Back to Journals » Veterinary Medicine: Research and Reports » Volume 5
Identification of canine platelet proteins separated by differential detergent fractionation for nonelectrophoretic proteomics analyzed by Gene Ontology and pathways analysis
Authors Trichler S, Bulla S, Mahajan N ![]() , Lunsford K, Pendarvis K, Nanduri B, McCarthy F, Bulla C
, Lunsford K, Pendarvis K, Nanduri B, McCarthy F, Bulla C
Received 24 April 2013
Accepted for publication 19 June 2013
Published 21 January 2014 Volume 2014:5 Pages 1—9
DOI https://doi.org/10.2147/VMRR.S47127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Shauna A Trichler,1,* Sandra C Bulla,1,* Nandita Mahajan,1 Kari V Lunsford,2 Ken Pendarvis,3 Bindu Nanduri,4,5 Fiona M McCarthy,3 Camilo Bulla1
1Department of Pathobiology and Population Medicine, 2Department of Clinical Sciences and Animal Health Center, College of Veterinary Medicine, Mississippi State University, Mississippi State, MS, 3Department of Veterinary Science and Microbiology, University of Arizona, Tucson, AZ, 4Department of Biological Sciences, College of Veterinary Medicine, Mississippi State University, Mississippi State, MS, 5Institute for Genomics, Biocomputing and Biotechnology, Starkville, MS, USA
*These authors contributed equally to this work
Abstract: During platelet development, proteins necessary for the many functional roles of the platelet are stored within cytoplasmic granules. Platelets have also been shown to take up and store many plasma proteins into granules. This makes the platelet a potential novel source of biomarkers for many disease states. Approaches to sample preparation for proteomic studies for biomarkers search vary. Compared with traditional two-dimensional polyacrylamide gel electrophoresis systems, nonelectrophoretic proteomics methods that employ offline protein fractionation methods such as the differential detergent fractionation method have clear advantages. Here we report a proteomic survey of the canine platelet proteome using differential detergent fractionation coupled with mass spectrometry and functional modeling of the canine platelet proteins identified. A total of 5,974 unique proteins were identified from platelets, of which only 298 (5%) had previous experimental evidence of in vivo expression. The use of offline prefractionation of canine proteins by differential detergent fractionation resulted in greater proteome coverage as compared with previous reports. This initial study contributes to a broader understanding of canine platelet biology and aids functional research, identification of potential treatment targets and biomarkers, and sets a new standard for the resting platelet proteome.
Keywords: proteome, differential detergent fractionation, dog, functional analysis, protein
Introduction
The roles that platelets play in blood hemostasis, wound healing, inflammation, and thrombus formation are well described, and more recently, platelets have also been recognized for their interactions with tumor cells.1 During platelet development, proteins necessary for the many functional roles of the platelet are stored within cytoplasmic granules. Platelets have been shown to take up and store many plasma proteins into granules. This makes the platelet a potential novel source of biomarkers for many disease states.2 Naturally occurring disease in the dog, a companion animal of veterinary importance, is gaining recognition as a valuable model of human disease.3,4 Translational studies focusing on identification of novel platelet protein markers for disease using the dog first require a description of the basal canine platelet proteome. This will allow for the necessary comparison of the canine and human platelet proteome.
Approaches to sample preparation for proteomic studies vary. Compared with traditional two-dimensional polyacrylamide gel electrophoresis systems, nonelectrophoretic proteomics methods that employ offline protein fractionation methods, such as the differential detergent fractionation (DDF) method described by McCarthy et al,5 have clear advantages. This technique, for instance, increases the yield of hydrophobic membrane proteins due to protease digestion based on the physicochemical properties of peptides. Because the DDF method assigns GO (Gene Ontology) cellular component terms to proteins based on the detergent fraction they were extracted in, as well as the number of transmembrane domains the protein contains, it allows identification of the subcellular localization of proteins expressed within the cell. Here we report a proteomic survey of the canine platelet proteome using DDF, mass spectrometry, and subsequent functional modeling. The canine platelet proteome described here provides evidence for in vivo expression or uptake of these proteins in the canine platelet. Additionally, since the method used for protein fractionation could predict subcellular localization of the proteins, we could also add to the “cellular component” of protein functional annotation based on subcellular localization as described below.
Materials and methods
Platelet isolation
Normal resting platelets were purified from the blood (collected in ethylenediaminetetraacetic acid) of three adult healthy female Walker hounds. Whole blood (3 mL) was layered on top of 3 mL of a 1.077 g/mL density gradient and spun at 400 g for 30 minutes at room temperature. The resulting top fraction was washed with HEPES-NaCl, layered over a combination of diluted 60% iodixanol 1.32 g/mL density barriers (10% and 13% stock solution, from top to bottom), and spun at 300 g for 20 minutes. The top fraction was washed, pelleted, and resuspended in HEPES-NaCl. Cell counts were performed manually with a hemocytometer and samples were found to have an average platelet purity of 99.98% ± 0.02%. The sample was then pelleted again and stored at −80°C. Platelet pellets from a total of 20 mL of blood from each dog were subjected to protein extraction (average of 4.7 ± 4.1 × 108 total platelets).
Protein extraction and identification
Proteins were isolated as previously described.5 Briefly, four different extractions using digitonin, Triton X-100, deoxycholate plus Tween 40, and sodium dodecyl sulfate were used to sequentially isolate proteins based upon decreasing solubility. Each of the fractions was precipitated with 25% trichloroacetic acid to remove salts and detergents, and trypsin digested. Peptides were desalted and then cleaned using a reverse phase macrotrap and strong cation exchange macrotrap (Michrom Bioresources Inc., Auburn, CA, USA), respectively, according to the manufacturer’s instructions. Samples were then dried and resuspended in 2% acetonitrile, 0.1% formic acid.
Peptide mass spectrometry was accomplished using an EASY-nLC (Thermo Scientific, Rockford, IL, USA) high performance liquid chromatography machine coupled with an LTQ Velos linear ion trap mass spectrometer (Thermo Scientific). The Easy-nLC was configured for reverse phase chromatography using a Hypersil Gold KAPPA C18 column (Thermo Scientific) with a flow rate of 333 nanoliters per minute. Peptides were separated for mass spectrometry analysis using an acetonitrile gradient starting at 2% acetonitrile, 0.1% formic acid and reaching 50% acetonitrile, 0.1% formic acid in 120 minutes, followed by a 15-minute wash of 95% acetonitrile, 0.1% formic acid. Column equilibration was handled automatically using the EASY-nLC. The eluate from the high performance liquid chromatography was fed directly to the LTQ Velos for nanospray ionization followed by tandem mass spectrometry analysis of detected peptides. The LTQ Velos was configured to perform one mass spectrometry scan followed by 20 tandem mass spectrometry scans of the 20 most intense peaks repeatedly over the 172-minute duration of each high performance liquid chromatography run. Dynamic exclusion was enabled with a duration of 5 minutes, repeat count of one, and a list length of 500. The collected spectra were subsequently analyzed using the X!Tandem6 (Open-source software, The Global Proteome Machine Organization) search algorithm.
Raw spectral data from the LTQ Velos were converted to mzML format using the msConvert tool from the ProteoWizard (Open-source software) software project7 because X!Tandem cannot read the Thermo raw format directly. The FASTA database used for peptide spectrum matching (target database) was the Canis familiaris RefSeq protein database from the National Center for Biotechnology Information. X!Tandem was configured to use tryptic cleavage sites with up to two missed cleavages. Precursor and fragment mass tolerance were set to 1000 ppm and 500 ppm, respectively. Four amino acid modifications were included in the database search, ie, single and double oxidation of methionine and both carboxymethylation and carboxamidomethylation of cysteine. A decoy search was also performed using a randomized version of the target database with the same search parameters as before. The search results were filtered using the methods described previously.8,9 A decoy score distribution of X!Tandem hyperscores and e-values was created and each match from the target database was evaluated as a possible outlier and assigned a probability of being such. Peptides from the target database were accepted if the probability of being an outlier was 95% or higher. A list of proteins and identified peptides was generated for each experimental sample.
Functional analysis of platelet proteome
Platelet proteins identified using mass spectrometry were analyzed using AgBase tools (Open-source software, Agbase, Mississippi State University).10 GORetriever (Open-source software, Agbase, Mississippi State University) was used to obtain GO annotation and these results summarized using GOSlimViewer with the Generic Slim set.11 We compared the function of the platelet proteome against the GO for the entire GO proteome by downloading the dog gene association file from AgBase (December 14, 2012) and comparing the relative proportions for each slimmed GO term.
Platelet proteins identified by mass spectrometry were also analyzed using DDF2GO,12 a tool that groups proteins into GO cellular component categories based on the fraction they were extracted in as well as their number of transmembrane domains, predicted using PSORTII13 (Open-source software, Human Genome Center, Tokyo, Japan) and TMpred14 (Open-source software, TMbase) online prediction softwares. DDF2GO uses a GO CC mapping table, previously established by studies from McCarthy et al using proteins’ DDF fractions and transmembrane domains, which dictates GO CC assignment conditions based on DDF fraction percentages and average transmembrane domains. Since platelets are anucleate, the annotations to the GO term “nucleus” were not included in the results.
Pathways analysis
We used Ingenuity Pathways Analysis (Ingenuity Systems Inc., Redwood City, CA, USA) to identify significant molecular functions and canonical pathways represented by platelet proteins as described earlier.15 A right-tailed Fisher’s Exact test was used to calculate a P-value determining the probability that each biological function assigned to that data set is due to chance alone. All molecular functions and metabolic and signaling pathways identified at a P-value of ≤0.05 were considered to be statistically significant.
Results
Protein identification
We identified 5,974 unique proteins from platelets collected from three healthy dogs (https://www.dovepress.com/get_supplementary_file.php?f=supplementary_file_47127.pdf). Only 298 (5%) of these identified proteins had previous experimental evidence of in vivo expression. The remaining 5,676 (95%) of the identified proteins are computationally predicted based on homology to known proteins in related species.
Functional analysis
Our GO analysis of the proteins identified by mass spectrometry indicated that 86% of the 5,974 proteins we identified had GO annotation. Using the generic GO Slim to summarize the GO function for our proteins, we identified the general functions represented in our data set. The most under-represented and over-represented GO functions in the canine platelet proteome dataset compared with the canine genome are shown in Figure 1, and the top ten GO terms for each ontology are shown in Figure 2.
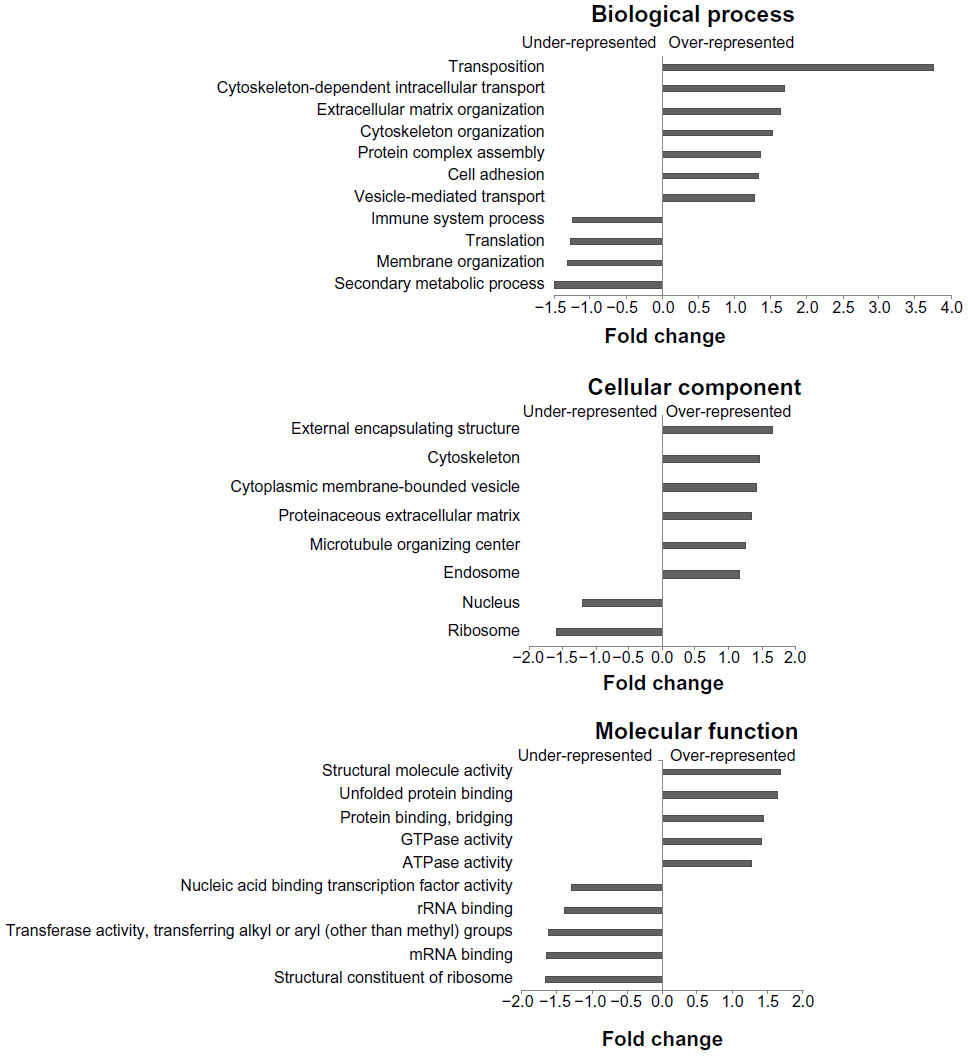 | Figure 1 Canine platelet proteome GO categories compared with the canine genome. The graphs show important under-represented and over-represented GO functions in canine platelets. The y-axis shows the categories within functional groups and the x-axis displays the relative difference between the percentages of the categories in the platelet proteome and the percentages in all canine GO annotations. |
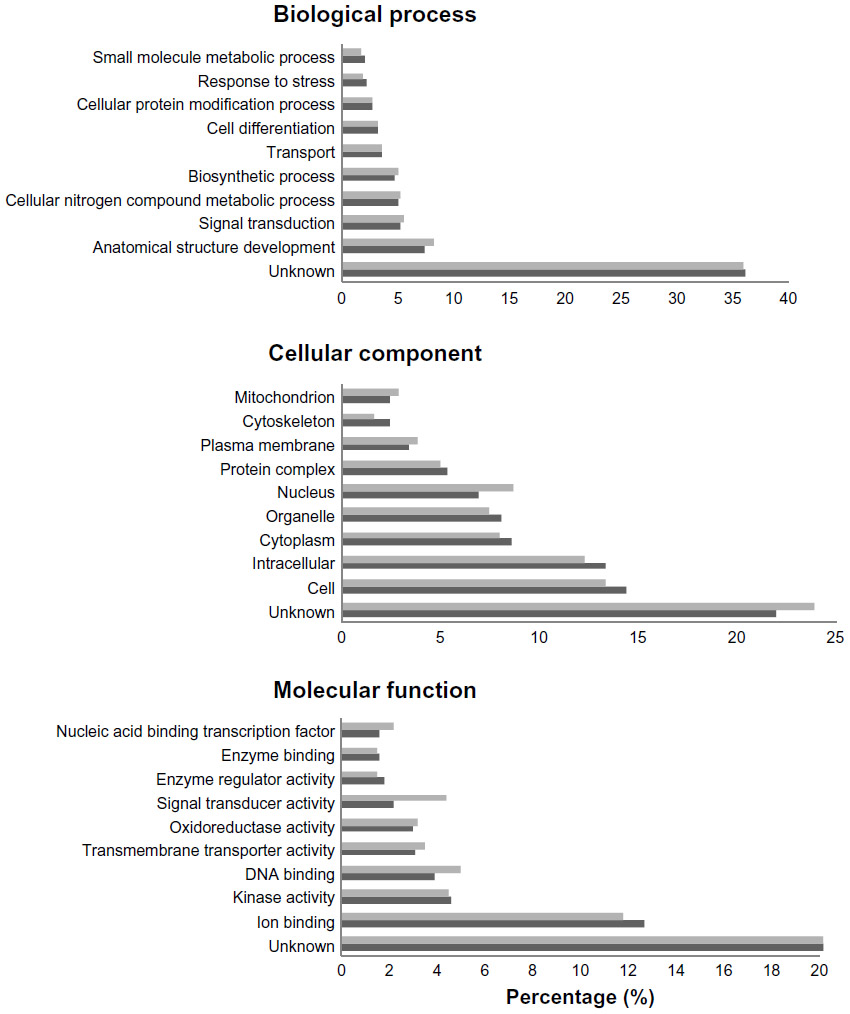 | Figure 2 Top ten GO categories in resting canine platelets compared with all canine annotations. Top ten GO categories in resting canine platelets (dark gray bars) compared with all canine annotations (light gray bars) for the three gene ontologies: biological process, cellular component, and molecular function. Categories are represented as percentages of the total. |
Compared with the canine genome, our canine platelet proteome was highly enriched in the GO biological process terms “cytoskeleton organization”, “cell recognition”, “cellular component organization”, “cell communication”, “cell-to-cell signaling”, and “response to endogenous/exogenous stimulus”, which can be related to important functions in platelets. The GO terms “translation” and “secondary metabolic process” were found to be under-represented in the canine platelet compared with the genome as would be expected considering the platelets’ lack of nuclei. The most represented GO biological process terms were “unknown” (36.20%), “anatomical structure development” (7.33%), and “signal transduction” (5.15%). The last two terms reflect the importance of intracellular signaling and structural changes in platelet function. In hemostasis, platelet surface receptors will bind to exposed subendothelial proteins upon vascular injury, which leads to platelet activation and morphologic change from a discoid to a spherical shape.
In comparison with the dog genome, our platelet proteome was highly enriched in the GO cellular component terms “cytoplasmic membrane-bound vesicle”, “endosome”, “proteinaceous extracellular matrix”, “organelle”, “plasma membrane”, and “cytoskeleton”. All of these GO terms highly enriched in the platelet are important to platelet function and morphology in normal hemostasis. Some under-represented GO terms in the platelet versus the genome included “nuclear”, “ribosome”, and “lipid particle”, which is not surprising considering platelets are anucleate and lack normal nucleated cell translational levels and membrane assembly. The most represented GO terms were “unknown” (22.03%), “cell” (14.49%), “intracellular” (13.46%), “cytoplasm” (8.73%), and “organelle” (8.28%). These localization categories are rather general and only indicate that within the proteins with known localization, the majority are annotated as being anywhere in the cell, including the membrane (cell) or not (intracellular).
Highly enriched protein GO molecular function terms present in the platelet compared with the dog genome included “protein binding”, “carbohydrate binding”, “enzyme regulator activity”, “cytoskeletal protein binding”, “actin binding”, and “receptor binding”, all of which are related to important platelet functions. Some of the most under-represented GO terms in this category are “structural constituent of ribosome” and “mRNA binding” which, again, make sense considering the anucleate nature of platelets and their limited protein translation functions. The most represented GO molecular function terms were “unknown” (49.16%), “ion binding” (12.63%), “kinase activity” (4.59%), “DNA binding” (3.92%), and “transmembrane transporter activity” (3.13%), which emphasize the importance of signaling in platelets.
In order to evaluate pathways and processes further, we used Ingenuity Pathway Analysis (Ingenuity Systems Inc.) software to obtain the significantly represented functions and pathways in the canine platelet proteome, as shown in Figures 3 and 4. In the analysis of the canine platelet proteome, “hematological system development and function”, “inflammatory response”, “cancer”, and “cellular movement” were among the top represented functional categories found in the canine platelet. These categories indicate the multifunctionality of platelets, which play important roles in inflammation and cancer development in addition to their hemostasis function.1 The three most represented functions include “cellular assembly and organization”, “cellular function and maintenance”, and “cell morphology”, which illustrates functions that are important during platelet activation and hemostatic function.
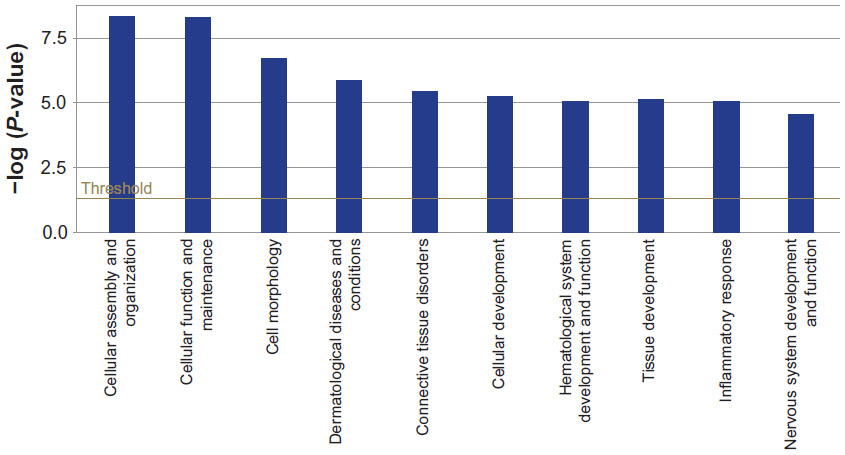 | Figure 3 Ten most significant biological functions represented in resting canine platelets proteome. The chart displays biological functions along the x-axis and significance in y-axis. The threshold line demonstrates the limit for significance (P = 0.05). |
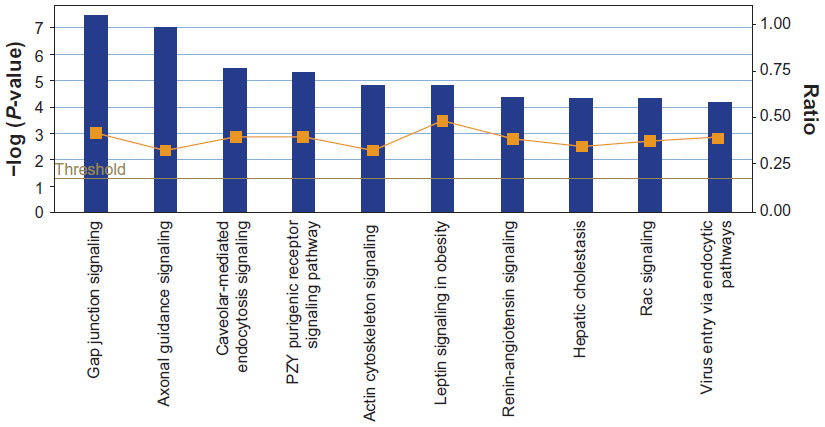 | Figure 4 Ten most significant canonical pathways represented in resting canine platelet proteome. Pathways are shown along the x-axis and the significance in the left y-axis. The threshold indicates the limit for significance (P = 0.05). The ratio is the number of proteins that meet the cutoff criteria divided by the total number of proteins in the pathway. |
The most significant canonical pathways are shown in Figure 4. “Thrombin signaling”, “HGF signaling”, “EGE signaling”, “PDGF signaling”, “integrin signaling”, and “thrombopoietin signaling” were among the top represented canonical pathways listed for the canine platelet. The proteins in these canonical pathways have significant roles in hemostasis, clot formation, wound healing, and angiogenesis. The presence of the “actin cytoskeleton” pathway as being the fifth most significant corroborates with the over-representation of cytoskeleton proteins indicated by the GO analysis results.
Subcellular localization of proteins
Platelet proteins were extracted using DDF, a multistep protein extraction method that uses four detergents (digitonin, Triton X-100, deoxycholate plus Tween 40, and sodium dodecyl sulfate) to separate proteins into four protein fractions based on decreasing solubility. Subsequent analysis of proteins extracted using DDF was performed using DDF2GO12 software which assigns cellular component-specific GO terms to each protein based on the detergent fraction the protein was extracted in and the number of transmembrane domains in the protein, if any.
Three healthy dog platelet samples were used for this proteomic study with an average of 2,668 proteins identified from each dog. We identified 1,803 proteins common to more than one dog which we then further analyzed to identify where these proteins were located in the platelet based on GO terms from DDF2GO.
The DDF method we used isolates proteins from throughout the entire platelet, as graphically illustrated in Figure 5 using cellular component terms from GOSlimViewer (Open-source software, Agbase, Mississippi State University) for proteins found in more than one of the three dogs. About 42% of the protein annotations were related to membranes, approximately 17% of which were to the plasma membrane in particular. Nearly 36% were related to organelles, with 10%, 3%, and 2% belonging to the mitochondrion, vesicle, and endoplasmic reticulum categories, respectively. About 22% of annotations related to the cytoplasm. These annotations indicate that the proteins found in more than one dog are located in the same subcellular regions.
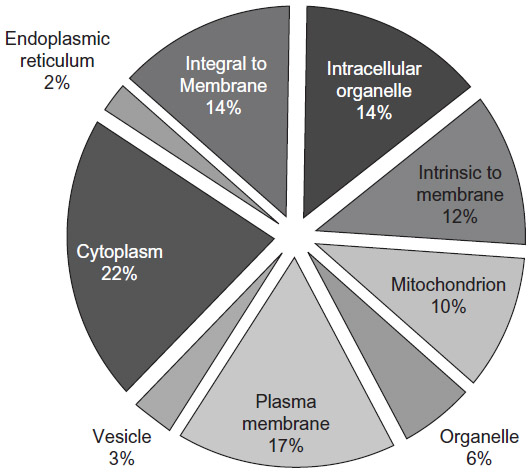 | Figure 5 Subcellular localization of the platelet proteins found in more than one dog by DDF2GO. The complete list of 1,803 platelet proteins from this study was used in determining the subcellular localization of the identified proteins using DDF2GO software. This figure shows the results for each of the identified categories as a pie chart. Each category and the relative percentage of total proteins present in that category are shown in the graph. |
In our platelet protein analysis we observed up to a 4.9-fold increase in information about subcellular localization using DDF2GO for protein functional analysis compared with AgBase’s GORetriever. DDF2GO provides protein annotation outputs that are different from traditionally used software like GORetriever. Tools such as GORetriever assign protein annotations in light of what is already known about the proteins. In addition to what is already known about the proteins, DDF2GO allows us to look at proteins based on the detergent fraction(s) that the protein was extracted in as well as the number of transmembrane domains the protein has. Proportionally, we observed a 4.0-fold increase in protein annotations related to “plasma membrane” and a 2.3-fold increase related to “cytoplasm”. The “mitochondrion” and “endoplasmic reticulum” GO cellular component categories had 4.9-fold and 1.6-fold increases in annotations for proteins in their categories, respectively. Use of DDF2GO software for protein annotation adds experimentally derived functional information, improving our functional analysis of the canine platelet proteome.
Discussion
In this report, we describe the canine platelet proteome and provide the first in vivo expression evidence of these proteins in the dog. We identified 2.4-fold more proteins than the most comprehensive platelet proteome previously reported.16 The number of proteins we found is also higher than previous human platelet predictions of 2,000–3,000 proteins17 and the most recent estimate of 5,000 proteins.18 This is likely due to the fact that previous studies used two-dimensional electrophoresis for protein separation. This technique is commonly used for proteomics but has the disadvantage of low yields of membrane, basic, high molecular weight and some signaling proteins.19,20 Our non-gel-based mass spectrometry coupled with DDF ensured a comprehensive proteome.
The fact that the majority of the proteins are categorized as “unknown” in all three ontologies reflects that there are still a significant number of proteins for which we have little or no functional information. This is a recognized problem across a large range of species;21 however, tissue expression data frequently provides the first clues to protein function and we report that these proteins are expressed in platelets.
The DDF technique offers unique advantages to protein extraction in that it does not disrupt cellular architecture. The first detergent used in DDF, ie, digitonin, efficiently isolates soluble cytosolic proteins due to how it interacts with cholesterol to form pores in the cell membrane. Triton X-100, the second sequential detergent used, solubilizes membrane and organelle proteins, while the third detergent used, ie, a combination of deoxycholate and Tween 40, extracts the soluble nuclear fraction. Sodium dodecyl sulfate is the final detergent used in DDF which primarily isolates the more insoluble proteins as well as nuclear matrix proteins.5 Because of this, proteins isolated in different detergent fractions prior to mass spectrometry analysis can allow identification of protein subcellular localization.
In our platelet protein analysis, we observed up to a 4.9-fold increase in information about subcellular localization using DDF2GO for protein functional analysis compared with AgBase’s GORetriever. Traditional protein functional analyses employ software such as GORetriever, which annotates proteins based on existing annotations for a given list of proteins. Not only does DDF2GO software factor in a protein’s existing annotation, but also considers the detergent fraction(s) that the protein was extracted in as well as the number of transmembrane domains if the given protein is a membrane protein. These additional considerations involved in annotation assignment by DDF2GO allow addition of knowledge as to where the protein came from and the proportion found in that subcellular location. This addition of experimentally derived functional information not only improves our functional analysis of the canine platelet proteome, but also confirms the suspicion that the knowledge of protein function and subcellular localization is incomplete. Use of DDF2GO allowed us to have improved coverage of subcellular localization data throughout the platelet.
Interestingly, there was a relatively high amount (7.15%) of proteins categorized as “nuclear”, which at first seems counterintuitive, since platelets are anucleate, although this finding is in accordance with reports from other studies.22–24 The presence of nuclear protein in platelets is not well understood and their possible role in platelet function is yet to be determined.22,25 One of the explanations for this finding takes into account the nature of the GO terms and how functional modeling relies on biological knowledge; while proteins are annotated to the nucleus, they also have other subcellular localizations and the GO annotation is not context-dependent. In our data, 71% of the proteins annotated as “nuclear” were also found to have other annotations in the GO cellular component category. Moreover, nuclear proteins may represent, in part, contamination by residual leukocytes. However, we believe that was not an issue in our analysis, since our samples were of high purity (99.98% by manual count), in accordance with some of the few studies that actually address sample contamination.16,23,24 Lastly, it has been stated that dog platelets can sometimes present fragments of nuclear material.26 Thus, considering this affirmation, the presence of nuclear components in platelet proteome analysis would be normal and expected.
Using DDF2GO software for platelet protein annotation analysis, the annotations we add are specific for proteins expressed in platelets. The nuclear category of GO terms and annotations are excluded from the output production. For the currently described rat and human platelet proteomes,16–18,24 the technique for platelet protein analysis described here has not been used.
Conclusion
This initial study contributes to a broader understanding of canine platelet biology and aids functional research, identification of potential treatment targets and biomarkers, and sets a new standard for the resting platelet proteome.
Acknowledgments
The authors would like to acknowledge Tony Arick and Lakshmi Pillai, who developed the pipeline and GO CC mapping script, and Allen Shack for assistance with the DDF technique.
Disclosure
The authors report no conflicts of interest in this work.
References
Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost. 2011;105 Suppl 1:13–33. | |
Cervi D, Yip TT, Bhattacharya N, et al. Platelet-associated PF-4 as a biomarker of early tumor growth. Blood. 2008;111:1201–1207. | |
MacEwen EG. Spontaneous tumors in dogs and cats: models for the study of cancer biology and treatment. Cancer Metastasis Rev. 1990;9:125–136. | |
Withrow SJ, Wilkins RM. Cross talk from pets to people: translational osteosarcoma treatments. ILAR J. 2010;51:208–213. | |
McCarthy FM, Burgess SC, van der Berg BH, Koter MD, Pharr GT. Differential detergent fractionation for non-electrophoretic eukaryote cell proteomics. J Proteome Res. 2005;4:316–324. | |
Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. | |
Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. | |
Rousseeuw PJ, Van Driessen K. A fast algorithm for the minimum covariance determinant estimator. Technometrics. 1999;41:212–223. | |
Filzmoser P, Garrett RG, Reimann C. Multivariate outlier detection in exploration geochemistry. Comput Geosci. 2005;31:579–587. | |
McCarthy FM, Gresham CR, Buza TJ, et al. AgBase: supporting functional modeling in agricultural organisms. Nucleic Acids Res. 2011;39:D497–D506. | |
McCarthy FM, Wang N, Magee GB, et al. Agbase: a functional genomics resource for agriculture. BMC Genomics. 2006;7:229. | |
Pillai LR, Arick T, Burgess SC, McCarthy FM. A high-throughput experimentally based method for identifying subcellular localization. (Manuscript in preparation.) | |
Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and detecting their subcellular localization. Trends Biochem Sci. 1999;24:34–36. | |
Cserzo M, Wallin E, Simon I, von Heijne G, Elofsson A. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng. 1997;10:673–676. | |
Peddinti D, Nanduri B, Kaya A, Feugang JM, Burgess SC, Memili E. Comprehensive proteomic analysis of bovine spermatozoa of varying fertility rates and identification of biomarkers associated with fertility. BMC Syst Biol. 2008;2:19. | |
O’Neill EE, Brock CJ, von Kriegsheim AF, et al. Towards complete analysis of the platelet proteome. Proteomics. 2002;2:288–305. | |
Dittrich M, Birschmann I, Mietner S, Sickmann A, Walter U, Dandekar T. Platelet protein interactions: map, signalling components, and phosphorylation groundstate. Arterioscler Thromb Vasc Biol. 2008;28:1326–1331. | |
Burkhart JM, Vaudel M, Gambaryan S, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120:73–82. | |
Maguire PB, Fitzgerald DJ. Platelet proteomics. J Thromb Haemost. 2003;1:1593–1601. | |
Garcia A, Watson SP, Dwek RA, Zitzmann N. Applying proteomics technology to platelet research. Mass Spectrom Rev. 2005;24:918–930. | |
Hanson AD, Pribat A, Waller JC, de Crecy-Lagard V. ‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list – and how to find it. Biochem J. 2009;425:1–11. | |
Martens L, Van Damme P, Van Damme J, et al. The human platelet proteome mapped by peptide-centric proteomics: a functional protein profile. Proteomics. 2005;5:3193–3204. | |
Qureshi AH, Chaoji V, Maiguel D, et al. Proteomic and phosphor-proteomic profile of human platelets in basal, resting state: insights into integrin signalling. PLoS One. 2009;4:7627. | |
Yu Y, Leng T, Yun D, et al. Global analysis of the rat and human platelet proteome – the molecular blueprint for illustrating multi-functional platelets and cross-species function evolution. Proteomics. 2010;10:2444–2457. | |
Gnatenko DV, Perrotta PL, Bahou WF. Proteomic approaches to dissect platelet function: half the story. Blood. 2006;108:3983–3991. | |
Stockham SL, Scott MA. Platelets. In: Fundamentals of Veterinary Clinical Pathology. 2nd ed. Ames, IA: Blackwell Publishing; 2008. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.