Back to Journals » International Journal of Nanomedicine » Volume 17
Hybrid Cell Membrane-Functionalized Biomimetic Nanoparticles for Targeted Therapy of Osteosarcoma
Authors Cai J, Liu J, Wu J, Li Y ![]() , Qiu X, Xu W, Xu P, Xiang D
, Qiu X, Xu W, Xu P, Xiang D
Received 28 October 2021
Accepted for publication 1 February 2022
Published 22 February 2022 Volume 2022:17 Pages 837—854
DOI https://doi.org/10.2147/IJN.S346685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Farooq A. Shiekh
Jia-Xin Cai,1– 3,* Ji-Hua Liu,1– 3,* Jun-Yong Wu,1– 3 Yong-Jiang Li,1– 3 Xiao-Han Qiu,1– 3 Wen-Jie Xu,1– 3 Ping Xu,1,2 Da-Xiong Xiang1– 3
1Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, 410011, Hunan, People’s Republic of China; 2Institute of Clinical Pharmacy, Central South University, Changsha, 410011, Hunan, People’s Republic of China; 3Hunan Provincial Engineering Research Center of Translational Medicine and Innovative Drug, Changsha, Hunan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ping Xu; Da-Xiong Xiang, Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, 410011, Hunan, People’s Republic of China, Email [email protected]; [email protected]
Purpose: In order to prepare a biomimetic nano-carrier which has inflammatory chemotaxis, homologous targeting and reduce immune clearance, for targeted chemotherapy of osteosarcoma, we fabricated the paclitaxel-loaded poly(lactic-co-glycolic) acid (PLGA) nanoparticles coated with 143B-RAW hybrid membrane (PTX-PLGA@[143B-RAW] NPs) and evaluate its anti-cancer efficacy in vitro and vivo.
Methods: PTX-PLGA@[143B-RAW] NPs were prepared by the ultrasonic method and were characterized by size, zeta potential, polymer dispersion index (PDI), Coomassie bright blue staining, transmission electron microscopy (TEM) and high performance liquid chromatography (HPLC). Cellular uptake, cell viability assay, flow cytometry and chemotactic effect of PTX-PLGA@[143B-RAW] NPs were evaluated in vitro. Biodistribution, anti-cancer therapeutic efficacy and safety of PTX-PLGA@[143B-RAW] NPs were evaluated in 143B osteosarcoma xenograft mice.
Results: The hybrid membrane successfully coated onto the surface of PLGA nanoparticles. PTX-PLGA@[143B-RAW] NPs had a drug loading capacity of 4.24 ± 0.02% and showed targeting ability to osteosarcoma. PTX-PLGA@[143B-RAW] NPs showed high cellular uptake and improved anti-cancer efficacy against 143B cells. More importantly, PTX-PLGA@[143B-RAW] NPs treatment suppressed tumor growth in tumor-bearing mice with minimal damage to normal tissues.
Conclusion: PTX-PLGA@[143B-RAW] NPs could be used for targeted drug delivery and osteosarcoma therapy.
Keywords: biomimetic nano-drug delivery system, osteosarcoma, paclitaxel, targeted therapy
Introduction
Osteosarcoma is a primary malignant tumor of the bone, which spreads in blood and is highly metastatic and have high risk of relapse.1 With the development of neoadjuvant chemotherapy and surgical techniques, amputation therapy, the treatment osteosarcoma has been gradually replaced by chemotherapy for limb salvage therapy.2 However, the high local aggressiveness and the potential for rapid metastasis present challenges for the treatment of osteosarcoma. For patients with osteosarcoma who are not suitable for surgical resection, stereotactic radiotherapy, radiofrequency ablation, or cryotherapy may be an alternative,3 but it still lacks an effective way for osteosarcoma targeting therapy.
In recent years, nano-drug delivery systems have been extensively studied in tumor targeting therapy because of their potential to enhance and maintain the clinical efficacy of chemotherapeutic drugs with low side effects. However, it still has drawbacks, such as being removed by the reticulo-endothelial system (RES) and low diffusion efficiency to tumor cells.4,5 It has been found that the surface properties of nanoparticles can be easily modified with membranes from different types of cells or biological vesicles to improve their biocompatibility and targeting efficiency. Membrane-coated nanomedicines can effectively avoid the surveillance of the immune system, prolong the blood circulation of drug, and increase the drug accumulation in the tumor or inflammatory sites. For example, CD47 is a special protein with immune escape signals, which is highly expressed on the membrane surface of red blood cells (RBCs).6 Nanoparticle coating with the RBC membrane can avoid being recognized by immune cells.7 Therefore, cell membrane-based nanoplatforms have a promising clinical application prospect for tumor therapy.8
The tumor microenvironment (TME) of osteosarcoma comprises various types of stromal cells, which secrete humoral factors such as inflammatory cytokines to establish an inflammatory environment.9 As an important immune cell and antigen presenting cell, tumor-associated macrophage (TAM), as the largest immune cell in TME, can internalize large particles, such as fragments, apoptotic cells and pathogens, to maintain homeostasis in human body.10 Due to its ability to respond to cytokine signals released by inflammatory cells, it has inflammatory chemotaxis.11 Therefore, macrophages not only have a high degree of infiltration in the tumor, but also can specifically bind to the tumor tissue. Macrophage membranes have also been further developed for use in cancer therapy. In addition, it has been reported that nanoparticles coated with macrophage cell membranes generally have more prolonged blood circulation, significantly improving tumor therapy.12,13
Cell membranes extracted from tumor cells can express “self-markers” and “self-identifying molecules” that can be used to coat various nanoparticles and provide homologous targeting.14 In addition, tumor membrane-coated nanoparticles showed binding ability and selective uptake in homologous tumor cells, resulting in reduced immune clearance after systematic administration compared with uncoated nanoparticles.15 The unique biological functions displayed by tumor cell membranes have tremendous clinical potential for enhanced drug delivery, local light therapy, enhanced imaging, or more effective immunotherapy. Hybrid membrane combine the advantages of multiple cell membranes. The research showed that the targeting ability of hybrid membrane coated nanoparticles was enhanced and the circulation time is prolonged due to the tumor cell membrane and erythrocyte membrane fusion.16
In this study, we prepared PLGA nanoparticles coated with hybrid membrane from the osteosarcoma cell membrane and macrophage cell membrane as PLGA@[143B-RAW] NPs loaded with paclitaxel (PTX) for targeted chemotherapy of osteosarcoma (Figure 1A).
 |
Figure 1 Preparation and characterization of PLGA@[143B-RAW] NPs. (A) Preparation of paclitaxel (PTX)-loaded PLGA nanoparticles with 143B-RAW hybrid membrane coating (PTX-PLGA@[143B-RAW] NPs). (B) Membrane extraction process by gradient centrifugation. (C) The particle size and zeta potential of PLGA nanoparticles. (D) The particle size of the membrane-coated nanoparticles prepared by the two methods. (I) co-extrusion; (II) ultrasonication. (E) TEM images of (I) bare nanoparticles and (II) hybrid membrane coated nanoparticles. (F) Protein distribution of cell membrane. (I) PLGA nanoparticles; (II) 143B cell membrane; (III) RAW264.7 cell membrane; (IV) Hybrid membrane; (V) PLGA@143B NPs; (VI) PLGA@RAW NPs; (VII) PLGA@[143B-RAW] NPs. (G) Comparison of protein distribution of cell membrane coated nanoparticles prepared by different methods. (I) PLGA nanoparticles; (II) PLGA@143B NPs by co-extrusion; (III) PLGA@RAW NPs by co-extrusion; (IV) PLGA@[143B-RAW] NPs by co-extrusion; (V) PLGA@143B NPs by ultrasonication; (VI) PLGA@RAW NPs by ultrasonication; (VII) PLGA@[143B-RAW] NPs by ultrasonication. (H) Average size of membrane coated nanoparticles over 8 days in PBS. (I) PLGA@143B NPs; (II) PLGA@RAW NPs; (III) PLGA@[143B-RAW] NPs. (I) Western blot analysis of cell membrane proteins. (I) PLGA nanoparticles; (II) 143B cell membrane; (III) RAW264.7 cell membrane; (IV) Hybrid membrane; (V) PLGA@143B NPs; (VI) PLGA@RAW NPs; (VII) PLGA@[143B-RAW] NPs. Characteristic 143B membrane marker was MMP-2, and characteristic RAW264.7 membrane marker was integrin α4. (Na+-K+-ATPase was used as a reference protein) (J) PTX release from (I) PLGA NPs at pH 7.4; (II) PLGA@[143B-RAW] NPs at pH 7.4; (III) PLGA NPs at pH 5.3; (IV) PLGA@[143B-RAW] NPs at pH 5.3. |
Method
Materials
PTX was purchased from Shanghai Yuanye Bio-Technology Corporation. PLGA was purchased from Sigma-Aldrich, USA. Polyvinyl alcohol was obtained from Aladdin Industrial Corporation, USA. Acetone was obtained from Sinopharm Group, China. Acetonitrile and methanol (HPLC grade) were purchased from Tedia Company, USA. All other chemicals and reagents employed in this study were of analytical grade.
Cell Culture
The human osteosarcoma cell line 143B was obtained from the Cell Bank of the Beina Chuanglian Company (BNCC, Beijing, China). 143B cells were maintained in RPMI 1640 (Thermo Fisher Scientific, USA) supplemented with 10% FBS (fetal bovine serum, Thermo Fisher Scientific, USA) and 1% penicillin-streptomycin (Thermo Fisher Scientific, USA). Mice monocyte-macrophage cell line RAW264.7 was also obtained from the Cell Bank of the Beina Chuanglian Company (BNCC, Beijing, China). DMEM (Thermo Fisher Scientific, USA) supplemented with 10% FBS (fetal bovine serum, Thermo Fisher Scientific, USA) and 1% penicillin-streptomycin (Thermo Fisher Scientific, USA) was used to culture macrophages. The passage number of 143B and RAW264.7 are both 4–5. Endothelial cell line HUVEC was obtained from American Type Culture Collection (ATCC, USA). And the culture medium was the same as that of RAW264.7 cell. All types of cells were cultured at 37°C with 5% CO2.
Cell Membrane Extraction
Isolation buffer 1 (IB-1) and isolation buffer 2 (IB-2) were prepared according to the protocol.17 143B cells were trypsinized for 1 min by trypsin (NCM Biotech, China) according to the product introduction. And RAW264.7 cells were blowing down from culture dish by pipette according to the cell product introduction. Both types of cells were centrifuged and suspended in an appropriate amount of cold separation buffer IB-1 at 4 °C, and a protease inhibitors cocktail (NCM Biotech, China) was added. Then, the samples were ultrasonically crushed with an ultrasonic cell crushing machine (100 W, 20s/30s) for 14 minutes, and the mixture was centrifuged at 800 × g at 4°C for 10 minutes to remove the unbroken cells and nuclei. The supernatant was centrifuged at 10,000 × g at 4°C for 10 min and the mitochondria were removed by discarding particles. The supernatant of 100,000 × g was ultracentrifuged at 4°C for 1 hour to obtain membrane precipitation. Finally, it was suspended in 500 μL IB-2, lyophilized and stored at −80°C for later use.
Preparation and Characterization of PTX-PLGA@[143B-RAW] NPs
The nanoparticle was prepared by nano-precipitation method.18 A proper amount of PLGA and PTX were dissolved in a small amount of acetone with a mass ratio of 10:1 to form the organic phase, and a certain amount of polyvinyl alcohol (PVA) was dissolved in water to prepare 0.25% PVA solution to form the aqueous phase. The organic phase was slowly injected into the water phase under stirring (1200 r/min), and the volume ratio of the organic phase to the water phase was 1:10. The liquid was transferred to a 10 kDA ultrafiltration centrifuge tube and centrifuged at 2500 × g for 30 minutes to remove PVA and some water-soluble small molecular impurities. The precipitation of nanoparticles was resuspended into pure water or PBS to obtain PLGA-PTX nanoparticles with blue opalescence, which was stored at 4°C for future use.
PTX-PLGA@[143B-RAW] NPs were prepared by co-extrusion. The solution of 143B cell membrane was added to the solution of RAW264.7 cell membrane, of which the membrane protein concentration ratio was 1:1. The hybrid membrane was obtained by extruding over 400 nm polycarbonate membrane 21 times at 37°C. PLGA-PTX nanoparticles were added to the hybrid membrane solution and coextruded over the 400 nm polycarbonate membrane for 21 times to obtain PTX-PLGA@[143B-RAW] NPs, and then stored at 4°C. Meanwhile, the above method was used to prepare PTX-loaded PLGA NPs coated with 143B membrane (PTX-PLGA@143B NPs) and PTX-loaded PLGA NPs coated with RAW264.7 membrane (PTX-PLGA@RAW NPs).
PTX-PLGA@[143B-RAW] NPs were prepared by ultrasonic method. The solution of 143B cell membrane was added to the solution of RAW264.7 cell membrane with ultrasonic treatment at 37°C for 10 minutes, of which the membrane protein concentration ratio was 1:1. Then PLGA-PTX nanoparticles were added to the hybrid membrane solution, with ultrasonic treatment at 37°C for 2 minutes to coat hybrid membrane on the surface of the nanoparticles. The mixed solution was centrifuged at 10,000 × g for 5 minutes to remove the excess membrane, and PTX-PLGA@[143B-RAW] NPs, was obtained, stored at 4°C for reserve. The above ultrasonic method was also used to prepare PTX-PLGA@143B NPs and PTX-PLGA@RAW NPs.
The particle size and zeta potential of the nanoparticles coated by the hybrid film were measured by the Dynamic Light Scattering (DLS). The morphology of the nanoparticles in the hybrid film was investigated by transmission electron microscopy (TEM, 2100F, JEOL Ltd., Japan). Finally, the membrane proteins were investigated by polyacrylamide gel electrophoresis (SDS-PAGE) (Bosterbio, USA) with 5% stacking gel and 12% separating gel and Coomassie brilliant blue (Beyotime Biotech Corporation, Shanghai, China) was used to stain in order to observe the protein distribution. Specific protein markers were verified by Western blot analysis and Na+-K+-ATPase (Abcam, USA) was used as a reference protein. After transferring the proteins to a nitrocellulose membrane, the membranes were probed with antibodies against matrix metalloproteinase 2 (MMP-2) (EPR1184; Abcam, USA) and integrin alpha-4 (EPR1355Y; Abcam, USA). Anti-rabbit IgG (ZB-5301; ZSGB-BIO, China) were conjugated with horseradish peroxidase for signal visualization.
The Release of PTX-PLGA@[143B-RAW] NPs
Tumor microenvironment is an acidic environment with pH about to 5.3. To simulate physiological pH or tumor microenvironment, drug release was conducted using phosphate buffered saline (PBS) solutions containing 0.5% Tween-20 at pH 7.4 and 5.3, respectively. In short, free PTX, PLGA-PTX, PTX-PLGA@[143B-RAW] NPs 500 μL were added to the upper chamber of each ultrafiltration tube (10 kDa), and corresponding release buffer was added to the lower chamber to meet the leakage tank conditions (Vup /V down>3). The ultrafiltration tube was placed in a constant temperature water bath at 37°C. The concentration of PTX was measured by HPLC with a certain amount of lower chamber liquid at a fixed time point, and fresh PBS was added after sampling.
Cell Uptake Assay
PLGA nanoparticles were labeled with Dil dye (Yeasen Biotech Corporation, Shanghai, China). The 143B tumor cells were inoculated in 24-well plates. The cells were cultured overnight in a cell incubator for adherent treatment. The four groups PLGA NPs, PLGA@143B NPs, PLGA@RAW NPs and PLGA@[143B-RAW] NPs with Dil labeled were added to each well, respectively, and incubated in the cell incubator for specific time. After reaching the expected time point, DAPI staining (Yuheng Biotechnology Corporation, Suzhou, China) was performed according to the instructions, and then observed under a fluorescence microscope.
Cell Viability Assay
143B cells were evenly planted on a 96-well cell culture plate at a density of 5×104 cells per well. After growing for 24 h, PLGA-PTX NPs, PTX-PLGA@143B NPs, PTX-PLGA@RAW NPs and PTX-PLGA@[143B-RAW] NPs were added to the 96-well plate, respectively, according to the concentration gradient and incubated for 48 h. The cellular viability was determined using MTS Cell Proliferation Colorimetric Assay (Promega, USA) by measuring the absorbance at 490 nm using a Multiskan FC (Thermo Fisher Scientific, USA). Experiments were repeated three times, and the data are representative of three experiments. And the IC50 values of 143B in each preparation group were calculated and compared.
Apoptosis Assay
For investigation of the induction of apoptosis, 143B cells were treated with PLGA-PTX NPs, PTX-PLGA@143B NPs, PTX-PLGA@RAW NPs and PTX-PLGA@[143B-RAW] NPs, respectively. After incubated 48h, cells were then stained by Annexin V-FITC/PI (BD Pharmingen) according to the instructions of the product. The percentage of the apoptotic cells was analyzed using flow cytometry (BD Biosciences).
Chemotaxis Experiment
HUVEC cells were planted on 24-well plates at a cell density of 5000 cells per well and incubated for 24 hours. After that, these cells were stimulated with 3 ng/mL TNF-α for 8 h while those cells that were not stimulated by TNF-α were used as controls. Then HUVEC cells were added with equal amounts of PLGA NPs, PLGA@143B NPs, PLGA@RAW NPs and PLGA@[143B-RAW] NPs with Dil staining and incubated at 37°C for 2 hours. After reaching the expected time point, DAPI staining was performed according to the instructions. The fluorescent intensity of groups was observed with a fluorescence microscope. Fluorescence intensity was analyzed with Image J and bar charts were drawn for comparison.
In vivo Biodistribution
To study the biodistribution of PLGA@[143B-RAW] NPs, a xenograft tumor model was established by subcutaneous injection of 1×107 143B cells into the right flank region of male BALB/c nude mice. Briefly, 1 mg PLGA nanoparticles were labeled by 16 μg DiR dye (1 mg/mL, Yeasen Biotech Corporation, Shanghai, China), unbounded DiR dye was removed by ultrafiltration. When the tumors reached about 200 mm3, mice were randomly divided into five groups with the different treatment of free DiR, DiR labeled PLGA NPs, PLGA@RAW NPs, PLGA@143B NPs and PLGA@[143B-RAW] NPs via tail vein injection, respectively. Biofluorescence images were obtained with the help of an FX PRO in vivo imaging system (Bruker, Germany) at 24 h after treatment, then mice were euthanized, and the major organs and tumors were excised and immediately imaged to obtain ex vivo images.
In vivo Anti-Tumor Study
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC), The Second Xiangya Hospital, Central South University and all animals were treated following the IACUC approved procedures. Animals were obtained from HUNAN SJA LABORATORY ANIMAL CO., LTD and housed according to the regulations of the IACUC. Tumor-bearing mice were generated by injecting 1×107 143B cells subcutaneously into the right flank region of male BALB/c nude mice (6 weeks). After the tumor volume reached approximately 100 mm3, PBS, free PTX (5 mg/kg), PLGA-PTX NPs (5 mg/kg PTX), PTX-PLGA@143B NPs (5 mg/kg PTX), PTX-PLGA@RAW NPs (5 mg/kg PTX) and PTX-PLGA@[143B-RAW] NPs (5 mg/kg PTX) were administrated through tail vein injection for four times with an interval of 2 days. Tumor volume and mice weight were measured every day for 8 days from the first administration. The mice were sacrificed on the second day of the last dose, blood samples, major organs and tumors were collected. According to the manufacturer’s instructions, plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and creatinine (Cr) were measured using assay kits (Huili Biotech, China). For histopathological assessment, tumors and major organs were weighed and fixed in 4% paraformaldehyde (PFA) and then stained with haematoxylin and eosin (H&E). Tumors were also stained with terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL). The stained tissue sections were visualized by a microscope (OlympusIX51).
Statistical Analysis
Data were presented as mean values ± SD. T-Test and Two-way analysis of variance (ANOVA) were performed at the significance level α = 0.05.
Results
Characterization of PTX-PLGA@[143B-RAW] NPs
Gradient centrifugation was used to extract the cell membrane of 143B and RAW264.7, respectively. As the extraction process is shown in Figure 1B, the precipitation was removed successively. The supernatant containing the cell membrane presented milky white. The cell membrane precipitate was obtained by ultracentrifugation, closely attached to the bottom of the centrifugal tube. After membrane precipitate resuspended by special buffer IB-2, the protein concentration of cell membrane was quantified by bicinchoninic acid (BCA) and cell membrane was stored at −80°C after lyophilization.
PLGA nanoparticles were prepared by nano-precipitation method. Their particle size and potential were characterized by DLS (Figure 1C), which was 132.73±0.61 nm and −24.60±1.32 mV, respectively. The encapsulated efficiency of PLGA-PTX was 64.86±0.17%, while drug loading was 4.24%±0.02, shown in Table S1.
The hybrid membrane requires the fusion of two different cell membranes. At present, there are two methods of extracting hybrid membranes from cells. One is to fuse the two cells and then perform membrane extraction (first fusion and then membrane extraction), and the other is to extract and fuse each cell membrane (first membrane extraction and then fusion).19 Studies had shown that using the first method to prepare hybrid membrane, new membrane proteins were obtained compared with the membrane protein of the original cell.20 In the latter method, different membranes were fused by ultrasonic treatment. It showed that the composition of the hybrid membrane and the original membrane remained the same, which was easier to control. Therefore, 143B cell membrane was fused with RAW264.7 cell membrane by the latter method, with ultrasonic treatment promoting their fusion. The membrane proteins were characterized by Coomassie bright blue (Figure 1F). The protein distribution of 143B-RAW overlapped with 143B cell membrane and RAW264.7 cell membrane, respectively, demonstrating the successful fusion of the two cell membranes.
To form the biomimetic nanoparticles, the hybrid membrane needs to cover the surface of core nanoparticles in a specific way. At present, membrane extrusion and ultrasonic treatment are the two most used methods according to research.21 We prepared PLGA@ [143B-RAW] NPs by the two methods, respectively, and characterized their particle size, zeta potential and protein distribution. The size and potential of nanoparticles prepared by the two methods were compared in Table S2. The size of nanoparticles prepared by the ultrasonic method was smaller than that by the co-extrusion method. In addition, the size of nanoparticles encapsulated by the three membranes prepared by the co-extrusion method had a significant difference. In comparison, the size range of nanoparticles encapsulated by the three membranes prepared by the ultrasonic method remained consistent (Figure 1D), indicating that the ultrasonic method may be better and repeatable. The morphology of PLGA nanoparticles and PLGA@ [143B-RAW] NPs was observed by TEM. PLGA nanoparticles were a white spherical shape, while PLGA@ [143B-RAW] NPs displayed an obvious core-shell structure, indicating that PLGA nanoparticles were successfully coated by the hybrid membrane (Figure 1E). The result of Coomassie brilliant blue staining showed that PLGA nanoparticles had no bands, indicating that they did not carry any proteins. It also showed that the protein content in the cell membrane suspension was higher than that of the membrane coated nanoparticles, which meant a slight loss of membrane proteins during the preparation process, but most of the membrane proteins were still retained. The hybrid membrane and the nanoparticles covered by the hybrid membrane overlap with the first two bands, indicating they had proteins from two different cell membranes (Figure 1F). To analyze specific protein markers in the two cell membrane materials, Western blotting analysis was performed (Figure 1I). The MMP-2 was a invasion-associated protein and was a tumor target, with high expression in osteosarcoma.22,23 Stronger signals for MMP-2 was observed on the 143B membrane, 143B-RAW hybrid membrane, PLGA@143B NPs and PLGA@ [143B-RAW] NPs. The specific marker, integrin α4, was expressed on the RAW264.7 membrane, 143B-RAW hybrid membrane, PLGA@RAW NPs and PLGA@ [143B-RAW] NPs.
To compare the effects of the co-extrusion method and ultrasonic method on membrane proteins, the protein content of membrane-coated nanoparticles prepared by the two methods was compared. The results showed that the bands of membrane-coated nanoparticles prepared by the ultrasonic method were darker, indicating higher protein content (Figure 1G). Therefore, the ultrasonic method was selected as the following preparation method so that the loss of membrane protein was less than that of the co-extrusion method.
In order to investigate the stability of the membrane-coated nanoparticles prepared by ultrasonic method, the particle size of the membrane-coated nanoparticles prepared by this method in PBS was measured for 8 consecutive days. Figure 1H shows that the particle size of the three kinds of nanoparticles was stable at about 220 nm, with no significant change, indicating that the nanoparticles prepared by this method are relatively stable.
PTX release from PTX-PLGA@ [143B-RAW] NPs and PLGA-PTX NPs in different pH environments (pH 5.3 and 7.4) was analyzed (Figure 1J). The membrane coating does not change the release behavior of PTX from the PLGA which consists of burst release period and plateau release period. And the PTX release is a controlled slow release. It was found that PTX release was pH-dependent, and the cumulative release rate of PTX increased at a low pH condition as time going on. In the environment of pH 5.3, PTX of PTX-PLGA@[143B-RAW] NPs was released about 90% within 72 h, before which the release curve reached a plateau. And it was released about 70% in the environment of pH 7.4. Regarding the release rate of PTX-PLGA@[143B-RAW] NPs at pH 7.4 and 5.3 are relatively faster than others, it requires further research.
The Cellular Uptake of PLGA@[143B-RAW] NPs by 143B Cells in vitro
Fluorescence co-localization was used to observe the uptake of PLGA@[143B-RAW] NPs by 143B cells (Figure 2A). The result showed that PLGA nanoparticles were almost not taken up by 143B cells, while PLGA@143B NPs and PLGA@[143B-RAW] NPs were more taken up by 143B cells. The uptake of PLGA@RAW NPs was less.
 |
Figure 2 The uptake of PLGA@[143B-RAW] NPs. (A) 143B cells uptake of different nanoparticles. (B) 143B cells uptake of PLGA@[143B-RAW] NPs at different times. Nucleus was labeled by DAPI (Blue). PLGA NPs was labeled by Dil (Red). (C) Semi-quantitative analysis of 143B cells uptake of Dil stained nanoparticles. (D) Semi-quantitative analysis of 143B cells uptake of PLGA@[143B-RAW] NPs at different times. (I) PLGA NPs; (II) PLGA@143B NPs; (III) PLGA@RAW NPs; (IV) PLGA@[143B-RAW] NPs. Scale bar=50 μm. Each point represents the mean ± SD, ****P < 0.0001. |
Semi-quantitative analysis of the fluorescence intensity showed that the fluorescence intensity of PLGA@143B NPs and PLGA@[143B-RAW] NPs group was almost the same (Figure 2C), indicating that the increased uptake rate was mainly related to the modification of 143B cell membrane. The fluorescence intensity of PLGA@RAW NPs was significantly weaker than that of PLGA@143B NPs and PLGA@[143B-RAW] NPs, indicating the uptake of macrophage membrane-modified nanoparticles by tumor cells was significantly lower than that of tumor membrane-modified nanoparticles. To sum up, it was proved that the tumor cell membrane has homologous targeting. The fluorescence intensity of PLGA nanoparticles without membrane modification was the lowest, indicating that they were hardly taken up by tumor cells within one hour.
The uptake of PLGA@[143B-RAW] NPs by 143B cells at different times was investigated (Figure 2B). The result showed that the uptake of PLGA@[143B-RAW] NPs by 143B cells increased with time going by. Through the fluorescence semi-quantitative analysis, PLGA@ [143B-RAW] NPs had the higher fluorescence intensity when incubating with 143B cells longer, indicating that the uptake of PLGA@[143B-RAW] NPs by 143B cells was time-dependent (Figure 2D). After 4 hours, the increased fluorescence intensity rate slowed down, indicating that the uptake rate of PLGA@[143B-RAW] NPs gradually slowed down and reached saturation.
Anti-Tumor Effect of PTX-PLGA@[143B-RAW] NPs on Osteosarcoma Cells
The anti-tumor effect of PTX-PLGA@[143B-RAW] NPs was investigated in vitro firstly (Figure 3A). It was found that PLGA-PTX NPs had the weakest anti-tumor effect on 143B cells. The effect of PTX-PLGA@143B NPs was slightly stronger than that of PLGA-PTX NPs at low concentration, but there was no significant difference at high concentration. PTX-PLGA@RAW NPs and PTX-PLGA@[143B-RAW] NPs had stronger cytotoxicity on 143B cells. PTX-PLGA@RAW NPs showed the anti-tumor effect to 143B cells at low concentrations, while the effect of PTX-PLGA@[143B-RAW] NPs was the strongest at high concentrations. In general, the cytotoxic effect on osteosarcoma cells of PLGA nanoparticles with membrane modified was significantly enhanced compared with the unmodified PLGA nanoparticles.
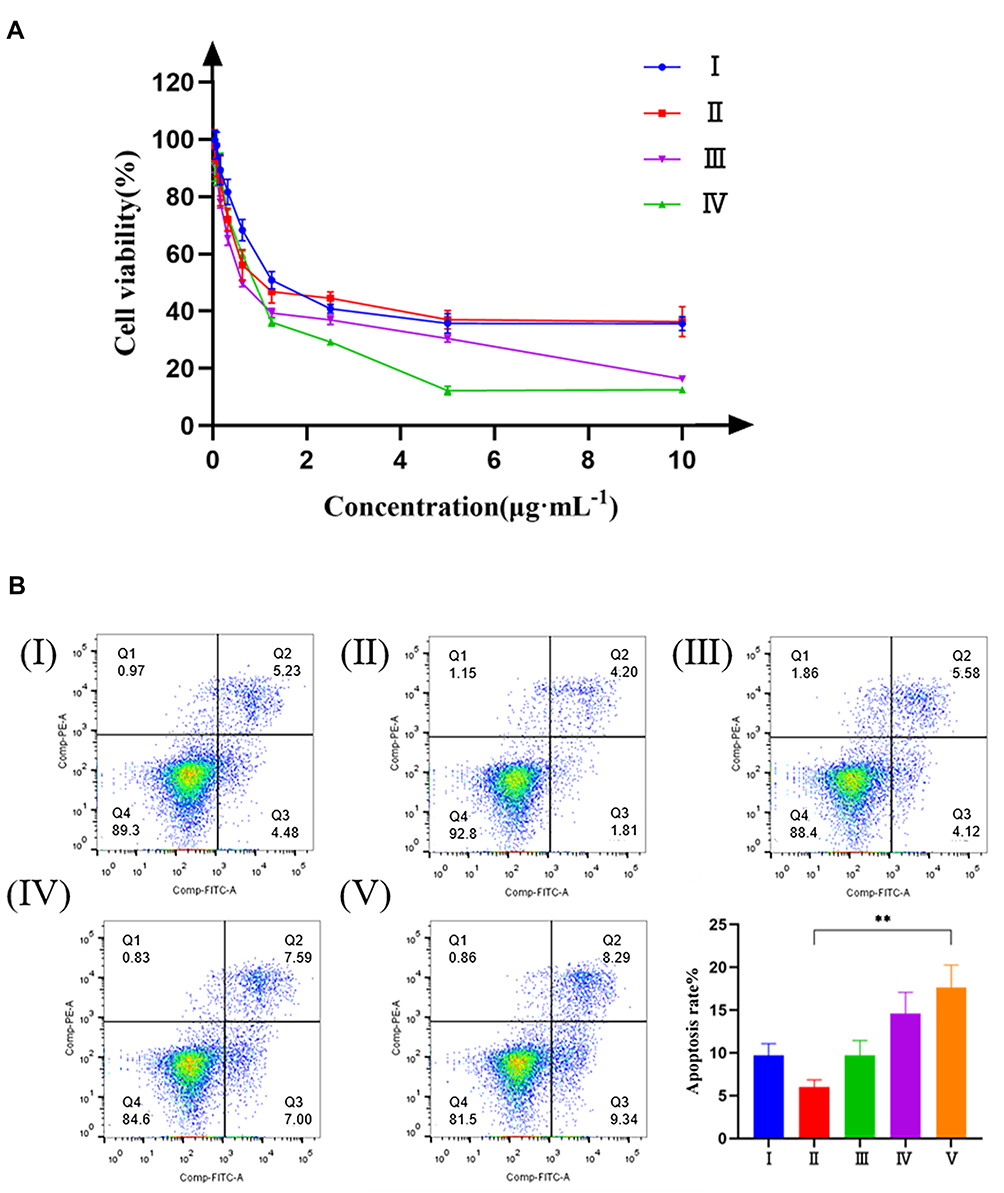 |
Figure 3 The cytotoxicity of PTX-PLGA@[143B-RAW] NPs in vitro. (A) Effects of different preparations on the viability of 143B cells. (I) PLGA-PTX NPs; (II) PTX-PLGA@143B NPs; (III) PTX-PLGA@RAW NPs; (IV) PTX-PLGA@[143B-RAW] NPs. (B) Apoptosis of 143B cells induced by different preparations. (I) Free PTX; (II) PLGA-PTX NPs; (III) PTX-PLGA@143B NPs; (IV) PTX-PLGA@RAW NPs; (V) PTX-PLGA@[143B-RAW] NPs. Each point represents the mean ± SD, **P < 0.01. |
The ability of PTX-PLGA@[143B-RAW] NPs to induce apoptosis was investigated further (Figure 3B). It was found that the percentages of apoptosis induced by PTX-PLGA@RAW NPs and PTX-PLGA@[143B-RAW] NPs were 14.59% and 17.63%, respectively, which meant PTX-PLGA@[143B-RAW] NPs had the most potent ability to induce apoptosis of 143B cells. And notably, PTX-PLGA@[143B-RAW] NPs and PTX-PLGA@RAW NPs significantly improved early apoptosis rate compared to other group, which was 9.34% and 7.00% respectively. The apoptosis rate was 9.70% in PTX-PLGA@143B NPs group and 9.71% in free PTX group, which accounted mainly for late phase. PLGA-PTX NPs had the weakest apoptosis induction, and also the weakest early apoptosis rate (1.81%). The reason why the free PTX group has a higher apoptosis-inducing rate than PLGA-PTX group may be that free PTX group can be taken up faster by tumor cells in vitro, so as to act on cells faster.
Chemotactic Effect of PLGA@[143B-RAW] NPs on Inflammatory Environment
In tumor microenvironment and pre-metastatic niche, the endothelium was often activated by the inflammatory cytokine and formed a inflammatory environment. To mimic the physical environment, HUVEC cells were stimulated with inflammatory factor TNF-α. Using HUVEC cells without TNF-α stimulation as control, the chemotactic effect of each group of nanoparticles on the inflammatory environment was investigated by fluorescence co-localization. As shown in Figure 4A, PLGA@[143B-RAW] NPs had the most substantial chemotactic effect on the inflammatory environment, followed by PLGA@RAW NPs. The chemotactic effect of PLGA@143B NPs on the inflammatory environment of HUVEC cells was weak, and PLGA nanoparticles had almost no chemotactic effect on HUVEC cells. The results showed that macrophage cell membrane could improve the chemotactic effect of nanoparticles on the inflammatory environment. According to the results of fluorescence co-localization, the fluorescence of each group of nanoparticles gathered around the cell. However, it did not enter the cell and surround the nucleus, indicating that the nanoparticles only had a chemotactic effect on inflammatory HUVEC cells, but would not be taken up by them.
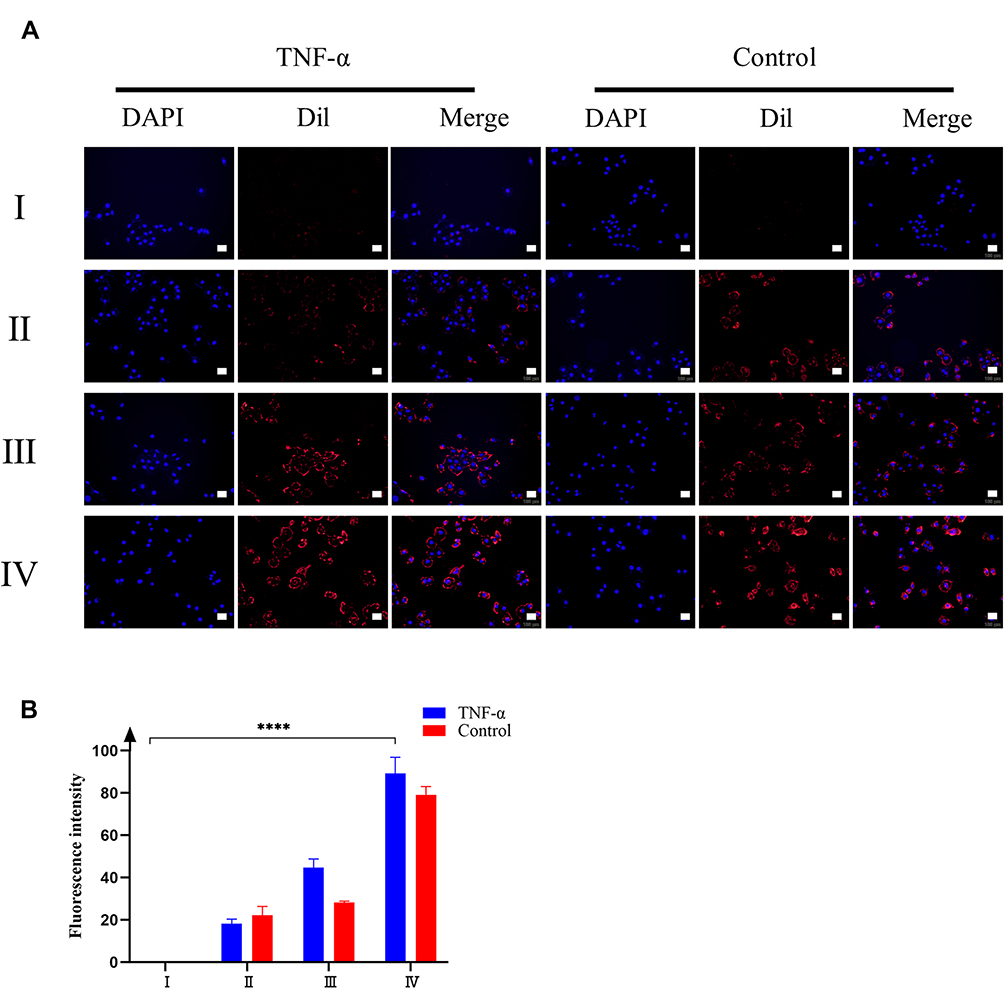 |
Figure 4 Chemotaxis of different nanoparticles on HUVEC cells. (A) The fluorescence co-localization of different preparations and HUVEC cells. The nucleus was labeled by DAPI (Blue). PLGA NPs were labeled by Dil (Red). (B) Semi-quantitative analysis of chemotaxis of HUVEC cells by different nanoparticles. The inflammatory factor TNF-α provided the inflammatory environment. (I) PLGA NPs; (II) PLGA@143B NPs; (III) PLGA@RAW NPs; (IV) PLGA@[143B-RAW] NPs. Scale bar=50 μm. Each point represents the mean ± SD, ****P < 0.0001. |
The fluorescence semi-quantitative results (Figure 4B) showed that for the PLGA@[143B-RAW] group and the PLGA@RAW group, the fluorescence intensity of the group stimulated by inflammatory factors was higher than that of the group without inflammatory factors stimulation, indicating that the nanoparticles modified by macrophage cell membrane had a stronger chemokine for the inflammatory environment. The fluorescence intensity of PLGA@143B NPs was not significantly affected by inflammatory factors, indicating that PLGA@143B NPs had no chemotactic effect on the inflammatory environment. PLGA nanoparticles showed very weak chemotaxis to HUVEC cells in both environments, with almost no chemotaxis. The result revealed that nanoparticles coating with macrophage membrane were more tend to inflammatory sites than nanoparticles without macrophage membrane modification.
Biodistribution
The in vivo biodistribution of PLGA@[143B-RAW] NPs was observed using a live imaging system. Tumor-bearing mice were administrated with free DiR, DiR labeled PLGA NPs, PLGA@RAW NPs, PLGA@143B NPs and PLGA@[143B-RAW] NPs via tail vein injection and were imaged at 6 h, 10 h and 24 h after administration. As shown in Figure 5A, similar to in vitro uptake experiments, free DiR and other four formulations accumulated in tumor in a time-dependent way, and they were also detected in other tissues. Tumors and major organs were excised after live mice imaging. The DiR labeled PLGA@143B NPs and PLGA@[143B-RAW] NPs could effectively reach the tumor site 24 h after administration (Figure 5B), and the fluorescence intensity of the DiR labeled PLGA@[143B-RAW] NPs was obviously higher than the DiR labeled PLGA@143B NPs (Figure 5C). In comparison, other formulations failed to reach the tumor site effectively over 24 h.
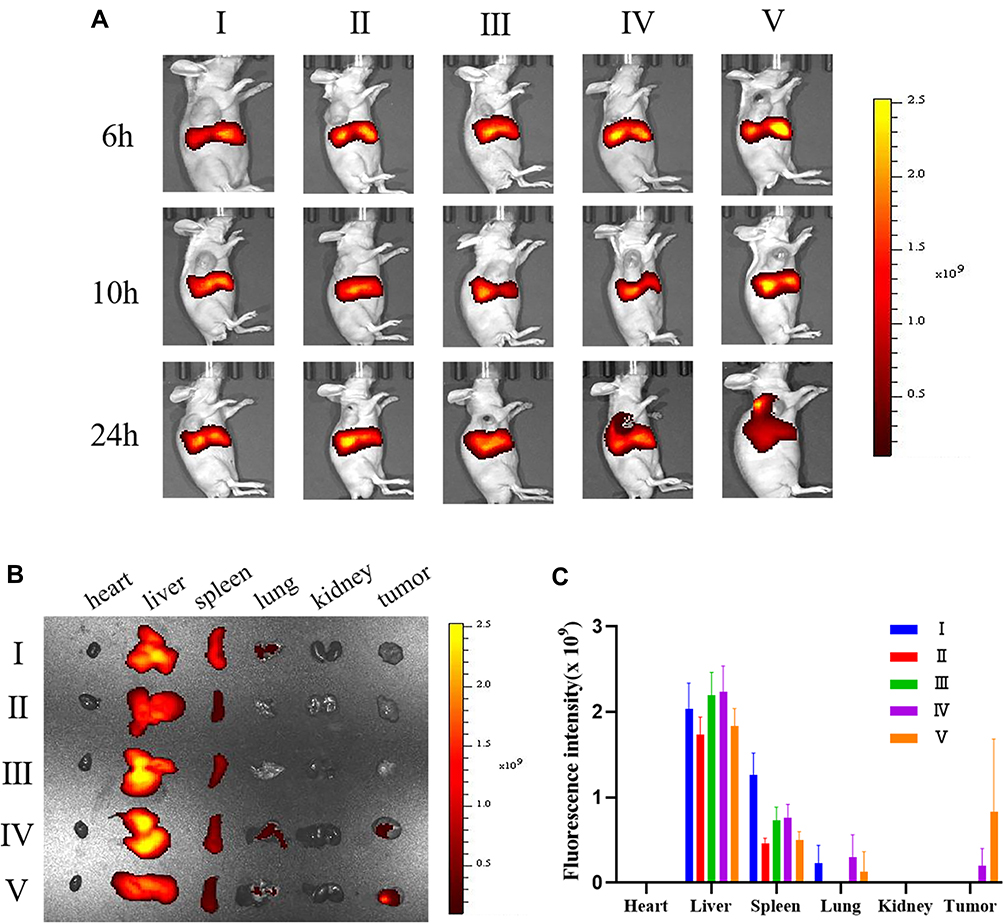 |
Figure 5 In vivo biodistribution of PLGA@[143B-RAW] NPs. (A) In vivo biodistribution of different formulations intravenously administered to 143B xenograft mice at different times. (B) Ex vivo images of major organs and tumors from 143B xenograft mice 24 h after intravenous injection of the five formulations. (C) Fluorescence intensity of the five formulations at tumors and major organs 24 h after intravenous injection. (I) DIR; (II) PLGA NPs; (III) PLGA@RAW NPs; (IV) PLGA@143B NPs; (V) PLGA@[143B-RAW] NPs. |
In vivo Anti-Tumor Effects of PTX-PLGA@[143B-RAW] NPs
For the investigation of the in vivo anti-cancer therapeutic efficacy of PTX-PLGA@[143B-RAW] NPs on osteosarcoma xenograft mice, mice were treated with different formulations intravenously four times with an interval of two days. The tumor of mice receiving PBS grew very fast as control, and mice receiving PLGA-PTX NPs and PTX-PLGA@RAW NPs showed similar tumor growth to that of the PBS group (Figure 6A and B, Figure S1). PTX-PLGA@143B NPs could suppress tumor growth to a certain extent, while free PTX or PTX-PLGA@[143B-RAW] NPs exhibited a greater decrease of tumor volume than PTX-PLGA@143B NPs, and PTX-PLGA@[143B-RAW] NPs was the most effective one. Excised tumors (Figure 6C) and tumor weight (Figure 6D) also confirmed the improved anti-cancer efficacy of PTX-PLGA@[143B-RAW] NPs as compared to free PTX. Additionally, the H&E staining and TUNEL immunohistochemistry showed clear cell death in the tumors of PTX-PLGA@[143B-RAW] NPs, PTX-PLGA@143B NPs, PTX-PLGA@RAW NPs and PLGA-PTX NPs treated mice (Figure 7). Blank areas of the PTX-PLGA@[143B-RAW] NPs group were the largest, with cell nuclear shrinkage, while the tumor cells in the PBS group were still full.
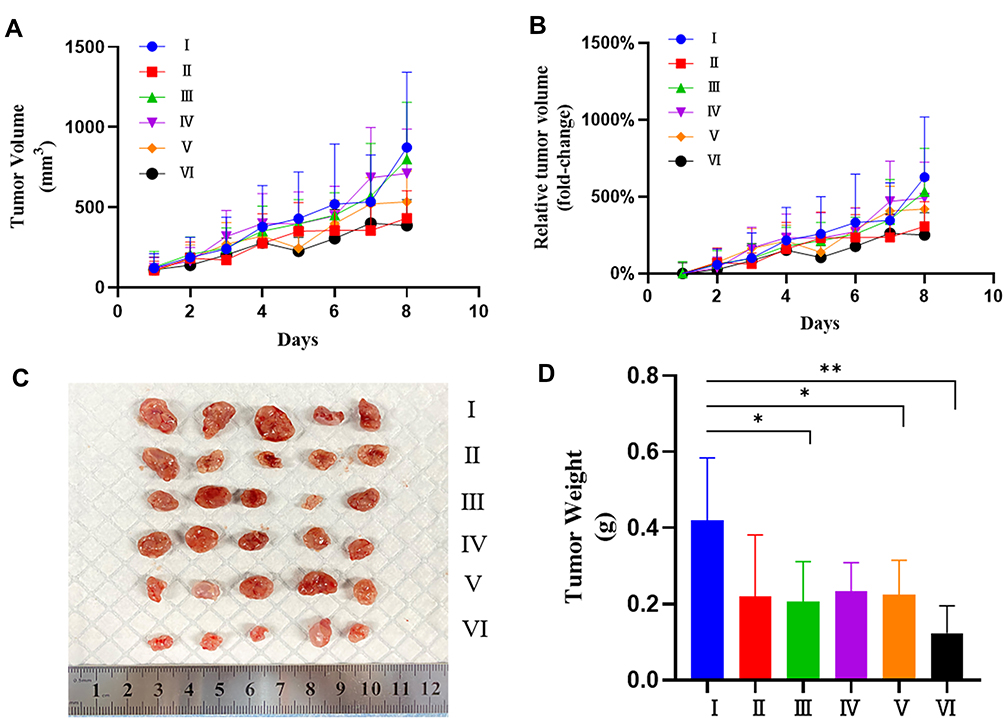 |
Figure 6 Therapeutic efficacy of PTX-PLGA@[143B-RAW] NPs against 143B tumors. (A) Absolute and (B) relative 143B tumor growth in mice during the experimental period. (C) Excised tumors at the end of experiments. (D) Tumor weight at the end of experiments. (I) PBS; (II) Free PTX; (III) PLGA-PTX NPs; (IV) PTX-PLGA@RAW NPs; (V) PTX-PLGA@143B NPs; (VI) PTX-PLGA@[143B-RAW] NPs. N = 5, each point represents the mean ± SD, *P < 0.05, **P < 0.01. |
 |
Figure 7 Histopathological confirmation of efficient therapeutic efficacy of PTX-PLGA@[143B-RAW] NPs against 143B tumor. H&E and TUNEL-stained tumors of mice after treatment. (I) PBS; (II) Free PTX; (III) PLGA-PTX NPs; (IV) PTX-PLGA@RAW NPs; (V) PTX-PLGA@143B NPs; (VI) PTX-PLGA@[143B-RAW] NPs. Scale bar = 200 μm. |
Safety Evaluation
To investigate the safety properties of PTX-PLGA@[143B-RAW] NPs, the change of mice body weight was recorded and evaluated. The body weight of mice receiving free PTX exhibited a significant reduction during treatment (Figure 8A and B). In contrast, the PTX-PLGA@[143B-RAW] NPs group did not occur significant body weight reduction during the whole treatment period, indicating the potential safety of PTX-PLGA@[143B-RAW] NPs. To further confirm its safety, the H&E staining immunohistochemistry of major organs was observed, which showed PTX group had the most damage in the liver and kidney, with inflammatory cell infiltration (Figure 8C). Moreover, mice receiving PTX-PLGA@[143B-RAW] NPs showed no negative effect on lung, heart, spleen and had slight injuries in the liver and kidney, but much less than the PTX group, suggesting its protection for organs. Additionally, we analyzed the relative levels of ALT and AST to reflect liver functions, and BUN and Cr to reflect renal functions. Briefly, the group with PTX treatment exhibited significantly increased levels of all measures, suggesting its toxicity to liver and renal. However, there was no significant difference between PTX-PLGA@[143B-RAW] NPs and PBS group in ALT, AST, BUN, and Cr levels, indicating PTX-PLGA@[143B-RAW] NPs had little effect on the liver and kidney (Figure 8D–G).
 |
Figure 8 Safety and biocompatibility evaluation of PTX-PLGA@[143B-RAW] NPs. (A) Absolute and (B) relative body weight change of tumor-bearing mice during the experimental period, n = 5. (C) H&E-stained hearts, livers, spleens, lungs and kidneys of mice after treatment. Scale bar = 200 μm. Plasma levels of (D) ALT, (E) AST, (F) BUN, (G) Cr in tumor-bearing mice two days after treatment for four times with an interval of two days, n = 5. (I) PBS; (II) Free PTX; (III) PLGA-PTX NPs; (IV) PTX-PLGA@RAW NPs; (V) PTX-PLGA@143B NPs; (VI) PTX-PLGA@[143B-RAW] NPs. Each point represents the mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
Discussion
Nano-drug delivery system has attracted more and more attention in recent years. Niosomes are widely used in the treatment of various diseases due to their unique advantages, such as outstanding stability and biocompatibility. For example, the novel drug delivery vehicle obtained by hybridizing bio-synthetic selenium nanoparticles with niosomes not only had high biocompatibility, could significantly induce tumor cell apoptosis and necrosis, but also protected normal cells from the toxic effects or other side effects of chemotherapy, thereby achieving the purpose of effective treating lung cancer.24 In addition, bovine serum albumin (BSA)-coated niosomes could also effectively encapsulate genetic drugs such as decoy oligodeoxynucleotides to inhibit the metastasis of glioblastoma multiforme, which could be a promising approach for targeted gene delivery in cancer therapy.25 The PLGA nanoparticles coated with hybrid membrane from the osteosarcoma cell membrane and macrophage cell membrane also show high biocompatibility and high targeting ability to osteosarcoma cells. In this study, we chose the human osteosarcoma cell line 143B and mice monocyte-macrophage cell line RAW264.7. Although 143B and RAW 264.7 from different species, they both belong to eukaryotic cells and have similarities in their membrane structure. They were used mostly in other related research and animal models. And RAW264.7 could stably grow and passage and the proliferation rate of 143B cells was high, which were convenient for cell culture and cell membrane extraction. In the present study, we successfully prepared PLGA@[143B-RAW] NPs to load PTX, and the drug loading was approximately 4%. The method we used to construct PTX-PLGA@[143B-RAW] NPs was the ultrasonic method.21 Compared with the co-extrusion method, the effect on protein content with the ultrasonic method was more negligible, which may be because mechanical squeezing would squeeze the membrane protein into fragments, thereby destroying the content and function of some membrane proteins. Therefore, the ultrasonic method was selected for further experiments and may be suitable for clinical application. In the drug release study, PTX released from PTX-PLGA@[143B-RAW] NPs was faster at 0–8 h, and then gradually slowed down. Compared with pH 7.4, PTX released faster under pH 5.3 because the acidic environment would cause the hydrolysis and degradation of the polymer, and the solubility of PTX at low pH was increased, which may promote PTX released and diffused into the surrounding medium faster.
For the cellular uptake assay, the uptake efficiency of PLGA@[143B-RAW] NPs and PLGA@143B NPs by osteosarcoma cells in vitro was significantly greater than that of unmodified PLGA NPs and PLGA@RAW NPs, suggesting that the targeting ability of PLGA@[143B-RAW] NPs to osteosarcoma cells mainly depended on the modification of the 143B cell membrane, which may be related to the homologous targeting of the tumor cell membrane.15 In addition, the cellular uptake of PLGA@[143B-RAW] NPs by osteosarcoma cells was time-dependent, indicating that PLGA@[143B-RAW] NPs could continue to release PTX into osteosarcoma cells through membrane fusion, thereby the accumulation of PTX in 143B cells increased, so as to achieve the purpose of killing tumor cells.
PTX can polymerize tubulin into stable microtubules and induce the formation of stable microtubule bundles in the G2/M phase, thereby promoting cell apoptosis.26 For the cell viability assay, we observed PTX-PLGA@[143B-RAW] NPs significantly improved the apoptosis of 143B cells compared with free PTX and PLGA-PTX NPs, because it was more taken up by osteosarcoma cells. Furthermore, compared with PTX-PLGA@RAW NPs and PTX-PLGA@[143B-RAW] NPs, the anti-tumor effect of PTX-PLGA@143B NPs was reduced, which may be because the tumor cell membrane carried a specific biomolecule that could inhibit tumor cell apoptosis or suppressed the anti-tumor effect of PTX on osteosarcoma cells to a certain extent. The mechanism needs to be further studied.
Macrophages, one of the most abundant cells in tumor microenvironments, also have the tendency to inflammatory sites.27 It was also reported that nanoparticles modified by macrophage membrane not only showed an immune escape ability and prolonged blood circulation time, but also had the chemotaxis on inflammatory environments.28 In this study, the chemotactic effect of PLGA@[143B-RAW] NPs on the inflammatory environment was also investigated. Compared with PLGA@143B NPs, PLGA@[143B-RAW] NPs and PLGA@RAW NPs showed a more substantial chemotactic effect on the inflammatory environment, which may be because inflammatory factor TNF-α could stimulate HUVEC cells to express ICAM-1, which could induce the adhesion of macrophages,29,30 so that the macrophage membrane coated nanocarriers could actively target the inflammatory sites, such as pre-metastatic niches and tumor sites.
In the biodistribution study, vigorous fluorescence intensity at liver, spleen and lung was detected, but there was a decrease in intensity later, suggesting that all four formulations firstly reached the liver, spleen, and lungs. After injection for 24 hours, we only observed fluorescence intensity at the tumor in PLGA@143B NPs and PLGA@[143B-RAW] NPs groups, which was because tumor membrane express CD47, the “don’t eat me” signal, so that PLGA@143B NPs and PLGA@[143B-RAW] NPs could escape from phagocytosis and arrived at tumor site gradually by CD47–SIRPα binding.31 Collectively, hybrid membrane-modified nanoparticles have a prolonged circulation time, and can increase the accumulation of PTX at the tumor site.
In the animal study, the five formulations were administered to mice at a dose of 5 mg/kg of PTX. We observed that the tumor volume and tumor weight of mice receiving PTX-PLGA@[143B-RAW] NPs was the smallest, and the other four groups had similar tumor weights, both higher than the hybrid membrane group. However, there was no significant difference between the free PTX and the PTX-PLGA@[143B-RAW] NPs, which may be because the actual drug loading of the nanoparticles was not as high as theoretically, so that its actual dose was not as much as that of free PTX. And a more considerable anticancer effect was feasible if higher PTX-PLGA@[143B-RAW] NPs were used. Although PTX-PLGA@[143B-RAW] NPs can effectively kill osteosarcoma cells, their effect on normal cells is also important for its clinical application. It is well known that PTX is toxic to tumor cells but also normal cells.32 In the safety evaluation, we observed that the body weight of mice receiving free PTX decreased rapidly after the first administration, and then dropped more later, while the body weight of the other groups did not change significantly. H&E staining of major organs and biocompatibility evaluation further proved that free PTX would cause obvious damage to the liver and kidney of mice, while the values of mice in the hybrid membrane group were all within the normal range. It may be because drugs were metabolized by the liver and the accumulation of a large amount of drug hurts the liver, and PTX-PLGA@[143B-RAW] NPs could reduce the uptake of PTX by other cells in normal tissues for its reduced toxicity. In conclusion, our study demonstrated that PLGA@[143B-RAW] NPs are safe and efficient delivery vehicles for the delivery of PTX to treat osteosarcoma.
Conclusion
In this study, PLGA@[143B-RAW] NPs were successfully prepared as biomimetic delivery platforms by coating hybrid membrane onto PLGA NPs. In vitro experiments showed that PTX-PLGA@[143B-RAW] NPs could promote the uptake of PTX by osteosarcoma cells, and induced apoptosis of osteosarcoma cells. It also had chemotactic effects on pre-metastatic niches in inflammatory environments. PLGA@[143B-RAW] NPs exhibited superior targeting efficacy to the tumor site. PTX-PLGA@[143B-RAW] NPs inhibited significantly the growth of tumor and also showed lower toxicity than free PTX. This study provided a drug delivery strategy for targeted therapy of osteosarcoma.
Compliance with Ethical Standards
The animal experiment was approved by the Institutional Animal Care and Use Committee (IACUC), The Second Xiangya Hospital, Central South University (Approval No.: 2021692) and all animal studies were carried out using the IACUC approved procedures.
Acknowledgment
This work was supported by the Fundamental Research Funds for the Central Universities of Central South University (grant number 2021zzts1072) and the Hunan Provincial Science and Technology Plan (grant number 2016TP2002). Many thanks to Dr X-B. H for technical support.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Picci P. Osteosarcoma (osteogenic sarcoma). Orphanet J Rare Dis. 2007;2:6. doi:10.1186/1750-1172-2-6
2. Casali PG, Bielack S, Abecassis N, et al. Bone sarcomas: ESMO-PaedCan-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl4):iv79–iv95. doi:10.1093/annonc/mdy310
3. Coleman R, Hadji P, Body JJ, et al. Bone health in cancer: ESMO clinical practice guidelines. Ann Oncol. 2020;31(12):1650–1663. doi:10.1016/j.annonc.2020.07.019
4. Wang SY, Hu HZ, Qing XC, Zhang ZC, Shao ZW. Recent advances of drug delivery nanocarriers in osteosarcoma treatment. J Cancer. 2020;11(1):69–82. doi:10.7150/jca.36588
5. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. doi:10.1016/j.jconrel.2016.01.020
6. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science (New York, NY). 2000;288(5473):2051–2054. doi:10.1126/science.288.5473.2051
7. Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108(27):10980–10985. doi:10.1073/pnas.1106634108
8. Zhai Y, Su J, Ran W, et al. Preparation and application of cell membrane-camouflaged nanoparticles for cancer therapy. Theranostics. 2017;7(10):2575–2592. doi:10.7150/thno.20118
9. Lei Y, Junxin C, Yongcan H, Xiaoguang L, Binsheng Y. Role of microRNAs in the crosstalk between osteosarcoma cells and the tumour microenvironment. J Bone Oncol. 2020;25:100322. doi:10.1016/j.jbo.2020.100322
10. Huang Q, Liang X, Ren T, et al. The role of tumor-associated macrophages in osteosarcoma progression - therapeutic implications. Cell Oncol. 2021;44(3):525–539. doi:10.1007/s13402-021-00598-w
11. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32(40):e2002054. doi:10.1002/adma.202002054
12. Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthcare Mater. 2015;4(11):1645–1652. doi:10.1002/adhm.201500129
13. Xuan M, Shao J, Dai L, Li J, He Q. Macrophage cell membrane camouflaged au nanoshells for in vivo prolonged circulation life and enhanced cancer photothermal therapy. ACS Appl Mater Interfaces. 2016;8(15):9610–9618. doi:10.1021/acsami.6b00853
14. Sun H, Su J, Meng Q, et al. Cancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumors. Adv Mater. 2016;28(43):9581–9588. doi:10.1002/adma.201602173
15. Harris JC, Scully MA, Day ES. Cancer cell membrane-coated nanoparticles for cancer management. Cancers. 2019;11(12):1836. doi:10.3390/cancers11121836
16. Wang D, Dong H, Li M, et al. Erythrocyte-cancer hybrid membrane camouflaged hollow copper sulfide nanoparticles for prolonged circulation life and homotypic-targeting photothermal/chemotherapy of melanoma. ACS nano. 2018;12(6):5241–5252. doi:10.1021/acsnano.7b08355
17. Suski JM, Lebiedzinska M, Wojtala A, et al. Isolation of plasma membrane-associated membranes from rat liver. Nat Protoc. 2014;9(2):312–322. doi:10.1038/nprot.2014.016
18. Lai P, Daear W, Löbenberg R, Prenner EJ. Overview of the preparation of organic polymeric nanoparticles for drug delivery based on gelatine, chitosan, poly(d,l-lactide-co-glycolic acid) and polyalkylcyanoacrylate. Colloids Surf B Biointerfaces. 2014;118:154–163. doi:10.1016/j.colsurfb.2014.03.017
19. Liao Y, Zhang Y, Blum NT, Lin J, Huang P. Biomimetic hybrid membrane-based nanoplatforms: synthesis, properties and biomedical applications. Nanoscale Horiz. 2020;5(9):1293–1302. doi:10.1039/D0NH00267D
20. Liu WL, Zou MZ, Liu T, et al. Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat Commun. 2019;10(1):3199. doi:10.1038/s41467-019-11157-1
21. Fang RH, Hu CM, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. doi:10.1021/nl500618u
22. Boonstra MC, de Geus SW, Prevoo HA, et al. Selecting targets for tumor imaging: an overview of cancer-associated membrane proteins. Biomark Cancer. 2016;8:119–133. doi:10.4137/BIC.S38542
23. Lu KH, Chen PN, Hsieh YH, et al. 3-Hydroxyflavone inhibits human osteosarcoma U2OS and 143B cells metastasis by affecting EMT and repressing u-PA/MMP-2 via FAK-Src to MEK/ERK and RhoA/MLC2 pathways and reduces 143B tumor growth in vivo. Food Chem Toxicol. 2016;97:177–186. doi:10.1016/j.fct.2016.09.006
24. Gharbavi M, Johari B, Mousazadeh N, et al. Hybrid of niosomes and bio-synthesized selenium nanoparticles as a novel approach in drug delivery for cancer treatment. Mol Biol Rep. 2020;47(9):6517–6529. doi:10.1007/s11033-020-05704-z
25. Gharbavi M, Johari B, Rismani E, Mousazadeh N, Taromchi AH, Sharafi A. NANOG decoy oligodeoxynucleotide-encapsulated niosomes nanocarriers: a promising approach to suppress the metastatic properties of U87 human glioblastoma multiforme cells. ACS Chem Neurosci. 2020;11(24):4499–4515. doi:10.1021/acschemneuro.0c00699
26. Horwitz SB. Mechanism of action of taxol. Trends Pharmacol Sci. 1992;13(4):134–136. doi:10.1016/0165-6147(92)90048-B
27. Zhang W, Wang M, Tang W, et al. Nanoparticle-laden macrophages for tumor-tropic drug delivery. Adv Mater. 2018;30(50):e1805557. doi:10.1002/adma.201805557
28. Hu C, Lei T, Wang Y, et al. Phagocyte-membrane-coated and laser-responsive nanoparticles control primary and metastatic cancer by inducing anti-tumor immunity. Biomaterials. 2020;255:120159. doi:10.1016/j.biomaterials.2020.120159
29. Miki I, Kusano A, Ohta S, et al. Histamine enhanced the TNF-alpha-induced expression of E-selectin and ICAM-1 on vascular endothelial cells. Cell Immunol. 1996;171(2):285–288. doi:10.1006/cimm.1996.0205
30. Usami Y, Ishida K, Sato S, et al. Intercellular adhesion molecule-1 (ICAM-1) expression correlates with oral cancer progression and induces macrophage/cancer cell adhesion. Int J Cancer. 2013;133(3):568–578. doi:10.1002/ijc.28066
31. Chao MP, Weissman IL, Majeti R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–232. doi:10.1016/j.coi.2012.01.010
32. Barkat MA, Beg S, Pottoo FH, Ahmad FJ. Nanopaclitaxel therapy: an evidence based review on the battle for next-generation formulation challenges. Nanomed. 2019;14(10):1323–1341. doi:10.2217/nnm-2018-0313
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.