Back to Journals » Drug Design, Development and Therapy » Volume 20
Identification of a Quinolone-Based Scaffold as a Dual SARS-COV-2 PLᵖʳᵒ and Mᵖʳᵒ Inhibitor: An Integrated Molecular Modeling and In-vitro Evaluation Approach
Authors Atatreh N, Bustanji L, Ferreira JC, Rabeh WM, Mahgoub RE ![]() , Ghattas MA
, Ghattas MA ![]()
Received 1 January 2026
Accepted for publication 15 April 2026
Published 4 May 2026 Volume 2026:20 593126
DOI https://doi.org/10.2147/DDDT.S593126
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Noor Atatreh,1,2 Lana Bustanji,1,2 Juliana C Ferreira,3 Wael M Rabeh,3 Radwa E Mahgoub,1,2 Mohammad A Ghattas1,2
1College of Pharmacy, Al Ain University, Abu Dhabi, United Arab Emirates; 2AAU Health and Biomedical Research Center, Al Ain University, Abu Dhabi, United Arab Emirates; 3Science Division, New York University, Abu Dhabi, 129188, United Arab Emirates
Correspondence: Mohammad A Ghattas, Email [email protected]
Introduction: The papain-like protease (PLᵖʳᵒ) plays a critical role in the SARS-CoV-2 life cycle by regulating both viral replication and immune evasion, making it an attractive target for antiviral drug development.
Methods: In this study, a virtual screening strategy combining pharmacophore modeling, molecular docking, enzymatic evaluation, and molecular dynamics (MD) simulations was employed to identify novel PLᵖʳᵒ inhibitors. From this process, 30 top ranked candidates were selected for experimental enzymatic evaluation.
Results: Among them, a quinolone-based scaffold compound ST50940260 demonstrated the highest inhibitory activity with an IC50 of 69.53 ± 4.4 μM, exhibiting favourable potency and lead like properties, with a docking score of − 10.215 kcal/mol. Notably, ST50940260 also showed inhibitory activity against Mᵖʳᵒ, suggesting dual target potential with an IC50 of 96.7 μM. To further explore structure activity relationships, a 2D similarity search was conducted, resulting in the identification and testing of seven structural derivatives, among which Z30231927 emerged as the most promising analogue with an IC50 of 124.8 μM against PLᵖʳᵒ. Molecular dynamics simulations of 100 ns revealed that the lead compound forms a stable complex with PLᵖʳᵒ, exhibiting interaction patterns comparable to those of the co-crystallized ligand. Furthermore, MM-GB/SA binding free energies were calculated from the MD trajectories for the lead (ST50940260), the derivative (Z30231927), and the co-crystallized ligand in 7JN2. The lead demonstrated favourable binding (− 27.33 kcal/mol) within a comparable range, while the derivative showed the most favourable binding (− 30.26 kcal/mol), followed closely by the co-crystallized reference (− 29.82 kcal/mol).
Conclusion: Overall, these findings highlight ST50940260 as a promising lead candidate for further optimization, with potential dual inhibitory activity against both proteases.
Keywords: docking, pharmacophore filtration, molecular dynamics, enzyme inhibition assay
Introduction
The year 2019 marked the emergence of a novel infectious disease, COVID-19, in Wuhan, China. Caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), this illness represents the third major coronavirus-related epidemic in the past twenty years, following the outbreaks of SARS-CoV in 2002 and MERS-CoV in 2012.1 On March 11, 2020, the World Health Organization (WHO) officially declared COVID-19 as a global pandemic due to its rapid and widespread transmission across countries.2 Five years later, COVID-19 continues to exert significant health and socio-economic burdens. According to data from the WHO, by early November 2024, approximately 777 million confirmed cases of COVID-19 and more than 7 million deaths had been reported worldwide.3 Notably, these figures were documented even after the WHO declared the end of the global health emergency on May 5, 2023,4 following nearly three years of significant disruption to international public health systems and the global economy.
COVID-19 has profoundly disrupted everyday life, overwhelmed public health systems, and shaken economies across the globe. In response, scientists worldwide have worked at an extraordinary pace to develop effective solutions, leading to the unusually rapid creation of vaccines and antiviral treatments.5–7 SARS-CoV-2 replication depends on several key viral enzymes, most notably the main protease (3CLpro/Mᵖʳᵒ), the RNA-dependent RNA polymerase (RdRp), and the papain-like protease (PLᵖʳᵒ).8 These proteins play central roles in the viral life cycle and have therefore become primary molecular targets in the development of antiviral agents. Currently small novel inhibitors approved treatments include the RdRp inhibitors remdesivir9 and molnupiravir,10 as well as the Mᵖʳᵒ /3CLpro inhibitor nirmatrelvir.11 Nevertheless, in-vitro studies, together with longitudinal sequencing of immunocompromised patients, have identified specific viral mutations associated with reduced sensitivity to these agents.12–14
Despite extensive efforts, PLᵖʳᵒ has not yet yielded inhibitors as potent as those for other SARS-CoV-2 targets, which is striking given its status as a highly promising antiviral target. Firstly, this antiviral target performs dual functions essential for SARS CoV 2 replication and immune evasion. It plays in a central role in the viral life cycle, as it processes the viral polyprotein into the functional components required for efficient replication. In addition, modulates the host immune response by removing ubiquitin and ISG15 from host proteins, an activity that suppresses innate immunity and supports viral replication. Secondly, PLᵖʳᵒ is highly conserved across SARS-CoV-2 variants and displays far less mutational flexibility than the spike protein or Mᵖʳᵒ It shares approximately 83% sequence identity with the SARS-CoV PLᵖʳᵒ,15 and a structural analyses study showed that residues within its core papain-like domain are largely intolerant to mutation.16
The structure of PLᵖʳᵒ consists of four principal domains: a ubiquitin like (Ubl) domain, and the thumb, palm, and fingers domains. Together, the thumb, palm, and fingers form the catalytic core responsible for substrate processing. The fingers domain houses a zinc binding site coordinated by four cysteine residues Cys189, Cys192, Cys224, and Cys226, which are essential for maintaining the enzyme’s structural integrity and catalytic function. The catalytic triad, composed of Cys111, His272, and Asp286, is positioned at the interface of the thumb and palm domains, forming the active center of the enzyme. Within this site, the terminal residues of ubiquitin Leu73, Arg74, Gly75, and Gly76 align precisely to interact with PLᵖʳᵒ during catalysis.17
Over the past few years, numerous inhibitors targeting SARS CoV 2 PLᵖʳᵒ and Mᵖʳᵒ have been reported. However, several studies have noted that many recently developed PLᵖʳᵒ inhibitors are structurally derived from the GRL0617 scaffold, resulting in limited scaffold diversity among current candidates. This has prompted researchers to emphasize the importance of identifying new chemical scaffolds for PLᵖʳᵒ inhibition. Relying heavily on closely related scaffolds may increase the risk that viral mutations affecting one class of inhibitors could compromise the activity of others with similar structures. Therefore, expanding the chemical diversity of PLᵖʳᵒ inhibitors is considered an important strategy to reduce the likelihood of cross resistance and to provide alternative starting points for further optimization.18
Dual inhibition of PLᵖʳᵒ and Mᵖʳᵒ represents an attractive antiviral strategy because it interferes with two essential proteolytic steps required for viral replication. By targeting both proteases simultaneously, such an approach can disrupt multiple stages of viral polyprotein processing and thereby reduce the efficiency of viral maturation. Consistent with this concept, cell-based studies have demonstrated that the combined inhibition of PLᵖʳᵒ and Mᵖʳᵒ produces additive antiviral effects compared with inhibition of either protease alone.19 In this context, the identification of a scaffold capable of inhibiting both proteases addresses an important unmet need for chemically distinct starting points that can be further optimized to improve potency, selectivity, and pharmacokinetic properties while maintaining a dual target profile.
This work focuses on discovering new lead like small molecule inhibitors that target the active site of PLᵖʳᵒ through an integrated approach that combines ligand based (LB) and structure based (SB) virtual screening with biological evaluation, structure-activity relationship (SAR) analysis, and molecular dynamics simulations.
Materials and Methods
Pharmacophore Generation and Validation
A ligand library was assembled by collating non-covalent SARS-CoV-2 PLᵖʳᵒ inhibitors reported in the literature and restricting the set to compounds with experimentally determined co-crystal structures deposited in the RCSB Protein Data Bank. 20 inhibitor–protein complexes meeting these criteria were retrieved, and the corresponding PDB entries were imported into the Molecular Operating Environment (MOE).20 Complexes were superposed using MOE’s Protein Alignment tool to align all co-crystallized ligands. Structures were then prepared in MOE (standard corrections and assignment of protonation states), after which protein structures and solvent molecules were removed, yielding a final ensemble of 20 aligned ligands.
From the compiled non-covalent PLᵖʳᵒ inhibitor library, five ligands were selected as a training set to span molecular weights of ~300–500 Da as shown in Figure 1. Rigidity (1–10 rotatable bonds) was not used as a selection criterion because the bioactive conformations were defined by the co-crystal structures. Pharmacophore Elucidation in MOE was applied to the aligned training set to identify shared multi-point features. During model generation, each ligand was kept in its co-crystallized (putative bioactive) conformation. The remaining 15 compounds from the non-covalent inhibitor library served as the positive test set. For the negative test set, decoy molecules were generated with the DUDE decoy generator using the active molecules as templates.21 The top-ranked pharmacophore hypotheses were validated using sensitivity and specificity22 (Supplementary Equations S1 and S2) using a test set comprising 15 known PLᵖʳᵒ inhibitors and 750 DUDE-generated decoys.
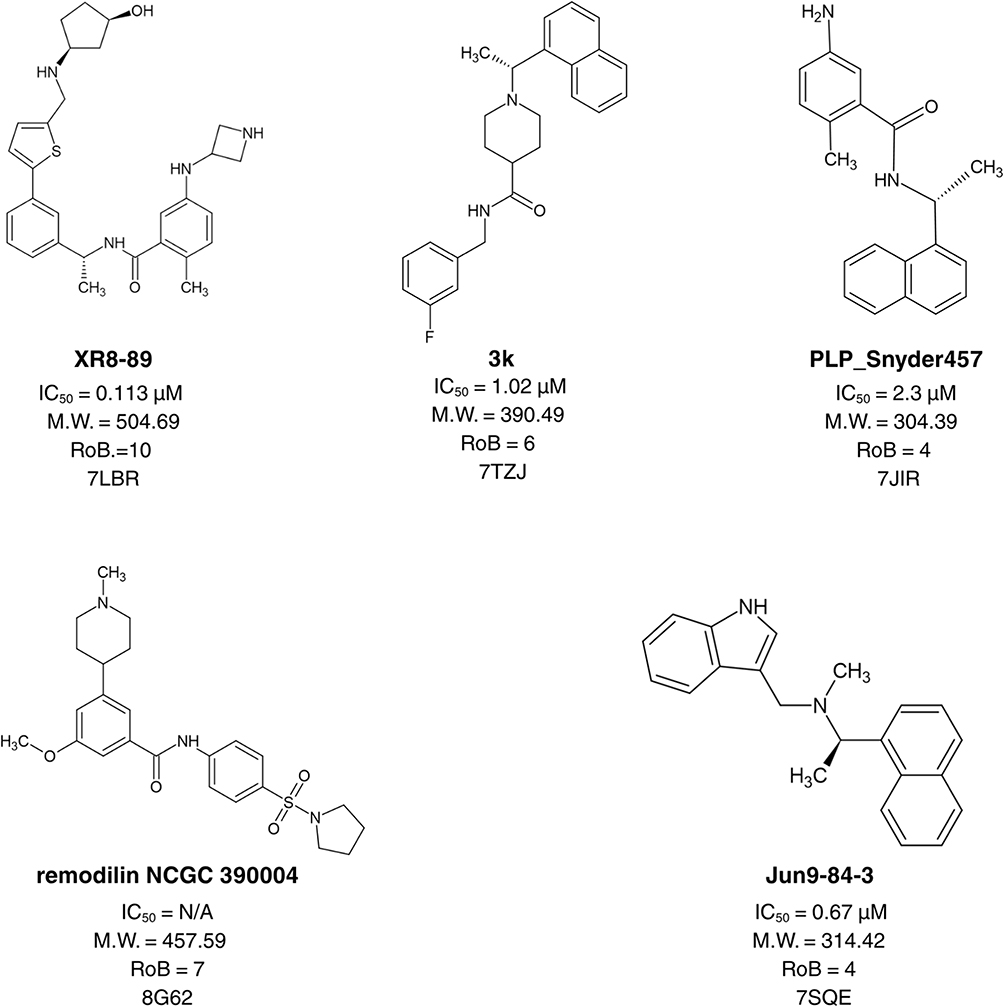 |
Figure 1 Compounds with crystal structures used as a training set for pharmacophore model construction. Compound names are shown in bold. |
Pharmacophore Performance in VS: Seeding Experiment
A dataset of 804 ligands was compiled (750 DUDE-generated decoys and 54 known actives collected from the literature). The compiled library was used to assess docking following pharmacophore filtration performance. Accordingly, two distinct workflows were employed to dock this dataset using the Glide-extra precision (XP) docking in Maestro from the Glide module:23 a pharmacophore-based filtration workflow (docking the filtered library of 183 ligands) and a conventional workflow (docking all 804 ligands). The performance of both workflows in correctly identifying and ranking the known positive inhibitors within the ligand dataset was assessed using a receiver operating characteristic curve (ROC).
Pharmacophore-Based and Druglike Library Filtration
A ligand database was constructed from the commercially available library ChemDiv24 and TimTec,25 making up a library of ~1.02 million ligands (1,021,102) pre-filtration. The compiled library was filtered as per a number of drug-like rules, namely Lipinski’s26 and Veber’s27 drug-like rules (molecular weight (M.W). ≤ 500 Da, LogP < 5, number of hydrogen bond acceptor (HBA) ≤ 10, number of hydrogen bond donor (HBD) ≤ 5, number of rotatable bond (RotB) ≤ 10, total polar surface area (TPSA) ≤ 140). The remaining ligands were then passed through a PAINS-Remover filter,28 to further filter our library removing potentially promiscuous compounds. The best-generated pharmacophore hypothesis was then used to narrow down the ligands to only those that passed the filter, resulting in a final library of ligands. Lastly, the pharmacophore filtered drug-like library was processed using the LigPrep module29 in Maestro.30
Protein Preparation and Grid Generation
The crystal structure of PLᵖʳᵒ (PDB: 7JN231) was downloaded from the RCSB PDB. To prepare the structure, all water molecules were initially removed. Subsequently, MOE’s protein preparation tool was used to model missing residues and incorporate hydrogen atoms.20 Further processing was performed using the Protein Preparation module in Maestro Schrödinger.30 At this stage, optimization of hydrogen atoms was performed, and refinement of the structure was performed through restrained minimization, achieving a maximum RMSD of 0.30 Å to reach a lower-energy conformation. The druggability score of ligand binding site was investigated using sitemap in Schrodinger Maestro and found to be 0.965. Following a 500 ns MD simulation and clustering, the top cluster of 7JN2 was found to have a better druggability score of 1.016 and hence was used for the VS.32
Generation of receptor grid for docking was performed around the centroid of the ligand found in the crystal structure. To ensure complete coverage of the binding cavity, the grid box was set to 15 Å and 26 Å, for the inner and outer boxes, respectively.
As part of the validation procedure preceding the virtual screening workflow, self-docking was carried out to assess the reproducibility of the ligand’s crystallographic binding orientation. The redocked pose generated using XP-docking was aligned with the original structure, yielding an RMSD of 0.71Å. This result confirms that the docking protocol is capable of accurately reproducing experimentally observed binding conformations within the PLᵖʳᵒ binding site.
Docking-Based VS
The promiscuous free, drug-like, pharmacophore-filtered library was docked into the PLᵖʳᵒ ‘s active site using the Glide workflow integrated within Maestro Schrödinger.33 The virtual screening workflow was conducted through three successive docking phases with progressively higher precision including high throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP). After each phase, approximately the top 20% of ligands with the highest scores from the previous stage were selected and advanced for further refinement in the next level of docking accuracy.
After the XP docking stage, the 500 ligands with the highest scores were carefully inspected to assess their binding poses, particularly their fit within the active pocket and their interactions with nearby crucial residues. Compounds that showed promising orientations were further screened using advanced aggregation prediction platforms such as Aggregator Advisor34 and BAD Molecule Filter.35
Molecular Dynamics (MD)
Molecular dynamics (MD) simulations were conducted on the lead compound, derivative, the co-crystallized reference ligand and apo protein to examine their stability within the PLᵖʳᵒ active site. All simulations were performed using the AMBER18 package.36 Protein partial charges and parameters were assigned using the ff19SB force field, while ligand parameters were generated with GAFF. Zn(II) coordination in PLᵖʳᵒ was modelled by applying the ZAFF parameter set to the zinc finger motif.37 System preparation was carried out in xle ap, after which the complex was neutralized with three Na⁺ ions and solvated in a TIP3P water model using a truncated octahedral box. Energy minimization was completed in two stages with the PMEMD engine. First, all solute atoms were restrained using a 500 kcal·mol−1·Å−2 force constant for 1000 cycles. This was followed by a second minimization step without restraints for another 1000 cycles.
The prepared system was gradually heated to a temperature of 300 K under the NVT ensemble conditions. During this phase, all hydrogen related bond lengths were fixed using the SHAKE algorithm, and temperature control was maintained through the Langevin thermostat with a collision frequency of 1.0 ps−1. A production simulation of 100 nanoseconds was then performed under the NPT ensemble, maintaining constant temperature and pressure at 300 K and 1.01×105 Pa, respectively. The binding free energies were determined using the MM-GB/SA approach,38 focusing on the last 30 nanoseconds of the trajectory to ensure ligand stabilization and reduce the influence of short-lived conformational variations within the active site. RMSD, RMSF, SASA and Rg values were reported for each system. Solvent-accessible surface area (SASA), radius of gyration (Rg), root-mean-square deviation (RMSD) values for both the backbone and ligand complexes, and root-mean-square fluctuation (RMSF) were calculated and recorded from each 100 ns MD simulation trajectory.
Enzyme Inhibition Assay
Enzyme purification was carried out as described in a previous publication.39 Assay conditions for PLᵖʳᵒ and Mᵖʳᵒ were optimized separately based on their enzymatic properties and previously reported protocols, resulting in the use of 1 µM enzyme and 5% DMSO for PLᵖʳᵒ and 20 µM enzyme with 20% DMSO for Mᵖʳᵒ measurements. The higher DMSO concentration (20%) was used in the Mᵖʳᵒ assay to maintain the solubility and stability of the hydrophobic peptide substrate, as lower DMSO concentrations promote substrate aggregation, whereas 20% DMSO prevents aggregation and enables reliable catalytic measurements, noting here that a control essay was used in all experiments.40 PLᵖʳᵒ inhibition assays were conducted using a Cytation 5 multi-mode microplate reader (BIOTEK Instruments, Winooski, VT, USA). The peptide substrate utilized in the assay contained the PLᵖʳᵒ cleavage site Leu-Arg-Gly-Gly (LRGG) and was flanked by a fluorescent 7-amino-4-methylcoumarin (AMC) tag and a corresponding carbobenzoxy (CBZ) quencher, forming the substrate Z-LRGG-AMC.
The proteolytic reaction was initiated by adding 1 µM of PLᵖʳᵒ enzyme to the peptide substrate in a reaction buffer composed of 50 mM HEPES (pH 7.5), 1 mM EDTA, and 0.5 mM TCEP, with 5% dimethyl sulfoxide (DMSO) included to enhance peptide solubility. The reaction was carried out in a 96-well microplate at 25 °C for 10 minutes, with fluorescence measurements recorded at excitation and emission wavelengths of 340 nm and 487 nm, respectively. Cleavage of the peptide substrate by PLᵖʳᵒ resulted in an increase in the AMC fluorescence signal, which was used to assess enzymatic activity.
The inhibitory effects of candidate compounds on PLᵖʳᵒ enzymatic activity were evaluated by determining their half-maximal inhibitory concentrations (IC50). The IC50 values were calculated at a fixed peptide substrate concentration of 200 µM, with serial 1:5 dilutions of the test inhibitors ranging from 0 to 400 µM. The dose-response data were analyzed using a four-parameter variable slope model in GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA). 3CLpro inhibition assays were conducted using a Cytation 5 multi-mode microplate reader (BIOTEK Instruments, Winooski, VT, USA). The peptide substrate utilized in the assay contained the PLᵖʳᵒ cleavage site Leu-Arg-Gly-Gly (LRGG) and was flanked by a fluorescent DABCY tag and a corresponding EDANS quencher, forming the substrate DABCYL-KTSAVLQSGFRKME-EDANS.
The proteolytic reaction was initiated by adding 20 µM of 3CLpro enzyme to the peptide substrate in a reaction buffer composed of 50 mM HEPES (pH 7.5), 1 mM EDTA, and 0.5 mM TCEP, with 20% dimethyl sulfoxide (DMSO) included to enhance peptide solubility. The reaction was carried out in a 96-well microplate at 30 °C for 10 minutes, with fluorescence measurements recorded at excitation and emission wavelengths of 340 nm and 485 nm, respectively. Cleavage of the peptide substrate by 3CLpro resulted in an increase in the DABCYL fluorescence signal, which was used to assess enzymatic activity.
The inhibitory effects of candidate compounds on 3CLpro enzymatic activity were evaluated by determining their half-maximal inhibitory concentrations (IC50). The IC50 values were calculated at a fixed peptide substrate concentration of 60 µM, with serial dilutions of the test inhibitors ranging from 0 to 400 µM. The dose-response data were analyzed using a four-parameter variable slope model in GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA). All assays were performed in triplicate in at least three independent experiments.
Lead Molecules Evaluation
Several tools were employed to assess unwanted properties, including nonspecific binding or a tendency to aggregate. Subsequently, pharmacokinetic properties and drug likeness were evaluated using SwissADME,34 and only those compounds that met all selection parameters were advanced for enzyme based experimental testing. The pKa values were predicted using the machine-learning-based tool MolGPKA.41
Results and Discussion
Pharmacophore Model Construction and Pharmacophore Performance Assessment
A library of previously reported PLᵖʳᵒ inhibitors, each accompanied by crystal structures and documented IC50 values, was compiled and utilized for pharmacophore model generation. From this collection, five representative compounds (PDB IDs: 7JIR, 7IZJ, 7SQE, 7LBR, and 8G62) were chosen as the training set based on their structural diversity, molecular weight range, and inhibitory potency. During pharmacophore modeling, the conformations of these ligands were retained exactly as observed in their crystallographic complexes. This approach eliminated the need to sample multiple conformations of flexible molecules, thereby minimizing potential sources of uncertainty. The selected compounds encompassed distinct chemical scaffolds and exhibited inhibitory activities within the low micromolar range. Using the MOE pharmacophore elucidation method, 166 pharmacophore hypotheses were generated and were evaluated for their performance and efficiency.
The pharmacophore assessment was initiated using 166 hypotheses generated by MOE, which were analyzed based on two primary parameters: sensitivity and specificity. The testing dataset consisted of 55 confirmed active inhibitors obtained from the compiled PDB database and 750 decoy molecules generated using the DUDE decoy online server to simulate inactive (false positive) compounds. After thorough analysis, the optimal pharmacophore model identified as the most suitable candidate exhibited a well-balanced performance, achieving 78.2% sensitivity in recognizing true active compounds and 83.2% specificity in excluding inactive or decoy molecules. The finalized hypothesis consisted of two hydrogen bond acceptor features (F1, F2: A), one aromatic ring feature (F3: ArH), a dual hydrogen bond donor/acceptor feature (F4: DA), and a cationic center (F5: A+). However, due to the initially low selectivity, the radius of Feature 2 was adjusted from 1.0 Å to 1.5 Å, which enhanced the model’s selectivity to 80% but resulted in a slight reduction in specificity to 81.5% as shown below in Figure 2. and (refer to Supplementary Table S1).
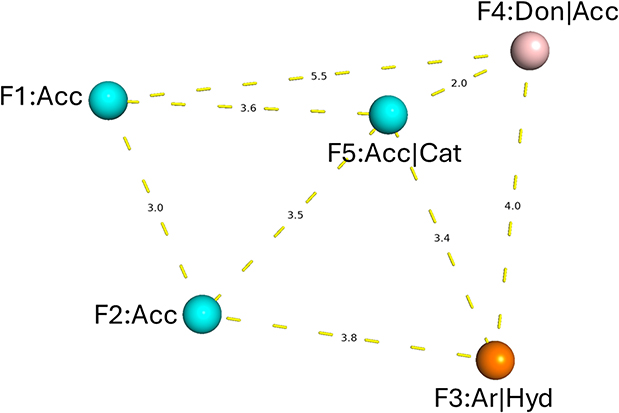 |
Figure 2 Key pharmacophoric features of the selected hypothesis in three-dimensional space, with distances reported in angstroms (Å). |
VS Protocol Validation: Seeding Experiment
Prior to its incorporation into the virtual screening workflow, the selected pharmacophore model underwent validation through a seeding experiment. A drug-like compound library comprising 805 ligands, including 54 confirmed active inhibitors, was assembled for this purpose. After ligprep the library had a total of 2927 ligands. Two independent Glide-XP docking strategies were implemented: the first utilized a pharmacophore-filtered approach, in which only the ligands that met the pharmacophore criteria of 627 ligands were subjected to docking, while the second employed a conventional non-filtered approach that docked the entire prepared library. The outcomes of both strategies were analyzed based on their efficiency in retrieving active compounds (true positives), and these values were subsequently used to generate the ROC curve as depicted in Figure 3.
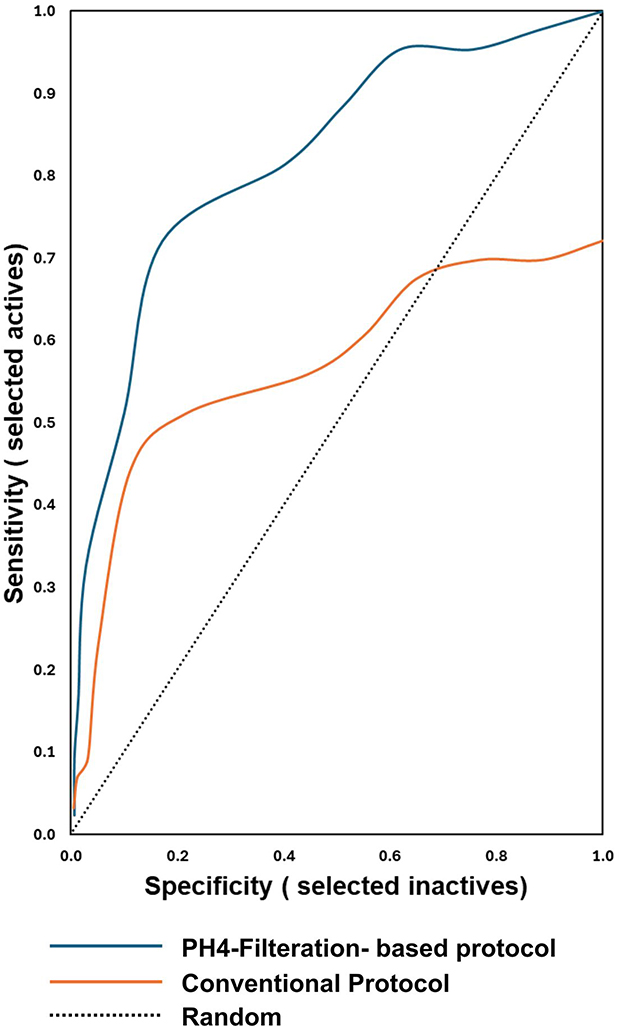 |
Figure 3 ROC curves comparing the conventional workflow and the pharmacophore filter–based (PH4) approach. |
The ROC curve illustrated the model’s capability to distinguish true positives (actives) from false positives (decoys), with actives plotted along the Y-axis and decoys along the X-axis. An ideal curve would rise sharply along the Y-axis, reaching a true positive rate of 1, signifying that all actives in the dataset were accurately identified, and would then extend horizontally, indicating that the resulting hit list contained only active molecules.42 Both virtual screening protocols were evaluated using the area under the curve (AUC), a widely accepted performance measure. AUC values between 0.9 and 1 indicate excellent performance, 0.80 to 0.9 represent good performance, 0.70 to 0.8 are considered fair, 0.50 to 0.7 indicate poor performance, and values below 0.5 denote failure.22 In this assessment, the pharmacophore-based filtration protocol demonstrated superior performance compared to the conventional non-filtered approach. In this study, the filtration-based approach demonstrated superior performance compared to the standard protocol, yielding an AUC of 0.82, which falls within the range considered good. In contrast, the conventional protocol produced an AUC of 0.57, reflecting a performance that is categorized as poor.
These findings demonstrate that the filtration-based protocol achieved markedly better discrimination between active and decoy compounds than the conventional workflow. Moreover, applying the pharmacophore filter prior to virtual screening effectively reduced the docking library by approximately 78.6% (from 2927 to 627 ligands), whereas the conventional method required processing the complete set of 2927 ligands, which is considerably more time- and resource-intensive. Collectively, these results underscore that integrating ligand-based and structure-based strategies can significantly enhance virtual screening performance by improving both computational efficiency and predictive accuracy.
Pharmacophore-Based and Druglike Filtration
Figure 4 provides an overview of the LB/SB workflow through which the initial set of 1,021,012 million compounds was screened against the SARS-CoV-2 PLᵖʳᵒ enzyme. A commercially available database was first used, and drug-likeness criteria were applied to filter the compounds.26,27 The resulting collection was then subjected to the PAINS-Remover filter,28 which was employed to exclude promiscuous or interference-prone structures narrowing down the initial set of approximately 1 million to 685,502 ligands. In the final step, the highest-performing pharmacophore model was applied, allowing the selection to be further refined to compounds exhibiting pharmacophoric features consistent with known PLᵖʳᵒ inhibitors. This process yielded a final set of 387,950 ligands.
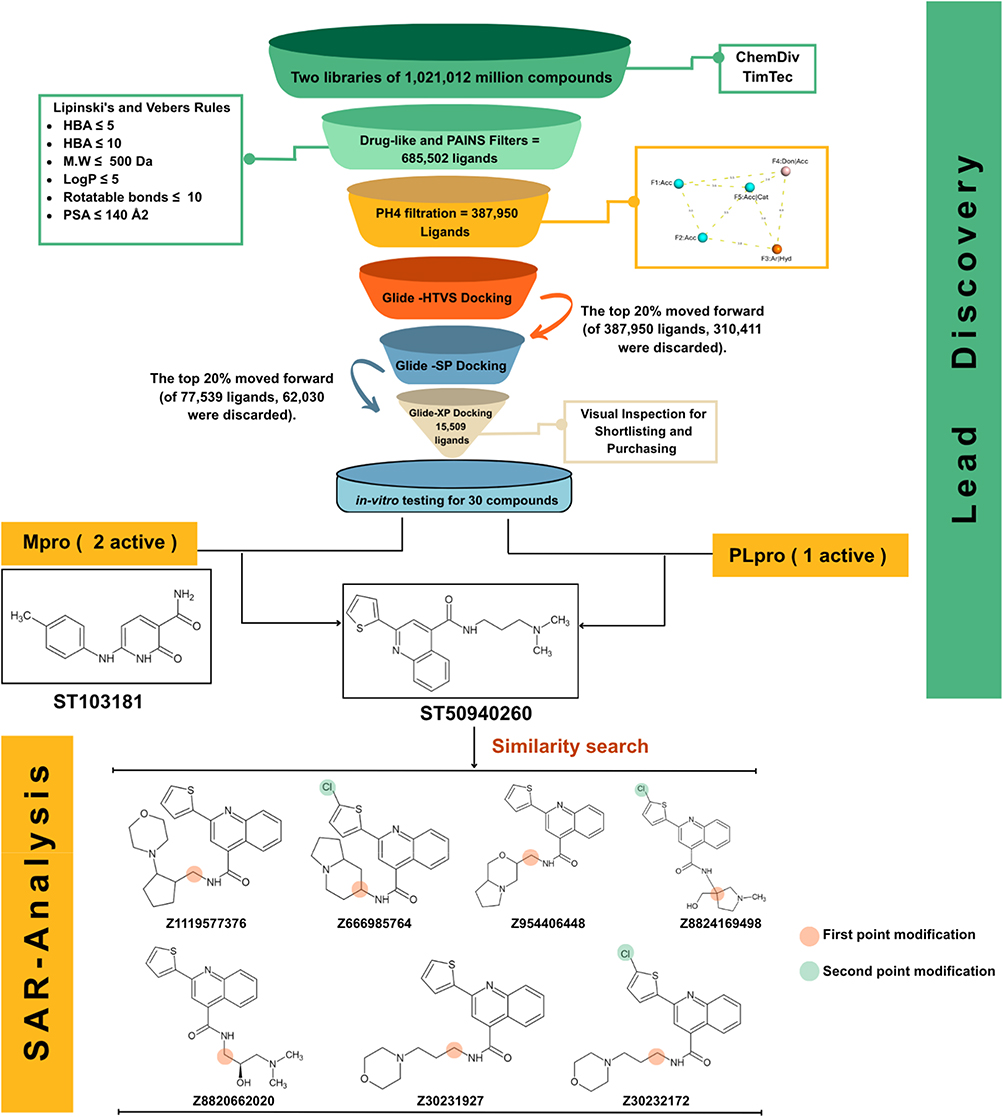 |
Figure 4 Workflow summary of the LB/SB virtual screening and subsequent structure activity analysis. |
Docking-Based VS
After ligand preparation, the virtual screening workflow was designed to further narrow down the drug-like and PH4-filtered ligands obtained from ChemDiv database. The curated library was subjected to molecular docking against the active site of the top cluster PLᵖʳᵒ (PDB ID: 7JN2). To balance accuracy and computational cost, a hierarchical three-stage docking strategy was implemented: initial high-throughput virtual screening (GLIDE-HTVS) of the entire dataset, followed by re-docking of the top 20% of hits using standard precision (GLIDE-SP), and a final refinement of the best candidates through extra precision docking (GLIDE-XP). This stepwise approach progressively enhanced docking accuracy while maintaining computational efficiency. By the end of the three-tier docking protocol, the library was filtered down to 15,509 ligands.
To validate the docking methodology and establish a selection benchmark, the MD-driven ligand conformation of the PLᵖʳᵒ complex was re-docked into its native active site, yielding a docking score of −6.590 kcal/mol. This score served as the cutoff criterion, with only compounds exhibiting docking energies equal to or lower (more favorable) than −6.590 kcal/mol retained for subsequent evaluation. From this refined subset, the top 500 ligands were shortlisted and visually inspected within the PLᵖʳᵒ binding pocket to assess spatial fit and the presence of key molecular interactions with surrounding residues. Compound selection was prioritized based on the ability to form key molecular interactions with residues identified from the reference co-crystallized ligand, particularly GLN267 and/or TYR266. To eliminate false positives arising from nonspecific aggregation, all shortlisted molecules were analyzed using the Aggregator Advisor and BAD molecule filters. Compounds that produced no alerts in either tool were considered reliable and advanced for biological testing, yielding a final selection of 30 ligands for in-vitro evaluation (details provided in Supplementary Table S2).
Enzyme Inhibition Assay
All purchased compounds were initially screened at a concentration of 100 µM against both PLᵖʳᵒ and Mᵖʳᵒ enzymatic assays, and only those exhibiting more than 50% inhibition were selected for further analysis. This subsequent evaluation included structure–activity relationship (SAR) analysis and IC50 determination. The lead compound was used as a query for a 2D similarity search, through which seven structurally related derivatives were identified, purchased, and tested for inhibitory activity against both SARS-CoV-2 proteases. The enzymatic assay results are summarized in Table 1, which shows that all tested derivatives were less active than the lead compound. Nevertheless, these findings provide valuable insight into the structure–activity relationship, as discussed in the following section.
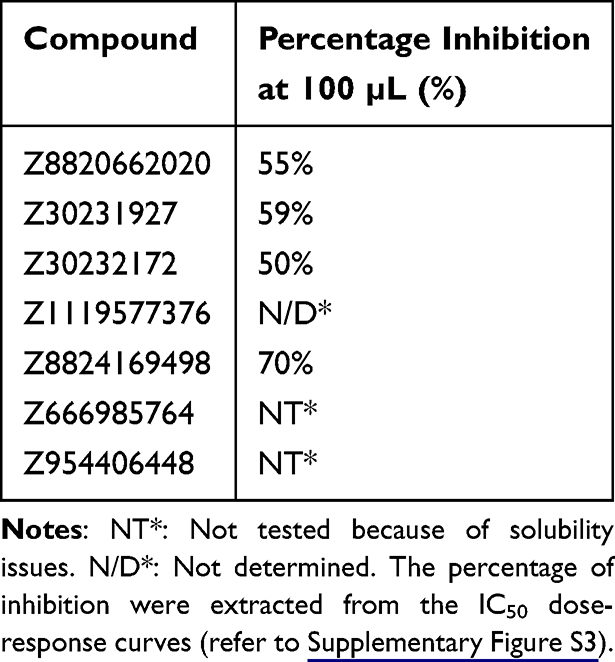 |
Table 1 Percentage Inhibition of PLᵖʳᵒ at 100 µm for the Seven Tested Derivatives |
Figure 5 presents the IC50 values for three compounds, including the lead compound ST50940260 and its most promising derivative, Z30231927, along with the Mᵖʳᵒ hit identified during the initial screening cycle. The lead compound exhibited the highest potency against PLᵖʳᵒ, with an IC50 value of 69.53 ± 4.4 µM (refer to Supplementary Figure S1). Interestingly, ST50940260 also demonstrated notable dual inhibitory activity against Mᵖʳᵒ, with an IC50 of 96.7 ± 12 µM (refer to Supplementary Figure S2). In contrast, the derivative Z30231927 showed reduced potency against PLᵖʳᵒ, with an IC50 of 124.82 ± 7.4 µM (refer to Supplementary Figure S3). Notably, the quinoline scaffold has been previously explored for its antiviral activity. The lead compound, ST50940260 is built around a quinoline-4-carboxamide core bearing a thiophene substituent, a scaffold that has been previously explored for antiviral activity. This lead compound has been investigated for its antiviral potential against Enterovirus D68 (EV-D68) a non-structural protein, where it demonstrated potent activity with an EC50 of approximately 0.7 µM.43
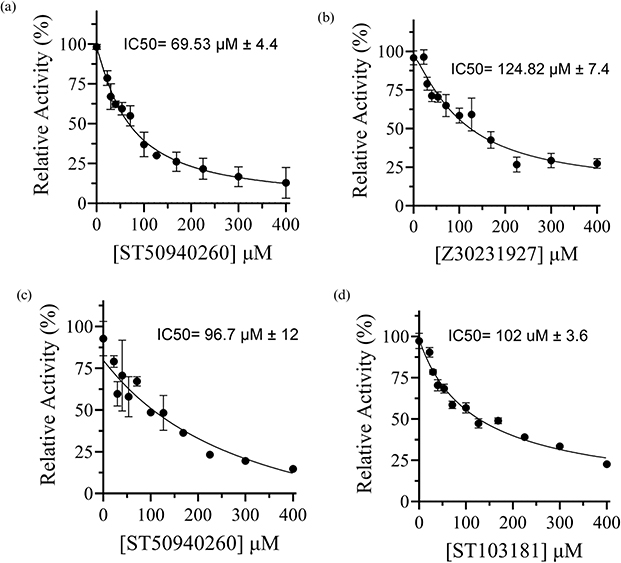 |
Figure 5 IC50 dose–response curves against SARS-CoV-2 proteases of: (a) lead compound ST50940260 against PLᵖʳᵒ, (b) derivative Z30231927 against PLᵖʳᵒ, (c) ST50940260 against Mᵖʳᵒ (d) ST103181 against Mᵖʳᵒ. |
Binding Mode Analysis
The docking strategy employed in this work utilized a flexible ligand–rigid receptor model, which did not account for the conformational adaptability of the protein’s active site. The binding interaction of the lead compound ST50940260, identified as the most promising PLᵖʳᵒ inhibitor with an IC50 value of 69.53 µM, was compared to that of the reference ligand from the PLᵖʳᵒ crystal structure (PDB ID: 7JN2) to evaluate relative binding efficiency and residue engagement within the active site. ST50940260 achieved a substantially more favorable docking score of −10.215 kcal/mol, representing an improvement of approximately 3.6 kcal/mol over the reference ligand’s previously reported value of −6.590 kcal/mol, suggesting a stronger predicted binding affinity. On the other hand, the derivative Z30231927 exhibited a markedly lower docking score (−6.704 kcal/mol) compared with the lead compound. As shown in Figure 6, ST50940260 adopts a well-accommodated binding pose within the PLᵖʳᵒ active site. The binding mode of the reference and the best derivative Z30231927 is also illustrated for comparison. Both ST50940260 and its derivative preserve a conserved hydrogen-bond interaction with Gln267 (labelled as Gln269 in our previous publication32), which is shared with the reference ligand. This interaction is particularly noteworthy, as Gln267 can adopt multiple rotamers that help regulate the conformation of the BL2 loop, in turn shaping how ligands are accommodated and stabilized within the binding site.32 This is particularly advantageous, as the involvement of this residue in interaction with both the lead and the reference ligand highlights that ST50940260 successfully engages key binding determinants within the active site.
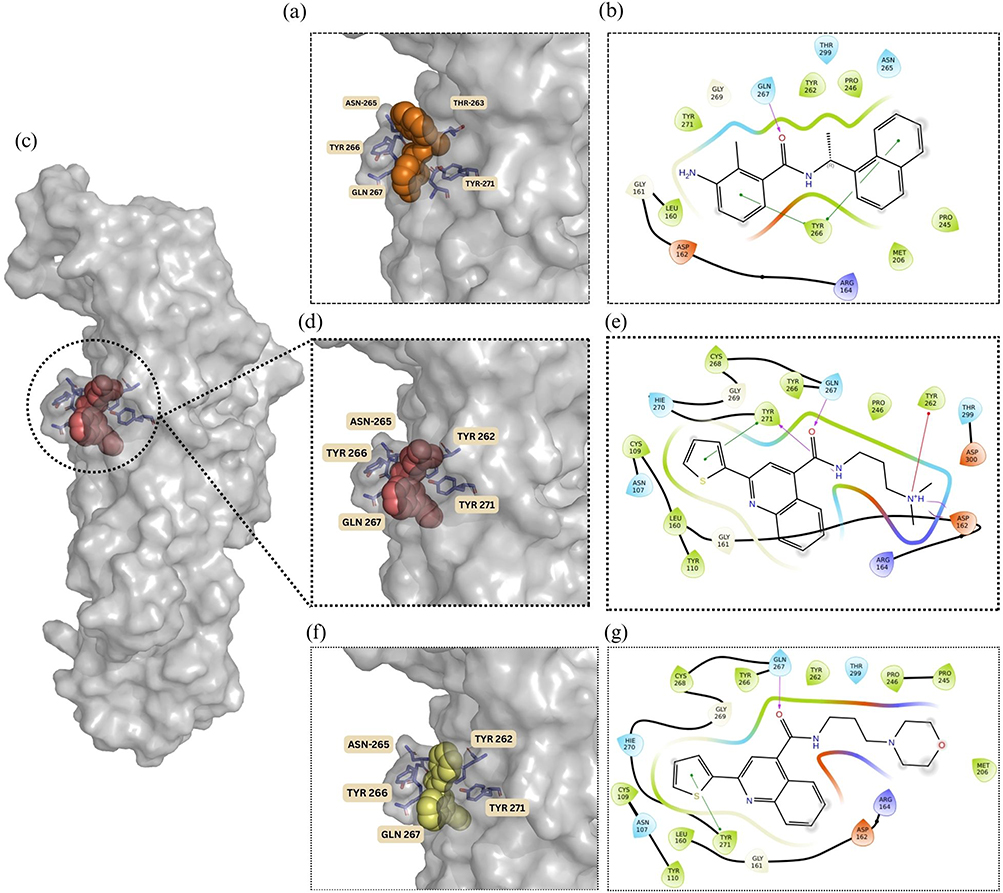 |
Figure 6 (a) Binding pose of the reference ligand within the enzyme’s active site (PDB: 7JN2). (b) 2D representation of the interactions between the reference ligand (Orange spheres) and the surrounding amino acid residues (c) Overall surface representation of the PLᵖʳᵒ protein highlighting the binding pocket occupied by the lead compound ST50940260 (d) zoomed in binding pose of the lead compound ST50940260 (red spheres) within the PLᵖʳᵒ enzyme active site. (e) 2D representation of the interactions between ST50940260 and the surrounding amino acid residues. (f) Binding pose of derivative Z30231927 (yellow spheres) within the PLᵖʳᵒ enzyme active site. (g) 2D representation of the interactions between Z30231927 and the surrounding amino acid residues. |
Also, Z30231927 and ST50940260 display highly similar orientations and share key interactions with neighboring residues, Gln267 and Tyr271, indicating that the quinoline-based core is consistently positioned within the binding pocket. Overall, both ligands exhibit good shape complementarity with the active site. Importantly, the lead compound forms an additional stabilizing interaction with Tyr262, which is enabled by the presence of its tertiary amine, and this interaction is absent in Z30231927. This additional contact likely contributes to the enhanced binding affinity observed for the lead compound. Furthermore, the higher inhibition potency of ST50940260 may be rationalized by its more basic tertiary amine, which has a higher pKa (≈9.3) compared with the morpholine nitrogen in Z30231927 (pKa ≈7.1) as shown in Table 2. At physiological pH, this difference increases the fraction of the protonated species, potentially strengthening interactions within the active site and thereby improving overall binding affinity.
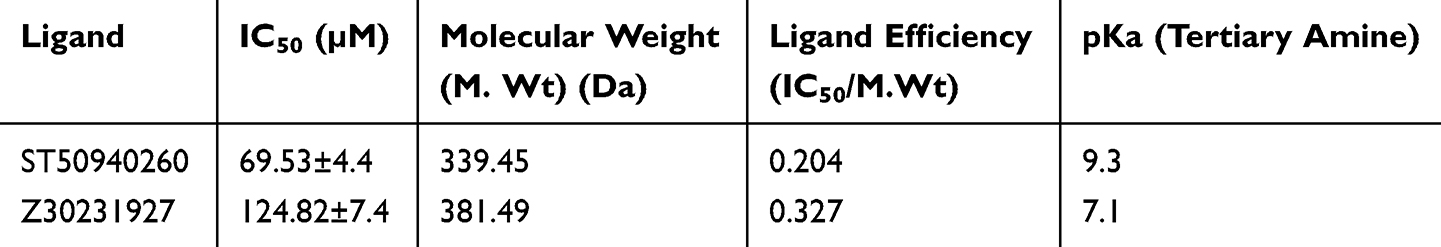 |
Table 2 Comparison of Ligand Efficiency and pKa of Lead Compound ST50940260 and Derivative Z30231927 |
Interestingly, our structure–activity relationship (SAR) analysis suggested that chlorination of the thiophene ring has a negative impact on inhibitory activity. Across the series, derivatives containing a chloro-substituted thiophene consistently showed weaker biological activity compared to those with an unsubstituted thiophene, indicating that this modification is not favorable for binding. This loss in activity may arise from subtle steric or electronic effects that interfere with optimal ligand positioning within the binding site.
Interestingly, the structure–activity relationship (SAR) analysis suggested that chlorination of the thiophene ring has a negative impact on inhibitory activity. Across the series, derivatives containing a chloro-substituted thiophene consistently showed weaker biological activity compared to those with an unsubstituted thiophene, indicating that this modification is not favorable for binding. This loss in activity may arise from subtle steric or electronic effects that interfere with optimal ligand positioning within the binding site.
This trend is clearly illustrated by comparing the two closely related derivatives Z30231927 and Z30232172, which differ only in the presence of a chloro substituent on the thiophene ring. Z30231927 achieved approximately 60% inhibition at 100 µM, whereas Z30232172 displayed less than 50% inhibition at the same concentration. Given the minimal structural difference between the two compounds, the reduced activity observed for Z30232172 can be directly attributed to thiophene chlorination. Overall, these findings highlight the importance of preserving an unsubstituted thiophene moiety for maintaining inhibitory potency and provide a clear direction for future structure optimization.
To gain deeper insight into the stability and interaction behavior of the docked complexes, 100 ns molecular dynamics (MD) simulations were conducted for the lead compound, derivative, co-crystallized reference ligand (PDB entry 7JN2), and apo protein, with the compounds positioned in the PLᵖʳᵒ active site according to their docked orientations. The co-crystallized ligand from PDB entry 7JN2 was included as a reference for comparison of binding stability and structural behavior. Throughout the simulations, root mean square deviation (RMSD) analyses were performed for both the ligands and protein backbones to monitor conformational stability and the persistence of key interactions over time. Radius of gyration (Rg), solvent accessible surface area (SASA), and root mean square fluctuation (RMSF) were also analyzed.
As shown in Figure 7, all four systems remained relatively stable over the 100 ns simulations, with backbone RMSD values within ~1.0–3.5 Å. The reference system backbone (7JN2) exhibited the highest fluctuations, exceeding 3.0 Å after 60 ns, indicating greater structural flexibility. The apo structure showed moderate stability (~1.2–2.5 Å), with a slight increase in fluctuations after 60 ns (Figure 7a). In contrast, the lead complex displayed the lowest and most stable RMSD (~1.0–2.0 Å), with minimal drift throughout the trajectory, suggesting enhanced stabilization of the protein backbone upon binding. The derivative complex showed intermediate behaviour (~1.2–2.5 Å), indicating partial stabilization. Overall, the lead compound provided the greatest structural stability compared to both the apo and reference systems.
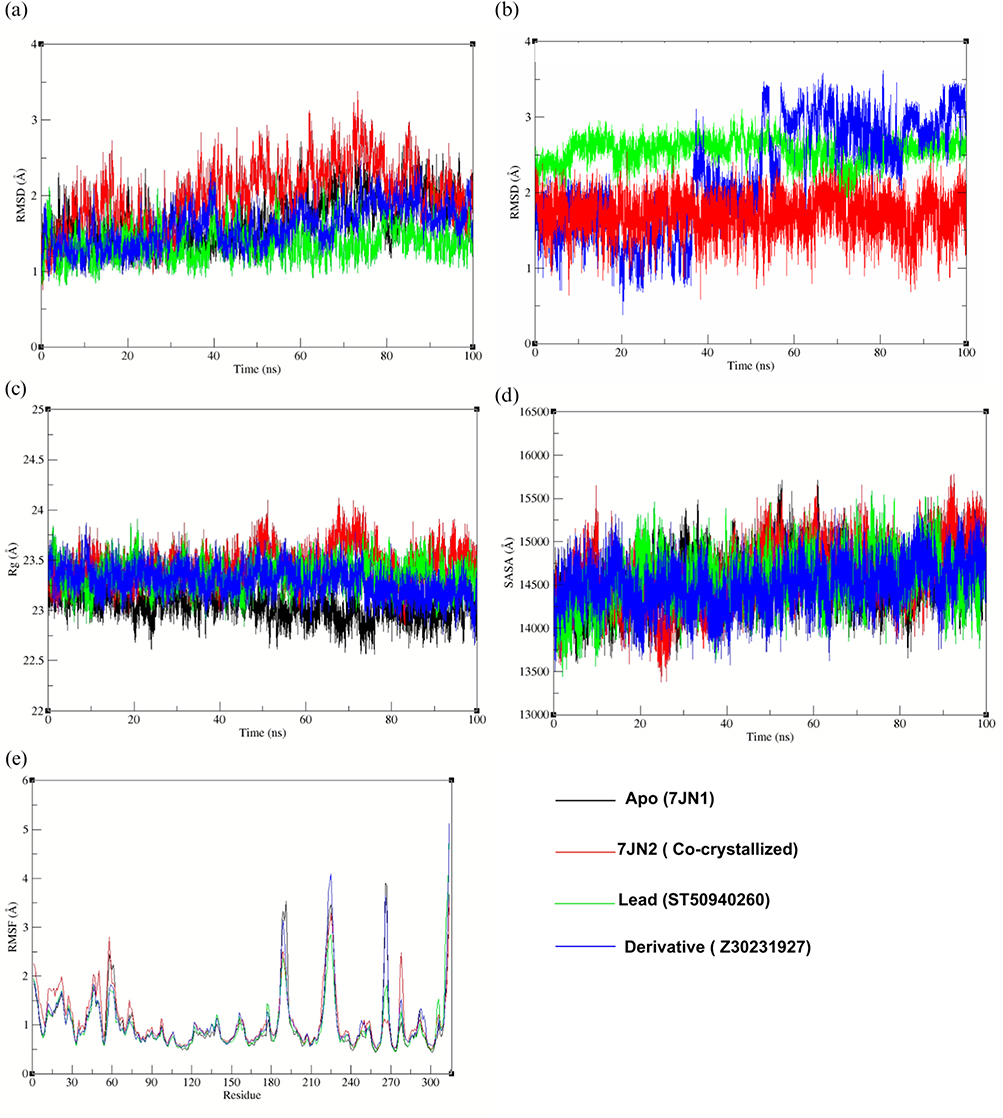 |
Figure 7 (a) Protein backbone RMSD, (b) ligand RMSD, (c) radius of gyration (Rg), (d) solvent-accessible surface area (SASA), and (e) root mean square fluctuation (RMSF) of PLᵖʳᵒ complexes over 100 ns molecular dynamics simulation. |
On the other hand, the ligand RMSD profiles indicate distinct dynamic behaviours across the 100 ns simulation (Figure 7b). The co-crystallized reference ligand maintains the lowest and most consistent RMSD (~1.5–2.0 Å), suggesting a stable binding conformation throughout the trajectory. The lead compound shows moderately higher RMSD values (~2.3–2.8 Å) but with relatively controlled fluctuations. In contrast, the derivative exhibits the largest fluctuations, particularly after ~50 ns, with RMSD values exceeding 3.0 Å, reflecting increased mobility within the binding site and indicating instability.
The radius of gyration (Rg) reflects how the atoms of a protein are distributed around its center and is commonly used as a measure of its compactness. Small changes in Rg over time can occur due to normal conformational fluctuations or folding events. However, a more stable and compact protein structure is typically indicated by minimal variation in Rg throughout the simulation.
In this study, the apo protein showed the lowest Rg values with minimal fluctuations, suggesting a slightly more compact structure in the absence of ligand binding. The co-crystallized reference ligand maintained consistent Rg values with only minor deviations. The lead compound exhibited slightly higher Rg values than the apo protein, yet remained stable, indicating preserved structural integrity (Figure 7c). Similarly, the derivative showed moderate fluctuations but remained within a comparable range to the reference system, with a slight decrease in Rg observed after 70 ns.
Protein structural properties can be influenced by ligand binding, which may in turn affect the SASA. An increase in SASA generally reflects protein expansion, whereas stable or lower fluctuations in SASA are typically expected during a well-equilibrated MD simulation. The derivative complex exhibited the most consistent SASA profile throughout the trajectory, remaining within 13,500–15,500 Å. The lead complex showed initial fluctuations but stabilized after ~20 ns, maintaining a relatively steady SASA thereafter (Figure 7d). The co-crystallized reference displayed moderate variability with noticeable fluctuations, while the apo system remained comparatively stable with no significant drift.
Figure 7e illustrates the RMSF profiles of the complexes as a function of residue number. RMSF analysis was performed to assess residue-level fluctuations and identify changes in protein flexibility upon ligand binding. Most residues exhibit low to moderate fluctuations (<2 Å), indicating overall structural stability across all systems. Some higher fluctuations are observed in specific regions, particularly around residues 180–200 and 220–240, which correspond to more flexible parts of the protein. To be more precise, the derivative displays slightly higher peaks in some regions, indicating greater local flexibility. In comparison, the lead and reference complexes show lower fluctuations in most regions, suggesting more stable residue behavior. The apo system follows a similar overall pattern.
The MM-GB/SA binding free energy results in Table 3. indicate that all systems exhibit favourable binding, as reflected by negative ΔG binding values. The derivative shows the most favourable binding (−30.26 kcal/mol), followed closely by the co-crystallized reference (−29.82 kcal/mol), while the lead also demonstrates favourable binding (−27.33 kcal/mol) within a comparable range.
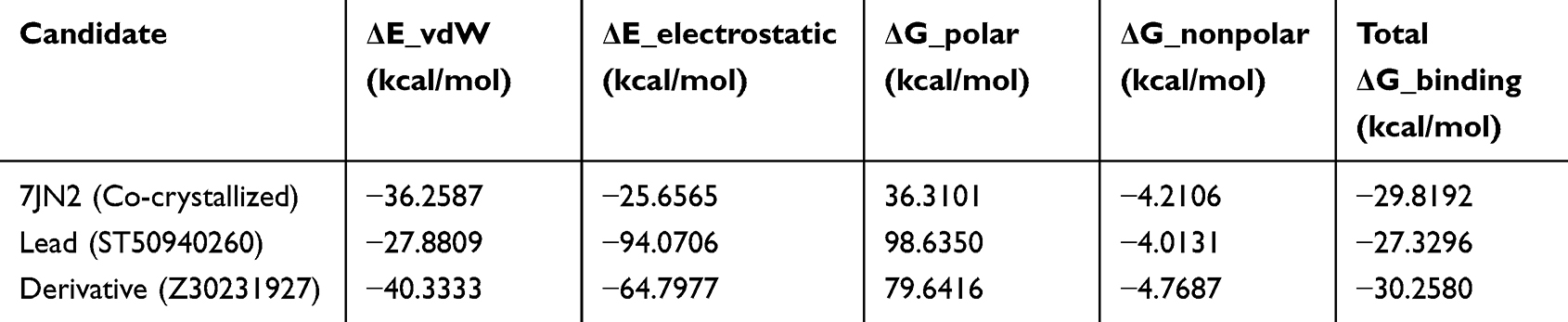 |
Table 3 MM-GB/SA Binding Free Energy (ΔG Binding) and Energy Decomposition Terms, Including van der Waals, Electrostatic, Polar Solvation, and Nonpolar Contributions, for the Lead, Derivative, and Co-Crystallized Reference Complexes |
Decomposition of the energy terms shows that both van der Waals and electrostatic interactions contribute to binding across all systems. The derivative displays a balanced contribution from these interactions, while the lead is characterized by a strong electrostatic component (−94.07 kcal/mol), supporting its interaction with the binding site, although this is partially offset by a higher polar solvation term. The co-crystallized reference exhibits more moderate contributions from both interaction types. The polar solvation term is unfavourable in all cases, reflecting the energetic cost of desolvation, whereas the nonpolar term provides a small favourable contribution.
A comparative analysis of the co-crystallized reference ligand (7JN2), the lead compound (ST50940260), and its derivative (Z30231927) reveals clear differences in binding behaviour and stability as depicted in Figure 8. Hydrogen bond analysis showed that the reference ligand maintained a persistent interaction with GLN267 throughout the trajectory and exhibited the most persistent hydrogen-bonding pattern across the simulation, maintaining approximately 1–2 hydrogen bonds for most frames (Figure 8d), while the lead compound formed sustained interactions with TYR262 (Figure 8a and b) and recurring hydrogen bonds throughout the simulation, although with greater fluctuation than the reference complex. This indicates stable anchoring in both systems. In contrast, the derivative lacked a consistent interaction with these key residues and instead formed transient contacts with TYR710, GLU590, and GLN267, suggesting a more dynamic and unstable binding mode (Figure 8c), and showed a less continuous hydrogen-bonding profile, with hydrogen bonds occurring less persistently and for shorter durations (Figure 8f). These trends are consistent with the MD results, where the reference and lead systems exhibited relatively stable RMSD and RMSF profiles, while the derivative showed greater fluctuations, particularly at later stages. While MM-GB/SA calculations showed that the derivative had the most favourable binding energy (−30.26 kcal/mol), this was mainly driven by stronger van der Waals and electrostatic contributions, despite a high polar solvation penalty. Overall, while the derivative is more energetically favourable, the reference and lead compounds display more stable and consistent interaction patterns, supporting a more reliable binding mode. This is further reflected in the experimental data, where the derivative displayed lower inhibitory potency than the lead, with an approximately 1.8-fold higher IC50 value.
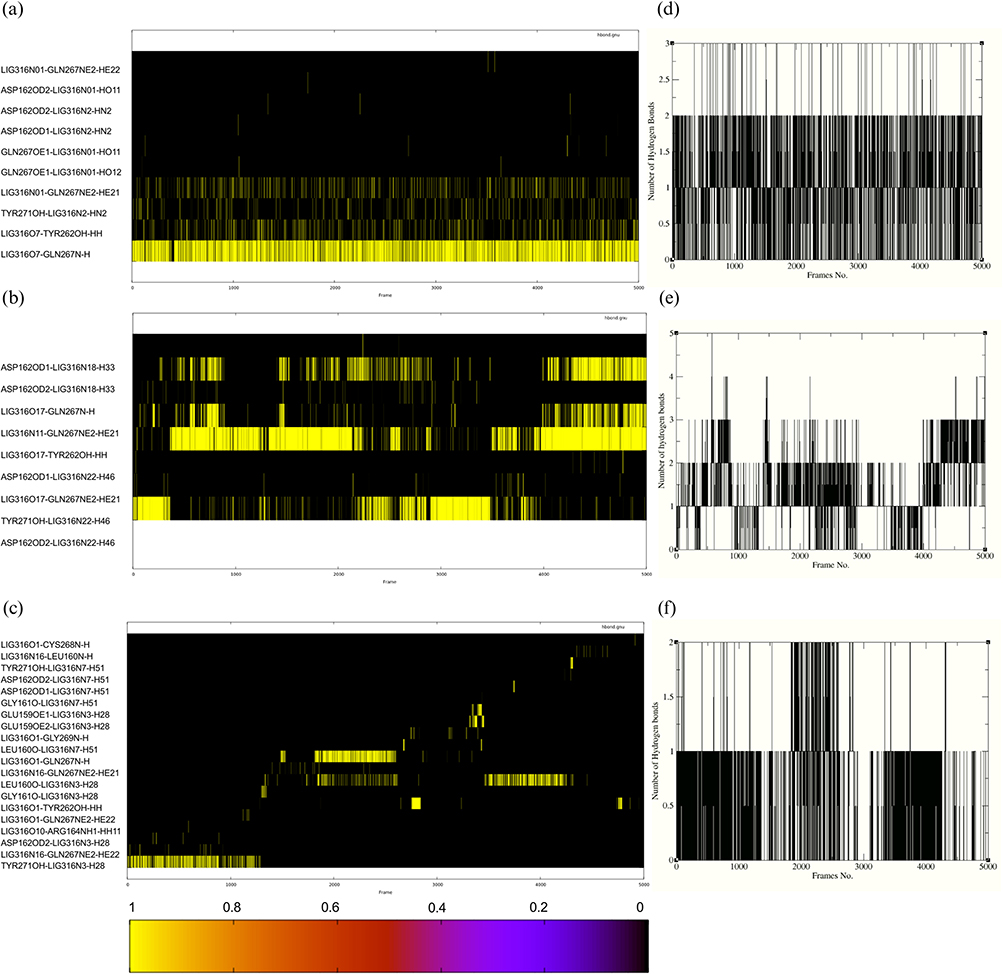 |
Figure 8 Hydrogen-bond analysis of the co-crystallized reference ligand (7JN2), lead compound (ST50940260), and derivative (Z30231927) during the molecular dynamics simulation. (a–c) show the hydrogen-bond occupancy maps for the co-crystallized reference, lead, and derivative complexes, respectively. (d–f) show the corresponding time evolution of the total number of hydrogen bonds for the same systems. In the occupancy maps, yellow indicates the presence of a hydrogen bond and black indicates its absence at a given frame. |
ADME Profile of ST50940260 and Z30231927
A pharmacokinetic and drug-likeness assessment was conducted to further characterize the lead compound ST50940260 and its most promising derivative, Z30231927. The predicted ADME properties of both compounds are summarized in Table 4. Both molecules showed high gastrointestinal absorption, suggesting good potential for oral administration. Interestingly, ST50940260 was predicted to be blood–brain barrier permeant, whereas Z30231927 was not, indicating a lower likelihood of central nervous system exposure for the derivative. In addition, Z30231927 was predicted to be a substrate of P-glycoprotein, an efflux transporter known to limit compound accumulation,44 while ST50940260 was not, which may further support its cellular retention.
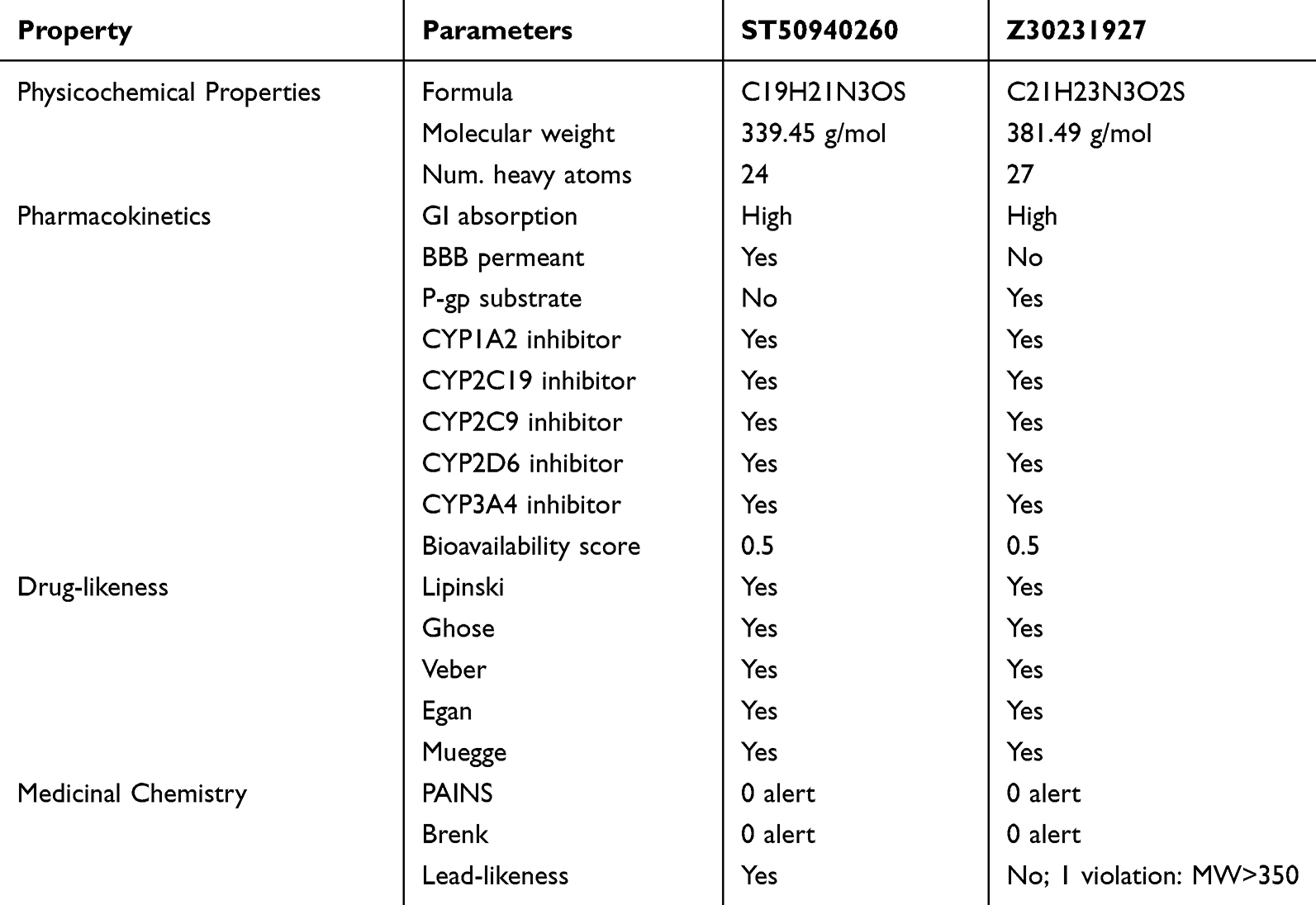 |
Table 4 Comparison of Physiochemical Properties, Pharmacokinetics and Drug-Likeness of Lead and Derivative Predicted by SwissADME |
Both compounds were predicted to inhibit several cytochrome P450 enzymes, including CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, suggesting a potential for drug–drug interactions that should be considered in future development. Evaluation of drug-likeness revealed that both ST50940260 and Z30231927 satisfied all major drug-likeness criteria, including Lipinski, Ghose, Veber, Egan, and Muegge rules. Importantly, neither compound triggered PAINS or Brenk alerts, supporting their suitability as chemically reliable candidates. However, lead-likeness analysis highlighted a distinction between the two, as ST50940260 complied with lead-like criteria, whereas Z30231927 showed a single violation due to its higher molecular weight. Overall, both compounds displayed favorable ADME and drug-like characteristics, with ST50940260 standing out as the more balanced lead candidate.
Conclusion
In this study, we identified two novel small-molecule inhibitors of the SARS-CoV-2 papain-like protease through an integrated ligand-based and structure-based virtual screening approach. Biological evaluation confirmed that both compounds, lead ST50940260 and derivative Z30231927 effectively inhibit PLᵖʳᵒ activity with IC50 values of 69.53 ± 4.4 µM and of 124.82 ± 7.4 µM. ST50940260 also showed inhibition on the main protease Mᵖʳᵒ, indicating a dual-target effect. Molecular dynamics simulations demonstrated that the lead compound binds stably within the PLᵖʳᵒ active site and form key interactions with critical active site residues with favourable binding (−27.33 kcal/mol) compared to the co-crystallized reference 7JN2. Collectively, these computational and preliminary experimental findings suggest that the identified lead is a promising PLᵖʳᵒ inhibitor for antiviral therapy. However, its current potency, with IC50 values in the micromolar range, reflects moderate activity and remains below that typically observed for clinically approved small molecule antivirals, which often exhibit low nanomolar potency. This underscores the need for further lead optimization prior to progression toward in-vivo or clinical evaluation. Future efforts should focus on structure-based optimization to enhance binding affinity and specificity, alongside systematic structure activity relationship studies and scaffold refinement to improve potency. Despite these limitations these results provide a strong foundation for developing new anti–COVID-19 therapies, especially since simultaneous inhibition of both PLᵖʳᵒ and Mᵖʳᵒ can more effectively suppress coronavirus replication. Nevertheless, further work is necessary to translate these findings into a viable therapy as detailed in-vivo efficacy studies and comprehensive pharmacokinetic and toxicity evaluations are required to confirm the compounds’ safety and efficacy. In addition, given the high conservation of coronavirus proteases, this approach may offer potential for broader antiviral applicability beyond SARS-CoV-2, although this remains to be validated in future studies. Overall, the dual PLᵖʳᵒ /Mᵖʳᵒ inhibitory activity exhibited by lead compound ST50940260 offers an especially promising starting point for the development of dual Mᵖʳᵒ and PLᵖʳᵒ inhibitors against COVID-19.
Disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–19. doi:10.1056/NEJMoa2001017
2. Coronavirus confirmed as pandemic by world health organization.
3. Soori H, Attah MOO. Evaluating community engagement strategies in COVID-19: insights from a national quasi-experimental intervention. BMC Public Health. 2025;25(1):2919. doi:10.1186/s12889-025-24403-7
4. World Health Organization. Statement on the fifteenth meeting of the IHR (2005) emergency committee on the COVID-19 pandemic. 2024.
5. Boras B, Jones RM, Anson BJ, et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat Commun. 2021;12(1):6055. doi:10.1038/s41467-021-26239-2
6. Jackson LA, Anderson EJ, Rouphael NG, et al. An mRNA vaccine against SARS-CoV-2 — preliminary report. N Engl J Med. 2020;383(20):1920–1931. doi:10.1056/NEJMoa2022483
7. Allais C, Connor CG, Do NM, et al. Development of the commercial manufacturing process for nirmatrelvir in 17 months. ACS Cent Sci. 2023;9(5):849–857. doi:10.1021/acscentsci.3c00145
8. V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155–170. doi:10.1038/s41579-020-00468-6
9. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of covid-19 — final report. N Engl J Med. 2020;383(19):1813–1826. doi:10.1056/NEJMoa2007764
10. Jayk Bernal A, Gomes da Silva MM, Musungaie DB, et al. Molnupiravir for oral treatment of covid-19 in nonhospitalized patients. N Engl J Med. 2022;386(6):509–520. doi:10.1056/NEJMoa2116044
11. Owen DR. An oral SARS-CoV-2 M ᵖʳᵒ inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593.
12. Nooruzzaman M, Johnson KEE, Rani R, et al. Emergence of transmissible SARS-CoV-2 variants with decreased sensitivity to antivirals in immunocompromised patients with persistent infections. Nat Commun. 2024;15(1):7999. doi:10.1038/s41467-024-51924-3
13. Zhu Y, Yurgelonis I, Noell S, et al. In vitro selection and analysis of SARS-CoV-2 nirmatrelvir resistance mutations contributing to clinical virus resistance surveillance. Sci Adv. 2024;10(30). doi:10.1126/sciadv.adl4013
14. Zhou S, Hill CS, Sarkar S, et al. β-d-N 4-hydroxycytidiNe INhibits SARS-CoV-2 through lethal MutageNesis but is also MutageNic To MammaliaN Cells. J Infect Dis. 2021;224(3):415–419. doi:10.1093/infdis/jiab247
15. Tan H, Hu Y, Jadhav P, Tan B, Wang J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J Med Chem. 2022;65(11):7561–7580. doi:10.1021/acs.jmedchem.2c00303
16. Wu X, Go M, Nguyen JV, et al. Mutational profiling of SARS-CoV-2 papain-like protease reveals requirements for function, structure, and drug escape. Nat Commun. 2024;15(1):6219. doi:10.1038/s41467-024-50566-9
17. Ratia K, Kilianski A, Baez-Santos YM, Baker SC, Mesecar A. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10(5):e1004113. doi:10.1371/journal.ppat.1004113
18. Wu X, Devine SM, Go M, et al. Divergent resistance pathways amongst SARS-CoV-2 PLpro inhibitors highlight the need for scaffold diversity. PLoS Pathog. 2025;21(9):e1013468. doi:10.1371/journal.ppat.1013468
19. Narayanan A, Narwal M, Majowicz SA, et al. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun Biol. 2022;5(1):169. doi:10.1038/s42003-022-03090-9
20. Molecular operating environment (MOE). (2022).
21. Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi:10.1021/jm300687e
22. Braga RC, Andrade CH. Assessing the performance of 3D pharmacophore models in virtual screening: how good are they? Curr Top Med Chem. 2013;13(9):1127–1138. doi:10.2174/1568026611313090010
23. Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. doi:10.1021/jm030644s
24. Chemdiv. Available from: https://www.chemdiv.com/catalog/screening-libraries/.
25. Available from: https://www.timtec.net/.
26. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:10.1016/S0169-409X(96)00423-1
27. Veber DF, Johnson SR, Cheng H-Y, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. doi:10.1021/jm020017n
28. Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740. doi:10.1021/jm901137j
29. Schrˆdinger. LigPrep | schrˆdinger. Schrˆdinger Release 2018-2. 2018.
30. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi:10.1007/s10822-013-9644-8
31. Osipiuk J, Tesar C, Endres M, et al. The crystal structure of papain-like protease of SARS CoV-2 in complex with PLP_Snyder441 inhibitor. BioRxiv. 2020. doi:10.2210/pdb7jn2/pdb
32. Atatreh N, Mahgoub RE, Ghattas MA. Results in chemistry evaluating the potential of PL pro as a drug target in SARS-CoV-2: MD simulations and druggability analysis. Results Chem. 2025;17:102565. doi:10.1016/j.rechem.2025.102565
33. Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi:10.1021/jm0306430
34. Irwin JJ, Duan D, Torosyan H, et al. An aggregation advisor for ligand discovery. J Med Chem. 2015;58(17):7076–7087. doi:10.1021/acs.jmedchem.5b01105
35. Abou Hajal A, Bryce RA, Amor BB, Atatreh N, Ghattas MA. Boosting the accuracy and chemical space coverage of the detection of small colloidal aggregating molecules using the BAD molecule filter. J Chem Inf Model. 2024;64(13):4991–5005. doi:10.1021/acs.jcim.4c00363
36. Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25(2):247–260. doi:10.1016/j.jmgm.2005.12.005
37. Peters MB, Yang Y, Wang B, et al. Structural survey of zinc containing proteins and the development of the Zinc AMBER force field (ZAFF). J Chem Theory Comput. 2010;6(9):2935–2947. doi:10.1021/ct1002626
38. Guimarães CRW, Cardozo M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J Chem Inf Model. 2008;48(5):958–970. doi:10.1021/ci800004w
39. Ferreira JC, Villanueva AJ, Al Adem K, et al. Identification of novel allosteric sites of SARS-CoV-2 papain-like protease (PLpro) for the development of COVID-19 antivirals. J Biol Chem. 2024;300(11):107821. doi:10.1016/j.jbc.2024.107821
40. Ferreira JC, Fadl S, Ilter M, et al. Dimethyl sulfoxide reduces the stability but enhances catalytic activity of the main SARS-CoV-2 protease 3CLpro. FASEB J. 2021;35(8):e21774. doi:10.1096/fj.202100994
41. Pan X, Wang H, Li C, Zhang JZH, Ji C. MolGpka: a web server for small molecule pKa prediction using a graph-convolutional neural network. J Chem Inf Model. 2021;61(7):3159–3165. doi:10.1021/acs.jcim.1c00075
42. Triballeau N, Acher F, Brabet I, Pin J-P, Bertrand H-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem. 2005;48(7):2534–2547. doi:10.1021/jm049092j
43. Musharrafieh R, Zhang J, Tuohy P, et al. Discovery of quinoline analogues as potent antivirals against enterovirus D68 (EV-D68). J Med Chem. 2019;62(8):4074–4090. doi:10.1021/acs.jmedchem.9b00115
44. Montanari F, Ecker GF. Prediction of drug–ABC-transporter interaction — recent advances and future challenges. Adv Drug Deliv Rev. 2015;86:17–26. doi:10.1016/j.addr.2015.03.001
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
De Novo Design of Cathepsin B1 Inhibitors as Potential Anti-Schistosomal Agents Using Computational Studies
Alzain AA, Elbadwi FA
Advances and Applications in Bioinformatics and Chemistry 2022, 15:29-41
Published Date: 1 August 2022