31–42. doi:10.1016/j.ces.2016.02.037
72. Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem Biol Interact. 2008;171:165–176. doi:10.1016/j.cbi.2006.12.006
73. Śledź P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Curr Opin Struct Biol. 2018;48:93–102. doi:10.1016/j.sbi.2017.10.010
74. Yuriev E, Agostino M, Ramsland PA. Challenges and advances in computational docking: 2009 in review. J Mol Recognit. 2011;24:149–164. doi:10.1002/jmr.1077
75. Luque I, Freire E. Structural stability of binding sites: consequences for binding affinity and allosteric effects. Proteins Suppl. 2000;41(S4):63–71.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.