Back to Journals » International Journal of General Medicine » Volume 16
Identification Invasion-Related Long Non-Coding RNAs in Lung Adenocarcinoma and Analysis of Competitive Endogenous RNA Regulatory Networks
Authors Mao Y ![]() , Cai F, Jiang T, Zhu X
, Cai F, Jiang T, Zhu X ![]()
Received 18 February 2023
Accepted for publication 1 May 2023
Published 15 May 2023 Volume 2023:16 Pages 1817—1831
DOI https://doi.org/10.2147/IJGM.S407266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Yuze Mao,1,* Fangyu Cai,2,* Tengjiao Jiang,1 Xiaofeng Zhu1
1Department of Cardio-Thoracic Surgery, First Affiliated Hospital of Jiamusi University, Jiamusi, Heilongjiang, 154000, People’s Republic of China; 2Department of Thoracic Surgery, Beidahuang Industry Group General Hospital, Harbin, Heilongjiang, 150088, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaofeng Zhu, Department of Cardio-Thoracic Surgery, First Affiliated Hospital of Jiamusi University, Jiamusi, Heilongjiang, 154000, People’s Republic of China, Tel +86-13845456700, Email [email protected]
Background: Cell invasion plays a vital role in cancer development and progression. Aberrant expression of long non-coding RNAs (lncRNAs) is also critical in carcinogenesis. However, the prognostic value of invasion-related lncRNAs in lung adenocarcinoma (LUAD) remains unknown.
Methods: Differentially expressed mRNAs (DEmRNAs), lncRNAs (DElncRNAs), and microRNAs (DEmiRNAs) were between LUAD and control samples. Pearson correlation analyses were performed to screen for invasion-related DElncRNAs (DEIRLs). Univariate and multivariate Cox regression algorithms were applied to identify key genes and construct the risk score model, which was evaluated using receiver operating characteristic (ROC) curves. Gene set enrichment analysis (GSEA) was used to explore the underlying pathways of the risk model. Moreover, an invasion-related competitive endogenous RNA (ceRNA) regulatory network was constructed. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed to detect the expression of prognostic lncRNAs in the LUAD and control samples.
Results: A total of 45 DElncRNAs were identified as DEIRLs. RP3-525N10.2, LINC00857, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3 were potential prognostic lncRNAs, the expression of which was verified by RT-qPCR in LUAD samples. Both the risk score model and nomogram used the prognostic lncRNAs. ROC curves showed the risk score model had moderate accuracy and the nomogram had high accuracy in predicting patient prognosis. GSEA results indicated that the risk score model was associated with many biological processes and pathways relevant to cell proliferation. A ceRNA regulatory network was constructed in which PDZRN3–miR-96-5p–CPEB1, EP300–AS1-miR-93-5p–CORO2B, and RP3– 525N10.2-miR-130a-5p–GHR may be key invasion-related regulatory pathways in LUAD.
Conclusion: Our study identified five novel invasion-related prognostic lncRNAs (RP3-525N10.2, LINC00857, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3) and established an accurate model for predicting the prognosis of patients with LUAD. These findings enrich our understanding of the relationships between cell invasion, lncRNAs, and LUAD and may provide novel treatment directions.
Keywords: lung adenocarcinoma, invasion, LncRNA, prognosis, CeRNA
Introduction
According to the World Health Organization, cancer is a leading cause of death worldwide.1 According to the International Agency for Research on Cancer Global Burden of Cancer Report 2020,2 an estimated 2.2 million new cancer cases and 1.8 million deaths, lung cancer is the second most commonly diagnosed and the leading cause of cancer death, representing approximately one in ten cancers diagnosed and one in five deaths. Non-small cell lung cancer accounts for approximately 85% of all lung cancers and is the leading subtype in morbidity and mortality.3 The annual incidence of lung adenocarcinoma (LUAD) is increasing.4 The major LUAD subclassifications are invasive, invasive non-mucinous, invasive mucinous, colloid, fetal, and enteric-type adenocarcinoma. Although surgical resection, chemotherapy, radiotherapy, immunotherapy, and targeted therapies have been used for treating LUAD for decades, patients still have a poor prognosis due to local recurrence or distant metastases.2,5 Therefore, there is an urgent need to identify new predictors of LUAD prognosis.
Metastasis is the process by which cancer cells from a primary lesion invade a distal organ or location and involves cell detachment and adhesion, cell invasion, and response to chemokine stimuli.6,7 Among these, invasion of cancer cells is one of the most important causes of metastasis. In recent years, markers of invasion-prone tumors have been identified. Liu et al constructed a prognostic stratification system based on invasion-related genes (IRGs) in colon cancer that could accurately predict patient outcomes.8 Zheng et al developed prognostic risk models based on differentially expressed IRGs in different molecular subtypes of LUAD.9 Therefore, studying invasion-related markers to ensure earlier treatment can potentially improve the prognosis and survival of cancer patients.
Long non-coding RNA (lncRNAs) are a class of 200 bp–100 kb non-coding RNA with unique sequences. Many non-coding RNAs are aberrantly expressed in tumors with high tissue specificity and may serve as prognostic biomarkers. For example, the lncRNA Neat1, a key molecule in cancer research, is elevated in breast,10 prostate,11 stomach,12 colorectal,13 liver,14 and ovarian alveolar cancer.15 lncRNA prognostic markers have also been reported in LUAD, such as immune- and autophagy-related lncRNAs.16,17 However, the prognostic value of invasion-associated lncRNAs in LUAD has not yet been investigated.
This study aimed to investigate invasion-related lncRNAs (IRLs) in LUAD. We screened IRLs, constructed a prognostic risk model, and investigated regulatory mechanisms in LUAD. We also verified their expression in patient samples. We identified that lncRNA–miRNA–mRNA relationships might play critical roles in the prognosis of LUAD. This study revealed IRLs and their role in LUAD. Our findings may help predict the prognosis of LUAD and distinguish the chemotherapeutic sensitivity of patients in low- and high-risk groups.
Materials and Methods
Data Acquisition
The lncRNA and mRNA expression data of 521 LUAD and 58 adjacent control samples and the microRNA (miRNA) expression data of 515 LUAD and 45 adjacent control samples were downloaded from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/). The GSE31210 dataset containing 226 LUAD and 20 adjacent control samples and GSE50081 dataset, containing overall survival (OS) information of 127 patients with LUAD, were downloaded from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) database. We used the clinical data of 497 patients with LUAD from TCGA-LUAD cohort to construct a prognostic model and nomogram. Moreover, 689 IRGs were obtained from the Molecular Signatures Database (MSigDB) (http://www.gsea-msigdb.org/gsea/msigdb/index.jsp) and the cancer single-cell state atlas (CancerSEA) (http://biocc.hrbmu.edu.cn/CancerSEA/home.jsp) database after de-duplicating. We randomly collected surgical specimens from ten groups of patients with LUAD who had signed informed consent to detect the expressions of prognostic lncRNAs in LUAD and control samples. Research was performed in accordance with the Declaration of Helsinki (as revised in 2013) and approval was obtained from the Ethics Committee of the First Affiliated Hospital of Jiamusi University (NO.20222001).
Differential Expression Analysis
The “DESeq2” R package was used to screen differentially expressed mRNAs (DEmRNAs), differentially expressed lncRNAs (DElncRNAs), and differentially expressed miRNAs (DEmiRNAs) between LUAD and control samples with |log2Fold change (FC)| >1 and adjusted p-value < 0.05.
Identification of Invasion-Related DElncRNAs
DEmRNAs and DElncRNAs were identified by overlapping TCGA-LUAD and GSE31210 datasets. The DEmRNAs were overlapped with IRGs to obtain DEIRGs. Next, Pearson correlation between DElncRNAs and DEIRGs were calculated to screen invasion-related DElncRNAs (DEIRLs) using |r >0.5| and p-value < 0.05. Overlapping DEIRLs between TCGA and GSE31210 were used for subsequent analyses.
Construction, Evaluation, and Validation of Risk Score Model
At a ratio of 7:3, 497 patients with LUAD in TCGA database were randomly segregated into either training or internal validation sets. Subsequently, univariate and multivariate Cox regression analyses were performed to filter OS-related DEIRLs for risk score model construction in the training set (p < 0.05). The risk score was calculated using the following formula:

where h0(t) is the baseline hazard function, β is the regression coefficient, and X is the normalized expression value of each prognostic IRL. Risk model establishment and validation were conducted according to Wang et al18 In brief, patients with LUAD in the TCGA training set were separated into low- and high-risk groups based on the optimal cut-off calculated by the surv_cutpoint function in the “survminer” R package. Kaplan–Meier (KM) analysis was used to analyze the OS of the low- and high-risk groups. Then, to evaluate the performance of the risk model, ROC curves were plotted using the R package “survivalROC.” The risk model was subsequently evaluated using the internal and external validation sets. Subsequently, the independent prognostic factors for patients with LUAD were identified by univariate and multivariate Cox regression analyses for plotting a nomogram, which was used to predict the survival of patients with LUAD at 1, 3, and 5 years. Finally, calibration and decision curves were constructed to assess nomogram performance.
Construction of ceRNA Regulatory Network
The miRanda database (http://cbio.mskcc.org/miRNA2003/miRanda.html) was used to screen for DEmiRNAs that interact with prognostic lncRNAs. The miRwalk (walk.umm.uni-heidelberg.de) and TargetScan (http://www.targetscan.org/) databases were used to predict the DEmRNAs targeted by the screened DEmiRNAs. A ceRNA regulatory network was constructed and visualized using the Cytoscape software.
RT-qPCR Analysis
We used reverse transcription-quantitative polymerase chain reaction (RT-qPCR) to detect the expression of the five potential prognostic lncRNAs in LUAD and normal lung tissues, according to Gan et al.19 Total RNA from LUAD samples (n = 10) and adjacent tumor control samples (n = 10) was extracted using Nuclezol LS RNA Isolation Reagent (ABP Biosciences Inc., China). After determining the concentration and purity of RNA, qualified RNA was used for reverse transcription using a SureScript-First-strand-cDNA-synthesis-kit (GeneCopoeia, USA). RT-qPCR was performed on a CFX96 Real-time PCR System (Bio-Rad, USA) using BlazeTaq SYBR Green qPCR Mix 2.0 (GeneCopoeia, USA) under the following thermal cycling conditions: 40 cycles at 95 °C for 30s, 95 °C for 10s, 60 °C for 20s, and 72 °C for 30s. The 2−ΔΔCt method was used to calculate gene fold expression. The primer sequences used in this study are listed in Table 1.
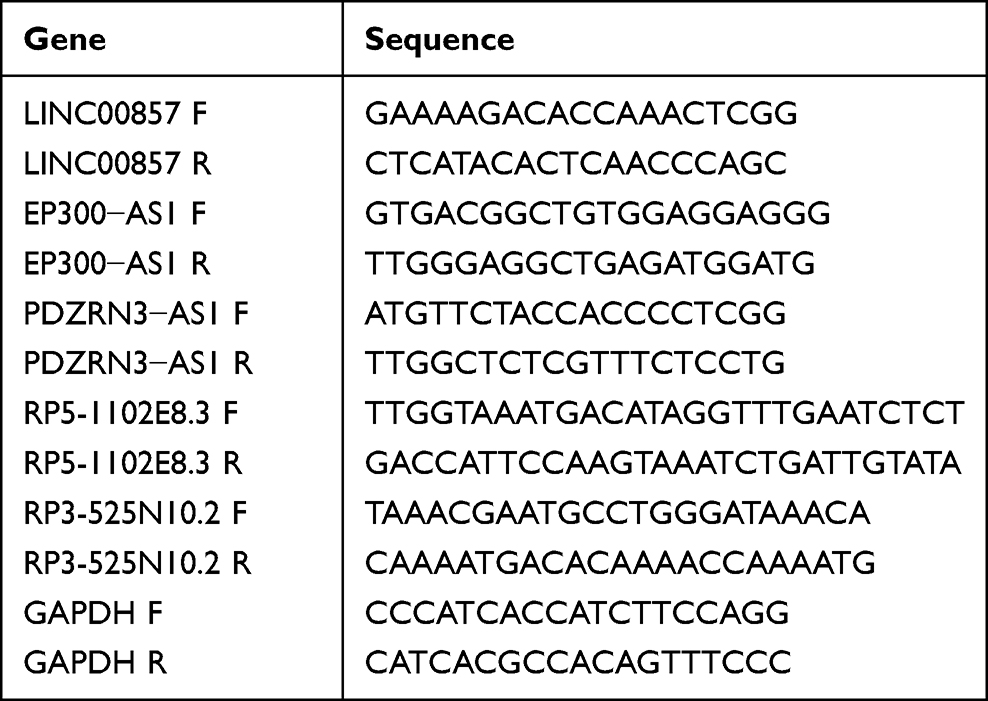 |
Table 1 RT-qPCR Primer Sequences Used in the Current Study |
Statistical Analysis
Principal component analysis (PCA) was performed for effective dimension reduction and pattern recognition of high-dimensional data in the risk model. Gene set enrichment analysis (GSEA) was performed for functional annotation of the low- and high-risk groups. The half-maximal inhibitory concentration of chemotherapy drugs was evaluated by the “pRRophetic” R package to predict the chemotherapeutic sensitivity of patients in low- and high-risk groups. Comparisons between two groups and among multiple groups were performed using the Wilcoxon and Kruskal–Wallis tests, respectively. All data were analyzed using R (version 4.0.0), and statistical significance was set at p < 0.05.
Results
Identification of 45 DEIRLs in LUAD
A total of 5353 DEmRNAs were identified between LUAD and control samples in the TCGA-LUAD cohort, including 3346 upregulated and 2007 downregulated genes (Figure 1A). The top 100 DEmRNAs are displayed in a heatmap (Figure 1B). A total of 3560 DElncRNAs were identified between LUAD and control samples in the TCGA-LUAD cohort, including 2489 upregulated and 1071 downregulated genes (Figure 1C). The top 100 DElncRNAs are depicted as a heatmap (Figure 1D). In the GSE31210 dataset, 1603 DEmRNAs and 104 DElncRNAs were identified between LUAD and control samples (Figure 1E), and the top 100 differentially expressed genes (DEmRNAs and DElncRNAs) are presented in a heatmap (Figure 1F). Then, 1362 common DEmRNAs were obtained by overlapping upregulated and downregulated DEmRNAs from TCGA-LUAD and GSE31210 datasets (Figure 1G). Next, 85 DEIRGs were screened by intersecting the common DEmRNAs with 689 IRGs (Figure 1H). Meanwhile, 58 common DElncRNAs were selected by intersecting upregulated and downregulated DElncRNAs from the TCGA-LUAD cohort and GSE31210 dataset (Figure 1I).
 |
Figure 1 Identification of common DEIRGs and DElncRNAs from TCGA-LUAD cohort and GSE31210 dataset. (A) The volcano map of the differentially expressed genes between tumor and control samples from TCGA-LUAD cohort; (B) The expressions of top 100 DEGs in heatmap from TCGA-LUAD cohort; (C) The volcano map of the differentially expressed lncRNAs between tumor and control samples; (D) The expressions of top 100 lncRNAs in heatmap. (E) The volcano map of the differentially expressed genes between tumor and control samples from GSE31210 dataset; (F) The expressions of top 100 DEGs in heatmap from GSE31210 dataset; (G) Venn plot of overlapping DEGs in TCGA-LUAD and GSE31210 datasets; (H) Venn plot of overlapped DEGs and IRGs; (I) Venn plot of overlapping DElncRNAs in TCGA-LUAD and GSE31210 datasets. Abbreviations: IRG, invasion-related gene; DE, differentially expressed; lncRNA, long non-coding RNA; LUAD, lung adenocarcinoma; DEG, differentially expressed gene. |
Next, we calculated the correlations between DEIRGs and common DElncRNAs in TCGA and GSE31210 datasets using Pearson correlation analysis. The correlation network of DEIRGs and common DElncRNAs from the TCGA cohort comprised 125 nodes and 987 edges (Figure 2A), and a correlation network from the GSE31210 dataset was composed of 136 nodes and 1827 edges (Figure 2B). Lastly, 45 common DEIRLs were obtained using a Venn algorithm for downstream analysis (Figure 2C).
 |
Figure 2 Identification of DEIRLs in LUAD. (A) The correlation network of DEIRGs and DElncRNAs in TCGA; (B) The correlation network of DEIRGs and DElncRNAs in GSE31210; (C) Venn plot of common DEIRLs in TCGA-LUAD and GSE31210. Abbreviations: lncRNA, long non-coding RNA; IRL, invasion-related lncRNA; DE, differentially expressed; LUAD, lung adenocarcinoma. |
Construction and Validation of the Risk Score Signature Based on Prognostic DEIRLs in LUAD
We explored the prognostic value of the 45 DEIRLs by performing univariate Cox regression in the training set. We observed that RP3-525N10.2, LINC00857, EP300-AS1, SBF2-AS1, PDZRN3-AS1, DEPDC1-AS1, and RP5-1102E8.3 were related to the OS of patients with LUAD (Figure 3A). Further multivariate Cox regression analysis revealed that RP3-525N10.2, LINC00857, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3 were potential prognostic lncRNAs in patients (Figure 3B). Moreover, RT-qPCR results revealed that LINC00857 expression was significantly elevated, and the expressions of RP3-525N10.2, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3 were significantly reduced in LUAD samples compared to those in controls (Figure 3C), which was consistent with the sequencing results. Subsequently, the risk score of each patient was calculated in the training, testing, GSE31210, and GSE50081 datasets. Next, the relationship between risk score and clinical features was explored in the TCGA-LUAD cohort. We observed significant differences in risk scores between groups stratified by gender, N staging, tumor staging, T staging, and smoking history (Figure 3D).
 |
Figure 3 Identificated prognostic DEIRLs and constructed a risk score model in LUAD from the training set. (A) Univariate COX analysis of DEIRLs in forest plot; (B) Identification of risk signature by multivariate COX analysis in forest plot; (C) The expression levels of IRLs signature in tumor and control samples by RT-qPCR; (D) Relationship between risk score and clinical features. (E) The risk curve and survival status of patients in TCGA-LUAD; (F) Kaplan-Meier curves of survival in low- and high-risk; (G) Heatmap of the expressions of LINC00857, RP5-1102E8.3, RP3525N10.2 in low- and high-risk score; (H) ROC curves of the risk score model for predicting the survival of 1-, 3- and 5-year. Abbreviations: lncRNA, long non-coding RNA; IRL, invasion-related lncRNA; DE, differentially expressed; LUAD, lung adenocarcinoma; ROC, receiver operating characteristic. *P<0.05, ***P<0.001, ****P<0.0001. |
Using the optimal cut-off value, we divided the patients into low- and high-risk groups (Supplementary Figure 1). The risk score and survival status distributions of patients with LUAD in the TCGA training set are presented in Figure 3E. KM analysis indicated that the high-risk group had worse clinical outcomes (Figure 3F). The expression levels of RP3-525N10.2, LINC00857, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3 are presented in Figure 3G. Moreover, the ROC curve indicated that the risk score model had moderate accuracy in predicting the 1-, 3-, and 5-year survival in the TCGA training set with an area under the curve (AUC) value > 0.6 (Figure 3H). Next, we validated the risk model in the TCGA testing set and the external GSE31210 and GSE50081 datasets. Consistent results were observed in the internal (Supplementary Figure 2A–D) and external (Supplementary Figure 2E–H and Supplementary Figure 2I–L) datasets, suggesting risk model reliability.
Development of a Nomogram for Predicting the Survival of LUAD Patients
Next, we explored whether the risk score was an independent prognostic factor using univariate and multivariate Cox regression analyses. Univariate Cox regression analysis revealed that the risk score was significantly associated with prognosis in both TCGA and GSE50081 datasets, and multivariate Cox regression analysis further confirmed that result (Table 2). Thus, we included risk score, N staging, and T staging to construct a nomogram to predict the 1-, 3-, and 5-year survival of patients with LUAD (Figure 4A). We noted good concordance between the observed and predicted 1-, 3-, and 5-year OS (Figure 4B). Moreover, the decision curves suggest that the nomogram has a better predictive value compared to a single risk score, T staging, and N staging (Figure 4C–E).
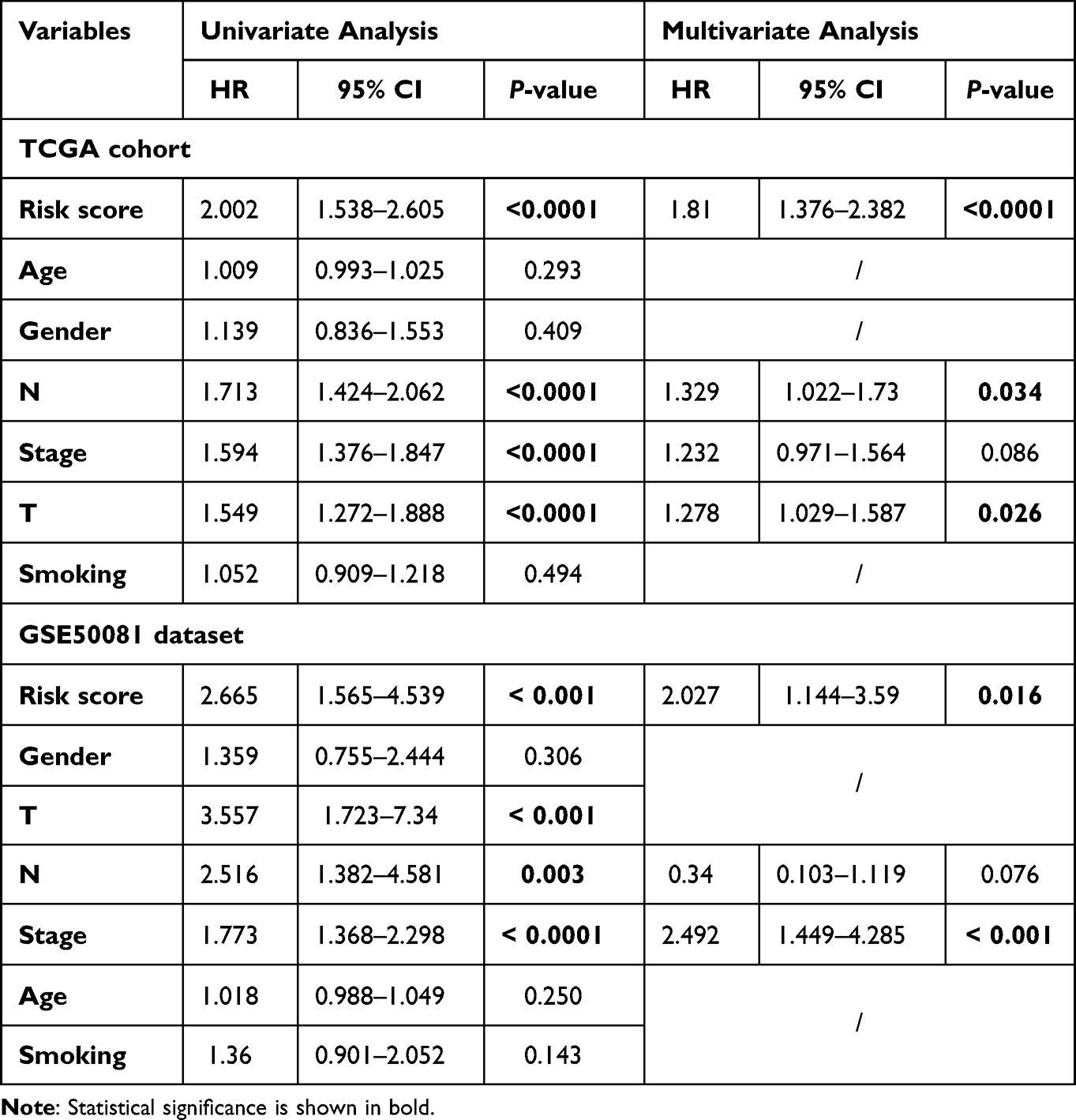 |
Table 2 Independent Prognostic Analyses |
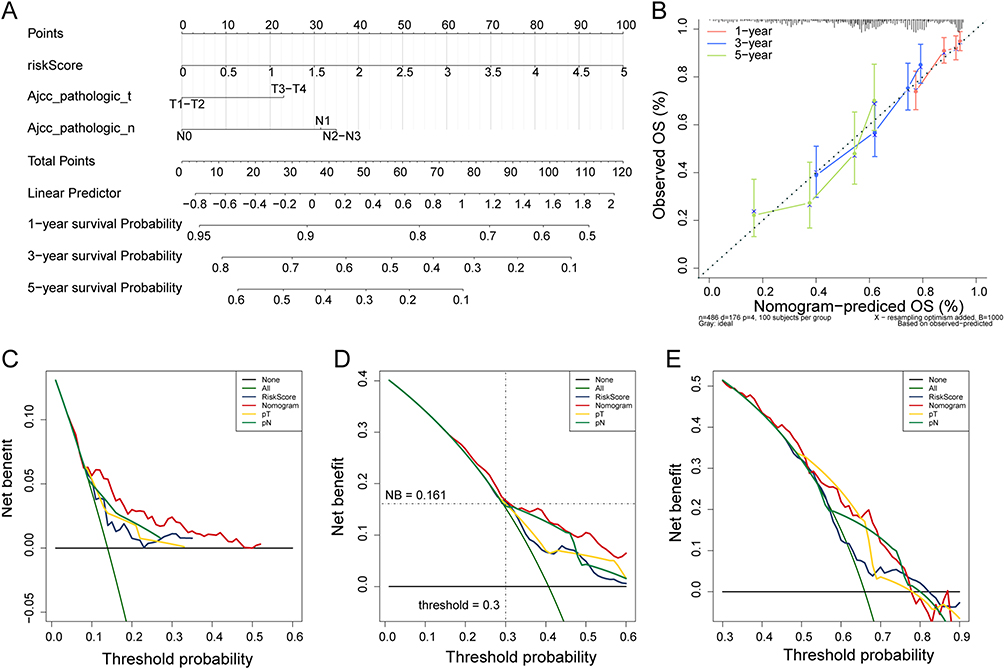 |
Figure 4 Construction of the nomogram. (A) Construction of the nomogram based on risk score, N staging and T staging for predicting the 1-, 3- and 5-year survival; (B) Calibration curves of the nomogram for 1-, 3- and 5-year; (C–E) Decision curve analysis for assessing the clinical use of nomogram in predicting the 1-, 3- and 5-year survival. |
Characterization of Low- and High-Risk Group
PCA was performed to compare the difference between low- and high-risk groups based on the risk model of the five prognostic IRLs (Figure 5A), 45 common DEIRLs (Figure 5B), and whole genome expression profiles (Figure 5C) from TCGA. The results suggested that the low-risk and high-risk groups were distinctly distributed (Figure 5A) prior to the other two patterns (Figures 5B and C). These results suggest that the risk model divided patients with LUAD into two groups and that the invasion propensity in the high-risk group differs from the low-risk group.
 |
Figure 5 PCA analysis and GSEA of the high and low risk groups. (A) PCA analysis based on the expression profile of 5 prognostic IRLs; (B) PCA analysis based on the expression profile of 45 common DEIRLs; (C) PCA analysis based on the expression profile of all genes in TCGA. (D) GSEA plot of the top 10 enriched biological processes; (E) Ridge plot of the top 10 enriched biological processes; (F) GSEA plot of the top 10 enriched KEGG pathways; (G) Ridge plot of the top 10 enriched KEGG pathways. Abbreviations: PCA, principal component analysis, GSEA, gene set enrichment analysis, lncRNA, long non-coding RNA; IRL, invasion-related lncRNA; DE, differentially expressed; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
We performed GSEA to identify the biological processes and pathways related to this risk model. The top ten enriched biological processes were mainly associated with cell proliferation, including mitochondrial translation, mitochondrial gene expression, chromosome segregation, meiotic cell cycle process, DNA-dependent DNA replication, meiotic cell cycle, DNA replication, cell cycle G2/M phase transition, microtubule-based movement, and cilium movement (Figure 5D and E). Similarly, cell cycle-related pathways, such as cell cycle and DNA replication, were also enriched. In addition, the risk score model was also relevant to proteasome, ribosome, spliceosome, steroid hormone biosynthesis, Parkinson’s disease, oxidative phosphorylation, asthma, and systemic lupus erythematosus (Figure 5F and G).
Additionally, we compared the sensitivity of patients in the low- and high-risk groups to drugs used in the treatment of LUAD, including docetaxel, gefitinib, paclitaxel, rapamycin, gemcitabine, and cisplatin. We observed that patients in the high-risk group were more sensitive to docetaxel, paclitaxel, and gemcitabine than those in the low-risk group (Figure 6).
 |
Figure 6 Comparison of chemotherapeutic sensitivity between the high- and low-risk groups. The IC50 of Docetaxel, Gefitinib, Paclitaxel, Rapamycin, Gemcitabine and Cisplatin were predicted and compared between low- and high-risk groups. ****P<0.0001, ns P>0.05. |
Construction of Invasion-Related ceRNA Network in LUAD
A total of 418 DEmiRNAs were identified between LUAD and control samples in TCGA, including 244 upregulated and 174 downregulated genes in the tumor samples (Figure 7A). The expression of the top 100 DEmiRNAs is depicted in a heatmap (Figure 7B). Using miRanda, we predicted 39 prognostic lncRNA–DEmiRNA pairs. Next, 506 DEmiRNA–DEmRNA pairs were predicted using miRWalk and TargetScan databases. Accordingly, we constructed a ceRNA network comprised 333 nodes and 545 edges (Figure 7C). In the ceRNA network, we observed that the DEmRNAs CPEB1, CORO2B, and GHR were IRGs. Thus, we extracted PDZRN3–miR-96-5p–CPEB1, EP300–AS1-miR-93-5p–CORO2B, and RP3–525N10.2-miR-130a-5p–GHR from the network (Figure 7D), which may play a role in critical invasion-related ceRNA regulatory relationships in LUAD. Figure 8A shows that the expression of CPEB1, CORO2B, and GHR was significantly lower in LUAD samples than in normal samples, which was contrary to the expression trend of their corresponding lncRNAs. The ROC curves of three lncRNAs and three DEmRNAs were plotted to explore the relevance of their expression to the survival of patients with LUAD. The results demonstrated that the survival rate of patients in the high- and low-expression groups of GHR, PDZRN3-AS1, and RP3–525N10.2 was notably different, and the prognosis was better in the high-expression group (Figure 8B).
 |
Figure 7 Build a ceRNA network. (A) The volcano map of the differentially expressed miRNA between tumor and control samples; (B) The expressions of top 100 miRNA in heatmap; (C) The prognostic lncRNA-related ceRNA network; (D) The prognostic lncRNA-invasion-related DEmRNA-DEmiRNA extracted from the ceRNA network. Abbreviations: miRNA, microRNA; mRNA, messenger RNA; lncRNA, long non-coding RNA; ceRNA, competitive endogenous RNA; DE, differentially expressed. |
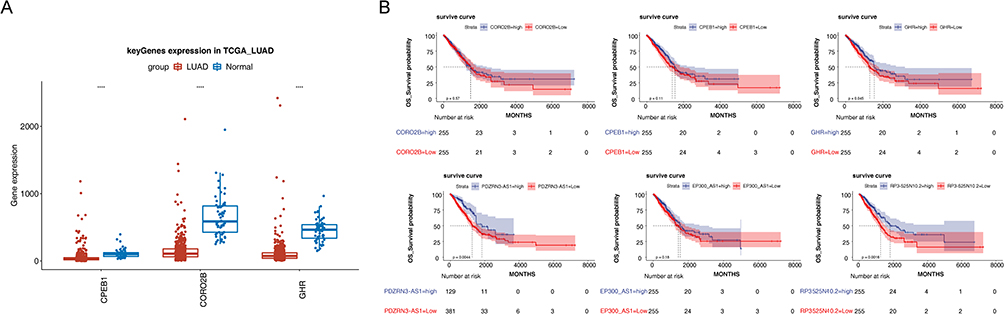 |
Figure 8 Analysis of ceRNA network. (A) Expression of CPEB1, CORO2B, and GHR in LUAD and NC samples. (B) Kaplan–Meier survival curves in the high- and low-expression groups of three prognostic lncRNAs and three DEmRNAs. Abbreviations: mRNA, messenger RNA; ceRNA, competitive endogenous RNA; DE, differentially expressed; LUAD, lung adenocarcinoma. ****P<0.0001. |
Discussion
In recent years, lung cancer incidence and mortality rates have been among the highest in the world and continue to increase. Non-small cell lung cancer is the most predominant subtype, and delayed diagnosis leads to patients with advanced disease,20 especially LUAD. To discover new biomarkers for diagnosis, the role of lncRNAs in lung cancer has been extensively investigated.21–23 In recent years, lung biopsies, electronic bronchoscopy, and mediastinum testing have been used. However, the screening process takes several weeks, and the condition of some patients deteriorates while they wait for hospital appointments.24 If specific and accurate biomarkers for diagnosis and prognosis are identified and applied, the survival and prognosis of patients with cancer will be improved through the use of corresponding therapy. Thus, we investigated the prognostic role of invasive markers in patients with LUAD.
In this study, we generated five potential prognostic lncRNAs for LUAD using univariate and multivariate Cox regression analyses. Based on these five lncRNAs, we constructed a risk model and nomogram that could accurately predict the prognosis of patients with LUAD. Lan et al25 reported that LINC00857 is a novel lncRNA signature for predicting the prognosis of patients with early-stage LUAD, and LINC00857 overexpression promotes cancer cell proliferation and invasion.26 Another study reported that LINC00857 promotes LUAD progression by targeting the miR-1179/SPAG5 axis, thus regulating cell proliferation, glycolysis, and apoptosis.27 In addition, LINC00857 is associated with invasion in colorectal and gastric cancer.28,29 No studies have reported the prognostic roles of RP3-525N10.2, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3 in cancer. Thus, further research is required to explore the mechanisms by which these lncRNAs promote invasion in LUAD and future research should focus broadly on the molecular pathways by which these lncRNAs act.
To identify the mechanism of these five lncRNAs in LUAD prognosis, we performed functional analysis using GSEA. This analysis indicated that these lncRNAs were significantly enriched in cell proliferation-related biological processes and cell cycle-related pathways, both of which play important roles in LUAD development. Several studies have also suggested that cell cycle and proliferation are regulated by lncRNAs. LINC00857 knockdown causes cell cycle arrest at G1/S via cyclin E1 (CCNE1) regulation, which decreases lung tumor cell growth.26 Yin et al observed that YT521-B homology domain family 2 (YTHDF2) deficiency inhibited lung cancer cell proliferation and migration.30 Yang et al reported that PTPRG-AS1 accelerated the cell cycle in LUAD cells.31 These findings indicate that the prognostic lncRNAs identified in the current study may be essential in LUAD pathogenesis and development by increasing the cell cycle rate and proliferation.Additionally, we compared the sensitivity of patients in low- and high-risk groups to drugs used in the treatment of LUAD and found that patients in the high-risk group were more sensitive to docetaxel, paclitaxel and gemcitabine than those in the low-risk group. Investigating these genes may provide new clues for LUAD prognosis.
Finally, we constructed an invasion-related ceRNA regulatory network and extracted three key sets of ceRNA regulatory relationships (CPEB1–hsa-miR-96-5p–PDZRN3-AS1, CORO2B–hsa-miR-93-5p–EP300-AS1, and GHR–hsa-miR-130a-5p–RP3-525N10.2). In miRNA-mRNA regulatory relationships, CPEB1, CORO2B, and GHR are differentially expressed IRGs. Nagaoka et al reported that reduced CPEB1 levels promote breast cancer cell metastasis to the lungs, whereas ectopic expression of CPEB1 inhibits this process.32 CORO2B is targeted by hsa-miR-93-5p and is involved in cigarette smoke-induced malignant transformation in BEAS-2B cells.33 In addition, GHR stimulation can promote cell invasion and metastasis,34 whereas its deficiency decreases fatal neoplasm incidence.35 In this study, we discovered that lncRNAs associated with LUAD metastatic propensity in the ceRNA network are indirectly associated with mRNA signaling. The data suggests that these lncRNA–miRNA–mRNA relationships may play critical roles in the prognosis of LUAD.
We analyzed the differences in the expression of CPEB1, CORO2B, and GHR in normal and tumor samples through bioinformatics methods and assessed the differences in survival between the high- and low-expression groups. Liu et al observed that MiR-96-5p overexpression promotes the proliferation, migration, and invasion of LUAD cells;36 Wang et al observed that upregulated CPEB 1 reversed the promoting effect of BMSC-Exos on lung cancer cell invasion, migration, and lung metastasis;37 Zhao et al observed that the expression of EP300-AS1 was downregulated in LUAD, which may play a tumor-suppressive role in LUAD.38 Pan et al reported that ALK rearrangements occurred in approximately 5% of non-small cell lung cancer, and GHR was identified as a novel ALK fusion partner in patients with non-small cell lung cancer.39 These findings further validate that PDZRN3-miR-96-5p-CPEB 1, EP300-AS1-miR-93-5p-CORO2B, and RP3-525 N 10.2 miR-130a-5p-GHR may be involved in a key regulatory pathway associated with LUAD invasion. In the future, studies of these pathways will further play positive roles.
Our study had some limitations. First, the reliability of this model needs to be iteratively improved in long-term clinical applications with larger sample sizes. Moreover, our gene signature was suited to the patients with surgical or biopsy specimens because this gene signature was based on gene expression data. Lastly, PDZRN3-miR-96-5p-CPEB 1, EP300-AS1-miR-93-5p-CORO2B, and RP3-525 N 10.2 miR-130a-5p-GHR may be involved in a regulatory pathway related to LUAD invasion, but we did not functionally validate this. This result will be verified in later studies using basic experiments. Nevertheless, despite these limitations, our study established an invasion-related prognostic lncRNAs signature that could be applied as an independent prognostic factor for LUAD and to predict response to prognosis. We will conduct in-depth research to validate our prognostic signature and strengthen its effectiveness and clinical practicability.
Conclusion
We identified five invasion-related prognostic lncRNAs (RP3-525N10.2, LINC00857, EP300-AS1, PDZRN3-AS1, and RP5-1102E8.3), established a risk model and nomogram that can accurately predict the survival of patients with LUAD, and preliminarily explored the molecular mechanism of prognostic lncRNAs in regulating LUAD. Thus, our study provides new insights into the prognostic indicators of LUAD.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of the First Affiliated Hospital of Jiamusi University. All participants provided signed informed consent in accordance with the Declaration of Helsinki.
Acknowledgments
We appreciate the support of all participators involved in this study.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. World Health Organization. Global health estimates 2020: deaths by cause, age, sex, by country and by region, 2000-2019. Available from: https://who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death.
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries [published correction appears in CA Cancer J Clin. 2020 Jul;70(4):313]. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Reck M, Rabe KF. Precision diagnosis and treatment for advanced non-small-cell lung cancer. N Engl J Med. 2017;377(9):849–861. doi:10.1056/NEJMra1703413
4. Nicholson AG, Tsao MS, Beasley MB, et al. The 2021 WHO classification of lung tumors: impact of advances since 2015. J Thorac Oncol. 2022;17(3):362–387. doi:10.1016/j.jtho.2021.11.003
5. Zheng Q, Min S, Zhou Q. Identification of potential diagnostic and prognostic biomarkers for LUAD based on TCGA and GEO databases. Biosci Rep. 2021;41(6):BSR20204370. doi:10.1042/BSR20204370
6. Suhail Y, Cain MP, Vanaja K, et al. Systems biology of cancer metastasis. Cell Syst. 2019;9(2):109–127. doi:10.1016/j.cels.2019.07.003
7. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020;5(1):28. doi:10.1038/s41392-020-0134-x
8. Liu J, Jiang C, Xu C, et al. Identification and development of a novel invasion-related gene signature for prognosis prediction in colon adenocarcinoma. Cancer Cell Int. 2021;21(1):101. doi:10.1186/s12935-021-01795-1
9. Zheng Z, Deng W, Yang J. Identification of 5-gene signature improves lung adenocarcinoma prognostic stratification based on differential expression invasion genes of molecular subtypes. BioMed Res Int. 2020;2020:8832739. doi:10.1155/2020/8832739
10. Park MK, Zhang L, Min KW, et al. NEAT1 is essential for metabolic changes that promote breast cancer growth and metastasis. Cell Metab. 2021;33(12):2380–2397.e9. doi:10.1016/j.cmet.2021.11.011
11. Chakravarty D, Sboner A, Nair SS, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun. 2014;5:5383. doi:10.1038/ncomms6383
12. Xu Y, Li Y, Qiu Y, et al. LncRNA NEAT1 promotes gastric cancer progression through miR-17-5p/TGFβR2 axis up-regulated angiogenesis. Front Cell Dev Biol. 2021;9:705697. doi:10.3389/fcell.2021.705697
13. Shen X, Ye Z, Wu W, et al. lncRNA NEAT1 facilitates the progression of colorectal cancer via the KDM5A/Cul4A and Wnt signaling pathway. Int J Oncol. 2021;59(1):51. doi:10.3892/ijo.2021.5231
14. Wang Z, Zou Q, Song M, Chen J. NEAT1 promotes cell proliferation and invasion in hepatocellular carcinoma by negative regulating miR-613 expression. Biomed Pharmacother. 2017;94:612–618. doi:10.1016/j.biopha.2017.07.111
15. Ding N, Wu H, Tao T, Peng E. NEAT1 regulates cell proliferation and apoptosis of ovarian cancer by miR-34a-5p/BCL2. Onco Targets Ther. 2017;10:4905–4915. doi:10.2147/OTT.S142446
16. Li Y, Shen R, Wang A, et al. Construction of a prognostic immune-related LncRNA risk model for lung adenocarcinoma. Front Cell Dev Biol. 2021;9:648806. doi:10.3389/fcell.2021.648806
17. Jiang A, Liu N, Bai S, et al. Identification and validation of an autophagy-related long non-coding RNA signature as a prognostic biomarker for patients with lung adenocarcinoma. J Thorac Dis. 2021;13(2):720–734. doi:10.21037/jtd-20-2803
18. Wang W, He L, Ouyang C, Chen C, Xu X, Ye X. Key common genes in obstructive sleep apnea and lung cancer are associated with prognosis of lung cancer patients. Int J Gen Med. 2021;14:5381–5396. doi:10.2147/IJGM.S330681
19. Gan BL, He RQ, Zhang Y, Wei DM, Hu XH, Chen G. Downregulation of HOXA3 in lung adenocarcinoma and its relevant molecular mechanism analysed by RT-qPCR, TCGA and in silico analysis. Int J Oncol. 2018;53(4):1557–1579. doi:10.3892/ijo.2018.4508
20. Sugawara S, Lee JS, Kang JH, et al. Nivolumab with carboplatin, paclitaxel, and bevacizumab for first-line treatment of advanced nonsquamous non-small-cell lung cancer. Ann Oncol. 2021;32(9):1137–1147. doi:10.1016/j.annonc.2021.06.004
21. Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96(4):1297–1325. doi:10.1152/physrev.00041.2015
22. Chen Z, Li JL, Lin S, et al. CAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J Clin Invest. 2016;126(6):2267–2279. doi:10.1172/JCI85250
23. Castellano JJ, Navarro A, Viñolas N, et al. LincRNA-p21 impacts prognosis in resected non-small cell lung cancer patients through angiogenesis regulation. J Thorac Oncol. 2016;11(12):2173–2182. doi:10.1016/j.jtho.2016.07.015
24. Navani N, Nankivell M, Lawrence DR, et al. Lung cancer diagnosis and staging with endobronchial ultrasound-guided transbronchial needle aspiration compared with conventional approaches: an open-label, pragmatic, randomised controlled trial. Lancet Respir Med. 2015;3(4):282–289. doi:10.1016/S2213-2600(15)00029-6
25. Mu L, Ding K, Tu R, Yang W. Identification of 4 immune cells and a 5-lncRNA risk signature with prognosis for early-stage lung adenocarcinoma. J Transl Med. 2021;19(1):127. doi:10.1186/s12967-021-02800-x
26. Wang L, He Y, Liu W, et al. Non-coding RNA LINC00857 is predictive of poor patient survival and promotes tumor progression via cell cycle regulation in lung cancer. Oncotarget. 2016;7(10):11487–11499. doi:10.18632/oncotarget.7203
27. Wang L, Cao L, Wen C, Li J, Yu G, Liu C. LncRNA LINC00857 regulates lung adenocarcinoma progression, apoptosis and glycolysis by targeting miR-1179/SPAG5 axis. Hum Cell. 2020;33(1):195–204. doi:10.1007/s13577-019-00296-8
28. Zhou D, He S, Zhang D, et al. LINC00857 promotes colorectal cancer progression by sponging miR-150-5p and upregulating HMGB3 (high mobility group box 3) expression. Bioengineered. 2021;12(2):12107–12122. doi:10.1080/21655979.2021.2003941
29. Zhang K, Shi H, Xi H, et al. Genome-wide lncRNA microarray profiling identifies novel circulating lncRNAs for detection of gastric cancer. Theranostics. 2017;7(1):213–227. doi:10.7150/thno.16044
30. Li Y, Sheng H, Ma F, et al. RNA m6A reader YTHDF2 facilitates lung adenocarcinoma cell proliferation and metastasis by targeting the AXIN1/Wnt/β-catenin signaling. Cell Death Dis. 2021;12(5):479. doi:10.1038/s41419-021-03763-z
31. Xue Y, Diao M, Lyu J, et al. Long noncoding RNAs PTPRG antisense RNA 1 targets cyclin D1 to facilitate cell proliferation in lung adenocarcinoma. Cancer Biother Radiopharm. 2021. doi:10.1089/cbr.2021.0168
32. Nagaoka K, Fujii K, Zhang H, et al. CPEB1 mediates epithelial-to-mesenchyme transition and breast cancer metastasis. Oncogene. 2016;35(22):2893–2901. doi:10.1038/onc.2015.350
33. Wang J, Yu XF, Ouyang N, et al. MicroRNA and mRNA interaction network regulates the malignant transformation of human bronchial epithelial cells induced by cigarette smoke. Front Oncol. 2019;9:1029. doi:10.3389/fonc.2019.01029
34. Herrington J, Carter-Su C. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol Metab. 2001;12(6):252–257. doi:10.1016/s1043-2760(01)00423-4
35. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13. doi:10.1126/scitranslmed.3001845
36. Liu Z, Cui Y, Wang S, et al. MiR-96-5p is an oncogene in lung adenocarcinoma and facilitates tumor progression through ARHGAP6 downregulation. J Appl Genet. 2021;62(4):631–638. doi:10.1007/s13353-021-00652-1
37. Wang G, Ji X, Li P, Wang W. Human bone marrow mesenchymal stem cell-derived exosomes containing microRNA-425 promote migration, invasion and lung metastasis by down-regulating CPEB1. Regen Ther. 2022;20:107–116. doi:10.1016/j.reth.2022.03.007
38. Zhao J, Li G, Zhao G, et al. Prognostic signature of lipid metabolism associated LncRNAs predict prognosis and treatment of lung adenocarcinoma. Front Oncol. 2022;12:986367. doi:10.3389/fonc.2022.986367
39. Pan X, Zhong A, Xing Y, Li X, Du H, Shi M. A novel GHR-ALK fusion gene in a patient with metastatic lung adenocarcinoma and its response to crizotinib: a case report. J Int Med Res. 2021;49(9):3000605211044652. doi:10.1177/03000605211044652
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Novel lncRNA Panel for Risk Stratification and Immune Landscape in Breast Cancer Patients

Li C, Wang X, Chen T, Li W, Yang Q
International Journal of General Medicine 2022, 15:5253-5272
Published Date: 27 May 2022
Comprehensive Analysis of the E2F Transcription Factor Family in Human Lung Adenocarcinoma
Wang Q, Liu J, Cheang I, Li J, Chen T, Li Y, Yu B
International Journal of General Medicine 2022, 15:5973-5984
Published Date: 2 July 2022
Expression, Clinical Significance, Immune Infiltration, and Regulation Network of miR-3940-5p in Lung Adenocarcinoma Based on Bioinformatic Analysis and Experimental Validation
Lin Z, Huang W, Xie Z, Yi Y, Li Z
International Journal of General Medicine 2022, 15:6451-6464
Published Date: 6 August 2022
Identification of Gefitinib Resistance-Related lncRNA-miRNA-mRNA Regulatory Networks and Corresponding Prognostic Signature in Patients with Lung Adenocarcinoma
Wang T, Wu Z, Li S, Chen Z, Chen Y, Yang Z
International Journal of General Medicine 2022, 15:7155-7168
Published Date: 11 September 2022
Comprehensive Analysis of circRNA-Related mRNAs as Prognostic Factors in Non-Smoking Women with Lung Adenocarcinoma
Wang H, Wei S, Wang L, Zhang Z
International Journal of General Medicine 2024, 17:5757-5771
Published Date: 3 December 2024