")
Back to Journals » International Journal of Nanomedicine » Volume 16
Hybrid Nanoparticles Modified by Hyaluronic Acid Loading an HSP90 Inhibitor as a Novel Delivery System for Subcutaneous and Orthotopic Colon Cancer Therapy
Authors Pan C, Zhang T, Li S, Xu Z, Pan B, Xu S, Jin S, Lu G , Yang S, Xue Z, Chen P, Shen X, Wang F , Xu C
Received 13 August 2020
Accepted for publication 26 January 2021
Published 2 March 2021 Volume 2021:16 Pages 1743—1755
DOI https://doi.org/10.2147/IJN.S275805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Chenwei Pan,1,* Tiaotiao Zhang,2,3,* Shaoxun Li,1,3,* Zhihua Xu,2,3 Binhui Pan,2,3 Sheng Xu,2,3 Shuanghong Jin,1,3 Guangrong Lu,2 Shouxing Yang,2 Zhanxiong Xue,2 Ping Chen,4 Xian Shen,5 Fangyan Wang,6 Changlong Xu2,7
1Department of Infectious Disease, Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 2Department of Gastroenterology, Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 3Second School of Medicine, Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 4Department of Infectious Disease, Shulan Hospital, Hangzhou, Zhejiang, 310012, People’s Republic of China; 5Department of Gastrointestinal Surgery, Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 6Department of Pathophysiology, School of Basic Medicine Science, Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 7Center for Diagnostics and Therapeutics, Institute for Biomedical Sciences, Georgia State University, Atlanta, GA, 30302, USA
*These authors contributed equally to this work
Correspondence: Fangyan Wang; Changlong Xu Email [email protected]; [email protected]
Background: As a therapeutic target for cancer treatment, HSP90 has been explored extensively. However, the significant side effects of the HSP90 inhibitor 17AAG have limited its clinical use.
Methods: In this study, we used hyaluronic acid (HA)–decorated DOTAP–PLGA hybrid nanoparticles (HA-DOTAP-PLGA NPs) as 17AAG-delivery carriers for targeted colon cancer therapy.
Results: Different methods were used to characterize the successful fabrication of these hybrid PLGA NPs. Our results demonstrated that internalization of HA-NPs in colon cancer cells was governed by CD44receptor–mediated endocytosis. Annexin V–propidium iodide staining experiments revealed that cell apoptosis induced by HA-NPs-17AAG in colon cancer cells was more efficient than free 17AAG. In two animal models used to screen anticancer efficacy (Luc-HT29 subcutaneous xenograft and AOM/DSS-induced orthotopic tumor model), HA-NPs-17AAG significantly inhibited xenograft and orthotopic tumor growth, demonstrating HA-NPs-17AAG had much better therapeutic efficiency than free 17AAG. It is worth noting that great biocompatibility of HA-DOTAP-PLGA NPs was observed both in vitro and in vivo.
Conclusion: Our research offers a preclinical proof of concept for colon cancer therapy with DOTAP-PLGA NPs as a creative drug-delivery system.
Keywords: cationic hybrid nanoparticles, hyaluronic acid, HA, HSP90 inhibitor, colon cancer, targeted delivery system
Introduction
Though significant progress has been achieved in controlling morbidity and mortality in colon cancer (sometimes called colorectal cancer), it is still the third–most common cancer worldwide.1,2 For colon cancer victims, unfortunately, traditional anticancer strategies, such as chemotherapy, provide only limited treatment efficacy in the clinic.3,4 To address this intrinsic limitation of chemotherapy, new therapeutic strategies should be investigated and developed to treat colon cancer.
To address this challenge in colon cancer, drug-delivery systems (DDSs) based on nanotechnology are opening up new avenues for DD vehicles.5,6 DDSs offer several advantages over conventional administration: improving drug concentration at desired locations, prolonging circulation,enhance pharmacological activity, protecting the drug against premature release, and reducing systemic side effects caused by dosing frequency. Many types of drug carriers with different morphologies can be synthesized with various materials, including polymers,7 inorganic substances,8 and combinations of the two.9,10 These DDSs usually take advantage of the EPR effect to deliver drugs to tumors. There are two major components involved in the EPR effect: is the nanoscale of the drug can alter its biodistribution, and the plasma half-life of a nanodrug can be increased by the EPR effect.11,12 However, the drawback of this passive approach is low bioavailability of drugs, since the targeting ability of DDSs is overdependence on the tumor microenvironment, such as in the case of compromised vasculature.13 In addition, the tumor microenvironment, eg, interstitial fluid pressure, also limits drug uptake and distribution to tumor cells.
Active targeting of DSSs might be an alternative strategy for efficient DD. Cancer cells or angiogenic endothelial cells always overexpress certain receptors by modification of DDSs with receptor ligands, such as aptamers, proteins, antibodies, and polysaccharides,14–18 which should be achieved active targeting to cancer with high efficiency. In the active approach, ligand receptors interact and are then internalized via receptor-mediated endocytosis, thereby enhancing the targeting ability of payload drugs to desired positions.
Among the active ligands, a natural polysaccharide, hyaluronic acid (HA), has aroused wide interest among researchers. HA has many advantages, such as active tumor targeting capacity, HAase-responsive capacity, and more importantly biocompatibility. HA can recognize its receptor, CD44, which is often overexpressed in tumor-cell membranes and has been determined to have a strong relationship with tumor progress, infiltration, and metastasis.19–21
Among the DDSs, PLGA-based nanoparticles (NPs) have received the most attention, due to their unique characteristics. PLGA is a polymer that is widely used in the biomedical field. When compared with other materials used in DDSs, PLGA possesses unparalleled benefits such as biodegradability, controlled release, and stability. Cationic lipid–PLGA hybrid NPs with biodegradable polymer as a core and cationic lipid as a versatile surface have been verified as a potential delivery system. Cationic lipid membranes, including 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) and 1,2-di-O-octadecenyl-3-trimethylammonium propane, have attracted great attention, because they can easily interact with anionic polysaccharides, peptides, proteins, and plasmid DNA encoding to form nanocomplexes for DD in vivo.
To achieve specific cancer therapy with minimal systemic toxicity from anticancer drugs, a considerable number of strategies have recently been investigated. Of these, molecule-targeted therapy, which often targets specific molecules involved in the growth and spread of cancer cells, has been developed. HSP90 is frequently overexpressed in tumor cells and has been considered a representative target.22 As a molecular chaperone, HSP90 is deemed to play a very important part in activating wide range of signaling proteins.26
Oxidative stress and inflammation, as with imbalance between antioxidants or radical scavengers and ROS, drive the upregulation of various HSPs, including HSP90. It has been reported that HSP90, with its capacity for regulating HSF1 and the HSF1–HSP90 heterocomplex in the cytoplasm, can prevent antiinflammatory cytokine, including IL10, transcription.25 Based on this, 17-allylaminogeldanamycin (17AAG), ane inhibitor of HSP90 that causes HSF1–HSP90 dissociation, should raise the expression of IL10 and at the same time decrease the expression of TNFα, IFNγ, and IL1β. As we know, intestinal inflammation, especially chronic inflammation, is thought to be a risk factor of colorectal cancer development, and this cascade could offer therapeutic benefits with regard to colon cancer.
In this study, we used single emulsion (oil in water) solvent evaporation to prepare DOTAP–PLGA hybrid NPs to load the HSP90 inhibitor 17AAG. Then, NPs were decorated with HA by electrostatic interaction (HA-NPs-17AAG). Given the advantages of the capacity of HA-coated cationic NPs for HA-CD44 receptor–medicated endocytosis and endosomal escape,antitumor effects were evaluated in both subcutaneous and orthotopic colon cancer models (Figure 1). Our findings indicated that the HA-NPs-17AAG developed may be potentially exploited as a novel DD carrier for treatment of colon cancer.
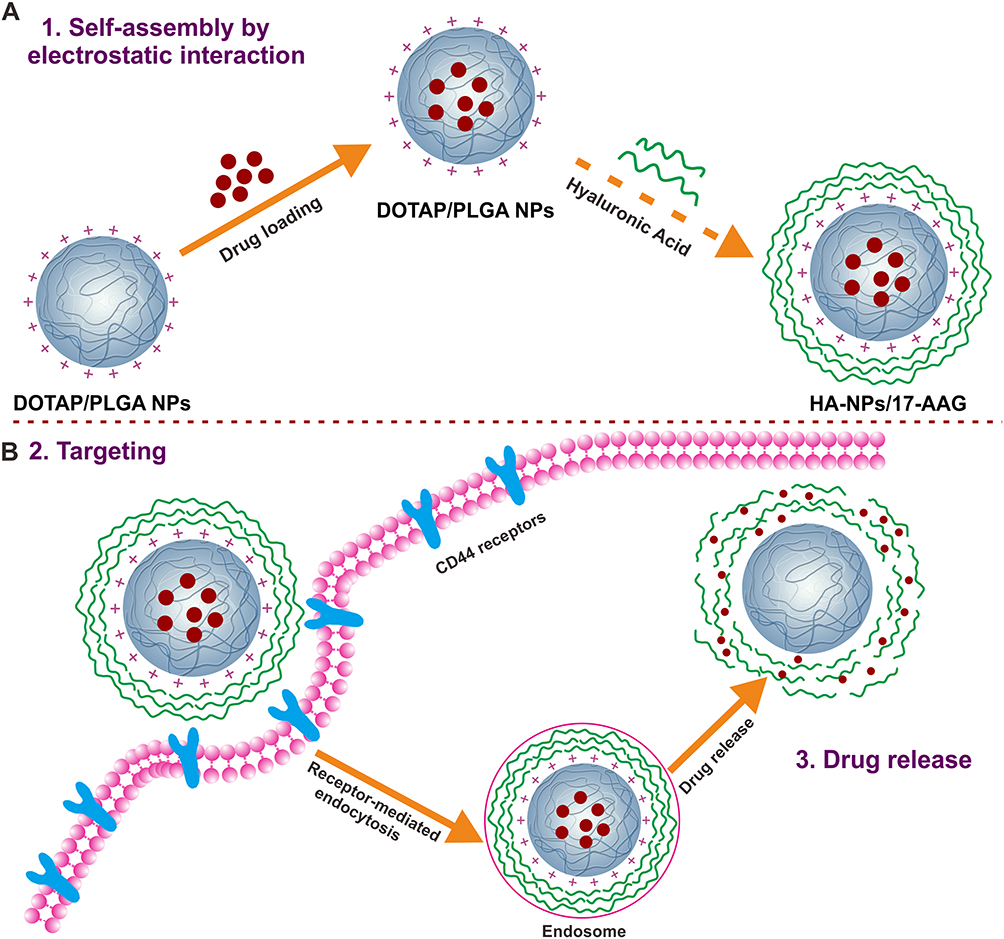 |
Figure 1 HA-DOTAP-PLGA NP–mediated transport of 17AAG to colon cancer cells. (A) Fabrication of HA-NPs-17AAG. (B) Drug delivery and release mediated by CD44 receptor. |
Methods
Materials and Reagents
Phalloidin–FITC, PLGA, PVA, and a cell death–detection kit were purchased from Sigma-Aldrich, fluorescent lipophilic dyes (DiL and DiR) from Promokine (Heidelberg, Germany),17AAG and standard from LC Laboratories (Woburn; MA, USA), HA from Shanghai Hanhong Technology, DOTAP from Avanti Polar Lipids, and cell-apoptosis and MTT cell-proliferation kits from Invitrogen.
Preparation of 17AAG-Loaded DOTAP–PLGA Hybrid NPs with or without HA Coating
Hybrid 17AAG-loaded DOTAP–PLGA NPs were prepared based on our previous methods, with slight modification. Briefly, PLGA (100 mg), DOTAP (21.6 mg), and 17AAG (10 mg) were first added to dichloromethane (DCM). Then, ultrasound was used to emulsify the DCM solution to get an oil-in-water emulsion. The NP emulsion was stirred, then evaporated to remove DCM. Finally, polymer NPs were purified with deionized water several times through centrifugation. For HA-DOTAP-PLGA NP preparation, HA solution at a concentration of 0.04% w:v was added dropwise to DOTAP-PLGA NPs solution, then the mixed solution was stirred for 4 hours. Finally, surplus HA was removed by threefold centrifugation and the HA-coated DOTAP-PLGA NPs obtained collected and lyophilized for subsequent use.
NP Characterization
A Malvern Zetasizer was used to measure size and ζ-potential of NPs using DLS. The morphology of NPs was characterized by TEM. High performance liquid chromatography (HPLC) was used to evaluate 17AAG-loading efficiency.
Loading efficiency of the drug in NPs was calculated:
loading content = (WT – WF)/WNP × 100%
where WT is the total weight of 17AAG fed, WF the weight of unencapsulated free 17AAG, and WNP the weight of NPs, and
encapsulation efficiency = (WT – WF)/WT × 100%
Slide-a-Lyzer mini dialysis devices (MW 20 kDa) were used to evaluate cumulative drug release of 17AAG from DOTAP-PLGA and HA-DOTAP-PLGA. Briefly, 10 mg of DOTAP-PLGA or HA-DOTAP-PLGA NPs were dispersed in PBS and then centrifuged in regular conditions. Aliquots were taken from the dialysis bag at designated time points, and 17AAG in buffer solution was quantified by HPLC.
Cell Culture
HCT116 and HT29 colon cancer cells were cultured in regulated condition. The cell-culture medium was DMEM, to which antibiotics (penicillin and streptomycin) and FBS were added. The two cell lines were purchased from ATCC. Both were tested and authenticated to eliminate any contamination via PCR and morphology.
Uptake of NPs by Colon Cancer Cells
Confocal imaging was used to evaluate the uptake of HA-NPs in vitro. Cells were cultured in slides and HA-NPs/Dil for designated periods. In the competition experiments, HA (2 mg/mL) was applied and incubated with colon cancer cells (HT29 and HCT116 cells) before HA-NPs/Dil NPs were added. Following fixation and dehydration with PFA and acetone, cells were then stained with phalloidin–FITC to visualize the cytoskeleton. Finally, cell nuclei were stained with DAPI and images captured on confocal microscopy.
Flow cytometry (FACS) was employed to count Dil+ cells taken up by HA-NPs in the presence of free HA. HT29 and HCT116 cells at a concentration of 105/well were seeded in six-well plates and then incubated with HA-NPs/Dil. Next, we washed and collected the cells. Finally, we used flow cytometry to analyze Dil+ cells.
Cellul-uptake efficiency was then analyzed. HT29 and HCT116 cells were seeded in six-well plates and incubated overnight. Cells were treated with NPs/Dil and HA-NPs/Dil, respectively (1 mg/mL). In this experiment, cells without treatment were used as controls. NPs were incubated with cells for, then fixed for FACS analysis.
Colocalization of HA-NPs
HT29 cells were cultured in chamber slides overnight, then NPs/Dil or HA-NPs/Dil were added and cultured for designated periods. To visualize recycled endosomes, FITC-labeled transferrin (Tf) at a concentration of 2 mg/mL stock (14 μL) was added to the cells and incubated for 15 minutes. At the end of the experiment, cells were fixed and washed for colocalization investigation.
Apoptosis Assays
Annexin V–FITC/propidium iodide (PI) experiments were performed to investigate the extent of cell apoptosis. Cells used in this experiment were on seeded slides. Free 17AAG (5 μg/mL) and HA-NPs-17AAG (equal to 5 μg/mL of 17AAG) were added. Then, the staining protocol was performed following the instructions. Images were captured by fluorescence microscopy. For flow-cytometry experiments, healthy, early apoptotic, and necrotic cells were detected with annexin V–FITC/PI double staining. All experiments were performed three times.
Animals
Mice involved in our research were housed under specific pathogen-free conditions. All were approved by the Institutional Animal Care and Use Committee (IACUC) of Wenzhou Medical University (Wenzhou, China).
In Vivo Antitumor Effects
To establish the subcutaneous tumor model, HT29-Luc cells were injected in the right flanks of BLAB/c nude mice to enable stable expression of luciferase. Then the mice were divided into four groups randomly: saline, free 17AAG, NPs-17AAG, and HA-NPs-17AAG. Mice were treated intravenously with 5 mg/kg 17AAG1 every 3 days for 15 days. Luciferase expression in Luc-HT29 cells was used to evaluate tumor growth. At the end of the experiment, (D18 from Luc-HT29 cells subcutaneous injection), tumor volume and body weight of mice were recorded for further analysis. Tumor volume was calculated with the formula V = 0.5LW2, where V represents volume, L the long diameter of the tumor and W the short diameter. Mice were monitored every day during the experiment.
To evaluate antitumor effects of HA-NPs-17AAG, an orthotopic colon cancer model was established. Briefly, mice were first injected intraperitoneally with azoxymethane (AOM) and fed normally for 7 days. Then, two cycles of DSS treatment were administered to the mice. It should be noted that each DSS treatment contained 7 days’ DSS water (3%) treatment and 14 days’ recovery, as illustrated in Figure 2A. Finally, mice loaded with orthotopic colon tumors were randomly divided into four groups: controls (saline, free 17AAG, NPs-17AAG, and HA-NPs-17AAG. At the end of experiments, colon tumors and tumor loading were counted and compared.
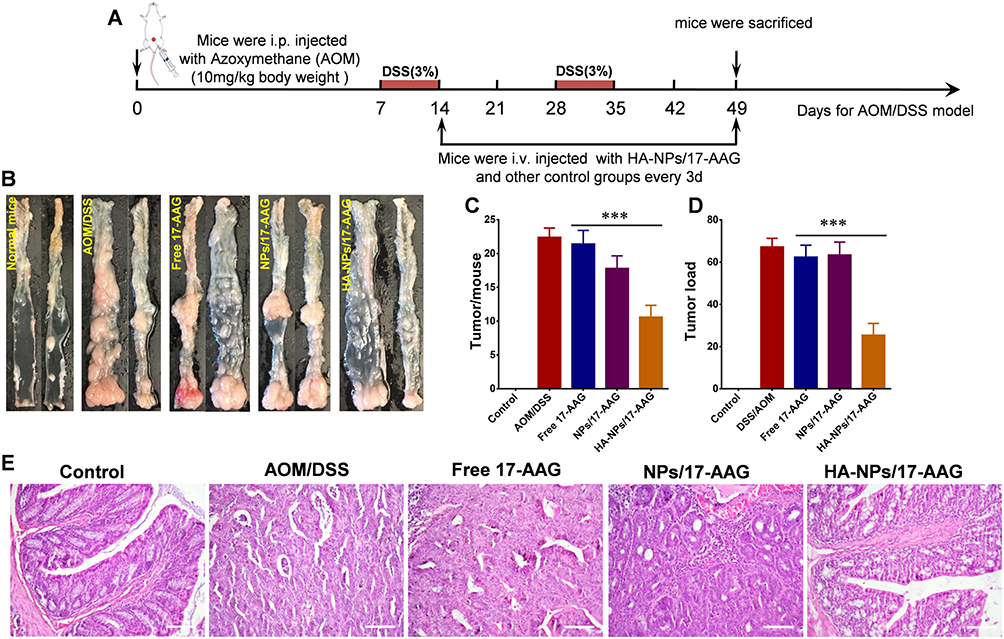 |
Figure 2 HA-NPs-17AAG showed antitumor effects in AOM/DSS-induced orthotopic tumor model. (A) Experimental design. (B) Typical tumor photos in the colon under different treatments. (C, D) Tumors/mouse and tumor load were determined (n=10). ***p<0.001. (E) H&E-stained colonic tumors. Bar 20 µm. |
H&E Staining and TUNEL Examination
H&E staining of heart, liver, spleen, lungs, kidneys, and tumors was performed according to standard procedures. Firstly, ultrathin sections (5 µm) were obtained using a paraffin microtome slicing machine, then stained with H&E. For TUNEL experiments, ultrathin sections of tumor tissue were stained with TUNEL reagent according to the manufacturer’s standard protocols, then nuclei were stained by DAPI. Histological and fluorescence images were captured by inverted microscopy via different channels.
Biocompatibility Assays
To compare the effects on cell viability of DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs in vitro, MTT assays were used. Briefly, the cell lines were seeded in plates overnight. Then DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs (10, 20, 50, 100, 200, 500 and 1,000 μg/mL) were added to the cell-culture medium for 1 and 2 days. The medium was then removed and MTT regent added to each well of the plates and cultured for 4 hours until purple precipitate. Finally, DMSO was added to dissolve blue formazan, and absorbance at 570 nm was measured for analysis. Cells without treatment were used as controls.
For colony-formation assays, cells were first incubated with NPs for more than a day, digested with EDTA, and around 500 continuously cultured for half a month. Cell media were changed regularly to maintain cell growth. Finally, cells were stained with glutaraldehyde (6%) and crystal violet (0.5%) for 1 hour. Colonies in the groups were calculated under microscopy.
Mice were administered HA-NPs or PBS (control) for 7 consecutive days and the biocompatibility of HA-NPs evaluated. At the end of the experiment, spleen/body weight was compared, cytokine-expression levels assessed with ELISA, and vital organs obtained and stained with H&E. Blood hemanalysis and biochemical analysis were performed using a hematology analyzer and a biochemical analyzer, respectively.
Statistical Analysis
Statistical significance was analyzed with ANOVAs and t-tests.
Results and Discussion
NP Preparation and Characterization
PLGA was used as the core of NPs for 17AAG delivery. As we know, PLGA has excellent properties, such as biodegradaility and biocompatibility. On account of these properties, PLGA has been widely studied as a delivery vehicle for drugs.26,27 A single emulsion (oil in water) method was adopted to prepare DOTAP-PLGA NPs using ultrasonic emulsification. DOTAP with positive charge was self-assembled on the surface of the PLGA core, bonding anionic HA via electrostatic interaction (Figure 1A). 17AAG is hydrophobic, and can be easily encapsulated into hydrophobic DOTAP-PLGA NPs. When the NPs interacted with CD44 receptors on cell surfaces, receptor-mediated endocytosis, endosomal escape, and drug release occurred in sequence (Figure 1B). TEM showed that both DOTAP-PLGA and HA-DOTAP-PLGA NPs had well-defined core–shell structures (Figure 3A, B [insert]). Particle size and ζ-potential of DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs were 149±1 nm, 46.43±2.97 mV and 232±3 nm, −15.60±1.55 mV, respectively (Figure 3C). These results indicated that by electrostatic interaction, HA had been successfully incorporated on the surface of DOTAP-PLGA NPs. HPLC revealed that drug loading of 17AAG in DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs was 42.7±3.2 μg/mg and 43.3±2.6 μg/mg and encapsulation efficiency 80%±2.3% and 82%±1.5%, respectively (Figure S1).
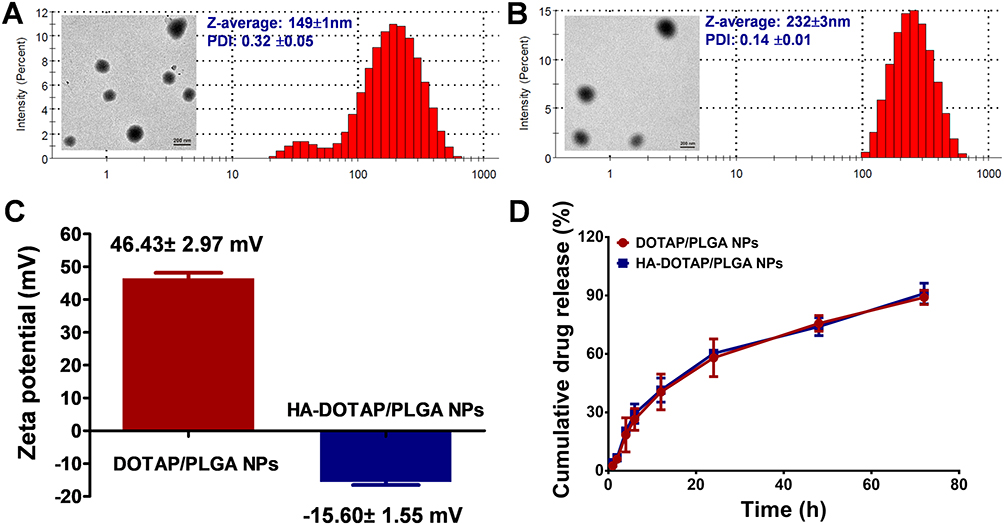 |
Figure 3 Material characterization. (A) TEM and DLS were used to characterize the morphology and size of DOTAP-PLGA NPs, respectively (n=3). (B) TEM and DLS were used to characterize the morphology and size of HA-DOTAP-PLGA NPs, respectively (n=3). (C) ζ-potentials of DOTAP-PLGA and HA-DOTAP-PLGA NPs (n=3). (D) Cumulative 17AAG-release profiles were evaluated with HPLC (n=3). |
Cumulative release of 17AAG from NPs was evaluated by HPLC. As shown in Figure 3D, 17AAG in both types of NPs revealed very similar drug-release profiles: during the initial 12 hours: around 40% of 17AAG was released, reaching percentage reach ~90% at 72 hours. These results indicated that 17AAG was released from the core of PLGA NPs with a sustained-release profile and achieved controlled release.
CD44 Receptor–Medicated Endocytosis Involved in Cellular Uptake
Achieving effective intracellular uptake is very important for the therapeutic efficacy of DDSs.28 We incubated HA-DOTAP-PLGA NPs (labeled with DiL) with two colon cancer cell lines to verify targeting ability. The cell-surface glycoprotein CD44, involving cell adhesion and migration, is the main receptor of HA. Cancer cells overexpressing CD44 receptors on the cell surface are responsible for the interaction of HA.29 HA-DOTAP-PLGA NPs were incubated with colon cancer cells at regular cell-culture temperature for 4 hours with or without HA (as competitor). We then evaluated the endocytosis of NPs by confocal microscopy, and HA-DOTAP-PLGA NPs showed strong DiL fluorescence (Figure 4A-a, C-a). In contrast, less fluorescence was observed for free HA (Figure 4A-b, C-b). These results confirmed that endocytosis of HA-DOTAP-PLGA NPs was mediated by CD44 receptors. Flow cytometry was then used to assess the cell-uptake efficiency of NPs labeled with DiL. Consistently with the confocal microscopy results, in HA-free groups HT29 (Figure 4B) and HCT116 cells (Figure 4D) had much higher fluorescence then the HA-present group. Through modification with HA, effective intracellular uptake of HA-DOTAP-PLGA NPs was achieved by HA-CD44–mediated internalization.
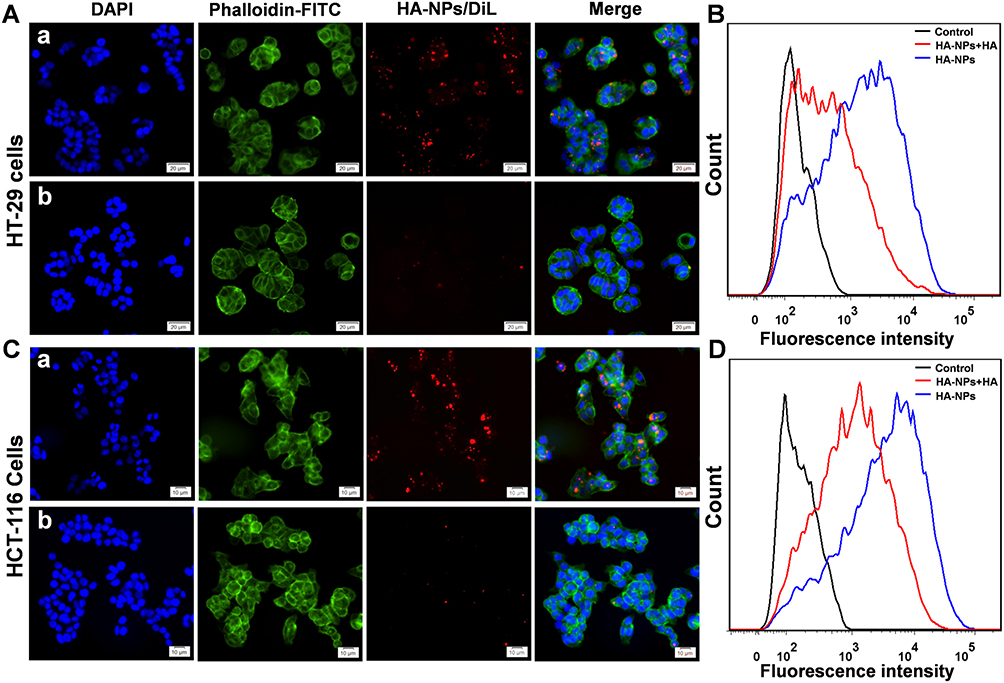 |
Figure 4 HA-DOTAP-PLGA NPs were taken up by colon cancer cells through HA-mediated endocytosis. (A) NPs incubated with HT29 cells: a, without competitor HA; b, with competitor HA. (B) NPs incubated with HT29 cells with or without competitor HA. Endocytosis efficiency was evaluated with FACS. (C) NPs incubated with HCT116 cells: a, without competitor HA; b, with competitor HA. (D) NPs incubated with HCT116 cells with or without competitor HA. Endocytosis efficiency was evaluated by FACS (n=5). |
Endocytosis is a cellular process in which large molecules or other cells enter the cell.With the help of clathrin, this process begins by caving in the cell membrane to form a vesicle that wraps around the material to get inside the cell. The Tf receptor is dimeric transmembrane glycoprotein that is internalized by clathrin-mediated endocytosis.30 We performed a colocalization experiment with HA-NPs/DiL and FITC-labeled Tf to further confirm HA-NPs/DiL internalization was receptor-mediated. As shown in Figure S2, HA-NPs/DiL and Tf-FITC showed a high degree of colocalization in HT29 cells based on fluorescence and FACS. CD44has been widely studied in cancer therapy due to its strong interaction with HA. As a natural ligand for CD44, HA has been used as an active targeting ligand by modifying the NP surface via chemical bonds or electrostatic interactions.There are two separate events in HA CD44–mediated internalization: binding of HA to CD44 and uptake of HA by CD44-mediated endocytosis. It has been confirmed that HA CD44–mediated endocytosis has a relationship of acylation of the CD44 cytoplasmic tail. More importantly, CD44 can also be recycled to the cell surface if the protein is not ubiquitinated. WCD44-mediated internalization of HA-DOTAP-PLGA NPs for colon cancer therapy is credible and has been widely reported.
We used flow cytometry to investigate the cellular uptake of DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs at different incubation times (Figure 5). At 1, 3, and 6 hours, both DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs showed increased internalization. With the same incubation periods, HA-DOTAP-PLGA NPs exhibited higher uptake efficiency than DOTAP-PLGA NPs, indicating receptor-medicated endocytosis had higher internalization efficiency. Our finding showed NPs decorated with HA can enhance cell-uptake efficiency, indicating HA is a distinguished target ligand.
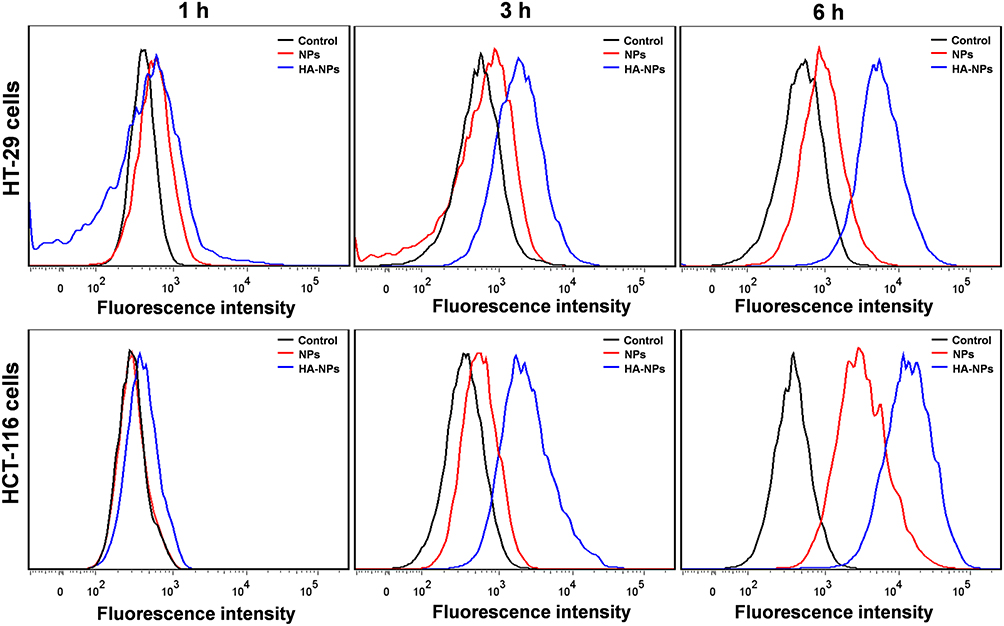 |
Figure 5 Uptake of NPs by colon cancer cells was time-dependent. DOTAP-PLGA and HA-DOTAP-PLGA NPs were coincubated with cells for designated periods (n=5). |
Biocompatibility of HA-DOTAP-PLGA NPs
For an ideal carrier, biocompatibility should be addressed for animal or clinical use.31 We used MTT assays to assess the biocompatibility of HA-DOTAP-PLGA NPs by quantifying the viability of cells treated with different concentrations of DOTAP-PLGA NPs and HA-DOTAP-PLGA NPs. Cells incubated with HA-DOTAP-PLGA NPs showed a similar cell-growth curve to those treated with the same concentrations of DOTAP-PLGA NPs, even at 1 mg/mL (Figure S3), indicating that HA modification did not change the biocompatibility of DOTAP-PLGA NPs. We further investigated the biocompatibility of HA-DOTAP-PLGA NPs using an extended cytotoxicity assay: the colony-formation assay.32 As shown in Figure S4, there was no significant change in colony formation ibetween the NP-treated and control groups in either cell line, indicating that HA-DOTAP-PLGA NPs had very high biocompatibility for cell-colony formation and did not alter the growth of the colon cancer cells we tested.
In vivo biocompatibility of HA-DOTAP-PLGA NPs was also explored. For mice administered HA-DOTAP-PLGA NPs for 7 days, HA-DOTAP-PLGA NP administration did not affectspleen/body weight (Figure S5A) or proinflammatory cytokines (TNFα, IL6, and IL1β; Figure S5B). On histological analysis, we found that there was no clear organ damage in the HA-DOTAP-PLGA NP group. The vital organs (heart, liver, spleen, lungs, and kidneys) were normal (Figure S5C). Along with as the hemanalysis and biochemical results, (Table S1), white blood cells, red blood cells, and hemoglobin were similar. Parameters reflecting hepatic and renal function, such as ALT, total bilirubin, total protein, and blood urea nitrogen, were also comparable. These results indicate that HA-DOTAP-PLGA NPs had very high biosecurity in vivo and did not affect of hepatic or renal function. Collectively, these results show that HA-DOTAP-PLGA NPs had acceptable biocompatibility both in vitro and in vivo and great potential as a safe platform for DD.
Enhanced Cell Apoptosis Caused by HA-NPs-17AAG
17AAG with high selectivity is used in clinical testing to treat different malignancies. 17‑AAG is able to inhibit cell proliferation, inducing apoptosis. We compared cell apoptosis induced by free 17AAG and HA-NPs-17AAG. Annexin V–FITC/PI experiments were utilized to test the two cell lines with different treatments. When cells undergo apoptosis, phosphatidylserine in the plasma membrane will translocate to the cell surface. In flow cytometry, annexin V, which has high affinity with phosphatidylserine, can be used to detect apoptotic cells.33 Fluorescence imaging showed free 17AAG- and HA-NPs-17AAG–treated HT29 cells were positively stained: both green and red, which represent live cells and apoptotic cells (Figure 6A). In contrast, there was no green or red signal in untreated control cells. Quantification analysis with flow cytometry showed PI and Aannexin V double-negative cell populations of 96.9%±2.1%, 31.6%±3.1%, and 14.1%±2.4% in HA-NPs-17AAG–treated, free 17AAG-treated, and control cells, respectively (Figure 6B and C). Similar results were obtained in HCT116 colon cancer cells, with PI and annexin V double-negative cell populations of 98.1%±1.4%, 27.7.2±4.0%, and 13.7%±1.5%, respectively (Figure S6). These fluorescence-imaging and flow-cytometry results demonstrated that HA-NPs-17AAG had outstanding therapeutic effects over free 17AAG. In addition, there were no side effects observed following intravenous injection of NPs. As such, HA-NPs-17AAG cansignificantly improve the antitumor activity of 17AAG and HA-DOTAP-PLGA NPs might be a good candidate for DD.
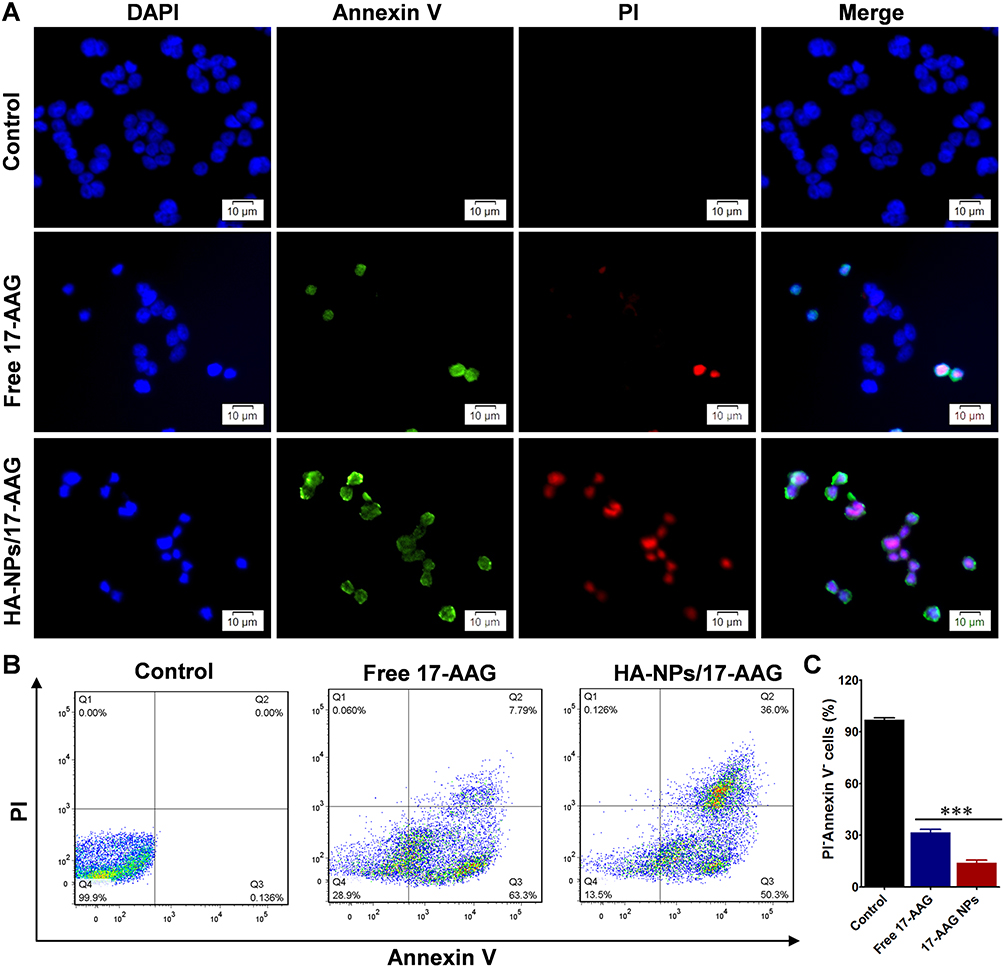 |
Figure 6 Apoptosis in HT29 cells was evaluated by fluorescence and FACS. (A) HT29 cells receiving different treatments, then stained by annexin V–FTIC/PI. Bar 10 μm. (B) Apoptosis was evaluated with FACS. (C) Quantitative analysis of FACS results from B. ***p<0.001 (n=5). |
HA-NPs-17AAG Exhibit Antitumor Effects in Subcutaneous Tumor Model
To evaluate the antitumor effects of HA-NPs-17AAG in vivo, immunodeficient mice bearing Luc-HT29 cells were used as a subcutaneous tumor model, as shown in Figure 7A. When tumor reched appropriate size, the four groups were divided and then administered saline, free 17AAG, NPs-17AAG, oand HA-NPs-17AAG, respectively. As shown in Figure 7B and, bioluminescence imaging of luciferase was used to monitor the growth of HT29 cells. Luciferase imaging in vivo is a molecular and gene-expression analysis and detection system developed in recent years that can be used to observe the growth and metastasis of cancer cells in living animals, the development of infectious diseases, the expression of specific genes, and other biological processes. Luciferase imaging of HT29 cells showed tha
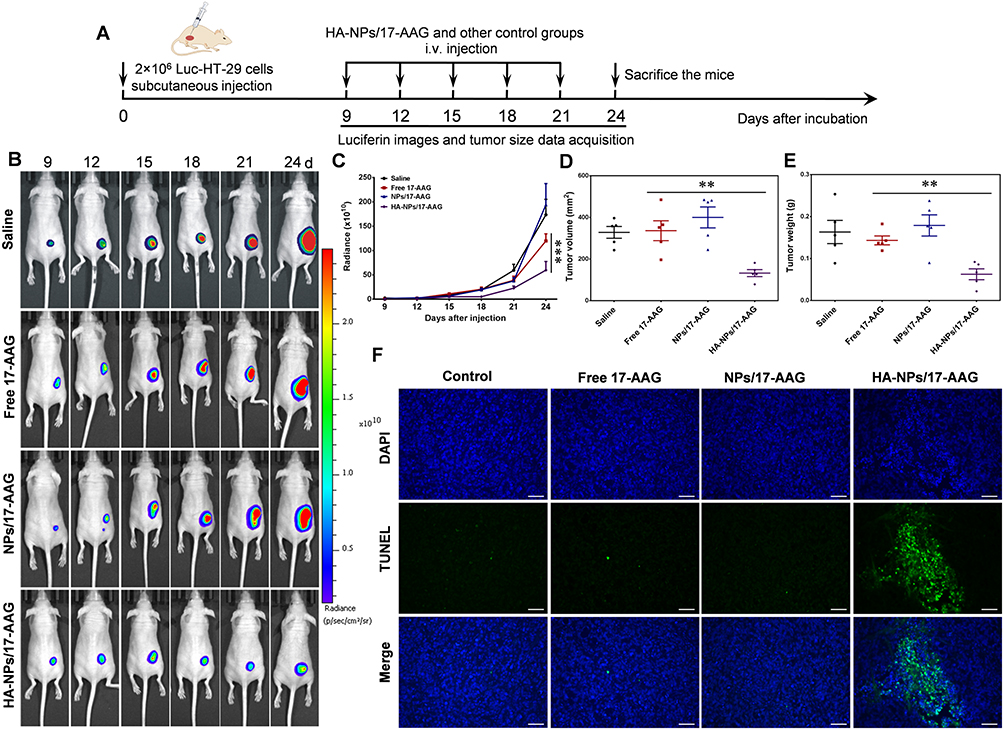 |
Figure 7 HA-NPs-17AAG had in vivo therapeutic efficacy in Luc-HT29 tumor-bearing mice. (A) Experimental design. (B) With administration of luciferin substrate, bioluminescence images of mice receiving different treatments for various periods were captured. (C) Tumor-growth curve in mice treated with HA-NPs-17AAG or control formulations. ***p<0.001. (D) Tumor volumes were measured and compared (n=5). **p<0.01. (E) Tumor weights were measured and compared (n=5). **p<0.01. (F) TUNEL assays were performed to assess cell apoptosis (n=5). Bar 20 μm. |
The HA-NPs-17AAG- group had the lowest tumor growth among the groups. When tumor volumes and weights were examined at the end of the experiment, the HA-NPs-17AAG group showed the lowest tumor volume and weight, and those values were remarkablly reduced when compared with the free 17AAG, saline, and NPs-17AAG control groups (Figure 7D and E, Figure S7). Consistently with in vitro apoptosis assays, our findings demonstrate that 17AAG loaded in NPs had better antitumor activity than 17AAG alone. As we know, free 17AAG can diffuse quickly into organs or tissue with intravenous administration,34 which will potentially affect healthy cells, and at the same time the amount of free drug delivered to the tumor site will decrease significantly. Free drugs usually have a short half-life in vivo and will be cleared from the body quickly; therefore, 17AAG loaded in NPs showed better antitumor activity than 17AAG alone. We did TUNEL apoptosis assays to explore this mechanism.TUNEL is a method of detecting DNA fragmentation by labeling blunt ends of double-stranded DNA. In Figure 7F, the saline, free 17AAG, and NPs-17AAG groups show few green dots, representing TUNEL-positive apoptotic cells. Significantly, in the HA-NPs-17AAG group, there are more green dots, representing severe apoptosis in this group. NPs-17AAG inhibited subcutaneous tumor growth by inducing tumor-cell apoptosis. We also performed histological analysis of tumor tissues to further investigate the enhanced antitumor activity of HA-NPs-17AAG. H&E staining indicated that large areas of tumor-cell necrosis were obtained in the HA-NPs-17AAG group when compared with control groups, which showed malignant tumor-cell hyperplasia (Figure 8). Treatment with HA-NPs-17AAG, inhibited the growth of colon cancer, suggesting HA-NPs-17AAG has a distinguished antitumor effect.
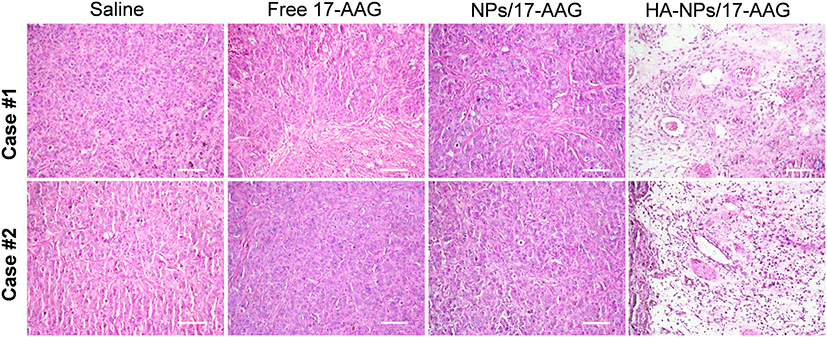 |
Figure 8 Evaluation of therapeutic efficacy of HA-NPs-17AAG by histological analysis. Bar 20 μm. |
HA-NPs-17AAG Also Exhibit Antitumor Effects in Orthotopic Tumor Model
The translatability of animal models is one of the greatest challenges for oncology studies at the preclinical stage. Xenograft tumor models are the most frequently used systems for the evaluation of in vivo efficacy of anticancer-drug candidates; however, these tumor models cannot mimic the actual tumor microenvironment.35 Therefore, more reliable orthotopic models are ideally suited to test the anticancer efficacy of drug candidates.36,37 The antitumor effects of HA-NPs-17AAG in the orthotopic tumor model caused by the combination of the carcinogenic agent AOM and an inflammatory agent (DSS) were then evaluated. As shown in Figure 2A, mice were treated with HA-NPs-17AAG or controls from D14-D49, then, killed for further analysis. As shown in Figure 2B, tumors appeared in the colon from the middle to distal portions, and it was obvious that the HA-NPs-17AAG group had fewer orthotopic tumors than the free 17AAG, saline, and NPs-17AAG groups. Tumors/mouse and tumor loading were markedly decreased by HA-NPs-17AAG treatment of AOM/DSS mice (Figure 2C and D). H&E staining indicated that AOM/DSS-treated mice had large flat adenomas, indicating high-grade dysplasia and carcinoma. In contrast, mice exhibited very low-grade, nonmalignant dysplastic lesions in the HA-NPs-17AAG group (Figure 2E). According to our previous research, apoptosis of intestinal epithelial cells plays a prominent but complicated role in colitis-associated cancer. We thereby evaluated intestinal epithelial cell apoptosis in this tumor model using TUNEL assays. As shown in Figure S8, the greatest degree of apoptosis was achieved in the HA-NPs-17AAG group, indicating a significant antitumor effect of HA-NPs-17AAG. Our study indicated that administration of HA-NPs-17AAG significantly suppressed colitis-associated cancer and might serve as a good DDS for colon cancer therapy.
Conclusion
Colon cancer is highly prevalent globally. In recent years, significant progress has been achieved in controlling its morbidity and mortality. However, colon cancer is still the fourth-leading cause of cancer-related deaths worldwide and the third–most commonly diagnosed malignancy.
Therefore, we still have a long way to go to control colon cancer. Promisingly, NP-based DDSs bring hope for colon cancer therapy. NPs decorated with targeted ligands that can recognize cell-surface receptors provide effective release of cytotoxic drugs.
In our research, single-emulsion (oil in water) solvent evaporation was used to prepare cationic lipid (DOTAP)–PLGA hybrid NPs to load the HSP90 inhibitor 17AAG. Then, NPs were decorated with HA with electrostatic interaction (HA-NPs-17AAG). Taking advantage of HA-coated cationic NPs with HA-CD44 receptor–medicated endocytosis and endosomal escape capacity, we investigated their potential efficacy in treating subcutaneous and orthotopic colon cancer (Figure 1).
An efficient HSP90 inhibitor, 17AAG is being studied widely in the treatment of cancers, as a single agent or combined with other anticancer drugs, in preclinical and clinical research.
However, such problems as degradation in the circulatory system and being off-target have limited its further use in the clinic. In this study, PLGA was used to load 17AAG to form HA-NPs-17AAG. PLGA is often used for DD, since it has been approved by the US Food and Drug Administration (FDA). In contrast to other types of DSS, PLGA has significant merits, eg, tailored biodegradation rate, FDA-approved for clinical use, biocompatibility, and great handling ability of the material surface. Because of these excellent properties, PLGA as a DDS has great potential for clinical use.
In summary, we constructed HA-decorated DOTAP–PLGA hybrid NPs that served as a new DDS to enhance therapeutic efficiency (HA-DOTAP-PLGA NPs-17AAG). Colon cancer cells, both HT29 and HCT116, took up HA-DOTAP-PLGA NPs via HA–CD44 interaction. HA-DOTAP-PLGA NPs-17AAG had greater efficiency for cell apoptosis and had better antitumor results in subcutaneous and orthotopic tumor models, indicating that HA-DOTAP-PLGA NPs could be explored as a novel DDS and have great potential in colon cancer therapy.
Ethics Approval
This research was approved by the Institutional Animal Care and Use Committee (IACUC) of Wenzhou Medical University (SYSK-2015-0009). All were performed according to accepted standards of the Guide for the Care and Use of Laboratory Animals.
Acknowledgments
This work was supported by grants from the Zhejiang Provincial Natural Science Foundation of China (LY20H180010), Wenzhou Science and Technology Bureau (Y20180142), and the Mega-Project for National Science and Technology Development under the 13th Five-Year Plan of China (2017ZX10105001-003)
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fortina P, Kricka LJ, Graves DJ, et al. Applications of nanoparticles to diagnostics and therapeutics in colorectal cancer. Trends Biotechnol. 2007;25(4):145. doi:10.1016/j.tibtech.2007.02.005
2. Zhang M, Yang C, Yan X, Sung J, Garg P, Merlin D. Highly biocompatible functionalized layer-by-layer ginger lipid nano vectors targeting p-selectin for delivery of doxorubicin to treat colon cancer. Adv Therapeutics. 2019;2(12):1900129. doi:10.1002/adtp.201900129
3. Hassan S, Prakash G, Ozturk AB, et al. Evolution and clinical translation of drug delivery nanomaterials. Nano Today. 2017;15:91. doi:10.1016/j.nantod.2017.06.008
4. Xu JQ, Zhang YL, Xu JC, et al. A biodegradable MnSiO3@ Fe3O4 nanoplatform for dual-mode magnetic resonance imaging guided combinatorial cancer therapy. Biomaterials. 2019;216.
5. Zhang MZ, Xu CL, Wen LQ, et al. A hyaluronidase-responsive nanoparticle-based drug delivery system for targeting colon cancer cells. Cancer Res. 2016;76(24):7208. doi:10.1158/0008-5472.CAN-16-1681
6. An J, Hu YG, Li C, et al. A pH/ultrasound dual-response biomimetic nanoplatform for nitric oxide gas-sonodynamic combined therapy and repeated ultrasound for relieving hypoxia. Biomaterials. 2020;230.
7. Zheng B, Chen L, Pan CC, et al. Targeted delivery of miRNA-204-5p by PEGylated polymer nanoparticles for colon cancer therapy. Nanomed Uk. 2018;13(7):769. doi:10.2217/nnm-2017-0345
8. Liu T, Wang C, Gu X, et al. Drug Delivery with PEGylated MoS2 Nano-sheets for Combined Photothermal and Chemotherapy of Cancer. Adv Mater. 2014;26(21):3433. doi:10.1002/adma.201305256
9. Ali A, Ahmed S. A review on chitosan and its nanocomposites in drug delivery. Int J Biol Macromol. 2018;109:273. doi:10.1016/j.ijbiomac.2017.12.078
10. Yang M, Zhang F, Yang C, et al. Oral Targeted Delivery by Nanoparticles Enhances Efficacy of an Hsp90 Inhibitor by Reducing Systemic Exposure in Murine Models of Colitis and Colitis-Associated Cancer. J Crohns Colitis. 2020;14(1):130. doi:10.1093/ecco-jcc/jjz113
11. Kobayashi H, Watanabe R, Choyke PL. Improving Conventional Enhanced Permeability and Retention (EPR) Effects; What Is the Appropriate Target? Theranostics. 2013;4(1):81. doi:10.7150/thno.7193
12. Kalyane D, Raval N, Maheshwari R, Tambe V, Kalia K, Tekade RK. Employment of enhanced permeability and retention effect (EPR): nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater Sci Eng C Mater Biol Appl. 2019;98:1252. doi:10.1016/j.msec.2019.01.066
13. Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135. doi:10.1016/j.jconrel.2010.08.027
14. Chen SQ, Song YQ, Wang C, et al. Chitosan-modified lipid nanodrug delivery system for the targeted and responsive treatment of ulcerative colitis. Carbohydr Polym. 2020;230:115613. doi:10.1016/j.carbpol.2019.115613
15. Marcandalli J, Fiala B, Ols S, et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell. 2019;176(6):1420. doi:10.1016/j.cell.2019.01.046
16. Yu KK, Li K, Lu CY, et al. Multifunctional gold nanoparticles as smart nanovehicles with enhanced tumour-targeting abilities for intracellular pH mapping and in vivo MR/fluorescence imaging. Nanoscale. 2020;12(3):2002. doi:10.1039/C9NR06347A
17. Xiao ZY, Levy-Nissenbaum E, Alexis F, et al. Engineering of Targeted Nanoparticles for Cancer Therapy Using Internalizing Aptamers Isolated by Cell-Uptake Selection. ACS Nano. 2012;6(1):696. doi:10.1021/nn204165v
18. Farokhzad OC, Cheng JJ, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. P Natl Acad Sci USA. 2006;103(16):6315. doi:10.1073/pnas.0601755103
19. Liang XY, Li XL, Duan JW, et al. Nanoparticles with CD44 Targeting and ROS triggering properties as effective in vivo antigen delivery system. Mol Pharmaceut. 2018;15(2):508. doi:10.1021/acs.molpharmaceut.7b00890
20. Yoo J, Rejinold NS, Lee D, et al. CD44-Mediated Methotrexate Delivery by Hyaluronan-Coated Nanoparticles Composed of a Branched Cell-Penetrating Peptide. ACS Biomater Sci Eng. 2020;6(1):494. doi:10.1021/acsbiomaterials.9b01724
21. Byeon Y, Lee JW, Choi WS, et al. CD44-targeting PLGA nanoparticles incorporating paclitaxel and FAK siRNA overcome chemoresistance in epithelial ovarian cancer. Cancer Res. 2018;78:6247.
22. Collins CB, Strassheim D, Aherne CM, Yeckes AR, Jedlicka P, de Zoeten EF. Targeted Inhibition of Heat Shock Protein 90 Suppresses Tumor Necrosis Factor–α and Ameliorates Murine Intestinal Inflammation. Inflamm Bowel Dis. 2014;20(4):685. doi:10.1097/01.MIB.0000442839.28664.75
23. Dudeja V, Vickers SM, Saluja AK. The role of heat shock proteins in gastrointestinal diseases. Gut. 2009;58(7):1000. doi:10.1136/gut.2007.140194
24. Collins CB, Aherne CM, Yeckes A, et al. Inhibition of N-terminal ATPase on HSP90 attenuates colitis through enhanced Treg function. Mucosal Immunol. 2013;6(5):960. doi:10.1038/mi.2012.134
25. Devulapally R, Sekar NM, Sekar TV, et al. Polymer Nanoparticles Mediated Codelivery of AntimiR-10b and AntimiR-21 for Achieving Triple Negative Breast Cancer Therapy. ACS Nano. 2015;9(3):2290. doi:10.1021/nn507465d
26. Mir M, Ahmed N, Rehman AU. Recent applications of PLGA based nanostructures in drug delivery. Colloid Surface B. 2017;159:217. doi:10.1016/j.colsurfb.2017.07.038
27. Donahue ND, Acar H, Wilhelm S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv Drug Deliver Rev. 2019;143:68.
28. Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol. 2015;6.
29. Puri V, Watanabe R, Singh RD, et al. Clathrin-dependent and -independent internalization of plasma membrane sphingolipids initiates two Golgi targeting pathways. J Cell Biol. 2001;154(3):535. doi:10.1083/jcb.200102084
30. Liu D, Yang F, Xiong F, Gu N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics. 2016;6(9):1306. doi:10.7150/thno.14858
31. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315. doi:10.1038/nprot.2006.339
32. Kumar G, Degheidy H, Casey BJ, Goering PL. Flow cytometry evaluation of in vitro cellular necrosis and apoptosis induced by silver nanoparticles. Food Chem Toxicol. 2015;85:45. doi:10.1016/j.fct.2015.06.012
33. Mellatyar H, Talaei S, Pilehvar-Soltanahmadi Y, et al. Targeted cancer therapy through 17-DMAG as an Hsp90 inhibitor: overview and current state of the art. Biomed Pharmacother. 2018;102:608. doi:10.1016/j.biopha.2018.03.102
34. Scholz A, Detjen K, Schulz P, et al. Development of orthotopic mouse models of pancreatic cancer, hepatocellular carcinoma and neuroendocrine tumors for preclinical evaluation of new substances. Ann Oncol. 2007;18:37.
35. Chu PY, Tsai SC, Ko HY, Wu CC, Lin YH. Co-Delivery of Natural Compounds with a Dual-Targeted Nanoparticle Delivery System for Improving Synergistic Therapy in an Orthotopic Tumor Model. ACS Appl Mater Inter. 2019;11(27):23880. doi:10.1021/acsami.9b06155
36. Shen Z, Liu T, Yang Z, et al. Small-sized gadolinium oxide based nanoparticles for high-efficiency theranostics of orthotopic glioblastoma. Biomaterials. 2020;235:119783. doi:10.1016/j.biomaterials.2020.119783
37. Zhang M, Viennois E, Rrasad M, Zhang Y, Wang L, Zhang Z. Edible ginger-derived nanoparticles: a novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials. 2016;101:321–340. doi:10.1016/j.biomaterials.2016.06.018
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.