Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Identification of Crucial Genes and Pathways Associated with Atherosclerotic Plaque in Diabetic Patients
Authors Li YY, Zhang S, Wang H, Zhang SX, Xu T, Chen SW, Zhang Y, Chen Y
Received 16 October 2020
Accepted for publication 26 December 2020
Published 4 February 2021 Volume 2021:14 Pages 211—220
DOI https://doi.org/10.2147/PGPM.S281705
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Yuan-Yuan Li,* Sheng Zhang,* Hua Wang, Shun-Xiao Zhang, Ting Xu, Shu-Wen Chen, Yan Zhang, Yue Chen
Department of Endocrinology, Baoshan Branch, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, 201999, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Zhang; Yue Chen
Department of Endocrinology, Baoshan Branch, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, No. 181 You-Yi Road, Shanghai, 201999, People’s Republic of China
Tel +86 13818856902; +86 13701994461
Email [email protected]; [email protected]
Background: Patients with diabetes have more calcification in atherosclerotic plaque and a higher occurrence of secondary cardiovascular events than patients without diabetes. The objective of this study was to identify crucial genes involved in the development of diabetic atherosclerotic plaque using a bioinformatics approach.
Methods: Microarray dataset GSE118481 was downloaded from the Gene Expression Omnibus (GEO) database; the dataset included 6 patients with diabetic atherosclerotic plaque (DBT) and 6 nondiabetic patients with atherosclerotic plaque (Ctrl). Differentially expressed genes (DEG) between the DBT and Ctrl groups were identified and then subjected to functional enrichment analysis. Based on the enriched pathways of DEGs, diabetic atherosclerotic plaque-related pathways were screened using the comparative toxicogenomics database (CTD). We then constructed a protein–protein interaction (PPI) network and transcription factor (TF)–miRNA–mRNA network.
Results: A total of 243 DEGs were obtained in the DBT group compared with the Ctrl group, including 85 up-regulated and 158 down-regulated DEGs. Functional enrichment analysis showed that up-regulated DEGs were mainly enriched in isoprenoid metabolic process, DNA-binding TF activity, and response to virus. Additionally, DEGs participating in the toll-like receptor signaling pathway were closely related to diabetes, carotid stenosis, and insulin resistance. The TF–miRNA–mRNA network showed that toll-like receptor 4 (TLR4), BCL2-like 11 (BCL2L11), and glutamate-cysteine ligase catalytic subunit (GCLC) were hub genes. Furthermore, TLR4 was regulated by TF signal transducer and activator of transcription 6 (STAT6); BCL2L11 was targeted by hsa-miR-24-3p; and GCLC was regulated by nuclear factor, erythroid 2 like 2 (NFE2L2).
Conclusion: Identification of hub genes and pathways increased our understanding of the molecular mechanisms underlying the atherosclerotic plaque in patients with or without diabetes. These crucial genes (TLR4, BC2L11, and GCLC) might function as molecular biomarkers for diabetic atherosclerotic plaque.
Keywords: diabetes, atherosclerotic plaque, differentially expressed genes, bioinformatics analysis
Introduction
Atherosclerosis is a chronic inflammation disease and the leading cause of morbidity and mortality globally.1 Atherosclerosis is a slowly progressive process, characterized by an accumulation of lipid in the arterial wall accompanied by multifocal structural alterations, leading to atheromatous plaque formation.2,3 Over time, the large necrotic lipid core is covered by a fibrous cap until, in advanced stages, the stability of the cap is destroyed, inducing plaque rupture and thrombosis, which can manifest as stroke or myocardial infarction. Cardiovascular disease is the leading cause of death in patients with diabetes.4 There is increasing evidence that diabetes induces hypercoagulability, which has a role in plaque rupture and increases the incidence and severity of clinical events.4
Previous researchers have reported the relationship between plaque characteristics and patients with and without diabetes. Burke et al indicated that total plaque in diabetic patients was significantly greater than that of nondiabetic individuals; in addition, the inflammatory response of diabetic plaques was stronger than that of nondiabetic plaque.5 An optical coherence tomography imaging study by Kato et al revealed that plaques in diabetic patients had a higher incidence of calcification and thrombus.6 Furthermore, van Haelst et al found that patients with diabetes had more calcification in atherosclerotic plaque and a higher occurrence of secondary cardiovascular events than patients without diabetes.7 Even though we can distinguish atherosclerotic plaque in diabetic and nondiabetic patients from morphologic fields, the effect of diabetes on gene expression in atherosclerotic plaque is not fully understood.
Macrophage accumulation plays a vital role in both plaque progression and stability, which can promote inflammation and aggravate disease.8,9 Thus, we selected a gene expression dataset (GSE118481) containing diabetic plaque macrophage (DBT) and nondiabetic plaque macrophage (Ctrl) for analysis. Differentially expressed genes (DEGs) between the DBT and Ctrl groups were identified, functional enrichment analysis of the DEGs was performed, and disease-related pathways were screened. We then constructed a protein–protein interaction (PPI) network and sub-network. Subsequently, microRNA (miRNA) and transcription factors (TFs) of DEGs were predicted and an integrated TF–miRNA–mRNA network was constructed. The analysis process of this study is shown in Supplementary Figure 1. We aimed to further understand the molecular mechanism by which diabetes promotes the formation of atherosclerotic plaque and to determine potential gene targets for personalized diagnosis and treatment strategies of diabetic atherosclerotic plaque.
Materials and Methods
Data Sources
The gene expression profile GSE118481 based on the GPL10558 Illumina HumanHT-12 V4.0 expression BeadChip platform was downloaded from the Gene Expression Omnibus (GEO) database (website: http://www.ncbi.nlm.nih.gov/geo/).10 This microarray data set included 16 nondiabetic samples (6 asymptomatic and 10 symptomatic) and 8 diabetic plaque samples (6 asymptomatic and 2 symptomatic). In order to study the effect of diabetes on atherosclerotic plaque, we selected asymptomatic samples for subsequent analysis. Therefore, 12 samples (6 DBT and 6 Ctrl) were included, and the clinical characteristics of these patients are listed in Supplementary Table 1. There were no significant differences in age (P = 0.24) and sex (P > 0.05) between the two groups.
Data Preprocessing and Identification of DEGs
The series matrix file for GSE118481 dataset was obtained from the GEO database,10 and the expression data of 6 DBT and 6 Ctrl macrophage samples were extracted for further analysis. Microarray expression profiling was standardized by Bioconductor bead array package,11 and the distribution of expression in each sample was visualized by boxplots. The probe ID was converted to a gene symbol using the annotation file, and probes that did not mapped to gene symbols were removed. If multiple probes matched the same gene, the mean value of probes was calculated. Empirical Bayes moderated t-test in the limma package (version 3.40.6)12 was used to identify DEGs between DBT and Ctrl samples. DEGs with P < 0.05 and |log fold change (FC)| >0.585 were considered statistically significant. The ggplot2 and heatmap of R (http://www.R-project.org/) were utilized to visualize the DEGs.
Disease-Related Co-Expression Network Analysis
Using Pearson correlation coefficients (r) in the stats of R package (version 3.6.1; http://www.R-project.org/), the co-expression of DEGs in DBT and Ctrl samples was, respectively, analyzed. Pairs with r > 0.95 and P < 0.05 were selected for further study. Cytoscape was applied to construct a co-expression network of Ctrl and DBT groups, and then the topological properties of the two networks were analyzed by using CytoNCA in Cytoscape.13 Furthermore, t-tests were used to calculate the difference between Ctrl and DBT networks. The sub-networks of DBT group were structured using the Cytoscape MCODE plugin,14 and networks with a score >3 were selected.
Functional Enrichment Analysis of DEGs
To understand the major biological functions of DEGs, we analyzed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of up- and down-regulated DEGs using the clusterProfiler package. Significant enrichment was defined by P < 0.05 and count >2.
Disease-Related Pathway Screening
Diabetes, carotid stenosis, and insulin resistance have effects on the development of diabetic atherosclerotic plaque. Therefore, to further identify diabetic plaque-related pathways, we screened pathways relevant to these diseases from the comparative toxicogenomics database (CTD), and these pathways were integrated with the KEGG pathways in the previous step.
PPI Network Construction
To determine the relationships among DEGs, we mapped the DEGs to the Search Tool for the Retrieval of Interacting Genes (STRING, version 11.0, http://www.string-db.org/) database, and only interactions with a combined score >0.7 were selected. Then, Cytoscape software was used to establish the PPI network, and hub nodes in the network were identified by CytoNCA. The PPI network was further analyzed by MCODE to explore functional modules, and score >5 was selected as the threshold.
TF–miRNA–mRNA Regulatory Network Analysis
To further understand the regulatory mechanism of DEGs, miRNAs of target genes were predicted using four available databases: miRWalk3.0,15 TargetScan,16 MiRDB,17 and MirTarBase.18 Putative miRNAs with score >0.95 and supported by at least two databases were selected; additionally, TF–target interactions were predicted by Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining (TRRUST) (https://www.grnpedia.org/trrust/).19 Subsequently, miRNA–target pairs and TF–target pairs were integrated to construct the TF–miRNA–mRNA regulatory network.
Results
Identification and Analysis of DEGs
The raw data were processed and the boxplots showed good normalized properties (Figure 1A). A total of 33,984 probes were obtained after annotation. A total of 243 DEGs were identified between the DBT and Ctrl groups, including 85 up-regulated and 158 down-regulated DEGs. The heat map and volcano plot of DEGs are shown in Figure 1B and C.
 |
Figure 1 Gene expression profile data analysis. (A) Boxplot of gene expression data after normalization. (B) Heat map of DEGs between Ctrl and DBT groups; green indicates Ctrl group and red indicates DBT group. (C) Volcano plot of DEGs between Ctrl and DBT groups. |
Disease-Related Co-Expression Network Analysis
Co-expression network analysis showed that 144 pairs and 162 DEGs were identified in the Ctrl group (Figure 2A), and 191 relationships and 170 DEGs were screened in the DBT group (Figure 2B). The topological properties of the Ctrl and DBT networks indicated that betweenness, closeness, and degree values were significantly higher in the DBT group than in the Ctrl group (Figure 2C), suggesting that co-expression of DEGs was more abundant in the DBT group than in the Ctrl group. The sub-network (score = 3.333) of the DBT group was constructed and included serine/threonine kinase 32B (STK32B), tripartite motif containing 22 (TRIM22), ninjurin 2 (NINJ2), and transmembrane protein 114 (TME114) (Figure 2D).
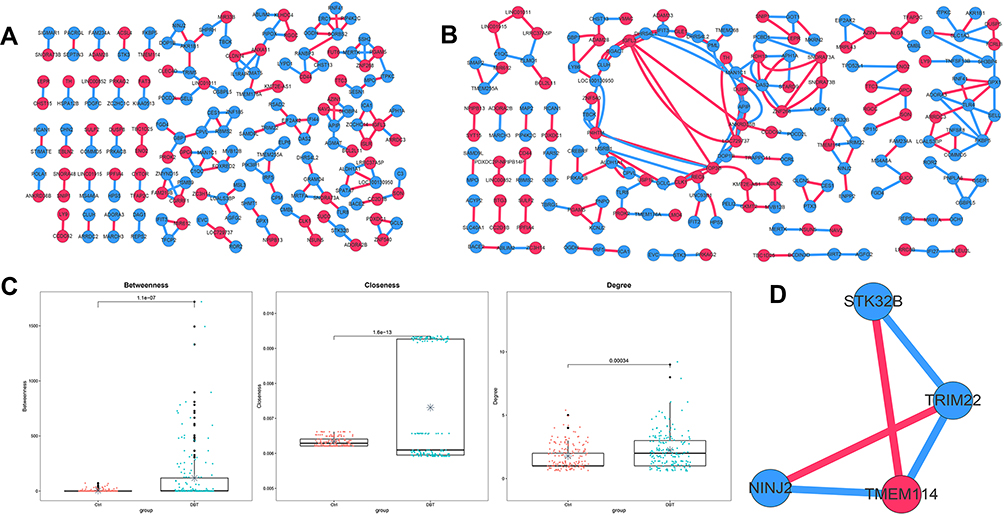 |
Figure 2 Disease-related co-expression network. (A) Co-expression network of Ctrl group. (B) Co-expression network of DBT group. (C) Topology properties of Ctrl and DBT group co-expression network. (D) Sub-network of DBT group. Red nodes indicate up-regulated DEGs, blue nodes indicate down-regulated DEGs, red lines represent positive correlation, and blue lines represent negative correlation. *Indicates the average of the data in each group. |
Functional Enrichment Analysis
Functional enrichment analysis of up-regulated and down-regulated DEGs was performed using the clusterProfiler tool. The top 10 significantly enriched GO terms and KEGG pathways are shown in Figure 3A and B. The up-regulated DEGs were significantly enriched in GO terms related to isoprenoid metabolic process, response to estradiol, and retinal metabolic process, and the down-regulated DEGs were markedly associated with regulation of DNA-binding TF activity, alpha-amino metabolic process, and response to virus (Figure 3A). For the KEGG pathway analysis, up-regulated DEGs were primarily involved in adipocytokine signaling pathway, cosphingolipid biosynthesis-lacto and neolacto series, and non-alcoholic fatty liver disease; in addition, down-regulated DEGs were mainly involved in folate biosynthesis, cysteine and methionine metabolism, and Chagas disease (American trypanosomiasis) pathways (Figure 3B).
 |
Figure 3 Functional enrichment analysis of DEGs. (A) GO analysis of the DEGs. (B) KEGG pathway analysis of the DEGs. The y-axis represents the GO terms or KEGG pathways, and the x-axis represents up-regulated and down-regulated DEGs. The size of bubbles represents the number of assigned genes, and the color of bubbles represents the adjusted P-value. The greater the number of DEGs associated with the term or pathway, the larger the bubble. |
Disease-Related Pathway Analysis
A total of 13 pathways were closely related to diabetes mellitus, one pathway was associated with carotid stenosis, and 10 pathways were associated with insulin resistance (Table 1). All three of these diseases were relevant to the toll-like receptor signaling pathway. Genes such as toll-like receptor 8 (TLR8), toll-like receptor 4 (TLR4), mitogen-activated protein kinase 4 (MAP2K4), and interferon regulatory factor 5 (IRF5) were involved in this pathway. In addition, 10 pathways were related to diabetes mellitus and insulin resistance, including acute myeloid leukemia, influenza A, non-alcoholic fatty liver disease (NAFLD), and adipocytokine signaling.
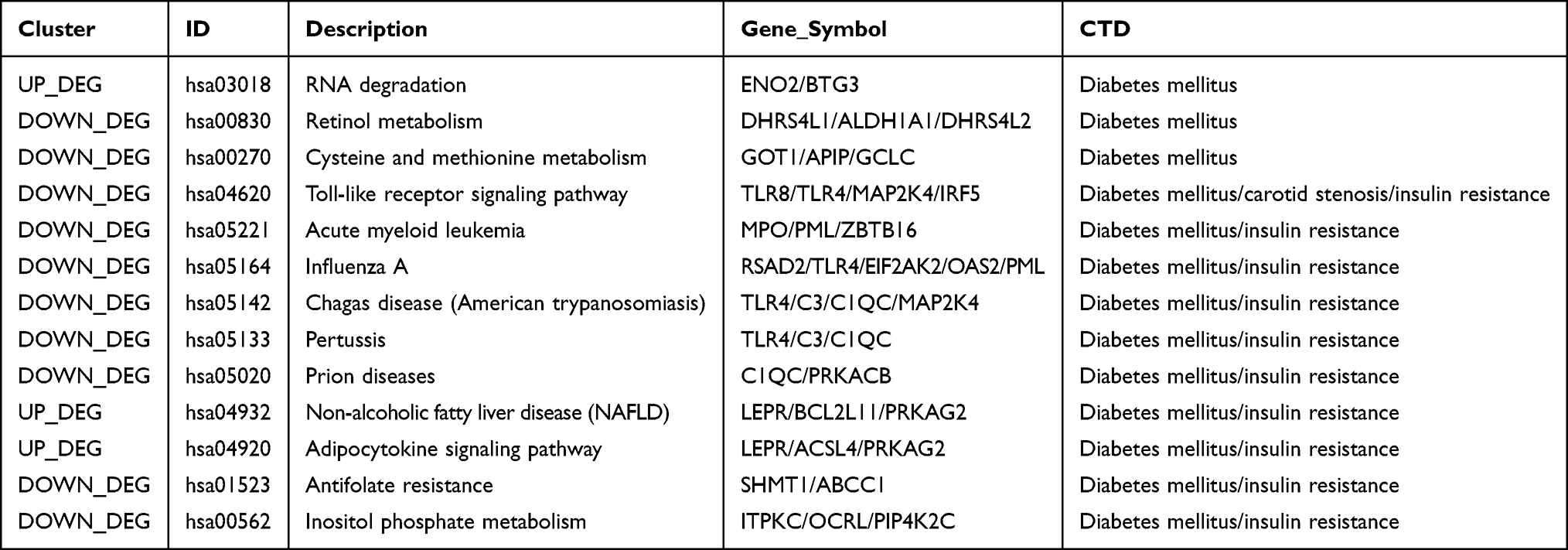 |
Table 1 Disease-Related Pathways Analysis |
PPI Network and Module Screening in DBT
The 225 proteins encoded by DEGs were searched in the STRING database and then used to construct the PPI network, which included 76 nodes and 114 pairs of edges (Figure 4A). Among these, several nodes with a higher degree [2′-5′-oligoadenylate synthetase 2 (OAS2, degree = 15), IRF5 (degree = 11), guanylate binding protein 1 (GBP1, degree = 11), and interferon-induced protein with tetratricopeptide repeats 3 (IFIT3, degree = 11)] could be considered hub proteins. Additionally, a module with score >5 was identified using the MCODE plugin. This sub-network was composed of 12 nodes and 43 pairs (Figure 4B). OAS2 (degree = 5), radical s-adenosyl methionine domain containing 2 (RSAD2, degree = 6), and eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2, degree = 5) were involved in diabetes and insulin resistance-related pathways.
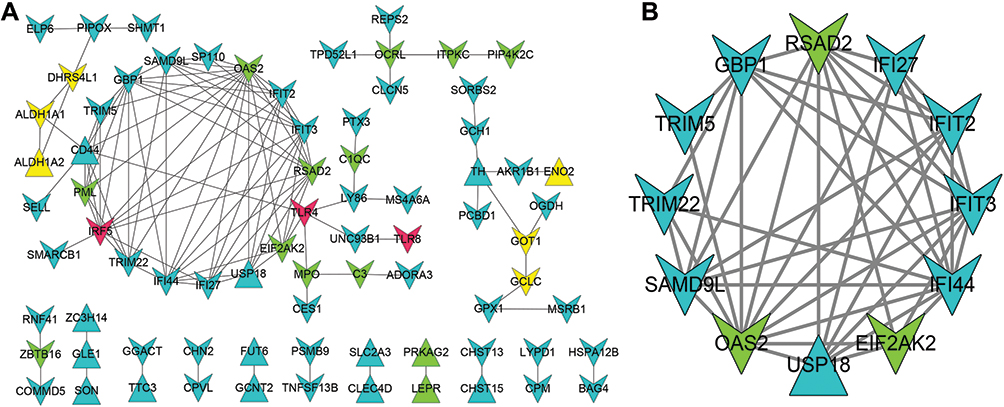 |
Figure 4 PPI network. (A) PPI network composed of 76 nodes and 114 edges. (B) Sub-network consisted of 12 nodes and 43 pairs. Triangle indicates up-regulated DEG, V-shape indicates down-regulated DEGs. Blue represent genes not involved in diseases-related pathways, yellow represent genes involved in diabetes-related pathways, green indicate genes enriched in diabetes and insulin resistance pathways, and red indicates genes participated in diabetes, insulin resistance, and plaque pathways. |
TF–miRNA–mRNA Regulatory Network Analysis
After screening, 102 miRNA–mRNA pairs and 114 TF-mRNA pairs were predicted, and then these pairs were integrated to structure a TF–miRNA–mRNA regulatory network. A total of 154 interactions were identified, involving 57 genes, 30 miRNA, and 75 TFs (Figure 5). In this regulatory network, we noted glutamate-cysteine ligase catalytic subunit (GCLC), BCL2 like 11 (BCL2L11), and TLR4 had higher degrees. GCLC was targeted by the TF nuclear factor, erythroid 2 like 2 (NFE2L2); BCL2L11 was targeted by hsa-miR-24-3p, and regulated by TF forkhead box O3 (FOXO3), and TLR4 was related to the process of three diseases and regulated by TF signal transducer and activator of transcription 6 (STAT6).
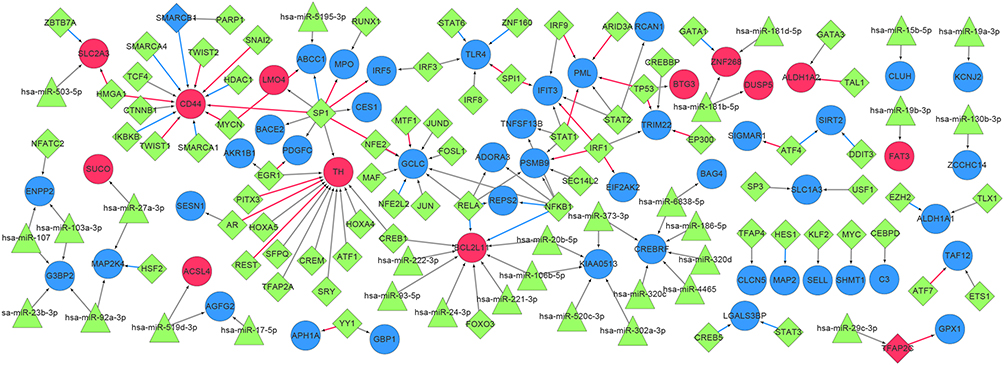 |
Figure 5 The TF–miRNA–mRNA regulatory network. Red nodes represent up-regulated DEGs, blue nodes represent down-regulated DEGs, green triangles represent miRNAs, green diamonds represent TF, and blue and red diamonds represent both TF and DEGs. Red lines indicate activation relationships and blue lines indicate inhibitory relationships. Abbreviations: TF, transcription factor; miRNA, microRNA; mRNA, messenger RNA; DEG, differentially expressed gene. |
Discussion
Diabetes is known to be associated with atherosclerotic plaque; however, the underlying molecular mechanism of the effect of diabetes on atherosclerotic plaque has not been fully elucidated. We analyzed gene expression patterns involved in diabetic atherosclerotic plaque using the dataset GSE118481. The results revealed that the toll-like receptor signaling pathway was associated with the pathogenesis of diabetic atherosclerotic plaque. Additionally, TLR4, BCL2L11, and GCLC were potential biomarkers for atherosclerotic plaque in patients with diabetes.
The formation and progression of atherosclerotic plaque is related to the accumulation of monocyte-derived macrophage in the arterial wall. Compared with patients without diabetes, the plaques in the coronary arteries of patients with diabetes gene generally exhibit larger necrotic cores and significantly greater inflammation, mainly composed of macrophages and T lymphocytes.20 Based on analysis of disease-related pathways, we observed that the toll-like receptor signaling pathway was significantly associated with diabetes mellitus, carotid stenosis, and insulin resistance; additionally, hub gene TLR4 was involved in this pathway. Madhur et al21 indicated that inflammation response could reduce the stability of atherosclerotic plaques in animal models. It is reported that the TLR signaling pathway is associated with systemic inflammation and immune response, and participates in angiogenesis, survival, and repair.22,23 Meanwhile, TLR4, as a member of the TLR family, is believed to activate nuclear factor-κB in response to short-chain fatty acids, triggering further activation of the immune system.24 Thus, TLR4 induced inflammation plays an important role in atherosclerotic plaque stability. Xu et al demonstrated that TLR4 was preferentially expressed by macrophages in human lipid-rich atherosclerotic lesions, where it might play a role to enhance and maintain the innate immunity and inflammation. In addition, the up-regulation of TLR4 in macrophages by oxidized low-density lipoprotein (LDL) suggested that TLR4 might provide a potential pathophysiological link between lipids as well as inflammation and atherosclerosis.25 In this analysis, we also found the relationship between TLR4 and diabetes. Devaraj et al demonstrated that expression of TLR4 was significantly increased in patients with type 1 diabetes, suggesting that TLR4 contributes to the pro-inflammatory state in diabetes.26 Moreover, knockout of TLR4 might alleviate inflammation in diabetic rats27 and TLR4 antagonist could attenuate atherogenesis in mice with diabetes.28 The antidiabetic drug class thiazolidinediones (TZDs) have been reported to reduce the risk of atherosclerosis in patients with type 2 diabetes, which might have an anti-atherosclerotic effect by inhibiting the TLR4 signaling pathway.29 These findings emphasized the importance of TLR4 in plaque formation of patients with diabetes. In the present study, we found that TLR4 was regulated by the TF STAT6. STAT6 has a major role in the immune system30 and is associated with macrophage polarization, which is critically involved in atherosclerosis progression and regression.31 Based on our results, we speculated that TLR4 and STAT6 might participate in the pathogenesis of diabetic atherosclerotic plaque via the TLR signaling pathway.
In the regulatory network, BCL2L11 had higher degree and was considered a hub gene. BCL2L11 encodes BCL-2 protein family, and its members participate in various cellular activities as anti- or proapoptotic regulators.32 A previous study revealed that BCL2L11 was connected to apoptosis of podocytes in diabetes.33 However, there are few reports about the association between BCL2L11 and carotid plaque. Our analysis showed that BCL2L11 was targeted by hsa-miR-24-3p. Erener et al observed that miR-24-3p was an effective biomarker to predict and diagnose diabetes.34 Moreover, miR-24-3p was found to limit macrophage vascular inflammation and slow the progression of atherosclerotic plaque.35 Taken together, hsa-miR-24-3p might affect the progression of diabetic atherosclerotic plaque by directly targeting BCL2L11. However, the specific regulatory mechanism of BCL2L11 in diabetic atherosclerotic plaque needs further elaboration.
We also found that GCLC was closely related to diabetic atherosclerotic plaque. GCLC is a rate-limiting enzyme of glutathione synthesis, and it is involved in susceptibility to myocardial infarction.36 Callegari et al found that the gain and loss of the ability to synthesize glutathione especially in macrophages had reciprocal effects on the initiation and progression of atherosclerosis at multiple sites in apoE-/- mice.37 Jain et al reported that the plasma level of GCLC was lower in diabetic patients than in healthy controls.38 Moreover, the GCLC polymorphism was associated with cellular redox imbalances and modulate the risk for diabetic nephropathy.39 In the TF–miRNA–mRNA network, GCLC was regulated by NFE2L2 (also known as NRF2), which is considered to be a master regulator of the antioxidant response.40 It regulates the expression of several genes including Phase II metabolic and antioxidant enzymes, and therefore plays an important role in preventing oxidative stress-mediated diabetes and related complications.41 In addition, overexpression of Nrf2 could protect pancreatic cells from oxidative damage in diabetes.42 Furthermore, a study by Harada et al revealed that activation of Nrf2 was observed in advanced atherosclerotic plaques, suggesting that Nrf2 might influence the inflammatory reactions in the plaques.43 Thus, we speculated that GCLC targeted by NFE2L2 might participate in the pathogenesis of diabetic atherosclerotic plaque.
Some limitations should be noted in the current study. First, the sample size of this study was small; further investigations based on a larger sample should be performed. Second, hub genes were identified using bioinformatics analysis; thus, experimental studies are needed to validate our results. Despite these limitations, this study provided some new insights into the pathogenesis and treatment of diabetic atherosclerotic plaques. Further large-scale studies are needed to corroborate these findings and investigate the potential underlying mechanisms involved. Meanwhile, clinical trials with more detailed investigation are also warranted before genes such as TLR4, BCL2L11, GCLC can be used in clinical setting.
Conclusion
In summary, we have conducted a comprehensive bioinformatics analysis of DEGs between diabetic and nondiabetic atherosclerotic plaque. Several genes have been identified with different expression patterns in diabetic and non-diabetic atherosclerotic plaque, such as TLR4, BCL2L11, GCLC, STAT6, and NFE2L2, as well as hsa-miR-24-3p. Meanwhile, pathway analysis showed that these genes were involved in the toll-like receptor signaling pathway. These findings provided better understanding of the underlying molecular mechanisms of diabetic atherosclerotic plaque. However, further research of these candidate genes was needed to confirm their effects in diabetic atherosclerotic plaque.
Acknowledgments
We thank Louise Adam, ELS(D), from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac) for editing the English text of a draft of this manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work; Yan Zhang and Yue Chen are the co-corresponding authors of this study.
Funding
This study was supported by the National Natural Science Foundation of China (82004117), Baoshan medical speciality project (BSZK-2018-A02), and the Cultivation Fund of Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine for NSFC (Grant Number: GZRPYJJ-201803).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci. 2018;132(12):1243–1252. doi:10.1042/CS20180306
2. Wierer M, Prestel M, Schiller HB, et al. Compartment-resolved proteomic analysis of mouse aorta during atherosclerotic plaque formation reveals osteoclast-specific protein expression. Mol Cell Proteomics. 2018;17(2):321–334. doi:10.1074/mcp.RA117.000315
3. Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017;12:18–34. doi:10.1016/j.redox.2017.01.007
4. de Gaetano M, McEvoy C, Andrews D, et al. Specialized pro-resolving lipid mediators: modulation of diabetes-associated cardio-, reno-, and retino-vascular complications. Front Pharmacol. 2018;9:1488.
5. Burke AP, Kolodgie FD, Zieske A, et al. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol. 2004;24(7):1266–1271. doi:10.1161/01.ATV.0000131783.74034.97
6. Kato K, Yonetsu T, Kim S-J, et al. Comparison of nonculprit coronary plaque characteristics between patients with and without diabetes: a 3-vessel optical coherence tomography study. JACC Cardiovasc Interv. 2012;5(11):1150–1158. doi:10.1016/j.jcin.2012.06.019
7. van Haelst ST, Haitjema S, de Vries J-P-P, et al. Patients with diabetes differ in atherosclerotic plaque characteristics and have worse clinical outcome after iliofemoral endarterectomy compared with patients without diabetes. J Vasc Surg. 2017;65(2):414–21. e5. doi:10.1016/j.jvs.2016.06.110
8. Tang J, Lobatto ME, Hassing L, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1(3):e1400223. doi:10.1126/sciadv.1400223
9. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12(1):10. doi:10.1038/nrcardio.2014.173
10. Barrett T, Suzek TO, Troup DB, et al. NCBI GEO: mining millions of expression profiles—database and tools. Nucleic Acids Res. 2005;33(suppl_1):D562–D6. doi:10.1093/nar/gki022
11. Dunning MJ, Smith ML, Ritchie ME, Tavaré S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics. 2007;23(16):2183–2184. doi:10.1093/bioinformatics/btm311
12. Smyth GK, Ritchie M, Thorne N, Wettenhall J. LIMMA: linear models for microarray data. in bioinformatics and computational biology solutions using R and bioconductor. Stat Biol Health. 2005.
13. Tang Y, Li M, Wang J, Pan Y, Wu F-X. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi:10.1016/j.biosystems.2014.11.005
14. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4(1):2.
15. Dweep H, Sticht C, Pandey P, Gretz N. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44(5):839–847. doi:10.1016/j.jbi.2011.05.002
16. Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. elife. 2015;4:e05005. doi:10.7554/eLife.05005
17. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2014;43(D1):D146–D52. doi:10.1093/nar/gku1104
18. Chou C-H, Shrestha S, Yang C-D, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2017;46(D1):D296–D302. doi:10.1093/nar/gkx1067
19. Han H, Cho J-W, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2017;46(D1):D380–D6. doi:10.1093/nar/gkx1013
20. Yahagi K, Kolodgie FD, Lutter C, et al. Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2017;37(2):191–204. doi:10.1161/ATVBAHA.116.306256
21. Madhur MS, Funt SA, Li L, et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31(7):1565–1572. doi:10.1161/ATVBAHA.111.227629
22. Kutikhin AG. Association of polymorphisms in TLR genes and in genes of the Toll-like receptor signaling pathway with cancer risk. Hum Immunol. 2011;72(11):1095–1116. doi:10.1016/j.humimm.2011.07.307
23. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi:10.3389/fimmu.2014.00461
24. Kim YM, Romero R, Oh SY, et al. Toll-like receptor 4: a potential link between “danger signals,” the innate immune system, and preeclampsia? Am J Obstet Gynecol. 2005;193(3 Pt 2):921–927. doi:10.1016/j.ajog.2005.07.076
25. Xu XH, Shah PK, Faure E, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104(25):3103–3108. doi:10.1161/hc5001.100631
26. Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. 2008;93(2):578–583. doi:10.1210/jc.2007-2185
27. Devaraj S, Tobias P, Jialal I. Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine. 2011;55(3):441–445. doi:10.1016/j.cyto.2011.03.023
28. Lu Z, Zhang X, Li Y, Lopes-Virella MF, Huang Y. TLR4 antagonist attenuates atherogenesis in LDL receptor-deficient mice with diet-induced type 2 diabetes. Immunobiology. 2015;220(11):1246–1254. doi:10.1016/j.imbio.2015.06.016
29. Jia S-J, Niu -P-P, Cong J-Z, Zhang B-K, Zhao M. TLR4 signaling: a potential therapeutic target in ischemic coronary artery disease. Int Immunopharmacol. 2014;23(1):54–59. doi:10.1016/j.intimp.2014.08.011
30. Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. 2011;50(1):87. doi:10.1007/s12026-011-8205-2
31. Peled M, Fisher EA. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front Immunol. 2014;5:579. doi:10.3389/fimmu.2014.00579
32. Luo S, Rubinsztein DC. BCL2L11/BIM: a novel molecular link between autophagy and apoptosis. Autophagy. 2013;9(1):104–105. doi:10.4161/auto.22399
33. Chuang PY, Dai Y, Liu R, et al. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS One. 2011;6(8):e23566. doi:10.1371/journal.pone.0023566
34. Erener S, Marwaha A, Tan R, Panagiotopoulos C, Kieffer TJ. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight. 2017;2(4). doi:10.1172/jci.insight.89656
35. Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nat Rev Cardiol. 2015;12(3):135. doi:10.1038/nrcardio.2014.207
36. Erdmann J, Stark K, Esslinger UB, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. 2013;504(7480):432. doi:10.1038/nature12722
37. Callegari A, Liu Y, White CC, et al. Gain and loss of function for glutathione synthesis: impact on advanced atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2473–2482. doi:10.1161/ATVBAHA.111.229765
38. Jain SK, Micinski D, Huning L, Kahlon G, Bass P, Levine SN. Vitamin D and L-cysteine levels correlate positively with GSH and negatively with insulin resistance levels in the blood of type 2 diabetic patients. Eur J Clin Nutr. 2014;68(10):1148. doi:10.1038/ejcn.2014.114
39. Vieira SM, Monteiro MB, Marques T, et al. Association of genetic variants in the promoter region of genes encoding p22phox (CYBA) and glutamate cysteine ligase catalytic subunit (GCLC) and renal disease in patients with type 1 diabetes mellitus. BMC Med Genet. 2011;12(1):129. doi:10.1186/1471-2350-12-129
40. Xu X, Luo P, Wang Y, Cui Y, Miao L. Nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) is a novel therapeutic target for diabetic complications. J Int Med Res. 2013;41(1):13–19. doi:10.1177/0300060513477004
41. Bhakkiyalakshmi E, Shalini D, Sekar TV, Rajaguru P, Paulmurugan R, Ramkumar KM. Therapeutic potential of pterostilbene against pancreatic beta-cell apoptosis mediated through Nrf2. Br J Pharmacol. 2014;171(7):1747–1757. doi:10.1111/bph.12577
42. Yagishita Y, Fukutomi T, Sugawara A, et al. Nrf2 protects pancreatic β-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes. 2014;63(2):605–618. doi:10.2337/db13-0909
43. Harada N, Ito K, Hosoya T, et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic Biol Med. 2012;53(12):2256–2262. doi:10.1016/j.freeradbiomed.2012.10.001
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.