")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 15
De Novo Design of Cathepsin B1 Inhibitors as Potential Anti-Schistosomal Agents Using Computational Studies
Authors Alzain AA , Elbadwi FA
Received 22 February 2022
Accepted for publication 21 July 2022
Published 1 August 2022 Volume 2022:15 Pages 29—41
DOI https://doi.org/10.2147/AABC.S361626
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr James Briggs
Abdulrahim A Alzain, Fatima A Elbadwi
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Gezira, Gezira, Sudan
Correspondence: Abdulrahim A Alzain, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Gezira, Gezira, Sudan, Tel +249-511854501, Fax +249-511861180, Email [email protected]
Background: Schistosomiasis is the world’s second most devastating disease after malaria and the leading cause of disease and mortality for more than 200 million people in developing countries. Cysteine proteases, in particular SmCB1, are the most well-researched biological targets for this disorder.
Objective: To apply computational techniques to design new antischistosomal agents against SmCB1 protein with favorable pharmacokinetic properties.
Methods: The smCB1 receptor-based pharmacophore model was created and used to screen 567,000 fragments from the Enamine library. The best scoring fragments have been linked to build novel compounds that were subjected to molecular docking, MM-GBSA free energy estimation, ADME prediction, and molecular dynamics.
Results: A seven-point pharmacophore hypothesis ADDDRRR was created. The developed hypothesis was used to screen 1.3 M fragment conformations. Among them, 23,732 fragments matched the hypothesis and screened against the protein. The top 50 fragments were used to design new 7745 compounds using the Breed ligand panel which were subjected to docking and MMGBSA binding energy. This led to the identification of 10 compounds with better docking scores (− 8.033– − 7.483 kcal/mol) and lower-bound free energies (− 58.49 – − 40.02 kcal/mol) compared to the reference bound ligand. Most of the designed compounds demonstrated good drug-like properties. Concerning Molecular dynamics (MD) simulation results, a low root mean square deviation (RMSD) range (0.25– 1.2 Å) was found for the top 3 complexes which indicated their stability.
Conclusion: We identified compounds that could be potential candidates in the search for novel Schistosoma mansoni inhibitors by targeting SmCB1 utilizing various computational tools. Three newly designed compounds namely breed 1, 2, and 3 showed promising affinity to the target as well as favorable drug-like properties which might be considered potential anti-schistosomal agents.
Keywords: pharmacophore modeling, molecular hybridization, docking, molecular dynamics, pharmacokinetic properties
Introduction
Schistosomiasis is one of the most human serious parasitic diseases, caused by infections by helminth parasites of the genus Schistosoma. It is a water-borne disease that increases its incidence, as it needs to make more use of water and land resources.1,2 This trematode is now spread in 77 nations, infects about 230 M people worldwide, and brings about 732 M people with infection risk.3–6 Concerning death and morbidity, WHO considers schistosomiasis as the most serious human helminth disease.
Schistosoma mansoni is a prominent etiological agent of this illness among the 5 species of Schistosomas that infect people. Immune-pathological reactions towards the trematodes eggs that aggregate in multiple organs are the cause of morbidity from this disease.7–11 Praziquantel (PZQ) is widely used in the present treatment of schistosomiasis. The mature worms are paralyzed by this oral anthelminthic, which has an effect of 85–90%.12–14 Unfortunately, it is risky to have only one effective drug, as the praziquantel resistance has been experimentally documented, as well as the PZQ curative percentages in the field, are low.16–18 It reaches only 13% of the target group, and it is typically not advisable for children under the age of 6 due to the bitterness and the big pill size.18,19 In addition, praziquantel cannot protect against reinfection, which requires additional therapy, and its efficacy is reduced in cases of severe infections.15,18,20 According to a recent analysis from an MDA program in Uganda, excessive drug administration may decrease the medication’s efficacy.21 Therefore, new medicines are urgently needed, but these significant diseases are often “ignored”, as they mainly affect poor and marginalized people.22 The WHO proposed a 40% reduction in worm loads in an experimental host as a criterion for financing Schistosoma vaccines for preclinical studies several decades ago, and only a few vaccines have crossed this fairly low threshold.15,16
Peptidases, especially those implicated in the nutritional breakdown of blood proteins, had long been known to be significant targets.23,24 Those enzymes are involved in parasite feeding, cytokine secretion, cellular invasion, and other processes.23–30 S. mansoni Cathepsin B “SmCB” is the most common cysteine protease identified in the intestines and cytoplasmic extracts of schistosomula and mature worms. It participates in the degradation of host blood molecules as well as nutritional intake.23–26,31,32 According to RNA interference experiments 35, worms exhibit a considerable impairment of growth at reduced SmCB1 in contrast to control parasites.33 One group has observed a decreased survival of the oocytes by inhibiting SmCB1.34,35 Indeed, SmCB1 is a proven biological target, and various medical chemistry studies have been conducted that produce trypanocidal antagonists in both parasite cultures and experimental animals.36–50
Vaccine production is a decades-long procedure, but advances in many emerging technologies, such as genomics, microarrays, transcriptomics, immunogenic profiles, and computational methods, have helped produce a promising target against Schistosoma antigens and candidate drugs against Schistosoma mansoni.51 Efforts have been made to develop vaccines to challenge this disorder, for example, Sm-p80-based vaccine that showed good efficacy in study,52,53 also a chimeric antigen was developed by another group that had a promising effect as a vaccine for this parasite, also a chimeric antigen was developed by another group which had a promising effect as a vaccine for this parasite.54
The X-ray structure of SmCB1 in combination with WRR-391 was recently determined by Jlkova group.55 Structure-based drug discovery is among the most popular and commonly utilized methods in the drug discovery process.56 As an outcome, the length and expense of medication research and development can be reduced.57 It allows the evaluation of activity in a diverse range of compounds and can assist in the development of new molecules with increased activity.58,59
In this study, we aimed to design novel SmCB1 inhibitors for schistosomiasis treatment using various in silico techniques including e-pharmacophore-based screening, molecular hybridization, molecular docking, MM-GBSA calculations, ADME (absorption, distribution, metabolism, and excretion) prediction, and molecular dynamics.
Materials and Methods
All computational studies were carried out using Maestro v 12.8 of the Schrodinger suite and academic Desmond v6.5 by D.E. Shaw Research for molecular dynamics. The workflow of this study is summarized in Figure 1.
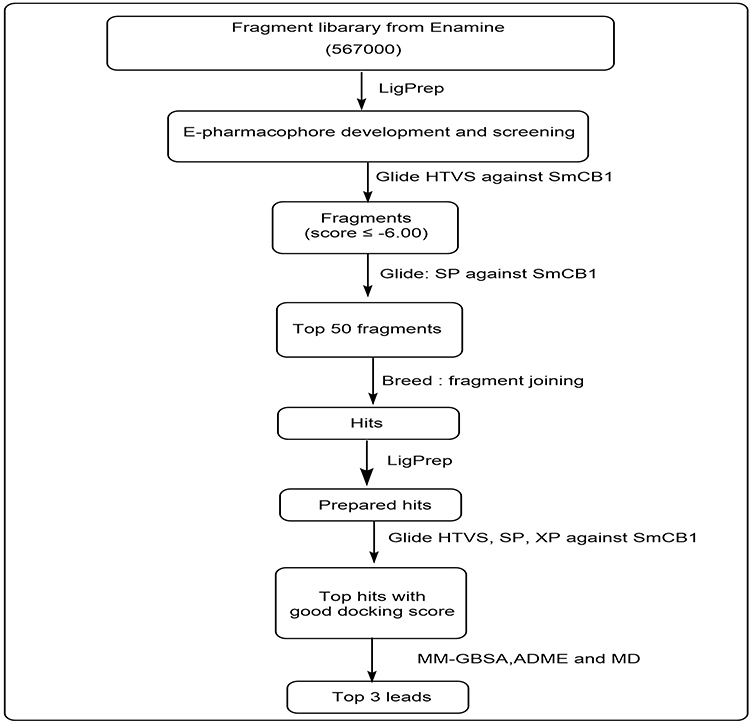 |
Figure 1 Study workflow. |
Preparation of the Protein
SmCB1 (PDB ID: 5OGQ) crystal structure was retrieved from the PDB.60 It was resolved at 1.91 Å and complexed with the ligand WRR391.55,61 The protein crystal structure was prepared using the Protein Preparation wizard tool of Schrodinger before docking to solve many issues related to protein.62 This procedure results in reassignment bond orders, the addition of hydrogens, recognition of disulfide bonds, filling of missing loops and side chains, and correction of any misidentified components. The protein structure was also subjected to constrained minimization. In which, heavy atoms in the structure are constrained to decrease torsional tension during this minimization process, while hydrogens are left unrestrained.
Retrieval and Preparation of Fragments
567,000 fragments were retrieved from the Enamine database63 and prepared with the LigPrep tool of Schrodinger.62 LigPrep ensures that ligand geometry is optimized and a high-energy 3D structure with optimal chiralities is created for each fragment.
Grid Generation of Protein Receptor
The active site of the SmCB1 protein was determined using Schrodinger’s Receptor Grid Generation tool. It selects the coordinates of the bound ligand to the receptor to produce a 3D grid with precise dimensions to reflect the active region of the receptor.
E-Pharmacophore Generation and Screening
The SmCB1-WRR391 complex was used as input for pharmacophore modeling using Schrodinger’s Phase64 to create structural features for pharmacophore-based screening.
The hypothesis was then used as a 3-dimensional search query to screen 1.3 M fragment conformations of the Enamine database for matching pharmacophore properties. Throughout the screening, the Phase module evaluates the fitness of compounds with respect to the query hypothesis and arranges them according to fitness scores. To generate SmCB1 inhibitors with the appropriate chemical properties, the compounds were required to match 4 features in the developed model. The top fragments were chosen and docked into SmCB1 protein utilizing Glide HTVS and SP docking modes, which helps to validate the developed pharmacophore model to identify potential inhibitors.
Fragment Joining and de Novo Compounds Design
The top 50 fragments with the best docking scores were used to construct new compounds with an increased binding affinity towards SmCB1 using Schrodinger’s Breed Ligand Creation Panel Tool. The bond angle variation was set to 15 degrees. The largest atom-to-atom distance was 1 Å. The designed compounds were then prepared using the LigPrep tool for the docking studies.
Molecular Docking and Binding Free Energy Calculations
The docking analysis examines the chemical interactions of several possible geometries of molecules (poses) with the SmCB1ʹs surrounding active site and orders them according to docking scores. The docking procedures were carried out using 3 distinct modes of the Glide tool: HTVS, SP, and XP.65
The MM-GBSA binding energy calculation is significantly more accurate than docking; as it considers the solvent effect in ligand-receptor binding. It was carried out for the top-scoring compounds from docking studies using the Prime module of Schrodinger.
To assess their potential efficacy in comparison to previously reported drug-like compounds, all of the selected compounds were compared to currently known reference WRR391 in terms of dock score and MMGBSA binding energy.
ADMET Prediction
The Qikprop module of Schrodinger predicts the ADME properties of compounds.66 The ability to evaluate these properties helps in the evaluation of molecules and saves the time needed to evaluate substances experimentally.
MD Simulation
MD Simulation is the ideal technique for achieving protein-ligand equilibrium, therefore, the SmCB1 in complex with the newly designed compounds that showed good binding interactions were subjected to MD studies using academic Desmond for 100 ns.67 The system was neutralized by the addition of Na+ and Cl− ions, and immersed in an orthorhombic box (10 × 10×10) filled with TIP3P water molecules. The MD task was done using an NPT ensemble at a temperature [300 K] and pressure [1.01325 bar], which are maintained during the simulation. For each system, 1000 frames were captured during the simulations.
Results
E-Pharmacophore Modeling and Screening
Because of its ability to test huge hit libraries in a short time and with minimal processing power, pharmacophore model-based screening has evolved as an important tool in computer-aided drug design. The stereo-electronic characteristics of the ligand are combined with the energy of its interactions with the protein structure in energetically optimized pharmacophore models. The top-ranked pharmacophore site was chosen for this investigation to formulate the pharmacophore hypothesis based on the complex SmCB1-WRR391 (Figure 2). The e-pharmacophore models were created by mapping the key 3-dimensional chemical characteristics involved in biological action utilizing chosen features. We have created a seven-point pharmacophore hypothesis ADDDRRR with 3 donors, one acceptor, and 3 aromatic rings, and their distances are shown in Figure 3. Four of these pharmacophore characteristics were screened against the fragment’s library. Of Which, 23,732 fragments matched the screened hypothesis.
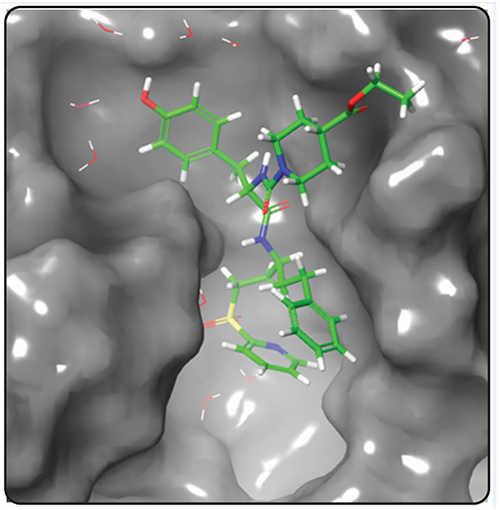 |
Figure 2 SmCB1 protein bound to the reference WRR391 (PDB ID: 5OGQ). |
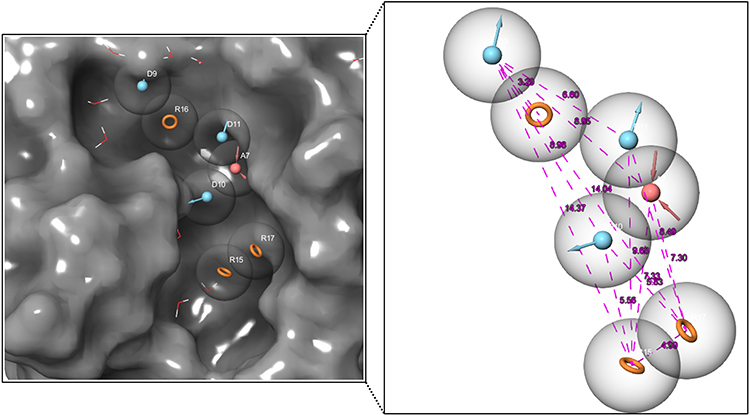 |
Figure 3 The developed Pharmacophore hypothesis using SmCB1-WRR391 complex (PDB ID: 5OGQ). |
Fragments Docking-Based Screening and Linking
The docking-based screening was used to examine and confirm the binding strength of fragments that matched the pharmacophore hypothesis against SmCB1 by docking them into SmCB1 receptors. The HTVS mode was utilized as the initial assessment level, and then the fragments with docking score ≤ −6.00 kcal/mol were docked in the SP mode. Following docking data analysis, the Breed Ligand Creation panel was utilized to link the top 50 fragments to build novel compounds. 7745 newly designed compounds were prepared for docking studies against SmCB1, as described in the next section.
Docking of the Newly Designed Compounds and MM-GBSA Calculations
The molecular docking estimates the interaction patterns of protein and ligands. The designed compounds were docked into SmCB1 protein using the HTVS, SP, and XP docking modes to estimate the binding affinity. As shown in Table 1 and Figure 4, 10 compounds exhibited better docking scores (−8.033– −7.483 kcal/mol) compared to the reference WRR391.
 |
Table 1 XP Docking Score, MM-GBSA Bind Free Energies, and ADME Properties of Top 10 Newly Designed Compounds SmCB1 Protein (PDB ID: 5OGQ) |
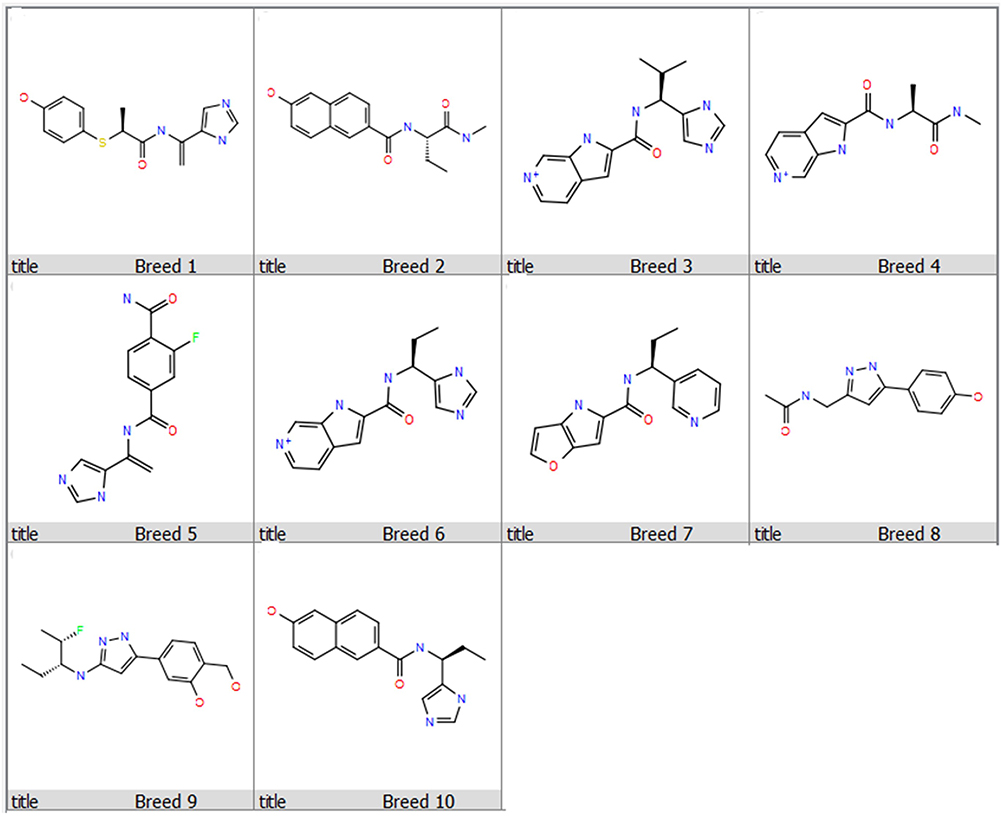 |
Figure 4 Chemical structures of the 10 compounds that showed promising binding affinity against SmCB1 protein. |
Using a graphical representation of the “Schrodinger ligand interaction module”, a detailed analysis of the molecular interactions between SmCB1 and the top 3 ligands was depicted in Figure 5. Breeds 1 and 3 formed 4 and 5 hydrogen bonds, respectively with GLY 144, GLY 269, and GLU 316. In which, breed 1 interacted with GLY 269 through two hydrogen bonds. Also, breed 1 formed a bridged hydrogen bond with GLY 244. Breeds 2 displayed five and 7 hydrogen bonds, respectively with GLN94, GLY 144, GLY 269, and GLU 316. While breed 2 interacted with GLY 269 through two hydrogen bonds.
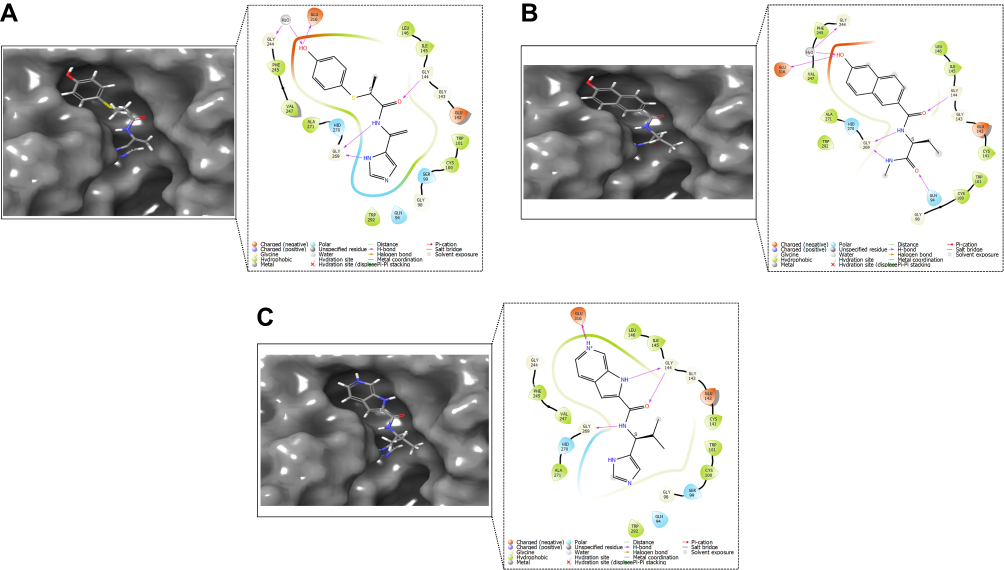 |
Figure 5 2D and 3D interaction of (A) Breed 1, (B) Breed 2 and (C) Breed 3 with SmCB1 protein (PDB ID: 5OGQ). |
The results of the docking showed that the ligands bind to the active region of the protein, but whether these relationships can be maintained enough to trigger any possible biological response since the biological response depends primarily on the free energy of the binding. As a result, the SmBC1-docked complexes in XP mode were subjected to binding MM-GBSA binding energy calculations. The results showed that 10 compounds had better free energies (−58.49 to −40.02 kcal/mol) in comparison with the reference WRR391.
ADMET Analysis
The Schrodinger’s Qikprop module was utilized to assess the drug-likeness (Lipinski’s rule of 5) and ADME evaluations of the designed SmCB1 inhibitors that exhibited the best binding affinity. All of the newly designed compounds did not violate the Lipinski’s rule of 5. Furthermore, the ADME properties of the compounds were studied; QPlogpo/w ranged from 0.646 to 2561, which is important for estimating the absorption and distribution of drugs throughout the body, and QPPCaco, a cellular permeability factor that influences the metabolic pathway, ranged from 110,617 to 459,646. The range of cellular membrane access was −0.512 to −3.221, while the range of QPlogPMDCK was 72.503 to 657.117. The % human oral absorption of these compounds ranged from 67 to 100%.
MD Simulation
MD simulations of selected protein-ligand complexes were performed to determine the appropriate dynamic state and estimate various forms of protein-ligand binding. The RMSD of Cα atoms was evaluated to examine the structural stability of the 3 protein-ligand complexes chosen. In addition, ligand-protein interactions were observed at various time intervals during the simulation experiment.
RMSD analysis (Figure 6) showed that SmCB1-breed 3 complex was the most stable among the 3 complexes and a medium fluctuation was observed for breed 2 and to a lower extent for breed 1. During the entire simulation, SmCB1-breed 1 complex has an RMSD range between 1.5–2.4 Å. Firstly, it had equilibrium for the first 40 ns, and for the remaining 60 ns it underwent small fluctuations with an RMSD range of 2.2–2.4 Å (Figure 6A). SmCB1-breed 2 complex showed an equilibrium till the 60 ns, then it displayed fluctuations around 1.8–2.4 Å until the end of the simulation (Figure 6B). SmCB1-breed 3 displayed the most stable interaction among the 3 compounds. It reached equilibrium for most of the entire simulation time with small fluctuations and an RMSD range of 1.5–2.1 Å (Figure 6C). We also performed MD stimulation for the reference ligand which had an RMSD range of 1.5–2.7 Å. For the first 40 ns, it showed high fluctuations. Lastly, it reached equilibrium for the remaining 60 ns (Figure 6D).
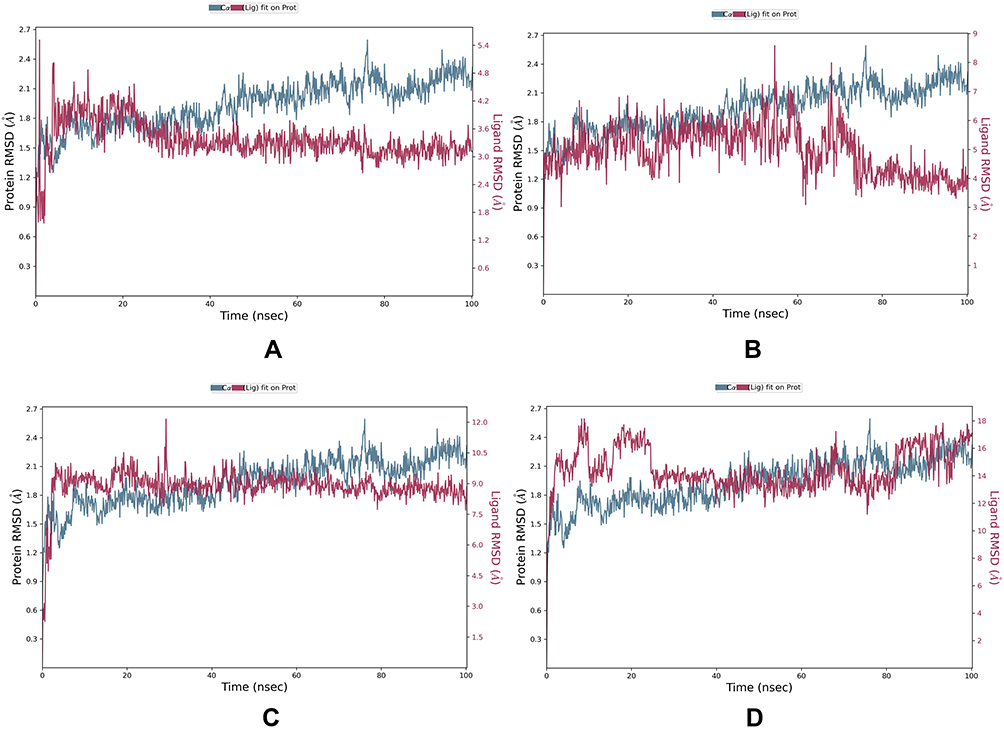 |
Figure 6 RMSD plot of SmCB1 protein-ligand interaction of top 3 complexes: (A) breed 1 RMSD, (B) breed 2 RMSD, (C) breed 3 RMSD, (D) reference. |
The interaction analysis (Figure 7) showed that the interactions responsible for maintaining the stability in breed 1 were direct and bridged hydrogen bond interactions mainly with HIS181 (80%), GLU242 (60%), GLY269 (70%), GLN94, CYS100, LEU267 and GLU316, and hydrophobic interactions with CYS100 and LEU146 (Figure 7A). In breed 2, there were 3 types of interactions, direct and bridged hydrogen bonds with GLY144 (50%) GLY143, GLU142, GLY138, GLU135, LEU146, and GLU142, and hydrophobic interactions with ILE193 and TRP101 (Figure 7B). In breed 3, there were only 2 types of interaction, direct and bridged H-bonds with GLU13, CYS311, SER315, THR246, and ASN223 (Figure 7C). Finally, the reference showed 3 types of interactions with LEU267 (70%), TRP292 (85%), GLY269, CYS100, ARG96, SER95, GLN94, HIS181, GLU265, and THR266 through direct and bridged hydrogen bonds as well as hydrophobic interactions with HIS180, HIS181, ILE193, LYS208, VAL247, PHE251, LEU252, TRP296, and LEU267 (Figure 7D). As we see they are many common interactions between the 3 complexes and the reference ligand.
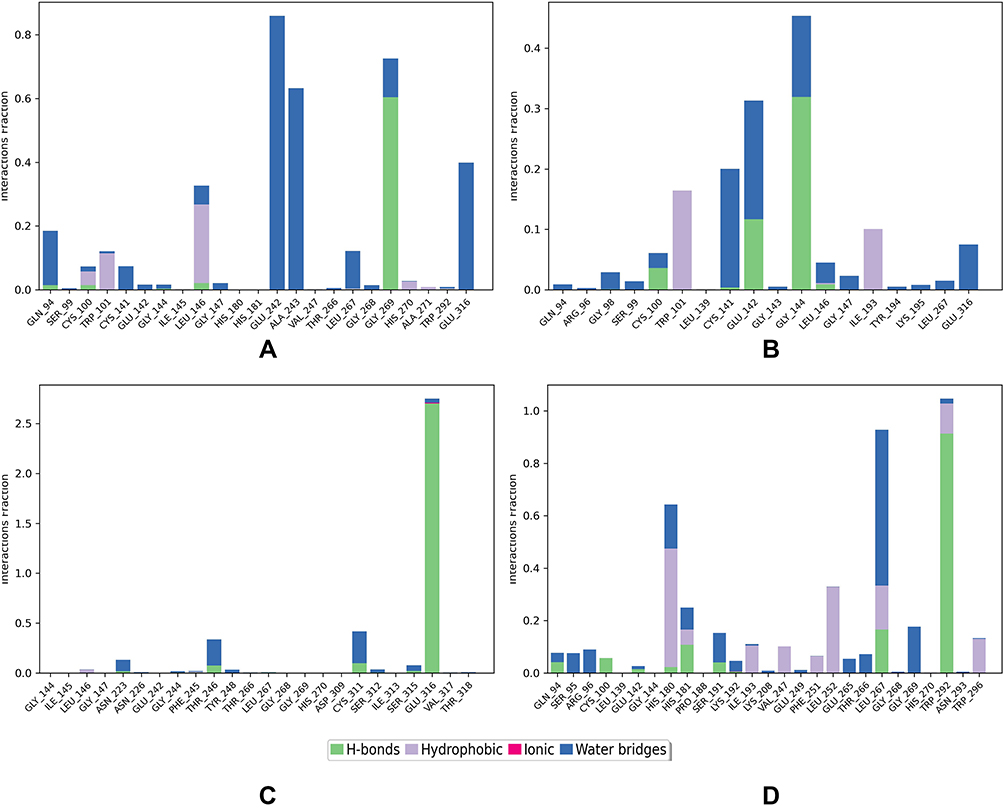 |
Figure 7 Histogram of ligand interaction of top complexes: (A) breed 1, (B) breed 2, (C) breed 3, (D) reference. |
Discussion
Poverty-related parasitic diseases affect millions of people in most developing countries and receive little attention in the research and development program of the pharmaceutical industry.55 Schistosomiasis is a worldwide health problem due to S. mansoni parasitic blood flows with more than 200 M infected individuals. Given our complete dependence on praziquantel and the absence of novel compounds entering the development area, there is an urgent need to discover novel schistosomal protein targets and active anti-schistosomal agents.68 Peptidases, especially those that catalyze the breakdown of blood proteins for nutritional purposes, have long been considered important targets for both medicines and vaccines.69 SmCB1, a peptidase released by the gastrodermis of schistosomes, is one of the most prominent and immunogenic peptidases.32,69 It is a protease found in the intestine that is essential for the digestion of host blood proteins as a source of nutrition. Reverse genetics and chemical tests indicate that it is essential for parasite multiplication and a promising target for the development of new anti-schistosomal medications.70 We were inspired to apply the potential of computational chemistry to the problem of this neglected disease, since only one drug is used for treatment, with significant concern about associated resistance, limited efficacy, and significant toxicity, which in some cases leads to treatment discontinuation. Computer chemistry has led to the creation of new techniques for predicting molecular properties related to pharmacological potential. As a result, the primary goal of this research is to use a de novo fragment-based design to discover new SmCB1 inhibitors as anti-schistosomal agents.
Firstly, 567,000 fragments were retrieved from the Enamine database and prepared using the LigPrep tool. Then the SmCB1-WRR391 complex was used as input for pharmacophore modeling to create structural features for pharmacophore-based screening. A seven-point pharmacophore hypothesis ADDDRRR has been established. The developed hypothesis was then used as a 3-dimensional search query to screen 1.3 M fragment conformations and looked for matching 4 pharmacophore features. To confirm the binding strength of the 23,732 fragments that matched the pharmacophore hypnosis we used docking-based screening. Following the screening data analysis, the top 50 fragments with the best docking scores were selected to build new compounds with increased SmCB1 binding affinity using the Breed ligand Creation panel. 77,45 compounds were designed and further subjected to docking to confirm their affinity to SmCB1. Molecular docking is commonly used to estimate ligand-protein complexes, screen compounds that will regulate the biological activity, and validate the docked complex’s expected geometries and interactions. The active site of SmCB1 and the top 3 ligands showed hydrogen bonds with GLN94, GLY 144, GLY 269, and GLU 316 residues. To assess their potential binding affinity in comparison to previously reported drug-like compounds, the designed compounds were compared to the reference WRR391 in terms of docking score and MMGBSA binding energy. The findings demonstrated that 10 compounds exhibited better docking scores (−8.033 – −7.483 kcal/mol) and lower MM-GBSA free energies (−58.49–-40.02 kcal/mol) compared to the reference WRR391.
Optimizing the ADMET properties, in addition to their pharmacological effects, increases drug discovery success. All of the 10 compounds were within the allowable limit of Lipinski’s rule of 5, absorption and distribution across the body, cell permeability, and cellular membranes access. Also, they did not cross the BBB. As a result, these compounds have the potential to behave as drug-like molecules, whereas reference WRR391 demonstrated weak cell permeability and a low % human oral absorption. These findings were additionally confirmed and validated by conducting MD studies. We investigated a dynamic change in the ligand-protein complex, which cannot be reliably detected by molecular docking but can be correctly characterized by MD simulations, which have the distinct advantage that both ligand and protein flexibility is made possible. For a given 100 ns time interval, the interactions between breeds 1,2,3, and the protein were measured. The RMSD is a prominent measure used to analyze the structural stability of the protein-ligand complex. RMSD analysis showed that SmCB1-breed 3 complex was more stable than the other 2 complexes and the reference ligand. But all the complexes showed a low RMSD range (1.5–2.4 Å) during the whole simulation time which suggested their stability.71
Lastly, Finally, we searched the literature and tried to validate and compare our results with other studies on this protein. Jlkova et al, conducted 3 studies on SmCB1 using docking and MM-GBSA free binding energy calculation studies. The top 10 resulting molecules have shown MM-GBSA free binding energy values in the range of −58.49 to −40.02 kcal/mol. With regard to the interaction analysis, we see that the results agreed in some residues, eg hydrogen bonds between ligand and GLY269, GLY144, and GLN94 of SmCB1.55,70,72 Jefferson group also carried out a study on SmCB1 using molecular docking. Similar interactions were reported such as hydrogen bonds between ligands and GLY269, GLY144, and GLN94 of SmCB1.73
Conclusion
Schistosomiasis is a severe and disregarded tropical disease caused by Schistosoma worms. The majority of global attempts to prevent schistosomiasis rely on the extensive administration of praziquantel to high-risk populations. Because of its involvement in Schistosoma survival, maturation, and replication, the SmCB1 protein is a key target of Schistosoma mansoni. Using in silico techniques, we tried to find new SmCB1 inhibitors. Here, an e-pharmacophore model was built and tested against a library of fragments from the Enamine database and supported by molecular hybridization, molecular docking, and MM-GBSA binding energy calculations. This led to the identification of 10 newly designed compounds with better docking scores and lower MM-GBSA binding free energies compared to the reference WRR391. Further, the active site of SmCB1 and the top 3 ligands displayed hydrogen bonds with GLN94, GLY 144, GLY 269, and GLU 316. The top 3 compounds (breeds 1, 2, and 3) were further subjected to molecular dynamics to verify their interactions stability. All the complexes showed a low RMSD values during the 100 ns simulation time which proven their stability. Also, the predicted ADME properties of these 3 compounds were within the acceptable ranges. This in silico study found that these compounds might be potential SmCB1 inhibitors and could be used as starting points for lead optimization. At present, we cannot examine these hits experimentally, we think these designed compounds can be valuable.
Acknowledgment
We acknowledge Mme Katia Dekimeche from Schrodinger for her support and help.
Disclosure
The authors declare that there are no conflicts of interest.
References
1. Schistosomiasis. Available from: https://www.who.int/en/news-room/fact-sheets/detail/schistosomiasis.
2. Soares Magalhães RJ, Biritwum N-K, Gyapong JO, et al. Mapping helminth co-infection and co-intensity: geostatistical prediction in Ghana. PLoS Negl Trop Dis. 2011;5(6):e1200. doi:10.1371/journal.pntd.0001200
3. Gryseels B, Polman K, Clerinx J, Kestens L. Human schistosomiasis. Lancet. 2006;368(9541):1106–1118. doi:10.1016/S0140-6736(06)69440-3
4. Colley DG, Bustinduy AL, Secor WE, King CH. Human schistosomiasis. Lancet. 2014;383(9936):2253–2264. doi:10.1016/S0140-6736(13)61949-2
5. Weerakoon KGAD, Gobert GN, Cai P, McManus DP. Advances in the Diagnosis of Human Schistosomiasis. Clin Microbiol Rev. 2015;28(4):939–967. doi:10.1128/CMR.00137-14
6. Mahmoud AA. Schistosomiasis. N Engl J Med. 1977;297(24):1329–1331. doi:10.1056/NEJM197712152972405
7. Abdulla M-H, Lim K-C, Sajid M, McKerrow JH, Caffrey CR. Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 2007;4(1):e14. doi:10.1371/journal.pmed.0040014
8. El-Garem AA. Schistosomiasis. Digestion. 1998;59(5):589–605. doi:10.1159/000007534
9. Burke ML, Jones MK, Gobert GN, Li YS, Ellis MK, McManus DP. Immunopathogenesis of human schistosomiasis. Parasite Immunol. 2009;31(4):163–176. doi:10.1111/j.1365-3024.2009.01098.x
10. McManus DP, Dunne DW, Sacko M, Utzinger J, Vennervald BJ, Zhou X-N. Schistosomiasis. Nat Rev Dis Prim. 2018;4(1):13. doi:10.1038/s41572-018-0013-8
11. Barakat RMR. Epidemiology of Schistosomiasis in Egypt: travel through Time: review. J Adv Res. 2013;4(5):425–432. doi:10.1016/j.jare.2012.07.003
12. Barda B, Coulibaly JT, Puchkov M, Huwyler J, Hattendorf J, Keiser J. Efficacy and Safety of Moxidectin, Synriam, Synriam-Praziquantel versus Praziquantel against Schistosoma haematobium and S. mansoni Infections: a Randomized, Exploratory Phase 2 Trial. PLoS Negl Trop Dis. 2016;10(9):e0005008. doi:10.1371/journal.pntd.0005008
13. Zwang J, Olliaro PL. Clinical efficacy and tolerability of praziquantel for intestinal and urinary schistosomiasis-a meta-analysis of comparative and non-comparative clinical trials. PLoS Negl Trop Dis. 2014;8(11):e3286. doi:10.1371/journal.pntd.0003286
14. Cioli D, Pica-Mattoccia L, Basso A, Guidi A. Schistosomiasis control: praziquantel forever? Mol Biochem Parasitol. 2014;195(1):23–29. doi:10.1016/j.molbiopara.2014.06.002
15. Merrifield M, Hotez PJ, Beaumier CM, et al. Advancing a vaccine to prevent human schistosomiasis. Vaccine. 2016;34(26):2988–2991. doi:10.1016/j.vaccine.2016.03.079
16. Wilson RA, Li X-H, Castro-Borges W. Do schistosome vaccine trials in mice have an intrinsic flaw that generates spurious protection data? Parasit Vectors. 2016;9:89. doi:10.1186/s13071-016-1369-9
17. Melman SD, Steinauer ML, Cunningham C, et al. Reduced susceptibility to praziquantel among naturally occurring Kenyan isolates of Schistosoma mansoni. PLoS Negl Trop Dis. 2009;3(8):e504. doi:10.1371/journal.pntd.0000504
18. Trainor-Moss S, Mutapi F. Schistosomiasis therapeutics: whats in the pipeline? Expert Rev Clin Pharmacol. 2016;9(2):157–160. doi:10.1586/17512433.2015.1102051
19. McManus DP, Loukas A. Current status of vaccines for schistosomiasis. Clin Microbiol Rev. 2008;21(1):225–242. doi:10.1128/CMR.00046-07
20. Siddiqui AA, Siddiqui SZ. Sm-p80-Based Schistosomiasis Vaccine: preparation for Human Clinical Trials. Trends Parasitol. 2017;33(3):194–201. doi:10.1016/j.pt.2016.10.010
21. Crellen T, Walker M, Lamberton PHL, et al. Reduced Efficacy of Praziquantel Against Schistosoma mansoni Is Associated With Multiple Rounds of Mass Drug Administration. Clin Infect Dis an off Publ Infect Dis Soc Am. 2016;63(9):1151–1159. doi:10.1093/cid/ciw506
22. Caffrey CR, Secor WE. Schistosomiasis: from drug deployment to drug development. Curr Opin Infect Dis. 2011;24(5):410–417. doi:10.1097/QCO.0b013e328349156f
23. Dalton JP, Clough KA, Jones MK, Brindley PJ. Characterization of the cathepsin-like cysteine proteinases of Schistosoma mansoni. Infect Immun. 1996;64(4):1328–1334. doi:10.1128/iai.64.4.1328-1334.1996
24. Tallima H, Dvořák J, Kareem S, et al. Protective immune responses against Schistosoma mansoni infection by immunization with functionally active gut-derived cysteine peptidases alone and in combination with glyceraldehyde 3-phosphate dehydrogenase. PLoS Negl Trop Dis. 2017;11(3):e0005443. doi:10.1371/journal.pntd.0005443
25. González AY, Sulbarán GS, Ballen DE, Cesari IM. Immunocapture of circulating Schistosoma mansoni cathepsin B antigen (Sm31) by anti-Sm31 polyclonal antibodies. Parasitol Int. 2016;65(3):191–195. doi:10.1016/j.parint.2015.12.008
26. Sajid M, McKerrow JH, Hansell E, et al. Functional expression and characterization of Schistosoma mansoni cathepsin B and its trans-activation by an endogenous asparaginyl endopeptidase. Mol Biochem Parasitol. 2003;131(1):65–75. doi:10.1016/s0166-6851(03)00194-4
27. McKerrow JH. Development of cysteine protease inhibitors as chemotherapy for parasitic diseases: insights on safety, target validation, and mechanism of action. Int J Parasitol. 1999;29(6):833–837. doi:10.1016/s0020-7519(99)00044-2
28. Renslo AR, McKerrow JH. Drug discovery and development for neglected parasitic diseases. Nat Chem Biol. 2006;2(12):701–710. doi:10.1038/nchembio837
29. Sajid M, McKerrow JH. Cysteine proteases of parasitic organisms. Mol Biochem Parasitol. 2002;120(1):1–21. doi:10.1016/s0166-6851(01)00438-8
30. McKerrow JH, Doyle PS, Engel JC, et al. Two approaches to discovering and developing new drugs for Chagas disease. Mem Inst Oswaldo Cruz. 2009;104 Suppl(1):263–269. doi:10.1590/s0074-02762009000900034
31. Stack CM, Dalton JP, Cunneen M, Donnelly S. De-glycosylation of Pichia pastoris-produced Schistosoma mansoni cathepsin B eliminates non-specific reactivity with IgG in normal human serum. J Immunol Methods. 2005;304(1–2):151–157. doi:10.1016/j.jim.2005.07.019
32. Kasný M, Mikes L, Hampl V, et al. Chapter 4. Peptidases of trematodes. Adv Parasitol. 2009;69:205–297. doi:10.1016/S0065-308X(09)69004-7
33. Correnti JM, Brindley PJ, Pearce EJ. Long-term suppression of cathepsin B levels by RNA interference retards schistosome growth. Mol Biochem Parasitol. 2005;143(2):209–215. doi:10.1016/j.molbiopara.2005.06.007
34. Hassan AS, Zelt NH, Perera DJ, Ndao M, Ward BJ. Vaccination against the digestive enzyme Cathepsin B using a YS1646 Salmonella enterica Typhimurium vector provides almost complete protection against Schistosoma mansoni challenge in a mouse model. PLoS Negl Trop Dis. 2019;13(12):e0007490. doi:10.1371/journal.pntd.0007490
35. Norbury L. Intranasal delivery of a formulation containing stage-specific recombinant proteins of Fasciola hepatica cathepsin L5 and cathepsin B2 triggers an anti-fecundity effect and an adjuvant-mediated reduction in fluke burden in sheep. Vet Parasitol. 2018;258:14–23. doi:10.1016/j.vetpar.2018.05.008
36. Mott BT, Ferreira RS, Simeonov A, et al. Identification and optimization of inhibitors of Trypanosomal cysteine proteases: cruzain, rhodesain, and TbCatB. J Med Chem. 2010;53(1):52–60. doi:10.1021/jm901069a
37. Brak K, Kerr ID, Barrett KT, et al. Nonpeptidic tetrafluorophenoxymethyl ketone cruzain inhibitors as promising new leads for Chagas disease chemotherapy. J Med Chem. 2010;53(4):1763–1773. doi:10.1021/jm901633v
38. Delcroix M, Sajid M, Caffrey CR, et al. A multienzyme network functions in intestinal protein digestion by a platyhelminth parasite. J Biol Chem. 2006;281(51):39316–39329. doi:10.1074/jbc.M607128200
39. Caffrey CR, Goupil L, Rebello KM, Dalton JP, Smith D. Cysteine proteases as digestive enzymes in parasitic helminths. PLoS Negl Trop Dis. 2018;12(8):e0005840. doi:10.1371/journal.pntd.0005840
40. Dos Santos Filho JM, Moreira DRM, Simone CA, et al. Optimization of anti-Trypanosoma cruzi oxadiazoles leads to identification of compounds with efficacy in infected mice. Bioorg Med Chem. 2012;20(21):6423–6433. doi:10.1016/j.bmc.2012.08.047
41. Abdulla M-H, O’Brien T, Mackey ZB, Sajid M, Grab DJ, McKerrow JH. RNA interference of Trypanosoma brucei cathepsin B and L affects disease progression in a mouse model. PLoS Negl Trop Dis. 2008;2(9):e298. doi:10.1371/journal.pntd.0000298
42. Doyle PS, Zhou YM, Engel JC, McKerrow JH. A cysteine protease inhibitor cures Chagas’ disease in an immunodeficient-mouse model of infection. Antimicrob Agents Chemother. 2007;51(11):3932–3939. doi:10.1128/AAC.00436-07
43. Bryant C, Kerr ID, Debnath M, et al. Novel non-peptidic vinylsulfones targeting the S2 and S3 subsites of parasite cysteine proteases. Bioorg Med Chem Lett. 2009;19(21):6218–6221. doi:10.1016/j.bmcl.2009.08.098
44. Ferreira RS, Dessoy MA, Pauli I, et al. Synthesis, biological evaluation, and structure-activity relationships of potent noncovalent and nonpeptidic cruzain inhibitors as anti-Trypanosoma cruzi agents. J Med Chem. 2014;57(6):2380–2392. doi:10.1021/jm401709b
45. Choe Y, Brinen LS, Price MS, et al. Development of alpha-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg Med Chem. 2005;13(6):2141–2156. doi:10.1016/j.bmc.2004.12.053
46. Ferreira RS, Bryant C, Ang KKH, McKerrow JH, Shoichet BK, Renslo AR. Divergent modes of enzyme inhibition in a homologous structure-activity series. J Med Chem. 2009;52(16):5005–5008. doi:10.1021/jm9009229
47. Ferreira RS, Simeonov A, Jadhav A, et al. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J Med Chem. 2010;53(13):4891–4905. doi:10.1021/jm100488w
48. Greenbaum DC, Mackey Z, Hansell E, et al. Synthesis and structure-activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J Med Chem. 2004;47(12):3212–3219. doi:10.1021/jm030549j
49. Magalhaes Moreira DR, de Oliveira ADT. Conformational restriction of aryl thiosemicarbazones produces potent and selective anti-Trypanosoma cruzi compounds which induce apoptotic parasite death. Eur J Med Chem. 2014;75:467–478. doi:10.1016/j.ejmech.2014.02.001
50. Mallari JP, Shelat A, Kosinski A, et al. Discovery of trypanocidal thiosemicarbazone inhibitors of rhodesain and TbcatB. Bioorg Med Chem Lett. 2008;18(9):2883–2885. doi:10.1016/j.bmcl.2008.03.083
51. Eyayu T, Zeleke AJ, Worku L. Current status and future prospects of protein vaccine candidates against Schistosoma mansoni infection. Parasite Epidemiol Control. 2020;11:e00176. doi:10.1016/j.parepi.2020.e00176
52. Zhang W, Ahmad G, Molehin AJ, et al. Schistosoma mansoni</em> antigen Sm-p80: prophylactic efficacy using TLR4 agonist vaccine adjuvant glucopyranosyl lipid A-Alum in murine and non-human primate models. J Investig Med. 2018;66(8):1124LP- 1132. doi:10.1136/jim-2018-000786
53. Siddiqui AJ, Molehin AJ, Zhang W, et al. Sm-p80-based vaccine trial in baboons: efficacy when mimicking natural conditions of chronic disease, praziquantel therapy, immunization, and Schistosoma mansoni re-encounter. Ann N Y Acad Sci. 2018;1425(1):19–37. doi:10.1111/nyas.13866
54. Pandya N, Kumar A. Immunoinformatics analysis for design of multi-epitope subunit vaccine by using heat shock proteins against Schistosoma mansoni. J Biomol Struct Dyn. 2022;1–20. doi:10.1080/07391102.2021.2025430
55. Jílková A, Rubešová P, Fanfrlík J, et al. Druggable Hot Spots in the Schistosomiasis Cathepsin B1 Target Identified by Functional and Binding Mode Analysis of Potent Vinyl Sulfone Inhibitors. ACS Infect Dis. 2021;7(5):1077–1088. doi:10.1021/acsinfecdis.0c00501
56. Idris MO, Yekeen AA, Alakanse OS, Durojaye OA. Computer-aided screening for potential TMPRSS2 inhibitors: a combination of pharmacophore modeling, molecular docking and molecular dynamics simulation approaches. J Biomol Struct Dyn. 2020:1–19. doi:10.1080/07391102.2020.1792346
57. Singh K, Sharma MC, Sharma S, Jain SV, Avchar MH. An approach to design potent anti-Alzheimer ’ s agents by 3D-QSAR studies on fused 5, 6-bicyclic heterocycles as c -secretase modulators using kNN – MFA methodology. Arab J Chem. 2013. doi:10.1016/j.arabjc.2013.02.002
58. Amnerkar ND, Bhusari KP. Synthesis, anticonvulsant activity and 3D-QSAR study of some prop-2-eneamido and 1-acetyl-pyrazolin derivatives of aminobenzothiazole. Eur J Med Chem. 2010;45(1):149–159. doi:10.1016/j.ejmech.2009.09.037
59. Bendix F, Wolber G, Seidel T. 3D Pharmacophore Elucidation and Virtual Screening Strategies for 3D pharmacophore- based virtual screening. J Med. 2010. doi:10.1016/j.ddtec.2010.11.004
60. Mendes V, Green SR, Evans JC, et al. Inhibiting Mycobacterium tuberculosis CoaBC by targeting an allosteric site. Nat Commun. 2021;12:1. doi:10.1038/s41467-020-20224-x
61. RCSB PDB - 5OGQ: structure of cathepsin B1 from Schistosoma mansoni in complex with WRR391 inhibitor. Available from: https://www.rcsb.org/structure/5OGQ.
62. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi:10.1007/s10822-013-9644-8
63. Databases - Enamine. Available from: https://enamine.net/11-databases.
64. Salam NK, Nuti R, Sherman W. Novel method for generating structure-based pharmacophores using energetic analysis. J Chem Inf Model. 2009;49(10):2356–2368. doi:10.1021/ci900212v
65. Lyne PD, Lamb ML, Saeh JC. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem. 2006;49(16):4805–4808. doi:10.1021/jm060522a
66. Papers JBC, Doi M, Matter ML, Ruoslahti E. A Signaling Pathway from the ␣ 5  1 and ␣ v  3 Integrins That Elevates bcl-2 Transcription. J Biol Chem. 2001;276(30):27757–27763. doi:10.1074/jbc.M102014200
67. Suryanarayanan V, Singh SK. Assessment of dual inhibition property of newly discovered inhibitors against PCAF and GCN5 through in silico screening, molecular dynamics simulation and DFT approach. J Recept Signal Transduct Res. 2015;35(5):370–380. doi:10.3109/10799893.2014.956756
68. Jílková A, Řezáčová P, Lepšík M, et al. Structural basis for inhibition of cathepsin B drug target from the human blood fluke, Schistosoma mansoni. J Biol Chem. 2011;286(41):35770–35781. doi:10.1074/jbc.M111.271304
69. Jensen KB, Driskell RR, Watt FM. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc. 2010;5(5):898–911. doi:10.1038/nprot.2010.39
70. Pavlína R, Leps M, Horn M, et al. Structural Basis for Inhibition of Cathepsin B Drug Target from the Human Blood Fluke, Schistosoma mansoni. J Biol Chem. 2011;286(41):35770–35781. doi:10.1074/jbc.M111.271304
71. Bochevarov AD, Harder E, Hughes TF, et al. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113(18):2110–2142. doi:10.1002/qua.24481
72. Jílková A, Horn M, Řezáčová P, et al. Activation route of the Schistosoma mansoni cathepsin B1 drug target: structural map with a glycosaminoglycan switch. Structure. 2014;22(12):1786–1798. doi:10.1016/j.str.2014.09.015
73. Rocha JA, Rego NCS, Carvalho BTS, et al. Computational quantum chemistry, molecular docking, and ADMET predictions of imidazole alkaloids of Pilocarpus microphyllus with schistosomicidal properties. PloS one. 2018;1–23.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.