Back to Journals » OncoTargets and Therapy » Volume 13
Identification of circRNA-miRNA-mRNA Networks for Exploring the Fundamental Mechanism in Lung Adenocarcinoma
Authors Liang L, Zhang L ![]() , Zhang J, Bai S, Fu H
, Zhang J, Bai S, Fu H
Received 22 October 2019
Accepted for publication 6 March 2020
Published 8 April 2020 Volume 2020:13 Pages 2945—2955
DOI https://doi.org/10.2147/OTT.S235664
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr XuYu Yang
Liming Liang,1 Liang Zhang,2 Jiguang Zhang,3 Shutang Bai,1 Hongdu Fu1
1No.2 Cardiothoracic Surgery, Central South University Xiangya School of Medicine Affiliated Haikou Hospital, Haikou City, Hainan Province 570208, People’s Republic of China; 2Department of Thoracic Surgery, The Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen City 518107, People’s Republic of China; 3Department of Thoracic Surgery, Fujian Provincial Hospital, Fuzhou City, Fujian Province 350001, People’s Republic of China
Correspondence: Liang Zhang
Department of Thoracic Surgery, The Seventh Affiliated Hospital, Sun Yat-sen University, No.628, Zhenyuan Road, Guangming (New) District, Shenzhen City 518107, People’s Republic of China
Tel/Fax +86-755-81206874
Email [email protected]
Background: Circular RNAs (circRNAs) were elucidated to act as competing endogenous RNAs (ceRNAs) and to play significant roles in cancer initiation and progression. We aimed to identify the important circRNAs in lung adenocarcinoma and intended to predict the functions of them via the bioinformatics analysis.
Methods: We extracted data from three Gene Expression Omnibus (GEO) datasets, GSE101586, GSE101684, and GSE104854, and identified the common differentially expressed circRNAs. Agarose gel electrophoresis and Sanger sequencing were used to verify these significant circRNAs. Then, qRT-PCR was performed to validate the expression through matched tissues and cell lines. Afterwards, a ceRNA network was constructed and functional analysis was performed to predict the potential mechanisms of circRNAs.
Results: Five circRNAs (hsa_circ_0072088, hsa_circ_0082564, hsa_circ_0008274, hsa_circ_0000519 and hsa_circ_0003528) were identified differentially expressed in the three datasets. Following the Agarose gel electrophoresis and qRT-PCR validation, hsa_circ_0072088 and hsa_circ_0008274 were chosen for further analysis. A ceRNA network of hsa_circ_0072088 and hsa_circ_0008274 was built based on the CSCD/TargetScan/MiRTarbase/StarBase 3.0. Then, the functional analysis showed that several meaningful terms were identified, such as “epithelial to mesenchymal transition (EMT)”, “Cellular response to TGF-β stimulus”, “MAPK signaling pathway”, and “PI3K-AKT signaling pathway”. Finally, several significant circRNA/miRNA/mRNA regulatory axes, which were predicted relating to cancer progress, were noted from the network.
Conclusion: We identified significant circRNAs and meaningful circRNA-miRNA-mRNA networks to provide novel insight into pathogenesis and therapy of lung adenocarcinoma.
Keywords: circRNAs, ceRNAs, GEO, lung adenocarcinoma, bioinformatics
Introduction
Pulmonary malignancy is the most commonly diagnosed cancer and ranks first among all cancers according to their incidence and mortality.1 Typically, lung cancer is classified into two major histologic types, non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), each has several subtypes. As a common form of NSCLC, lung adenocarcinoma (LUAC) accounts for approximately 50% and is increasing year by year, especially in young adults, women and never-smokers.2,3 Some serum tumor markers, such as squamous cell carcinoma antigen (SCCA) and carcinoembryonic antigen (CEA), and imaging modalities, like computed tomography (CT), are available for early-stage LUAC diagnosis. However, traditional tumor markers have relatively low specificity and sensitivity; meanwhile, CT might cause radiation damage on human body. Therefore, effective biomarkers are urgently needed for diagnosing early-stage LUAC. In recent years, with the development of molecular biology and the rise of translational medicine, novel markers and technologies are emerging for diagnosis and treatment of LUAC.4–6
Circular RNAs (circRNAs) are categorized as a type of endogenous non-coding RNAs (ncRNAs) and have been regarded as transcription errors during the splicing process. Recently, accumulating evidences have revealed that circRNAs, which formed a covalent closed continuous loop without a polyadenylated tail and 5ʹ to 3ʹ polarity, are of vital significance in various cellular processions, like gene expression regulation, cell cycle progression, chromatin modification, etc.7,8 Furthermore, circRNAs exhibit tissue-specific expression and incredible diversity, and they have stable structure and their abundance are associated with developmental age or stage. Moreover, many studies stated that aberrant expressions of circRNAs would play significant roles in the tumorigenesis and cancer progression.9 For instance, as shown in investigations, circPRKC1 was upregulated in lung adenocarcinoma and it could promote cell proliferation and migration by sponging miR545/589.10 Additionally, circRNA_102231, circFADS2, etc., have been proved to be candidate biomarkers and potential regulators for LUAC initiation and progression.11,12 Researches into circular RNAs are becoming a hotspot in the field of RNA for their various functions and specific properties. Nowadays, microarray and RNA-sequencing (RNA-seq) were widely used for measuring transcripts, discovering aberrantly expressed gene and identifying new transcriptional units; meanwhile, bioinformatics methods are usually used for processing these data. The Gene Expression Omnibus (GEO) database provides microarray and RNA-seq data for researchers around the world. Thus, GSE101586,13 GSE10168414 and GSE10485415 were utilized for discovering lung cancer-related circRNAs.
In this study, public datasets were employed for exploring the aberrant circRNAs and their potential molecular function. Also, we aimed to predict the potential mechanisms of these circRNAs by constructing a ceRNA network. Furthermore, several experiments, such as agarose gel electrophoresis and qRT-PCR, were performed. Above all, we provided powerful evidence for the preliminary study of lung cancer-related circRNAs. Naturally, further experiments were supposed to be performed in the coming exploration.
Methods
Specimen of LUAD Tissues
Forty LAC and corresponding adjacent normal tissues were obtained from 20 patients in the Seventh Affiliated Hospital, Sun Yat-sen University. The tissues were collected frozen in liquid nitrogen immediately after surgical resection and stored at −80°C until RNA extraction. None of these patients received radiotherapy, chemotherapy or target therapy before the surgery. All the patients had been informed; and written informed consents were obtained from them. The Ethics Committee of the Seventh Affiliated Hospital, Sun Yat-sen University approved this study.
Cell Culture
Two LUAD cell lines, A549 and H1299, and a normal cell line, 16HBE, were used in this study (Chinese Academic of Science). The 16HBE cells were cultured in DMEM, and A549 and H1299 cells were cultured in RPMI-1640 medium (Gibco, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. All the cell lines were maintained at 37°C in a humidified atmosphere with 5% CO2.
CircRNA Expression Profile Analysis
A flowchart of this study is shown in Figure 1. In considering the fewer samples explored in the single microarray, GEO database was used for searching any suitable datasets. Thus, GSE101586, GSE101684, and GSE104854 were selected (Details of these three datasets are shown in Table 1). Among them, GSE101586 and GSE101684 were processed in microarray, while GSE104854 was processed by high-throughput sequencing. We downloaded raw data and re-organized them as raw counts expression matrix. Then, limma R package was applied to normalize the raw counts (R code was provided in Table S2), plus, the fold-change and Student’s t testing were used for identifying differentially expressed circRNAs (DECs) in the two sets, respectively. The criterion for DECs was |fold-change|≧2 and P-value < 0.05. Furthermore, graphical heatmaps and RCircos plots were performed based on the circRNAs information.
 |
Table 1 Information of Expression Profiling Microarray Datasets |
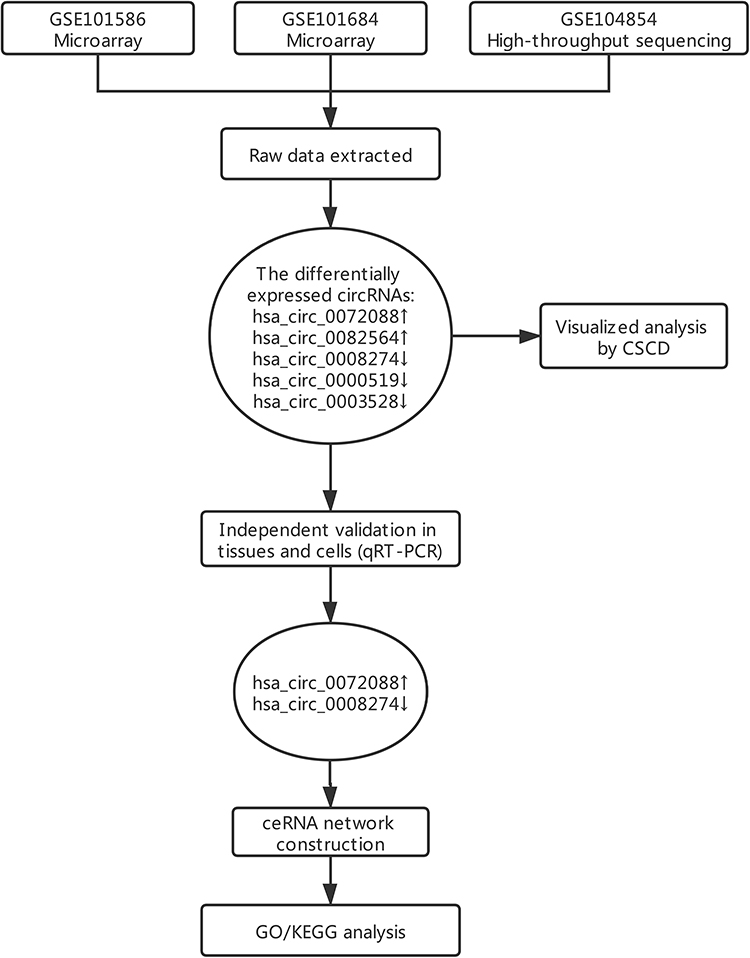 |
Figure 1 A flow chart of this study. |
Construction of circRNA-miRNA-mRNA Interaction Network
Cancer-Specific CircRNAs Database (CSCD) is an online tool for studying the structure model of cancer-specific circRNAs. In order to investigate the miRNAs sponged to DECs, CSCD was applied for searching the possible miRNA binding sites. Subsequently, the miRNA–mRNA interactions were obtained from MiRTarbase (http://miRTarBase.mbc.nctu.edu.tw/),16 TargetScan (http://www.targetscan.org/),17 and StarBase 3.0 (http://starbase.sysu.edu.cn/).18 Target mRNAs were intersected from the above.
Furthermore, the data of miRNAs and mRNAs, including their expression profile and clinical patient characteristics, were used for filtering the miRNAs and mRNAs which were significantly associated with patients’ survival. Thus, the key miRNAs and mRNAs were found by integrative analysis and the CircRNA-miRNA-mRNA network could be visualized by Cytoscape software.
Functional and Pathway Analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID)19 (http://david.abcc.ncifcrf.gov/) provides systematic and integrative annotation tools for predicting biological function of genes. We used the DAVID database to investigate the potential function of the key genes from the previous step to identify the underlying function of circRNAs. Significant results were obtained with P value < 0.05.
In addition, we analyzed the key genes related pathways according to the pathway analysis of KOBAS,20 an online database for functional gene set enrichment and functional annotation, and corrected P < 0.05 was considered statistically significant.
RNA Extraction and qRT-PCR
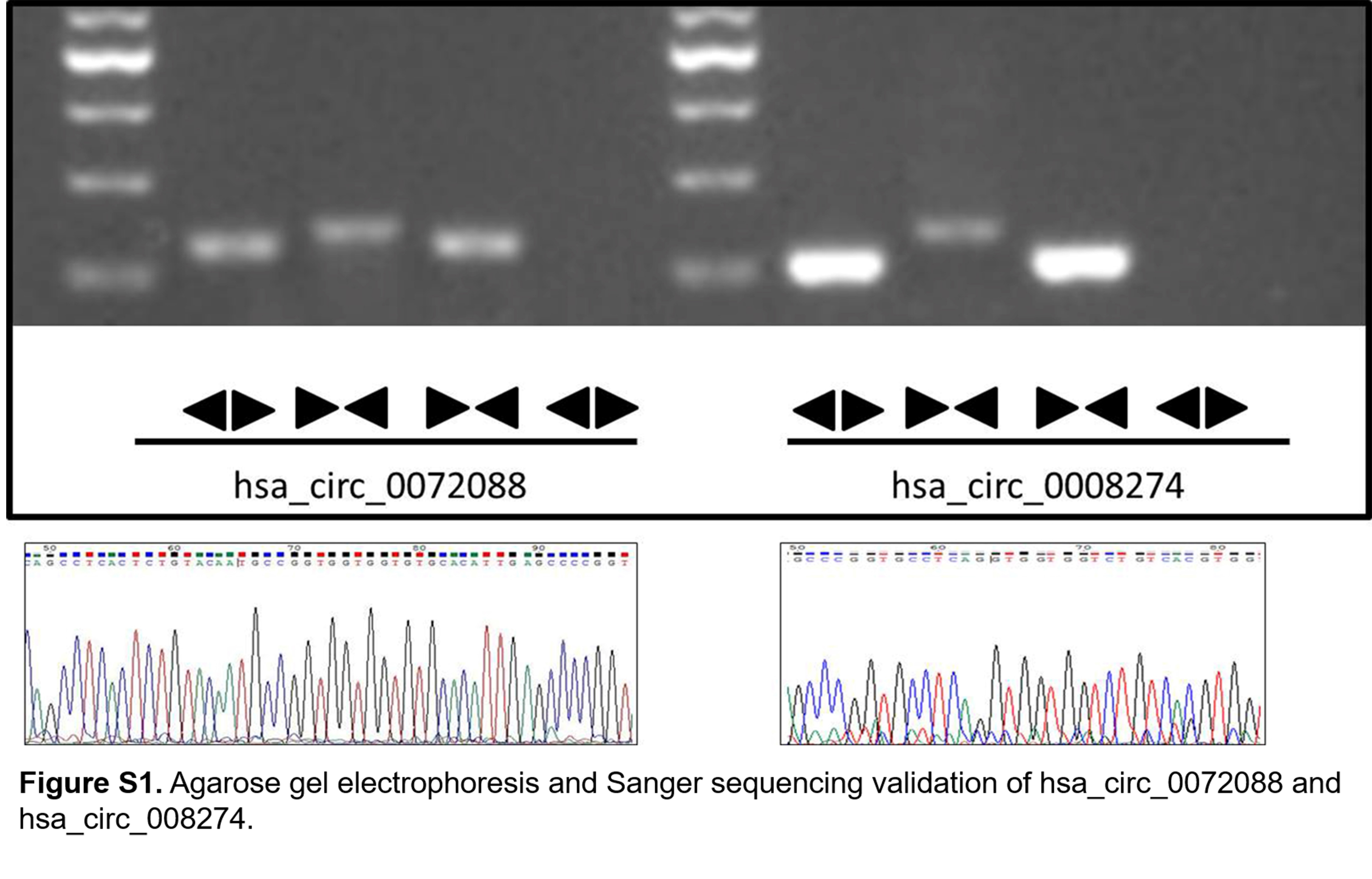
All the cell lines (16HBE, A549, and H1299) and 40 matched tissues were used to perform qRT-PCR. Total RNA of them was extracted by TRIzol Reagent (Invitrogen) and reversely transcribed with a reverse transcriptase kit from Takara Bio, Inc. In order to verify the circular form of these DECs, gel electrophoresis was performed and the PCR products were confirmed by Sanger sequencing. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control.
The relative expression levels of DECs were calculated using 2−ΔΔt, and GraphPad Prism 7.0 was used to visualize the results. The conditions and the primers used for the qRT-PCR reactions are described in Tables S1.
Results
Identification of DECs
Three microarray datasets, GSE101516, GSE101684, and GSE104854 were included in this study. Using the limma R package, we studied the DECs in these 3 datasets, respectively (logFC > 2 and p < 0.05). Furtherly, we summarized the general characteristics of the DECs obtained from the previous step. First, we took intersection of these 3 datasets (Figure 2), and the heatmaps are shown in Figure 3. The circRNA hsa_circ_0072088 and hsa_circ_0082564 were found upregulated, while the hsa_circ_0008274, hsa_circ_0000519, and hsa_circ_0003528 were identified downregulated in the results. Also, as can be seen from Figure 3, hsa_circ_0072088, hsa_circ_0082564, hsa_circ_0008274, hsa_circ_0000519, and hsa_circ_0003528 were located in different chromosomes (the basic information of these 5 circRNAs is shown in Table 2).
 |
Table 2 Basic Information of Differentially Expressed circRNAs |
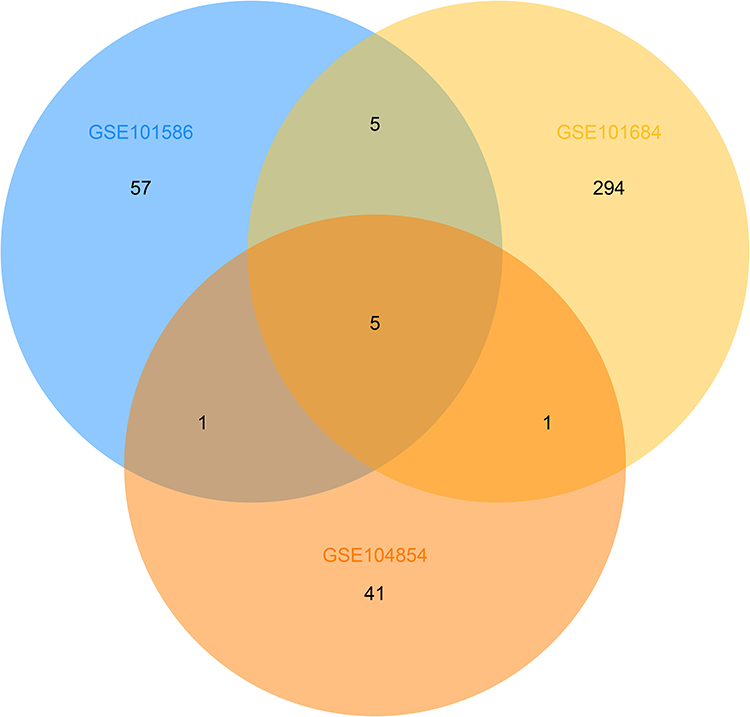 |
Figure 2 Identification of overlapping DECs in GSE101586, GSE101684 and GSE104854. |
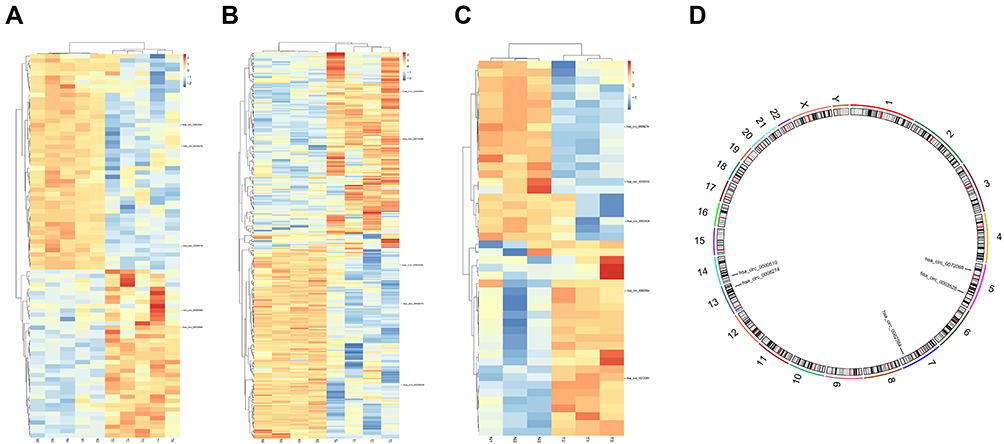 |
Figure 3 Heatmaps of differentially expressed circRNAs in 3 datasets (A–C) and the chromosomal location of 5 common circRNAs (D). |
Finally, we used CSCD, a comprehensive cancer-specific circRNA database and plays a vital role in the research for the function and regulation of cancer-associated circRNAs, to study the structure patterns of DECs (Figure 4).
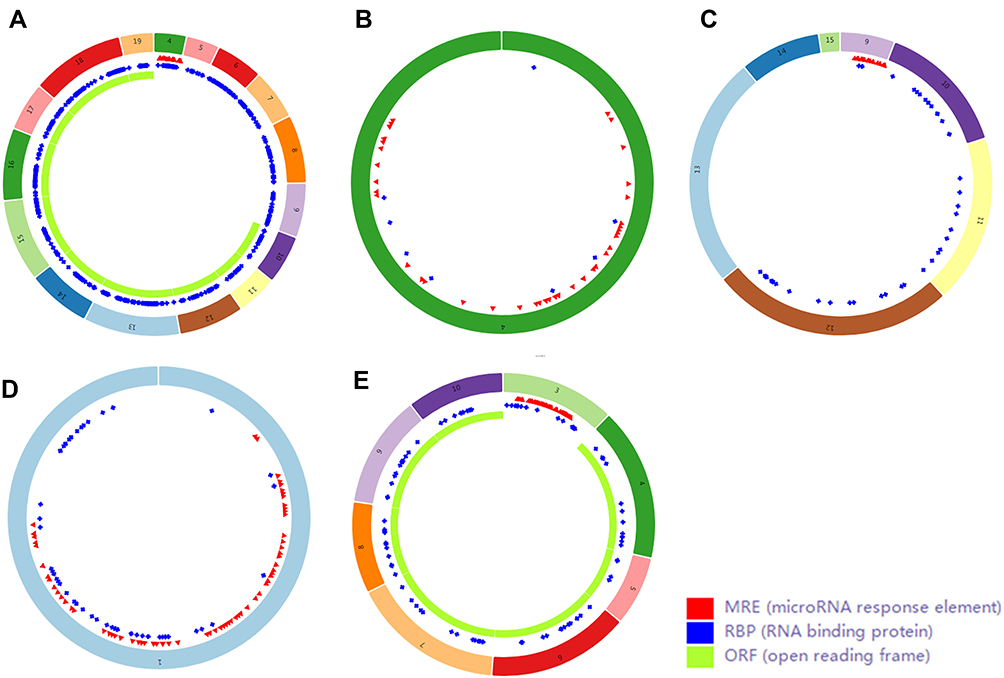 |
Figure 4 Structure patterns of hsa_circ_0072088 (A), hsa_circ_0082564 (B), hsa_circ_0008274 (C), hsa_circ_0000519 (D) and hsa_circ_0003528 (E) based on the CSCD database. |
Identification of DECs by qRT-PCR
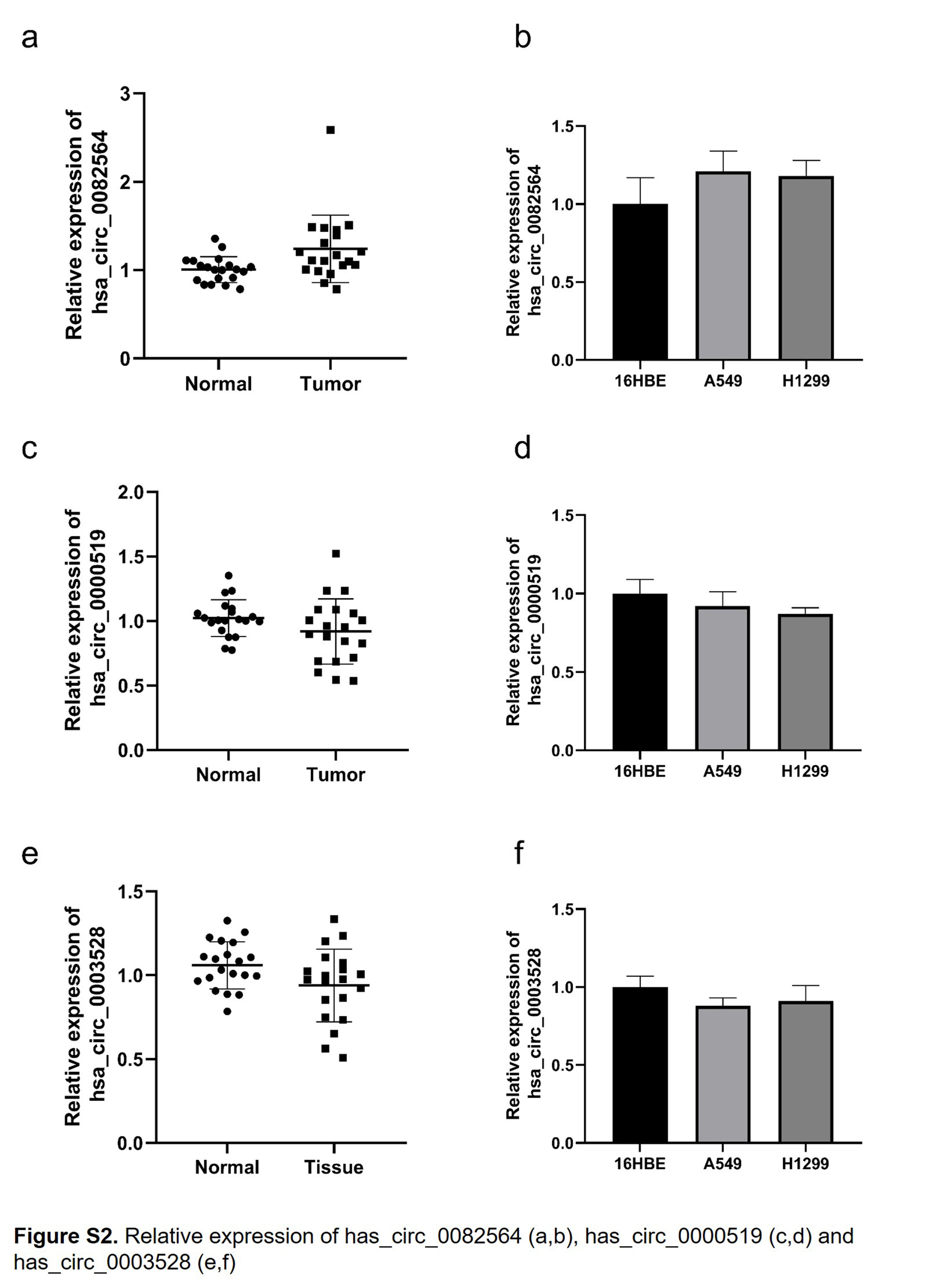
Considering the signature value of DECs, qRT-PCR was performed for validation of DECs expression. In this step, the cell lines (16HBE, A549, and H1299) and 20 matched tissues were used to perform qRT-PCR. In order to ensure the specialty and back-spliced junction of DECs, agarose gel electrophoresis and Sanger sequencing were performed (Figure S1). Then, qRT-PCR showed that hsa_circ_0082564 and hsa_circ_0000519 cannot be detected for their relatively low expression level in tumor tissue, and hsa_circ_0003528 presented the downregulated trend, with a P value > 0.05 (Figure S2). Finally, hsa_circ_0072088 and hsa_circ_0008274 were found differentially expressed in normal tissues compared to cancerous tissues with a P value < 0.05. Also, the circRNA expression validation was performed using cells with the same result (Figure 5).
{kind=link}
{kind=link}
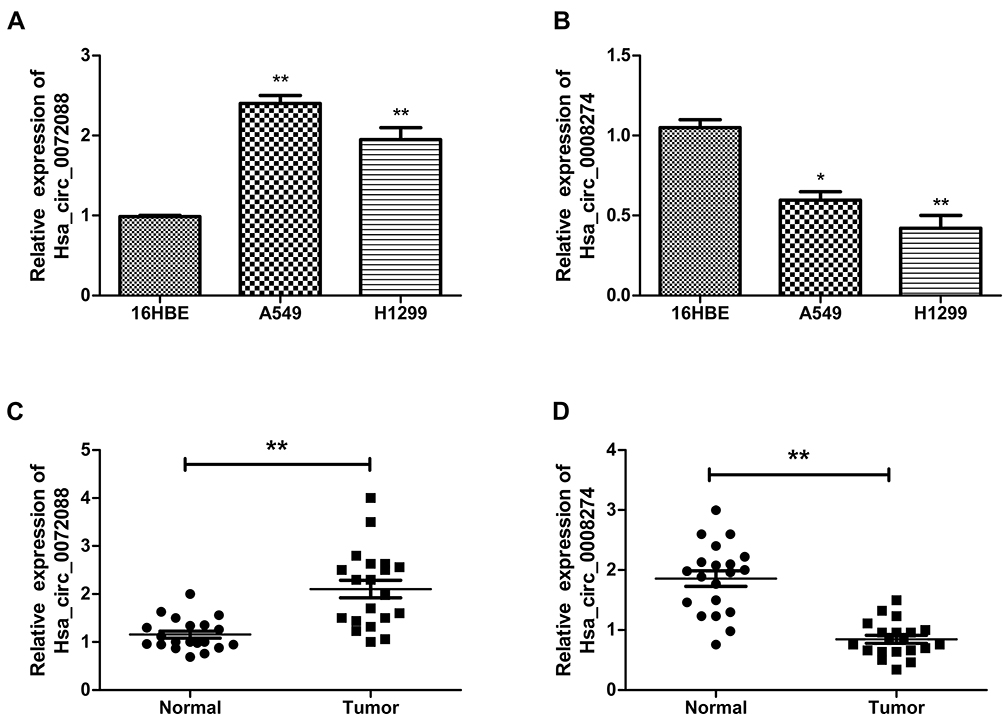 |
Figure 5 Expression of hsa_circ_0072088 in tissues and cell lines (A, C). Expression of hsa_circ_008274 in tissues and cell lines (B, D). **P<0.01.*P<0.05. |
Construction of a ceRNA Network
“miRNA sponge” has been largely acknowledged as a potential mechanism of circRNA working. Thus, a ceRNA network was constructed as per the DECs. CSCD database was used for circRNA-miRNA prediction, and the regulator relationships of miRNA-mRNA were obtained from MiRTarbase, TargetScan, and StarBase 3.0. The circRNA-miRNA-mRNA network was then constructed by Cytoscape (Figure 6). Interestingly, we found that miR-578 was predicted to be common downstream of hsa_circ_0072088 and hsa_circ_0008274, which indicated that there might be some functional connection between them.
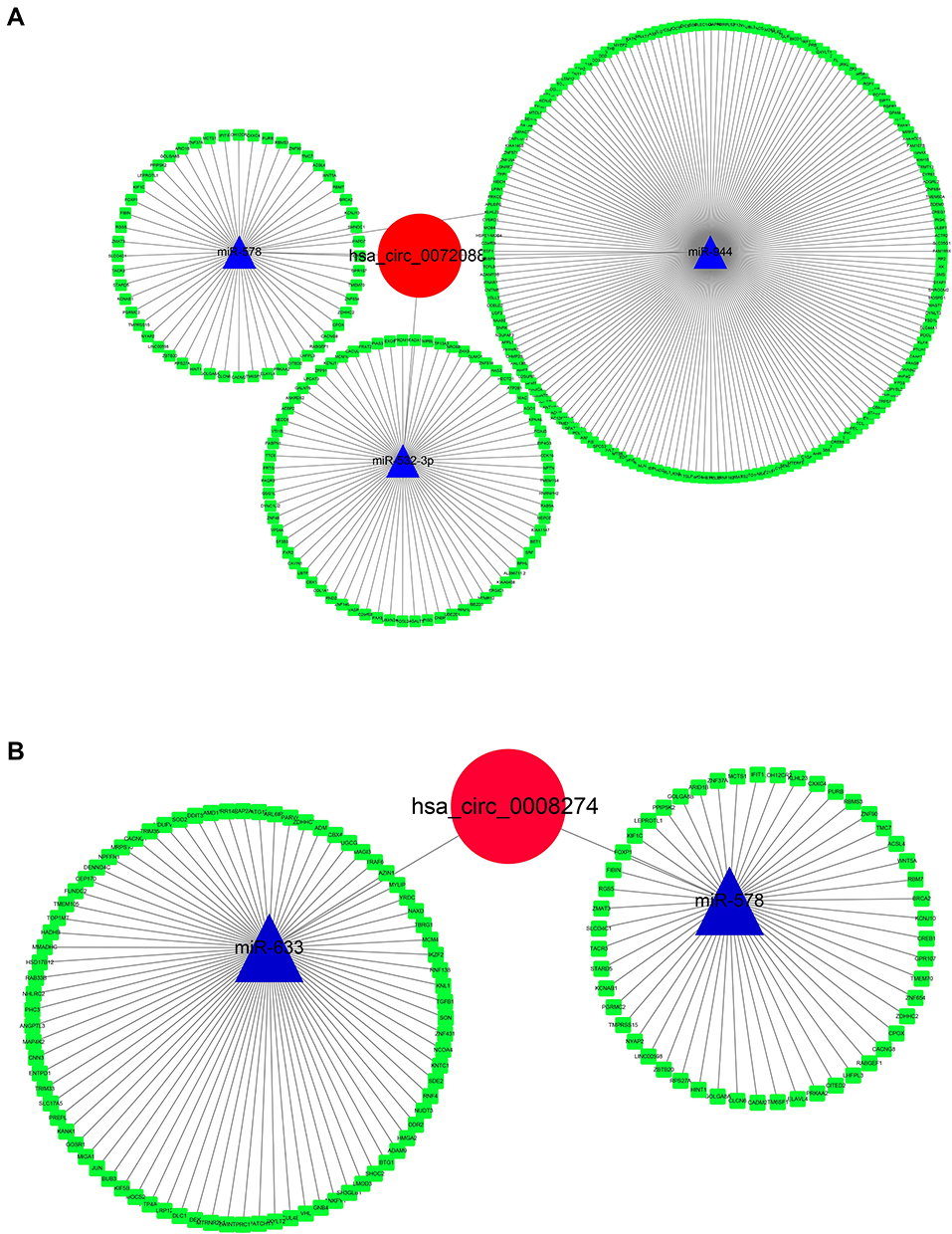 |
Figure 6 Construction of circRNA-miRNA-mRNA network of hsa_circ_0072088 (A) and hsa_circ_008274 (B). |
Functional Analysis of mRNAs
As a novel non-coding RNA, circRNA was still not well understood. Although the functional analysis was not directed on DECs, we considered that the mRNAs may play significant roles in ceRNA network and the analysis on mRNAs could reveal the potential mechanisms of DECs. Thus, DAVID and KOBAS online database were applied for further exploration. By submitting the mRNAs obtained above, GO and KEGG analysis were performed.
As can be seen from Figure 7, the GO analysis of hsa_circ_0072088 was mainly enriched in regulation of cellular response to growth factor stimulus, cellular response to transforming growth factor beta stimulus, response to transforming growth factor beta, etc., which revealed that the circRNA might play roles in tumor growth and apoptosis. Fewer cancer-related pathways were obtained from the KEGG analysis, and MAPK signaling pathway was worthy of special attention.
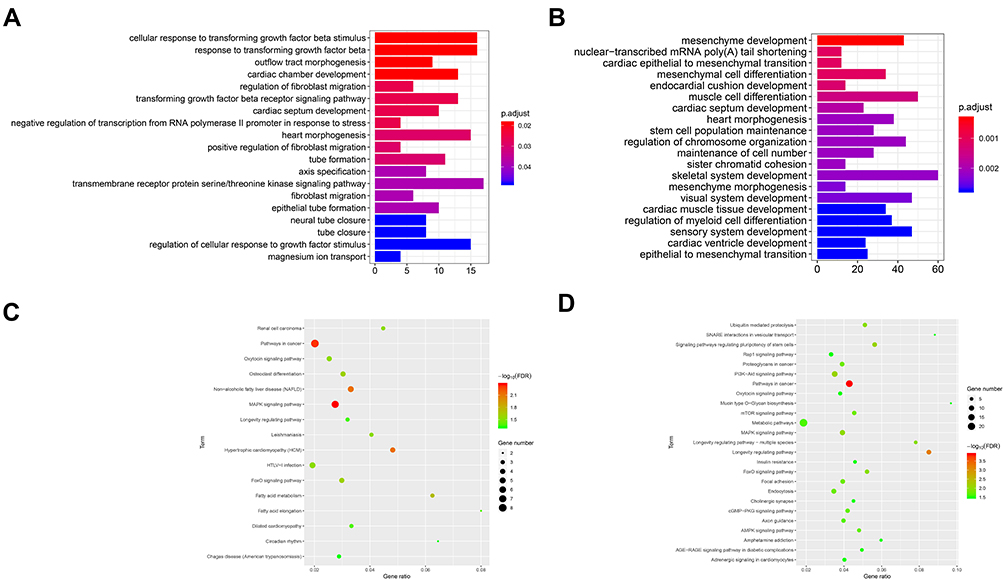 |
Figure 7 GO and KEGG analysis of hsa_circ_0072088 (A, C) and hsa_circ_008274 (B, D). |
As for hsa_circ_0008274, we noticed that the GO analysis was mainly enriched in skeletal system development, mesenchyme development, muscle cell differentiation, etc., and it seemed like there was a faint relationship between them and lung cancer. Then, we noticed that epithelial to mesenchymal transition (EMT) was obtained, which had been proved to play important roles in cancer invasion and metastasis. The KEGG analysis showed promising results that some famous pathways, like mTOR signaling pathway, MAPK signaling pathway, and PI3K−Akt signaling pathway were obtained, which provides more possibility on the assumption that hsa_circ_0008274 may play significant roles in cancer initiation and progression.
Key circRNA-miRNA-mRNA Axis Analysis
In order to further understand the potential function of circRNAs and to provide valuable information for experiments, we reorganized the circRNA-miRNA-mRNA axis according to the results of functional analysis.
From the previous steps, multiple GO terms and KEGG pathways were found, and mRNAs were clustered in any different functions. Next, we reorganized the mRNAs which were significantly associated with cancer progression and linked their upstream miRNA and circRNA (Table 3). We thought these axes could provide more information and more appropriate for specific experiments.
 |
Table 3 Cancer-Related circRNA/miRNA/mRNA Axis |
Discussion
CircRNAs, which lacked 5ʹ-caps and 3ʹ-polyadenylated tails, was first put forward by Sanger and had been ignored in traditional transcriptome studies for decades.21 Particularly, with next-generation sequencing and computational biology methods developed, a great deal of circRNAs was identified and found to play significant roles in multiple tissues and cells.22 Nowadays, exploring the underlying mechanisms and functions of circRNAs has attracted more and more attention. Among the studies, many researchers have indicated that circRNAs play significant roles in different malignant tumors. For instance, hsa_circ_0013958 was identified to be overexpressed in the plasma and tissue of non-small cell lung cancer, and its expression level was meaningfully related with the tumor-node-metastasis (TNM) and lymphatic metastasis.23 Also, hsa_circ_0058246 was found to be upregulated in gastric cancer (GC) and associated with tumor recurrence.24 In addition, increasing studies have shown that circRNAs may serve as important regulators in various cancers.25–27 However, the novel circRNAs and the function they played were still needed to explore.
In this study, the public database, GEO, was used to collect the circRNA expression. GSE101586, GSE101684, and GSE104854 were chosen for analyzing in lung adenocarcinoma. Considering for removing any other factor, we downloaded the raw data of these three datasets, and reorganized them into matrix file. Limma R package was used to explore the differentially expressed circRNAs, respectively. Also, in order to focus on the most significant circRNAs, we took the intersection of 3 datasets. Three heatmaps and a Circos plot were drawn to show relative expression and the location of DECs. In the result, hsa_circ_0072088 and hsa_circ_0082564 were found upregulated, while the hsa_circ_0008274, hsa_circ_0000519, and hsa_circ_0003528 were found downregulated in lung adenocarcinoma. Furthermore, qRT-PCR and agarose gel electrophoresis were performed, and the results were verified by Sanger sequencing. Hsa_circ_00720881 and hsa_circ_0008274 were then identified indeed different expressed in tissues and cells. As conserved endogenous RNAs, circRNAs could sponge the miRNAs and play significant roles in regulating miRNA target genes.28,29 In order to explore the aforementioned two circRNAs that sponged miRNAs, an online tool, CSCD, was used, and it could predict the MREs within 50bp downstream and upstream of junction point of circRNA.30 Then, miRNA-targeted mRNAs were predicted by using MiRTarbase, TargetScan, and StarBase 3.0; thus the ceRNA network was constructed.
Furthermore, the genes were input following the collection of then, and then GO and KEGG analysis were performed. As can be seen, the mRNAs, which were predicted the downstream of hsa_circ_0008274, were enriched in various functions. We highlighted the functions related to lung cancer, like “regulation of cellular response to growth factor stimulus”, “cellular response to TGF-β stimulus”, “response to transforming growth factor beta”, and “MAPK signaling pathway”. Also, we noticed the “EMT”, which was proved to be associated with cancer invasion and metastasis, were obtained in the hsa_circ_0008274. The pathways, such as mTOR signaling pathway, MAPK signaling pathway, and PI3K−Akt signaling pathway, give more enlightenment on researches into tumor growth and progression. Thus, we further reorganized the ceRNA network and selected the most interested circRNA/miRNA/mRNA axes, like “EMT”, “Cellular response to TGF-β stimulus”, “MAPK signaling pathway”, and “PI3K-AKT signaling pathway”, with details noted in Table 3.
Nowadays, biological big data was emerging, and bioinformatics was gaining more and more attention. Some public databases, like GEO and the cancer genome atlas (TCGA), were widely used for data analysis. Since the TCGA has no public circRNA data, we just chose the GEO database to study and validate with our own tissues and cell lines. As is known, the bioinformatics analysis provided guidance and suggestions for follow-up experiments, saved money for researches, and improved efficiency. Thus, a series of analysis were performed in this study. Besides, Sun et al and Guan et al also performed bioinformatical analysis for circRNAs in hepatocellular carcinoma and gastric cancer, which used different methods and studied the ceRNA in different angles.31,32 Thus, it would be reasonable to believe that bioinformatics provide guidance for our study. Certainly, further rigorous experiments are necessary to validate these findings. Given that the results are based on computational biology, further in-depth studies on circRNAs are indispensable to verify their possible roles in lung adenocarcinoma.
Funding
This study was supported by the Training Project of Middle and Young Cadre Talent in Fujian Provincial Health Planning (the role and mechanism of the EZH2/h3-k27 trimethylation/DAPK pathway in the regulation of proliferation, invasion and apoptosis of non-small cell lung cancer) (No. 2016-ZQN-2).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Hsu CL. Advanced non-small cell lung cancer in patients aged 45 years or younger: outcomes and prognostic factors. BMC Cancer. 2012;12(1):241. doi:10.1186/1471-2407-12-241
3. Torresdurán M, Barrosdios JM, Fernándezvillar A, Ruanoravina A. Residential radon and lung cancer in never smokers. A systematic review. Cancer Lett. 2014;345(1):21–26. doi:10.1016/j.canlet.2013.12.010
4. Lu T, Wang Y, Chen D, Liu J, Jiao W. Potential clinical application of lncRNAs in non-small cell lung cancer. Oncol Targets Ther. 2018;11:8045–8052. doi:10.2147/OTT.S178431
5. Weidle UH, Birzele F, Nopora A. MicroRNAs as potential targets for therapeutic intervention with metastasis of non-small cell lung cancer. Cancer Genom Proteom. 2019;16(2):99–119. doi:10.21873/cgp.20116
6. de Fraipont F, Gazzeri S, Cho WC, Eymin B. Circular RNAs and RNA splice variants as biomarkers for prognosis and therapeutic response in the liquid biopsies of lung cancer patients. Front Genet. 2019;10:390. doi:10.3389/fgene.2019.00390
7. Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96(4):1297–1325. doi:10.1152/physrev.00041.2015
8. Wang Y, Lu T, Wang Q, Liu J, Jiao W. Circular RNAs: crucial regulators in the human body (Review). Oncol Rep. 2018;40(6):3119–3135. doi:10.3892/or.2018.6733
9. Hu W, Bi ZY, Chen ZL, et al. Emerging landscape of circular RNAs in lung cancer. Cancer Lett. 2018;427:18–27. doi:10.1016/j.canlet.2018.04.006
10. Qiu M, Xia W, Chen R, et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 2018;78(11):
11. Zong L, Sun Q, Zhang H, et al. Increased expression of circRNA_102231 in lung cancer and its clinical significance. Biomed Pharmacother. 2018;102:639–644. doi:10.1016/j.biopha.2018.03.084
12. Zhao F, Han Y, Liu Z, Zhao Z, Li Z, Jia K. circFADS2 regulates lung cancer cells proliferation and invasion via acting as a sponge of miR-498. Biosci Rep. 2018;38(4):BSR20180570. doi:10.1042/BSR20180570
13. Chen T, Yang Z, Liu C, et al. Circ_0078767 suppresses non-small-cell lung cancer by protecting RASSF1A expression via sponging miR-330-3p. Cell Prolif. 2019;52(2):e12548. doi:10.1111/cpr.2019.52.issue-2
14. Zhao J, Li L, Wang Q, Han H, Zhan Q, Xu M. CircRNA expression profile in early-stage lung adenocarcinoma patients. Cell Physiol Biochem. 2017;44(6):2138–2146. doi:10.1159/000485953
15. Ding X, Zhang S, Li X, et al. Profiling expression of coding genes, long noncoding RNA, and circular RNA in lung adenocarcinoma by ribosomal RNA-depleted RNA sequencing. FEBS Open Bio. 2018;8(4):544–555. doi:10.1002/feb4.2018.8.issue-4
16. Chou CH, Shrestha S, Yang CD, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2017;46(Database issue):D296–D302.
17. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005. doi:10.7554/eLife.05005
18. Jun-Hao L, Shun L, Hui Z, Liang-Hu Q, Jian-Hua Y. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92. doi:10.1093/nar/gkt1248
19. Da WH, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009. doi:10.1038/nprot.2009.154
20. Wu J, Mao X, Cai T, Luo J, Wei L. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006;34(WebServer issue):W720–W724. doi:10.1093/nar/gkl167
21. Lei K, Bai H, Wei Z, et al. The mechanism and function of circular RNAs in human diseases. Exp Cell Res. 2018;368(2):147–158. doi:10.1016/j.yexcr.2018.05.002
22. Chen -L-L, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–388. doi:10.1080/15476286.2015.1020271
23. Zhu X, Wang X, Wei S, et al. hsa_circ_0013958: a circular RNA and potential novel biomarker for lung adenocarcinoma. FEBS J. 2017;284(14):2170–2182. doi:10.1111/febs.2017.284.issue-14
24. Fang Y, Ma M, Wang J, Liu X, Wang Y. Circular RNAs play an important role in late-stage gastric cancer: circular RNA expression profiles and bioinformatics analyses. Tumor Biol. 2017;39(6):1010428317705850. doi:10.1177/1010428317705850
25. Xuan L, Qu L, Zhou H, et al. Circular RNA: a novel biomarker for progressive laryngeal cancer. Am J Transl Res. 2016;8(2):932.
26. Xie H, Ren X, Xin S, et al. Emerging roles of circRNA_001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget. 2016;7(18):26680.
27. Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA‐9 to suppress hepatocellular carcinoma progression. Hepatology (Baltimore, Md). 2017;66(4):1151–1164. doi:10.1002/hep.29270
28. Zhang Y, Liang W, Zhang P, et al. Circular RNAs: emerging cancer biomarkers and targets. J Exp Clin Cancer Res. 2017;36(1):152. doi:10.1186/s13046-017-0624-z
29. Chen -L-L. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17(4):205. doi:10.1038/nrm.2015.32
30. Xia S, Feng J, Chen K, et al. CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res. 2017;46(D1):D925–D929. doi:10.1093/nar/gkx863
31. Sun X, Ge X, Xu Z. Identification of circular RNA–microRNA–messenger RNA regulatory network in hepatocellular carcinoma by integrated analysis. J Gastroenterol Hepatol. 2019.
32. Guan Y-J, Ma J-Y; International WSJCC. Identification of circRNA–miRNA–mRNA regulatory network in gastric cancer by analysis of microarray data. Cancer Cell Int. 2019;19(1):183
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.