Back to Journals » Journal of Inflammation Research » Volume 17
Identification and Validation of Hub Genes Related to Neutrophil Extracellular Traps-Mediated Cell Damage During Myocardial Infarction
Authors Ke D, Ni J, Yuan Y, Cao M, Chen S, Zhou H
Received 6 November 2023
Accepted for publication 23 January 2024
Published 1 February 2024 Volume 2024:17 Pages 617—637
DOI https://doi.org/10.2147/JIR.S444975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Da Ke,1 Jian Ni,1 Yuan Yuan,1 Mingzhen Cao,1 Si Chen,2 Heng Zhou1
1Department of Cardiology, Renmin Hospital of Wuhan University, Hubei Key Laboratory of Metabolic and Chronic Diseases, Wuhan, 430060, People’s Republic of China; 2Hubei Key Laboratory of Metabolic and Chronic Diseases, Wuhan, 430060, People’s Republic of China
Correspondence: Heng Zhou, Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, 430060, People’s Republic of China, Email [email protected]
Purpose: Studies have shown that neutrophil-mediated formation of neutrophil extracellular traps (NETs) leads to increased inflammatory response and cellular tissue damage during myocardial infarction (MI). We aimed to identify and validate possible hub genes in the process of NETs-mediated cell damage.
Methods: We performed an immune cell infiltration analysis of the MI transcriptome dataset based on CIBERSORT and ssGSEA algorithms. Gene expression profiles of NETs formation (GSE178883) were used to analyze the physiological processes of peripheral blood neutrophils after phorbol myristate acetate (PMA) stimulation. Bioinformatics and machine learning algorithms were utilized to find candidate hub genes based on NETs-related genes and transcriptome datasets (GSE66360 and GSE179828). We generated the receiver operating curve (ROC) to evaluate the diagnostic value of hub genes. Next, the correlation between hub genes and immune cells was analyzed using CIBERSORT, ssGSEA and xCell algorithms. Finally, we used quantitative real-time PCR (qRT-PCR) and immunohistochemistry to verify gene expression.
Results: Immune cell infiltration analysis revealed that inflammatory cells such as neutrophils were highly expressed in the peripheral blood of patients with MI. Functional analysis of differentially expressed genes (DEGs) in GSE178883 indicated that the potential pathogenesis lies in immune terms. Using weighted gene co-expression network analysis (WGCNA) and machine learning algorithms, we finally identified the seven hub genes (FCAR, IL1B, MMP9, NFIL3, CXCL2, ICAM1, and ZFP36). The qRT-PCR results showed that IL-1B, MMP9, and NFIL3 mRNA expression was up-regulated in the MI group compared to the control. Immunohistochemical results showed high MMP9, IL-1B, and NFIL3 expression in the infarcted area compared to the non-infarcted area and sham-operated groups.
Conclusion: We identified seven hub genes associated with NETs-mediated cellular damage during MI. Our results may provide insights into the mechanisms of neutrophil-mediated cell injury during MI.
Keywords: myocardial infarction, neutrophils, cell damage, neutrophil extracellular traps, bioinformatics, endothelial cells
Graphical Abstract:

Introduction
Myocardial infarction (MI), caused by thrombosis or arterial occlusion, is the leading cause of morbidity and mortality among all cardiovascular diseases.1 Many studies have shown that immune cells are involved in the inflammatory response to MI and heart failure.2–5 For example, myocardial injury triggers an infiltration of neutrophils and macrophages into the heart tissue. These immune cells play a crucial role in removing cellular debris, initiating inflammation through the secretion of pro-inflammatory cytokines, and attracting other pro-inflammatory cells.2,6
Neutrophils, the most predominant type of leukocyte, arrive at the site of inflammation in a cascade-like manner. This results in the activation of specific effector functions such as degranulation, phagocytosis, and the release of reactive oxygen species.7 In 2004, Brinkmann et al demonstrated that neutrophils stimulation with phorbol myristate acetate (PMA) release granule proteins and chromatin that together form extracellular fibers that bind Gram-positive and -negative bacteria.8 Subsequent studies by Fuchs et al showed that upon stimulation, the nucleus of neutrophils loses its shape, followed by disintegration of the nuclear envelope and granule membranes, allowing for the mixing of neutrophil extracellular traps (NETs) components, and finally, NETs are released with the rupture of the cell membrane.9 Distinct from apoptosis and necrosis, this cell death process is dependent on the creation of reactive oxygen species (ROS) by NADPH oxidase.9 This will eventually lead to the release of myeloperoxidase (MPO) and neutrophil elastase (NE) from cytoplasmic azurophilic granules.10 Except for degrading virulence factors and killing bacteria, there is growing evidence that NETs play a role in a variety of diseases, such as sepsis,11 acute respiratory distress syndrome (ARDS),12 chronic obstructive pulmonary disease (COPD),13 systemic lupus erythematosus (SLE),14 and atherosclerosis.15 Recently, many studies have shown that NETs play a damaging role in acute myocardial infarction (AMI).16 For example, one study found that in addition to providing a scaffold for thrombus formation, NETs formation also exacerbates endothelial cell injury, which may be an important contributor to the development of MI.17
Here, we conducted a comprehensive analysis of NETs-related genes in MI, with a particular focus on endothelial cell damage by NETs. We comprehensively analyzed the differentially expressed genes (DEGs) in endothelial cells stimulated by NETs and took the intersection of these DEGs with MI-related genes. We expect to discover biomarkers of MI associated with NETs formation, especially regarding endothelial cell injury by NETs. Our findings may provide a new direction for studying NETs-mediated cell injury, particularly endothelial cells during MI.
Materials and Methods
Data Source
All gene expression data (microarray data, RNA-seq data) were downloaded from the Gene Expression Omnibus (GEO) database. The datasets GSE48060 and GSE66360 were annotated by platforms of GPL57, the dataset GSE62646 was annotated by platforms of GPL6244, GSE178883 and GSE179828 were annotated by platforms of GPL2467. In addition, we used an online web server (https://tabula-muris.ds.czbiohub.org/) to analyze gene expression in single-cell RNA-Seq data from a set of healthy mouse hearts.18
Identification and Enrichment Analyses of DEGs
GEO2R is a tool for differential analysis of expression profiling microarrays in the GEO database, with which we can compare two or more datasets of samples from the GEO database to obtain the DEGs. DEGs of GSE178883 and GSE179828 were respectively identified using GEO2R. The volcano diagram of DEGs was drawn using the “ggplot2” package. Based on the DAVID database (https://david.ncifcrf.gov/), the functional and molecular biological properties of the overlap were analyzed by GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) annotations.
Protein-Protein Interaction (PPI) Network Construction and Gene Set Enrichment Analysis (GSEA)
Based on the STRING database (http://string-db.org), the PPI network of overlap DEGs was created and visualized using Cytoscape software (version 3.9.1). GSEA was performed using the GSEA software (version 4.3.2). GSEA is a threshold-free method that examines all genes on the basis of their differential expression rank or other score, without first filtering the genes based on a predetermined set of criteria.
Evaluation of Immune Cell Infiltration
CIBERSORT, is a method for characterizing the cell composition of complex tissues from their gene expression profiles.19 The correlation analysis of 22 infiltrating immune cell types was evaluated using the “corrplot” package in the R software. ssGSEA is an extension of the GSEA method that allows for the definition of enrichment scores that represent the absolute enrichment of genomes in each sample in a given dataset.20 xCell, based on the ssGSEA method, performed cell type analysis using gene expression data from 64 immune and stromal cells.21
Weighted Gene Co-Expression Network Analysis (WGCNA)
WGCNA is a computational approach in systems biology that designed to characterize the patterns of correlation between genes across several microarray datasets.22 WGCNA can be used to identify modules of highly correlated genes, to summarize these modules using the module eigengene or an intramodular hub gene, and to calculate module membership measures. In this study, the gene expression matrices were subjected to hierarchical cluster analysis in order to identify and remove any outliers. Subsequently, a correlation heatmap was generated to visualize the relationship between modules and traits, with the inclusion of P-values within the respective boxes. The relationship between gene expression and AMI was determined by evaluating gene significance and module membership.
Machine Learning
To find potential hub genes, three different machine learning algorithms were utilized. The Lasso regression analysis technique, performed through the “glmnet” R package, utilizes regularization to minimize prediction error. Support vector machine (SVM) is a supervised classification algorithm based on statistical learning theory. The random forest (RF) algorithm, implemented using the “randomForest” R package, was employed to identify a subset of candidate genes. This algorithm leverages ensemble learning by integrating numerous trees to enhance predictive accuracy. The overlapping genes identified by the Lasso model, SVM-RFE and random forest were defined as hub genes for subsequent research and validation.
Animals Experimental Model
All experimental animal procedures were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University, and were also in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The mice were placed on the heating operation pad in the right lateral decubitus position, near the left axilla, the mouse skin and the third fourth rib were cut, the heart and left anterior descending artery (LAD) were fully exposed, the blood vessels were ligated with 7–0 stitched sutures at approximately 2 mm from the lower margin of the left atrial appendage under a stereomicroscope, and the distal area of the ligature was whitish due to ischemia, which was indicated by successful surgery. Mice in the sham group took the same operation but without suture ligation to ligate the blood vessels, and then the muscle and skin at the incision were sutured by layer.
Echocardiography and Speckle-Tracking Analysis
The day before mice were sacrificed, they were anesthetized with isoflurane (1.5–2%) and underwent transthoracic echocardiography to assess cardiac function. Echocardiographic parameters, such as left ventricular internal dimension diastolic (LVIDd), left ventricular internal dimension systolic (LVIDs), left ventricular ejection fraction (LVEF), heart rate (HR) and fractional shortening (FS), were measured using M-mode tracings. These measurements were obtained by averaging data from a minimum of five consecutive cardiac cycles. To evaluate the impact of myocardial remodeling following MI injury on cardiac dynamics at both global and regional levels, high-frequency echocardiography was performed using the speckle-tracking algorithm. To assess radial and longitudinal strain, strain rate, displacement, and velocity, cardiac cycles were acquired from the parasternal long-axis view.
Immunohistochemistry (IHC)
Isolated mouse hearts were immediately placed in 10% KCl solution and fixed in 10% formalin for 12 hours, followed by gradient alcohol dehydration, xylene hyalinization and paraffin embedding, and made into sections (4–5 μm). The paraffin sections were fully immersed in xylene solution, and after the immersion was completed, they were removed and dehydrated in a gradient alcohol solution. Paraffin sections were heated for antigen recovery using the pressure cooker method, and the sections were blocked for 1 h and then incubated overnight in a refrigerator at 4°C with antibodies against IL-1β (Abmart, TA5103), MMP9 (ABclonal, A11521), and the NFIL3 (Proteintech, 11773-1-AP) receptor, followed by incubation of the anti-rabbit/mouse EnVisionTM +/horseradish peroxidase reagent at a temperature of 37 °C for an additional 1 h, and then detected using a DAB staining kit. Images were quantified with ImageJ software.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from the left ventricular tissue samples by grinding and lysis with TRIzol lysis solution, extracted by trichloromethane, precipitated by isopropanol and rinsed by 75% ethanol. The purity as well as the concentration of the proposed RNA was determined by NanoDrop One/Onec (Thermo Scientific), and the dilution volume was calculated and reverse transcription was performed. Light Cycler 480 SYBR Green 1 Master Mix was used to perform qRT-PCR. The data was analyzed using the 2^-(∆∆Ct) method and normalized to GAPDH. The details about all primer sequences are listed in Table S1.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism software (version 9.0) or R software. The experimental data is in the form of mean ± SD. Two independent sample t-tests are used for sample comparison between two groups, and one-way ANOVA is used for sample comparison between multiple groups. A P value less than 0.05 was considered statistically significant.
Results
Immune Cell Infiltration Analysis
The research flowchart is presented in Figure 1. Cardiac healing following MI involves the mobilization and activation of immune cells.23 For example, monocytes circulate the vasculature at a steady state and are recruited to sites of inflammation and engage in the development of cardiovascular disease.24 Therefore, we performed an immune infiltration analysis of the gene expression dataset GSE48060 using two algorithms, “CIBERSORT” and “ssGSEA” to determine whether activated neutrophils are linked to the different disease states. The correlations between the infiltrating immune cells of “CIBERSORT” are shown in Figure 2A. The immune cell composition of samples from the AMI and control groups was compared (Figure 2B). Boxplot was generated to visualize the differences in immune infiltrating cell types of “CIBERSORT” between the AMI and control groups (Figure 2C). The boxplot analysis of the differential in immune cell infiltration demonstrated that patients with AMI exhibited a significantly elevated amounts of neutrophils compared to the control group. We also used the ssGSEA algorithm to analyze the immune cell infiltration in GSE48060 (Figure 2D and E). This algorithm compares the expression of 28 immune cell subtypes between the AMI and control groups. Similar to the results of the CIBERSORT analysis, there were significant differences in neutrophil expression levels between the two groups.
 |
Figure 1 Flowchart of the study design. Abbreviations: AMI, acute myocardial infarction; PMA, phorbol myristate acetate; NETs, neutrophil extracellular traps; NRGs, NETs-related genes; DEGs, differentially expressed genes; HUVECs, human umbilical vein endothelial cells. |
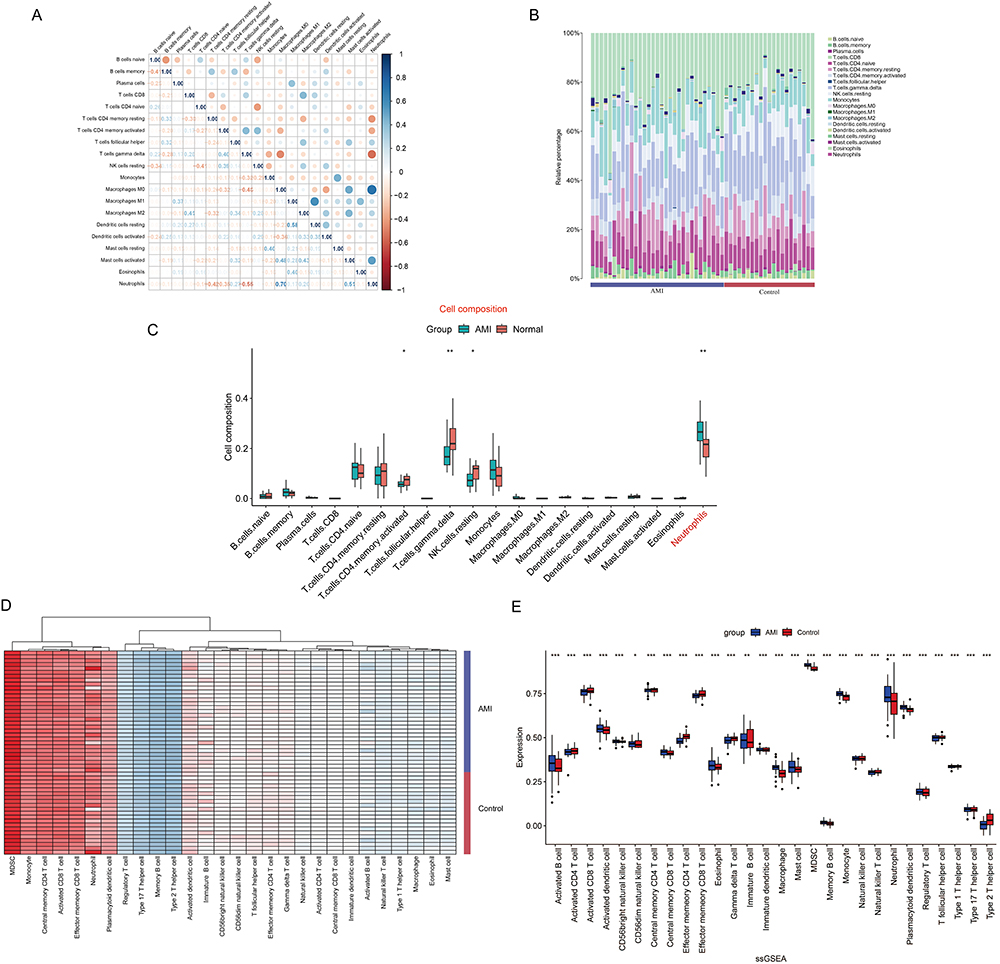 |
Figure 2 Immune cell infiltration analysis. (A) Correlation matrix of 20 immune cell subtype compositions. Blue represents positive correlation, Orange represents negative correlation, and the number in the square represents correlation. (B) Proportion of 20 subtypes of immune cells in different samples from AMI and control groups. (C) The proportion of the 20 immune cell subtypes in the AMI group and the control group is visualized in a barplot. (D) Heatmap of immune cell subtypes expression in AMI and control groups. (E) The barplot compares 28 immune cell subtypes proportion between AMI and control groups based on ssGSEA algorithm. *P < 0.05; **P < 0.01; ***P < 0.001. |
Functional Annotation of PMA-Stimulated Neutrophils
NETs are reticular structures released by neutrophil activation and are closely related to the subsequent damage after MI.17 NETs are involved in the inflammatory pathophysiology of a variety of illnesses, such as cardiovascular and infectious diseases.25 PMA can induce neutrophil aggregation and subsequent NETs release. Therefore, we analyzed differential gene expression after PMA stimulation of neutrophils. We selected human circulating neutrophils stimulated with 100 nM PMA or treatment with a control medium in GSE178883. Volcano plot showing the expression of differential genes between the two groups (Figure 3A). Figure 3B shows the top GO items under biological process (BP), molecular function (MF), and cellular component (CC). Next, functional enrichment analysis was performed based on the DEGs. The KEGG pathway enrichment analysis showed that the up-DEGs were mainly enriched in antigen processing and presentation, Th17 cell differentiation and Th1 and Th2 cell differentiation (Figure 3C). The down-DEGs were mainly enriched in neutrophil extracellular trap formation, Fc gamma R−mediated phagocytosis and cytokine−cytokine receptor interaction (Figure 3C). The results of GSEA analysis showed that HALLMARK_MYC_TARGETS_V2, HALLMARK_TNFA_SIGNALING_VIA_NFKB and HALLMARK_MYC_TARGETS_V1 were the most enriched gene sets in PMA group (Figure 3D). This confirmed that activation of inflammation-related pathways is an important feature of PMA-stimulated neutrophils.
 |
Figure 3 Functional annotation of PMA-stimulated neutrophils. (A) Volcano plot of the DEGs in GSE178883. (B) GO enrichment analysis of DEGs. (C) KEGG functional enrichment analysis of up- and down-regulated genes in GSE178883. (D) GSEA comparing PMA treatment with vehicle control. Enrichment plots of key pathways in PMA treatment neutrophils. Abbreviations: PMA, phorbol myristate acetate. NES, Normalized Enrichment Score. |
Significant Module Genes Identification in AMI via WGCNA
The WGCNA method was used to identify gene-gene modules associated with AMI and controls. We first eliminate the abnormal samples by setting a threshold (Figure 4A). A scale-free co-expression network was then established with the soft thresholding power to 14 (Figure 4B). A total of five modules were identified for future analysis. The primed modules were eventually displayed under the clustering tree (Figure 4C). We made a map of the connections between the identified modules, and the brown module showed a strong correlation with AMI (Figure 4D). The correlation between the module mean and gene significance (GS) for the brown module was found to be highly significant, providing further evidence suggesting that genes inside the brown module may possess functional significance related to AMI (Figure 4E). Figure 4F shows the heatmap of the selected gene. A total of 311 genes within the brown module were included in the subsequent study (Table S2).
 |
Figure 4 Significant module genes identification in AMI via WGCNA. (A) Sample clustering to detect outliers (B) Mean connectivity analysis and scale-free index analysis for different soft-threshold power. (C) A dendrogram illustrating the clustering of genes, with distinct modules represented by different colors. (D) Heatmap of the correlation between module characteristic genes and AMI (E) A scatter plot was generated to show the association between GS and module membership (MM) within the brown module. (F) Network heatmap plot of genes. |
Identification of Hub Genes Associated with AMI and NRGs Through Machine Learning
We next obtained NETs-related genes (NRGs) from the previous literature.26 After taking the intersection of NRGs and MI-related genes from the brown module, we finally obtained 28 Intersecting genes (AMI-NRGs) (Figure 5A). The KEGG pathway enrichment analysis showed that IL-17 signaling pathway, TNF signaling pathway, Toll-like receptor signaling pathway and NF-kappa B signaling pathway were significantly enriched (Figure 5B). The results of GO functional enrichment indicated that the AMI-NRGs were mainly enriched in “inflammatory response”, “positive regulation of inflammatory response”, “neutrophil chemotaxis” regarding BP; “extracellular region”, “extracellular space”, “specific granule membrane” regarding CC; “identical protein binding”, “chemokine activity”, “RAGE receptor binding” regarding MF (Figure 5C). We then imported the AMI-NRGs into the STRING database and built a PPI network via Cytoscape (Figure 5D). Subsequently, three machine learning algorithms, including SVM-RFE, random forest, and Lasso regression, were employed to identify and select potential hub genes (Figure 5E–H), and we finally got four hub genes (FCAR, IL1B, MMP9 and NFIL3) (Figure 5I).
 |
Figure 5 Identification of hub genes associated with AMI and NRGs through machine learning. (A) The 28 overlay genes between WGCNA analysis and NETs-related genes were presented by Venn diagram. (B) The most enriched KEGG pathways were presented by the Sankey diagram. (C) GO enrichment analysis of the AMI-NRGs. (D) PPI network of AMI-NRGs, isolated nodes were removed. (E and F) Visualization of Lasso regression. (G) The column shows that the random forest algorithm was used to order genes according to their importance score. (H) The hub genes were screened by the SVM-RFE algorithm. (I) The Venn diagram displays four common genes identified by Lasso, random forest and SVM-RFE algorithms, which were identified as the hub genes in AMI-NRGs. |
Screening of DEGs from GSE179828
Neutrophil-mediated endothelial cell activation and injury play important roles in multiple disease states.27 The release of NETs in blood vessels may trap red blood cells (RBCs), activate platelets, and damage the endothelium thereby promoting coagulation, vascular occlusion, and thrombosis.27 Therefore, we analyzed the RNA-sequencing data of NETs-stimulated endothelial cells. We finally identified a set of 985 genes that were differentially expressed in human umbilical vein endothelial cells (HUVECs), 623 genes are upregulated, and 362 genes are downregulated (Figure 6A). The KEGG pathway enrichment analysis showed that TNF signaling pathway, NF-kappa B signaling pathway and Osteoclast differentiation were significantly enriched (Figure 6B). The GO analysis showed that the differential genes were most significantly enriched in the regulation of transcription from RNA polymerase II promoter in BP, nucleus in CC, and protein binding in MF (Figure 6C–E, Table S3).
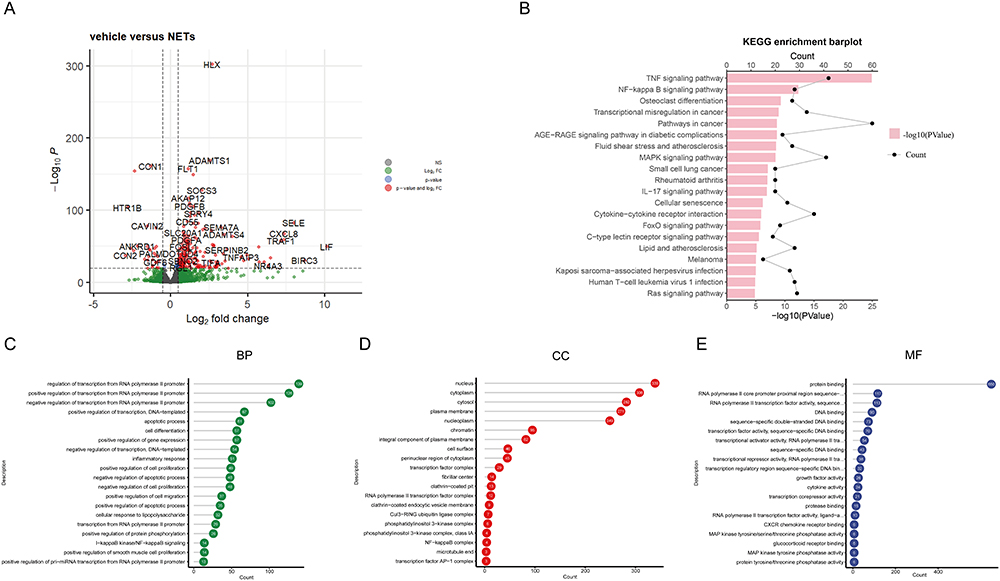 |
Figure 6 Screening of DEGs from GSE179828. (A) Volcano plot of GSE179828. (B) The KEGG enrichment barplot of the DEGs. (C–E) The top 20 GO items under BP, CC, and MF. |
Identification of Hub Genes Associated with AMI and NETs Stimulated Endothelial Cells
We next take the intersection of the endothelial cells-related genes acquired from GSE179828 with AMI-related genes from the brown module and finally obtained 59 intersecting genes (AMI-ECRGs) (Figure 7A). Functional enrichment analysis was performed based on the AMI-ECRGs (Figure 7B and C). The enrichment results are similar to the analysis outcome of AMI-NRGs, TNF signaling pathway, NF-kappa B signaling pathway and IL-17 signaling pathway are the top three enriched pathways (Figure 7B). We then imported the AMI-ECRGs into the STRING database and built a PPI network via Cytoscape (Figure 7D). The CytoHubba plugin’s four algorithms (Degree, Stress, Betweenness, and Radiality) were then utilized to identify common genes (Figure 7E). Finally, we screen out potential hub genes using three machine learning algorithms (random forest, Lasso regression, and SVM-RFE) (Figure 7F–I). CXCL2, ICAM1, and ZFP36 were identified as the intersection of common genes from SVM-RFE, random forest, and Lasso. (Figure 7J).
 |
Figure 7 Identification of hub genes associated with AMI and NETs stimulated endothelial cells. (A) The 59 overlay genes between WGCNA analysis and the differentially expressed gene of GSE179828 were presented by Venn diagram. (B) The most enriched KEGG pathways were presented by the Sankey diagram. (C) GO enrichment analysis of the AMI-ECRGs. (D) PPI network of AMI-ECRGs, isolated nodes were removed. (E) The intersection of the first 15 genes from each of the four algorithms (Degree, Stress, Radiality and Betweenness) in the cytoHubba plugin resulted in the identification of eleven hub genes. (F and G) Visualization of Lasso regression. (H) The column shows that the random forest algorithm was used to order genes according to their importance score. (I) The hub genes were screened by the SVM-RFE algorithm. (J) The Venn diagram displaying three common genes identified by Lasso, random forest and SVM-RFE algorithms, which were identified as the hub genes in AMI-ECRGs. |
Expression Profiles of Hub Genes in Different Cell Types of the Heart
The network of hub genes and their co-expression genes was created using the GeneMANIA platform, as shown in Figure 8A and B. Next, we used a publicly available single-cell RNA-Seq and FACS database of healthy mouse hearts to verify hub gene expression (Figure 8C and D). Because FCAR is not expressed in mice, we selected the remaining six hub genes for analysis. As shown in Figure 8D, Il1b, Cxcl2 and Mmp9 were primarily expressed in leukocytes and icam1 was mainly expressed in endothelial cells. Nfil3 is mainly expressed in fibroblasts and cardiomyocytes, whereas Zfp36 is ubiquitously expressed in a wide range of cells including endothelial cells, fibroblasts, and leukocytes. The dataset GSE95755 serves as a comprehensive transcription resource that encompasses diverse cardiac cell types throughout the processes of heart development, repair, and regeneration.28 We selected cells isolated from infarcted and non-infarcted areas of adult mouse hearts for our subsequent validation. Figure 8E shows the expression levels of hub genes in the four cell types (cardiomyocyte, fibroblast, endothelial cell and leukocyte) between the MI and sham groups. The expression levels of Il1b, Mmp9, Cxcl2 and Zfp36 were elevated in all four cell types after the MI operation compared to the sham group (Figure 8F–I).
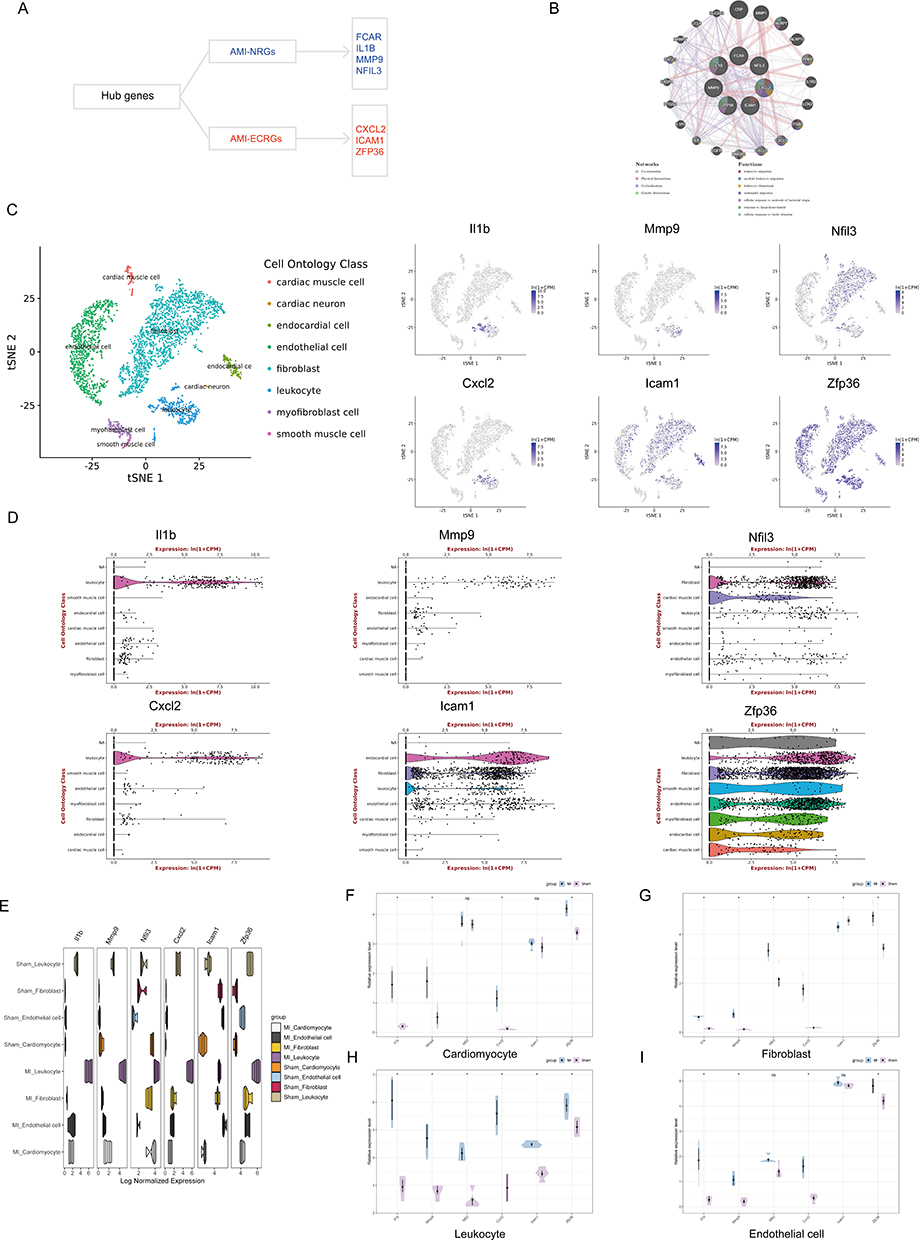 |
Figure 8 Expression profiles of hub genes in different cell types of the heart. (A) Hub genes include AMI-NRGs (FCAR, IL1B, MMP9 and NFIL3) and AMI-ECRGs (CXCL2, ICAM1 and ZFP36). (B) Hub genes and their co-expression genes were analyzed by GeneMANIA. (C and D) Expression of hub genes in different cell types of the healthy mouse heart. Measured by FACS in the Tabula Muris publically-available dataset. (E) Expression levels of hub genes in different cell types between MI and sham groups. (F–I) Expression levels of hub genes in (F) cardiomyocyte, (G) fibroblast, (H) leukocyte and (I) endothelial cells between MI and sham groups. |
The Diagnostic Value of Hub Genes
We evaluated the predictive accuracy of the hub genes (AMI-NRGs and AMI-ECRGs) in distinguishing normal individuals an AMI. Receiver operating curve (ROC) were used to assess the diagnostic value of each candidate hub gene. The normal individuals and MI patients in the dataset of GSE66360 could be accurately distinguished (Figure 9A–D and K–N). The AMI-NRGs and AMI-ECRGs also demonstrated significant accuracy in distinguishing AMI from normal individuals in GSE66360, with an AUC of 0.938 (95% CI 0.8911 to 0.9848) and an AUC of 0.909 (95% CI 0.8547to 0.9633) (Figure 9E and N). We also evaluated our hub genes in another dataset GSE62646 (Figure 9F–J and O–R) and the AMI-ECRGs got good results (Figure 9R). Additionally, we use several datasets including GSE66360, GSE62646 and GSE48060 to validate hub gene expression (Figure 9S). The relative expression levels of CXCL2, FCAR, ICAM1, MMP9, and NFIL3 were significantly higher in AMI than in the control groups in GSE66360 and GSE48060.
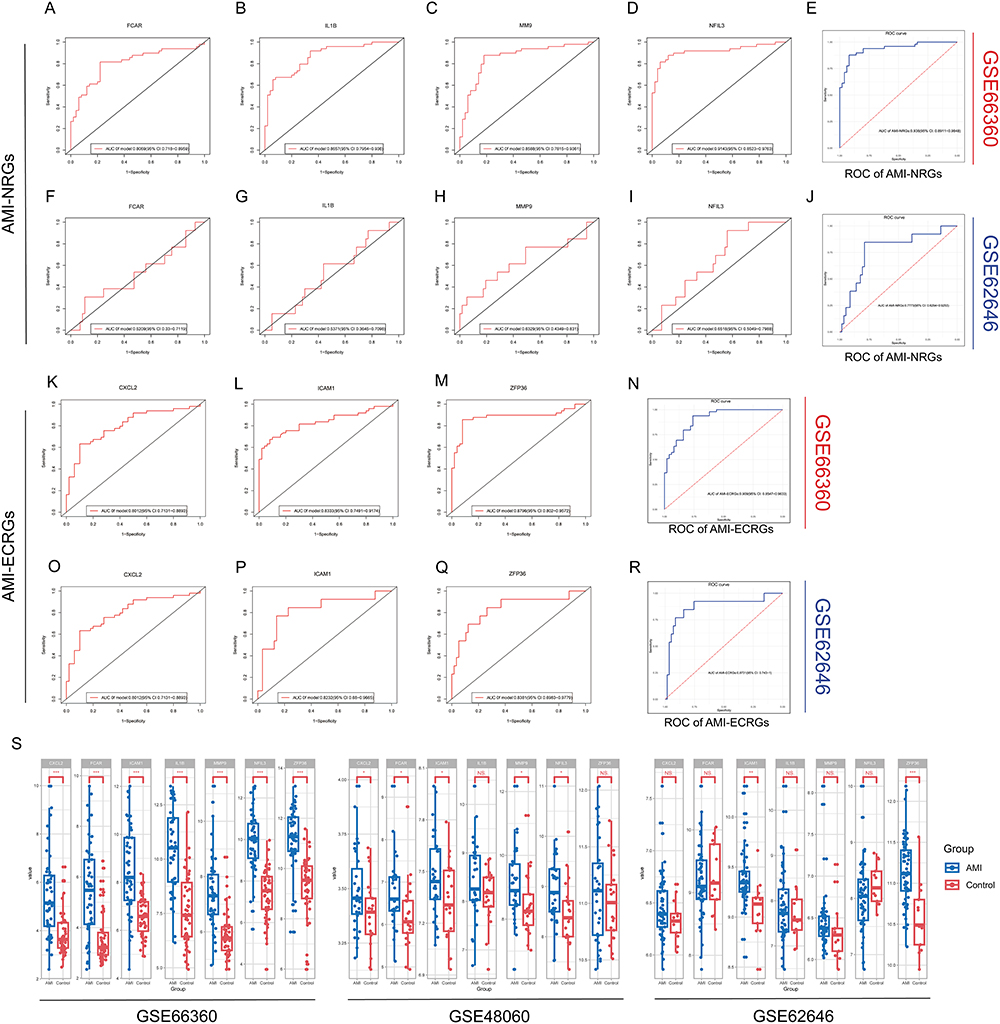 |
Figure 9 The diagnostic value of hub genes. (A–D and F–I) The ROC curve of FCAR, IL1B, MMP9 and NFIL3 in (A–D) GSE66360 and (F–I) GSE62646. (E and J) The ROC curve of AMI-NRG in (E) GSE66360 and (J) GSE62646. (K–M and O–Q) The ROC curve of CXCL2, ICAM1 and ZFP36 in (K–M) GSE66360 and (O–Q) GSE62646. (N and R) The ROC curve of AMI-ECRG in (N) GSE66360 and (R) GSE62646. (S) The expression of hub genes in GSE66360, GSE62646 and GSE48060. The comparison between the AMI and control groups used the mean t-test. P < 0.05 was considered to be statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001. |
Correlation Analysis Between Hub Genes and Infiltrating Immune Cell Types
To conduct a more comprehensive evaluation of the differences in immune cell infiltration and signature gene sets between AMI and control samples, the CIBERSORT, ssGSEA and xCell algorithms were employed. Based on the correlation study conducted using CIBERSORT, it was found that, except CXCL2, the remaining six hub genes exhibited a significant positive correlation with the infiltration of neutrophils and macrophages M0 (Figure 10A). Consistent with the results of the analysis using CIBERSORT, the ssGSEA results showed that except CXCL2 the other six hub genes were positively correlated with activated dendritic cell, eosinophil, macrophage, mast cell, neutrophil and plasmacytoid dendritic cell (Figure 10B). In the test dataset GSE62646, our analysis also showed that neutrophils and monocytes were positively correlated with hub genes (Figure 10C and D). Next, we analyzed the dataset using the xCell algorithm. The results showed a high positive correlation of genes with monocytes, NKT and neutrophils (Figure 10E and F).
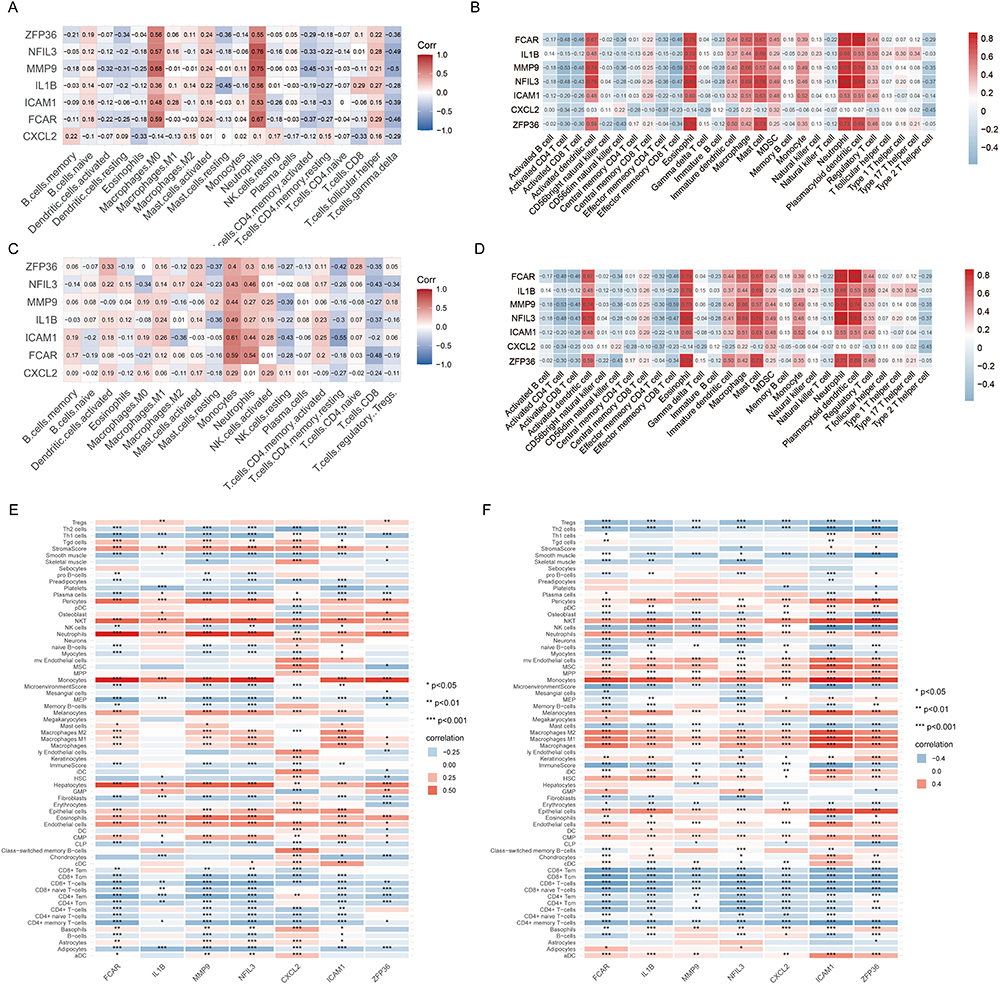 |
Figure 10 Correlation analysis between hub genes and infiltrating immune cell types. (A, B) Correlation heatmap showing the relationship between seven hub genes and the infiltrated immune cells based on CIBERSORT and ssGSEA algorithms in GSE48060. (C, D) Correlation heatmap showing the relationship between seven hub genes and the infiltrated immune cells based on CIBERSORT and ssGSEA algorithms in GSE62646. (E, F) Correlation heatmap showing the association between seven hub genes and the infiltrated immune cells based on the xCell algorithm. *P < 0.05, **P < 0.01, ***P < 0.001. |
Establishing a Model of MI
To verify the expression level of hub genes, we established a MI model using C57BL/6J mice. Echocardiography was conducted to evaluate the functional alterations of the mice in each experimental group. Figure 11A shows the representative B- and M-mode echocardiographic imaging of the heart. Mice after MI surgery exhibited significantly decreased LVEF and FS but exhibited significantly increased LVIDs and LVIDd compared with the sham controls (Figure 11B–F). Strain analysis and 2-dimensional speckle-tracking echocardiography were used to further evaluate the detrimental cardiac remodeling that occurs in mice following an MI. As shown in Figure 11G–J, an evaluation of the function of the left ventricular using longitudinal and radial strain analysis showed a notable decrease in global longitudinal strain (GLS) and lower radial strain in the anterior segments in post-MI hearts of mice compared to mice in the sham group.
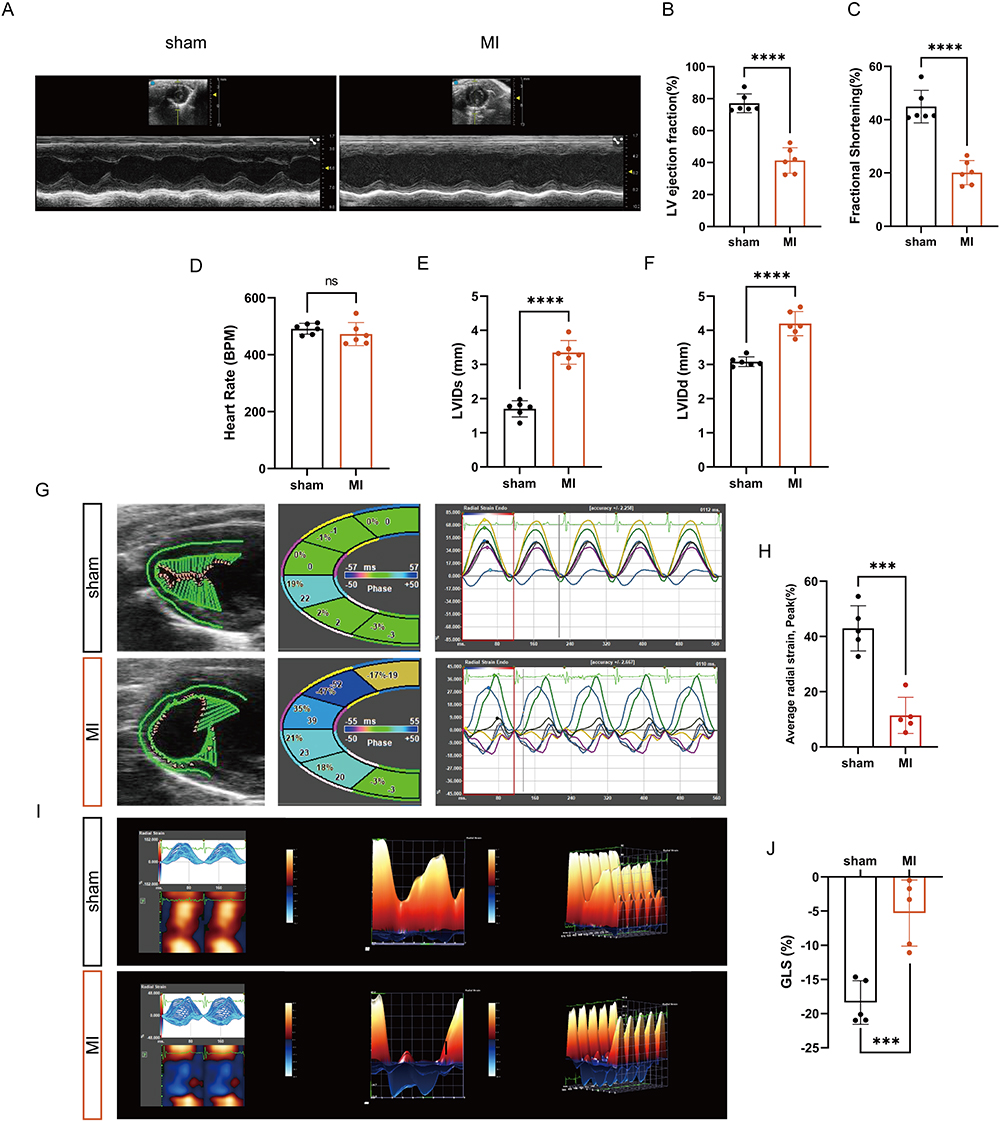 |
Figure 11 Establishing a model of myocardial infarction. (A) Representative B- and M-mode echocardiographic images of sham and MI mice. (B–F) Echocardiographic quantification of LVEF, FS, HR, LVIDd and LVIDs in the indicated groups (n=6). (G) Representative speckle-tracking echocardiography-based analysis of left ventricular function in sham and MI mice at day 7 post-MI. (H) Average radial strain was measured by spot tracking technology (n=5). (I) Representative long-axis radial myocardial strain maps deconvoluting infarction-induced regional myocardial wall deformation in sham and MI mice at day 7 post-MI. (J) GLS was measured by spot tracking technology (n=5). ***P < 0.001, ****P < 0.0001. |
Validation of Gene Expression
To further demonstrate the expression pattern of hub genes, qRT-PCR verification was performed in the left ventricular tissue samples of the sham and MI groups. The results indicated that IL-1β, MMP9 and NFIL3 were highly expressed in MI samples (Figure 12A and B). However, the expression levels of ICAM, ZFP36, and CXCL2 were not significantly different between the two groups, which may be due to our insufficient sample size and single time point validation after MI. Next, we used immunohistochemistry to further verify the gene expression, in parallel, IL-1β, MMP9 and NFIL3 showed substantial upregulation in the infarcted areas (Figure 12C). Statistical analysis showed a significant increase in positive areas in the infarcted area, but no significant difference between the non-infarcted area and the sham groups (Figure 12D-F).
 |
Figure 12 Validating gene expression. (A) The mRNA expression of AMI-NRGs (IL-1β, MMP9, NFIL3) was identified via qRT-PCR (n = 5–6). (B) The mRNA expression of AMI-ECRGs (ICAM1, ZFP36, CXCL2) were identified via qRT-PCR (n = 5–6). (C) Representative images of IHC staining of hearts from MI and sham groups mice. (D-F) Statistical analysis of IHC staining (n =3). Scale bar, 100μm. *P < 0.05, **P < 0.01, ***P < 0.001. |
Discussion
There is increasing evidence that supports the significant involvement of neutrophils in the progression of coronary artery disease (CAD), particularly in the subsequent problems that arise, such as acute coronary syndrome and heart failure.29 For example, activated lesional smooth muscle cells (SMCs) attract neutrophils, triggering the ejection of neutrophil NETs.30 Among them, the nuclear protein histone H4 binds to and lyses SMCs, this process can ultimately lead to the destabilization of plaques.30 Therefore, neutrophils may be an independent predictor for risk stratification in patients with ACS and/or cardiac revascularization when analyzed concomitantly with other markers of inflammation.31
In the present study, we first analyzed the immune infiltration of MI based on the CIBERSORT and ssGSEA algorithms. Similar to the results of previous studies,32 we found that neutrophils were higher in the AMI group. In a subsequent enrichment pathway analysis of the DEGs in GSE178883, we found that in addition to the formation of NETs, the down-regulated DEGs were also enriched in Fc gamma R−mediated phagocytosis and Cytokine−cytokine receptor interaction. This may reflect neutrophils as one of the major players during acute inflammation.33 We also observed that up-regulated DEGs were enriched in Th17 cell differentiation and Th1 and Th2 cell differentiation. This finding suggests that, apart from their inherent function in resisting infections and responding to injuries, neutrophils are increasingly recognized as crucial regulators of the adaptive immune system.34
Using three machine learning algorithms, we finally filtered out four hub genes in the AMI-NRGs (FCAR, IL1B, MMP9 and NFIL3). The FCAR gene has been identified as a member of the immunoglobulin gene superfamily and is responsible for encoding a receptor that specifically binds to the Fc region of immunoglobulin A (IgA). FcαRI/CD89 is unique in that it is neither expressed in mice nor is there any homolog in mice.35 The previous studies have shown that neutrophils possess natural anti-tumor properties and can induce potent tumor cell killing via targeting the immunoglobulin A Fc receptor.36 Here, our study suggests that neutrophils may mediate cellular injury during MI through the immunoglobulin A Fc receptor. Similar to FCAR, IL-1β, recognized as a member of a much larger IL-1 family of cytokines, is expressed primarily in myeloid cells.37 Large numbers of neutrophils can quickly accumulate at infection or tissue damage sites, and become the predominant local source of IL-1.38 IL-1β can be converted from an inactive precursor to a biologically active cytokine through inflammasome activation of the intracellular cysteine protease caspase-1 and caspase-1 independent cleavage mediated by serine proteases derived from neutrophils.39 Many studies have demonstrated the important role of IL-1 in MI.40 Thus, inhibition of IL-1β may reduce inflammatory injury in the acute phase of MI by inducing an anti-inflammatory effect.
In neutrophils, preformed MMP-9 is mainly stored in gelatinase granules and subsequently released in response to stimulation by inflammation or tissue injury.41 In the early stages after MI, MMP9 is mainly derived from neutrophils.42 Serum exosome MMP9 expression is elevated in patients with MI, and can be an effective biomarker for diagnosis of AMI.43 Nishiguchi et al demonstrated that local MMP-9 level could determine the early clinical presentation in patients with acute MI.44 Our experiments also confirmed that MMP9 is highly expressed in MI tissues. NFIL3, also known as Adenovirus E4 promoter-binding protein (E4BP4), is one of the mammalian basic leucine zipper transcription factors. Research has revealed that NFIL3 is linked to numerous physiological and biochemical processes.45 For example, NFIL3 has been found to be related to immune-mediated diseases, like SLE, arthritis and Crohn’s disease.46 In addition, some studies have demonstrated that NFIL3 may regulate the pathological process of heart failure through calcium signaling mechanisms, autocrine signaling, and insulin-like growth factor II receptor.47 Our experiments confirm that NFIL3 is highly expressed in ischemic myocardial tissue and may be involved in neutrophil-mediated cell damage. Next, further studies are needed to determine the specific mechanism of NFIL3 in NETs-mediated cell injury.
To investigate possible targets of NETs-mediated endothelial cell injury during MI, we took the intersection of the brown module with the DEGs of GSE179828, and finally get three hub genes (CXCL2, ICAM1 and ZFP36). Although our experimental validation did not reveal significant differences in the expression levels of these genes in the control and MI groups, previous studies have shown the importance of these genes in the development of many cardiovascular diseases. For example, several studies found that CXCL2 has an important role in cardiovascular disease, especially in the development of atherosclerosis. The study conducted by Chang et al provided evidence supporting the notion that the absence of Sirt4 contributes to the progression of atherosclerosis through the activation of the NF-κB/IκB/CXCL2/3 signaling pathway.48 Another study reveals that the joint suppression of TNFα, CXCL2, and CCL2 inhibits the ability of aged visceral fat transplants to promote the development of atherosclerosis.49 The expression of ICAM-1 is increased in the milieu of atherosclerotic plaques, where it is observed to be expressed by endothelial cells, macrophages, and smooth muscle cells. Gross et al demonstrate that soluble intercellular adhesion molecule 1 (sICAM-1) concentration may be an early biomarker that indicates changes in the artery wall that accompany atherosclerosis.50 ZFP36 is a small group of mRNA binding and destabilizing proteins expressed in almost all eukaryotes.51 A previous bioinformatics analysis study suggests that ZFP36 may be an endothelial cell senescence-related gene in patients with MI.52 Li et al also found significantly elevated ZFP36 protein levels in ischemic human heart tissue and knockdown of ZFP36 in HCMECs using siRNA significantly inhibited cell proliferation.53 Our analysis suggests that ZFP36 may be one of the targets of endothelial cell injury by NETs during MI and may be a promising novel target for the treatment of ischemic heart disease.
Our study has several limitations. First, we did not integrate more datasets to screen for the DEGs, and more advanced bioinformatics and machine learning methods may screen for hub genes more accurately. Second, clinical samples and data are needed to further validate gene expression levels and their diagnostic value. Finally, further in vivo and in vitro loss-and-gain-of-function experiments are necessary to investigate the mechanisms of hub genes in NETs-mediated cellular damage.
Conclusions
In conclusion, our study identified seven candidate diagnostic hub genes, including three genes associated with NETs-related endothelial cell injury. These hub genes are likely to serve as important markers for the diagnosis of MI. Furthermore, analyzing these hub genes and immune cells may provide new insights into potential therapeutic approaches for NETs-related cell injury during MI.
Abbreviations
MI, myocardial infarction; NETs, neutrophil extracellular traps; PMA, phorbol myristate acetate; ROC, receiver operating curve; MPO, myeloperoxidase; DEGs, differentially expressed genes; WGCNA, weighted gene co-expression network analysis; ARDS, acute respiratory distress syndrome; COPD, chronic obstructive pulmonary disease; SLE, systemic lupus erythematosus; TLR2, Toll-like receptor 2; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; HUVECs, human umbilical vein endothelial cells; LVIDd, left ventricular internal dimension diastolic; LVIDs, left ventricular internal dimension systolic; LVEF, left ventricular ejection fraction; HR, heart rate; FS, fractional shortening; ROS, reactive oxygen species; GLS, global longitudinal strain; sICAM-1, soluble intercellular adhesion molecule 1; IgA, immunoglobulin A; CAD, coronary artery disease; SMCs, smooth muscle cells; SVM, Support vector machine; RF, random forest; GS, gene significance.
Data Sharing Statement
The datasets used and analyzed in this study are available from the GEO database.
Ethics Approval and Informed Consent
The study was approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University (approval No. 20220304A), and were also in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The human data used in this study were approved by the Review Board of Renmin Hospital of Wuhan University.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81770399), Health Commission of Hubei Province scientific research project (WJ2021Q035).
Author Contributions
All authors have made significant contributions to the reported work, including contributions to the conception, study design, execution, data acquisition, analysis, and interpretation or in all these aspects. They have been involved in drafting, revising, and critically reviewing the manuscript. All authors have provided their final approval for the version to be published and reached an agreement on the journal to which the article has been submitted. All authors agree to take responsibility and be accountable for the contents of the article.
Funding
This work was supported by National Natural Science Foundation of China (81770399), Health Commission of Hubei Province scientific research project (WJ2021Q035).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yap J, Irei J, Lozano-Gerona J, Vanapruks S, Bishop T, Boisvert WA. Macrophages in cardiac remodelling after myocardial infarction. Nat Rev Cardiol. 2023;20(6):373–385. doi:10.1038/s41569-022-00823-5
2. Frantz S, Hundertmark MJ, Schulz-Menger J, Bengel FM, Bauersachs J. Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies. Eur Heart J. 2022;43(27):2549–2561. doi:10.1093/eurheartj/ehac223
3. Kumar V, Prabhu SD, Bansal SS. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front Cardiovasc Med. 2022;9:992653. doi:10.3389/fcvm.2022.992653
4. Kumar V, Rosenzweig R, Asalla S, Nehra S, Prabhu SD, Bansal SS. TNFR1 contributes to activation-induced cell death of pathological CD4(+) T lymphocytes during ischemic heart failure. JACC. 2022;7(10):1038–1049. doi:10.1016/j.jacbts.2022.05.005
5. Abplanalp WT, John D, Cremer S, et al. Single-cell RNA-sequencing reveals profound changes in circulating immune cells in patients with heart failure. Cardiovasc Res. 2021;117(2):484–494. doi:10.1093/cvr/cvaa101
6. Rurik JG, Aghajanian H, Epstein JA. Immune cells and immunotherapy for cardiac injury and repair. Circ Res. 2021;128(11):1766–1779. doi:10.1161/CIRCRESAHA.121.318005
7. Margraf A, Lowell CA, Zarbock A. Neutrophils in acute inflammation: current concepts and translational implications. Blood. 2022;139(14):2130–2144. doi:10.1182/blood.2021012295
8. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385
9. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi:10.1083/jcb.200606027
10. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. doi:10.1083/jcb.201006052
11. Sônego F, Castanheira FV, Ferreira RG, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. 2016;7:155. doi:10.3389/fimmu.2016.00155
12. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3(3). doi:10.1172/jci.insight.98178
13. Grabcanovic-Musija F, Obermayer A, Stoiber W, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. 2015;16(1):59. doi:10.1186/s12931-015-0221-7
14. Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153. doi:10.1038/nm.4027
15. Pertiwi KR, van der Wal AC, Pabittei DR, et al. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thromb Haemost. 2018;118(6):1078–1087. doi:10.1055/s-0038-1641749
16. Zhang Z, Ding S, Wang Z, et al. Prmt1 upregulated by Hdc deficiency aggravates acute myocardial infarction via NETosis. Acta Pharm Sin B. 2022;12(4):1840–1855. doi:10.1016/j.apsb.2021.10.016
17. Wu Y, Wei S, Wu X, Li Y, Han X. Neutrophil extracellular traps in acute coronary syndrome. J Inflamm. 2023;20(1):17. doi:10.1186/s12950-023-00344-z
18. Schaum N, Karkanias J, Neff NF, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562(7727):367–372. doi:10.1038/s41586-018-0590-4
19. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods. 2015;12(5):453–457. doi:10.1038/nmeth.3337
20. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi:10.1038/nature08460
21. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi:10.1186/s13059-017-1349-1
22. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
23. Weissman D, Maack C. Mitochondrial function in macrophages controls cardiac repair after myocardial infarction. J Clin Invest. 2023;133(4). doi:10.1172/JCI167079
24. Ruder AV, Wetzels SMW, Temmerman L, Biessen EAL, Goossens P. Monocyte heterogeneity in cardiovascular disease. Cardiovasc Res. 2023;119(11):2033–2045. doi:10.1093/cvr/cvad069
25. Shirakawa K, Kobayashi E, Ichihara G, et al. H(2) inhibits the formation of neutrophil extracellular traps. JACC. 2022;7(2):146–161. doi:10.1016/j.jacbts.2021.11.005
26. Wu J, Zhang F, Zheng X, et al. Identification of renal ischemia reperfusion injury subtypes and predictive strategies for delayed graft function and graft survival based on neutrophil extracellular trap-related genes. Front Immunol. 2022;13:1047367. doi:10.3389/fimmu.2022.1047367
27. Shi H, Gandhi AA, Smith SA, et al. Endothelium-protective, histone-neutralizing properties of the polyanionic agent defibrotide. JCI Insight. 2021;6(17):17. doi:10.1172/jci.insight.149149
28. Quaife-Ryan GA, Sim CB, Ziemann M, et al. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017;136(12):1123–1139. doi:10.1161/CIRCULATIONAHA.117.028252
29. Sreejit G, Johnson J, Jaggers RM, et al. Neutrophils in cardiovascular disease: warmongers, peacemakers, or both? Cardiovasc Res. 2022;118(12):2596–2609. doi:10.1093/cvr/cvab302
30. Silvestre-Roig C, Braster Q, Wichapong K, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019;569(7755):236–240. doi:10.1038/s41586-019-1167-6
31. Guasti L, Dentali F, Castiglioni L, et al. Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb Haemost. 2011;106(4):591–599. doi:10.1160/TH11-02-0096
32. Arruda-Olson AM, Reeder GS, Bell MR, Weston SA, Roger VL. Neutrophilia predicts death and heart failure after myocardial infarction: a community-based study. Circ Cardiovasc Qual Outcomes. 2009;2(6):656–662. doi:10.1161/CIRCOUTCOMES.108.831024
33. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi:10.1038/nri3399
34. Bert S, Nadkarni S, Perretti M. Neutrophil-T cell crosstalk and the control of the host inflammatory response. Immunol Rev. 2023;314(1):36–49. doi:10.1111/imr.13162
35. Koernig S, Campbell IK, Mackenzie-Kludas C, et al. Topical application of human-derived Ig isotypes for the control of acute respiratory infection evaluated in a human CD89-expressing mouse model. Mucosal Immunol. 2019;12(4):1013–1024. doi:10.1038/s41385-019-0167-z
36. Gruijs M, Sewnath CAN, van Egmond M. Therapeutic exploitation of neutrophils to fight cancer. Semin Immunol. 2021;57:101581. doi:10.1016/j.smim.2021.101581
37. Broderick L, Hoffman HM. IL-1 and autoinflammatory disease: biology, pathogenesis and therapeutic targeting. Nat Rev Rheumatol. 2022;18(8):448–463. doi:10.1038/s41584-022-00797-1
38. Dubyak GR, Miller BA, Pearlman E. Pyroptosis in neutrophils: multimodal integration of inflammasome and regulated cell death signaling pathways. Immunol Rev. 2023;314(1):229–249. doi:10.1111/imr.13186
39. Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol. 2015;33(1):49–77. doi:10.1146/annurev-immunol-032414-112306
40. Haybar H, Bandar B, Torfi E, Mohebbi A, Saki N. Cytokines and their role in cardiovascular diseases. Cytokine. 2023;169:156261. doi:10.1016/j.cyto.2023.156261
41. Becirovic-Agic M, Chalise U, Daseke MJ, et al. Infarct in the heart: what’s MMP-9 got to do with it? Biomolecules. 2021;11(4):491. doi:10.3390/biom11040491
42. Zhang N, Aiyasiding X, Li WJ, Liao HH, Tang QZ. Neutrophil degranulation and myocardial infarction. Cell Commun Signal. 2022;20(1):50. doi:10.1186/s12964-022-00824-4
43. Chen Z, Yan Y, Wu J, Qi C, Liu J, Wang J. Expression level and diagnostic value of exosomal NEAT1/miR-204/MMP-9 in acute ST-segment elevation myocardial infarction. IUBMB Life. 2020;72(11):2499–2507. doi:10.1002/iub.2376
44. Nishiguchi T, Tanaka A, Taruya A, et al. Local matrix metalloproteinase 9 level determines early clinical presentation of ST-segment-elevation myocardial infarction. Arterioscler Thromb Vasc Biol. 2016;36(12):2460–2467. doi:10.1161/ATVBAHA.116.308099
45. Cowell IG. E4BP4/NFIL3, a PAR-related bZIP factor with many roles. Bioessays. 2002;24(11):1023–1029. doi:10.1002/bies.10176
46. Chen Z, Fan R, Liang J, et al. NFIL3 deficiency alleviates EAE through regulating different immune cell subsets. J Adv Res. 2022;39:225–235. doi:10.1016/j.jare.2021.10.011
47. Velmurugan BK, Chang RL, Marthandam Asokan S, et al. A minireview of E4BP4/NFIL3 in heart failure. J Cell Physiol. 2018;233(11):8458–8466. doi:10.1002/jcp.26790
48. Chang S, Zhang G, Li L, et al. Sirt4 deficiency promotes the development of atherosclerosis by activating the NF-κB/IκB/CXCL2/3 pathway. Atherosclerosis. 2023;373:29–37. doi:10.1016/j.atherosclerosis.2023.04.006
49. Song J, Farris D, Ariza P, et al. Age-associated adipose tissue inflammation promotes monocyte chemotaxis and enhances atherosclerosis. Aging Cell. 2023;22(2):e13783. doi:10.1111/acel.13783
50. Gross MD, Bielinski SJ, Suarez-Lopez JR, et al. Circulating soluble intercellular adhesion molecule 1 and subclinical atherosclerosis: the Coronary Artery Risk Development in Young Adults Study. Clin Chem. 2012;58(2):411–420. doi:10.1373/clinchem.2011.168559
51. Snyder BL, Blackshear PJ. Clinical implications of tristetraprolin (TTP) modulation in the treatment of inflammatory diseases. Pharmacol Ther. 2022;239:108198. doi:10.1016/j.pharmthera.2022.108198
52. Xiang J, Shen J, Zhang L, Tang B. Identification and validation of senescence-related genes in circulating endothelial cells of patients with acute myocardial infarction. Front Cardiovasc Med. 2022;9:1057985. doi:10.3389/fcvm.2022.1057985
53. Li Z, Solomonidis EG, Berkeley B, et al. Multi-species meta-analysis identifies transcriptional signatures associated with cardiac endothelial responses in the ischaemic heart. Cardiovasc Res. 2023;119(1):136–154. doi:10.1093/cvr/cvac151
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Neutrophil Extracellular Traps Aggravate Contrast-Induced Acute Kidney Injury by Damaging Glomeruli and Peritubular Capillaries
Wang H, Gao T, Zhang R, Hu J, Gao S, Wang Y, Qi X, Zhou Y, Zheng G, Dong H
Journal of Inflammation Research 2023, 16:5629-5646
Published Date: 28 November 2023
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024
Neutrophil Extracellular Traps Enhance Procoagulant Activity and Predict Poor Prognosis in Patients With Metastatic Breast Cancer
Gong Y, Chen B, Tan Q, Wei W
International Journal of General Medicine 2025, 18:1247-1259
Published Date: 4 March 2025
Unveiling the Temporal Dynamics and Molecular Regulation Profiles of Neutrophil Extracellular Traps Following Spinal Cord Injury

Li J, Chang C, Li Y, Cui S, Bai J, Zhang C, Wang X, Li K, Jian F
Journal of Inflammation Research 2025, 18:10585-10608
Published Date: 6 August 2025
The eNAMPT-Integrin α5β1 Axis Mediates Neutrophil-Endothelial Cell Interactions Driving Inflammation in Ulcerative Colitis
Di Y, Ji W, Xiong W, Song W, Chen G, Li D, Qi F
Journal of Inflammation Research 2025, 18:18307-18321
Published Date: 30 December 2025