Back to Journals » Journal of Inflammation Research » Volume 17
Host Factors Modulate Virus-Induced IFN Production via Pattern Recognition Receptors
Authors Wang J, Dong Y, Zheng X, Ma H, Huang M, Fu D, Liu J ![]() , Yin Q
, Yin Q ![]()
Received 25 December 2023
Accepted for publication 28 May 2024
Published 12 June 2024 Volume 2024:17 Pages 3737—3752
DOI https://doi.org/10.2147/JIR.S455035
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Adam Bachstetter
Jingjing Wang,1 Yirui Dong,1 Xuewei Zheng,1 Haodi Ma,1 Mengjiao Huang,1 Dongliao Fu,1 Jiangbo Liu,2 Qinan Yin1,3
1School of Medical Technology and Engineering, Henan University of Science and Technology, Luoyang, People’s Republic of China; 2Department of General Surgery, First Affiliated Hospital, College of Clinical Medicine, Henan University of Science and Technology, Luoyang, People’s Republic of China; 3Henan Engineering Research Center of Digital Pathology and Artificial Intelligence Diagnosis, The First Affiliated Hospital of Henan University of Science and Technology, Luoyang, People’s Republic of China
Correspondence: Qinan Yin, School of Medical Technology and Engineering, Henan University of Science and Technology, No. 263 Kaiyuan Avenue, Luoyang, People’s Republic of China, 471003, Email [email protected] Jiangbo Liu, First Affiliated Hospital, College of Clinical Medicine, Henan University of Science and Technology, Guanlin Avenue, Luoyang, 471031, People’s Republic of China, Email [email protected]
Abstract: Innate immunity is the first line of defense in the human body, and it plays an important role in defending against viral infection. Viruses are identified by different pattern-recognition receptors (PRRs) that activate the mitochondrial antiviral signaling protein (MAVS) or transmembrane protein 173 (STING), which trigger multiple signaling cascades that cause nuclear factor-κB (NF-κB) and interferon regulatory factor 3 (IRF3) to produce inflammatory factors and interferons (IFNs). PRRs play a pivotal role as the first step in pathogen induction of interferon production. Interferon elicits antiviral activity by inducing the transcription of hundreds of IFN-stimulated genes (ISGs) via the janus kinase (JAK) – signal transducer and activator of transcription (STAT) pathway. An increasing number of studies have shown that environmental, pathogen and host factors regulate the IFN signaling pathway. Here, we summarize the mechanisms of host factor modulation in IFN production via pattern recognition receptors. These regulatory mechanisms maintain interferon levels in a normal state and clear viruses without inducing autoimmune disease.
Keywords: pattern recognition receptors, interferon-signaling pathway, host factors
Introduction
Viral infection is a serious threat to human health. The innate immune system is the first line of defense against virus invasion. Pattern recognition receptors (PRRs) of host cells recognize pathogen-associated molecular patterns (PAMPs) of viruses, which activates innate immune response signaling pathways and induces interferon and cytokine expression.1,2 Interferon is the main factor of host cell resistance to virus invasion, and it is divided into type I interferon (IFN-α, IFN-β), type II interferon (IFN-γ), and type III interferon (IFN-λ1, IFN-λ2, IFN-λ3, IFN-λ4).3,4 Interferon binds to specific IFN receptors on the cell surface and activates the JAK/STAT signaling pathway to induce the expression of interferon-stimulating genes (ISGs), which enhance antiviral abilities by regulating the activity of natural killer (NK) cells, macrophages, and T cells.5
Pattern recognition receptors of host cells primarily include Toll-like receptors (TLRs), RIG-I receptors (RLRs), and NOD-like receptors (NLRs), which recognize RNA viruses,6 and receptors that recognize DNA viruses, such as cyclic GMP-AMP synthase (cGAS), DEAD-Box helicase 41 (DDX41) and interferon gamma-inducible protein 16 (IFI16).3 TLR3 induces interferon expression via the toll/interleukin-1 receptor (TIR) domain-containing adaptor inducing IFN-β (TRIF)-TANK binding kinase 1 (TBK1)/inhibitor κB kinase ε (IKKε)-IRF3 pathway.2 Intracytoplasmic pattern recognition RLRs, such as retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), recognize intracytoplasmic viral dsRNA, activate the RIG-I/MDA5-MAVS-TBK1/IKK-ε-IRF3 signaling pathway, and induce interferon expression.6,7 By recognizing DNA viruses, the DNA pattern recognition receptor cGAS synthesizes the second messenger cGAMP, which binds to STING and activates the cGAS-STING-TBK1/IKK-IRF3 signaling pathway to induce interferon expression.8
Thorough research of interferon signaling pathway has discovered an increasing number of host factors that regulate interferon signaling pathways by regulating the activity of PRRs, adaptors, kinases and transcription factors. This approach enriches the understanding of the interferon signaling pathway and provides more targets for the development of antiviral drugs. In our immune system, pathogen-associated molecular patterns (PAMPs) of pathogens, which are distinct from the host, are detected by pattern recognition receptors (PRRs).9 This recognition activates downstream signaling pathways that the production of IFN and inflammatory factors.10 PRRs play a key role as the first step in pathogen-induced interferon production in host cells. This review highlights the host factors that regulate the production of IFNs induced by viral infection via PRRs (Table 1 and Table 2).
 |
Table 1 PRRs Affected by Positive Regulatory Factors |
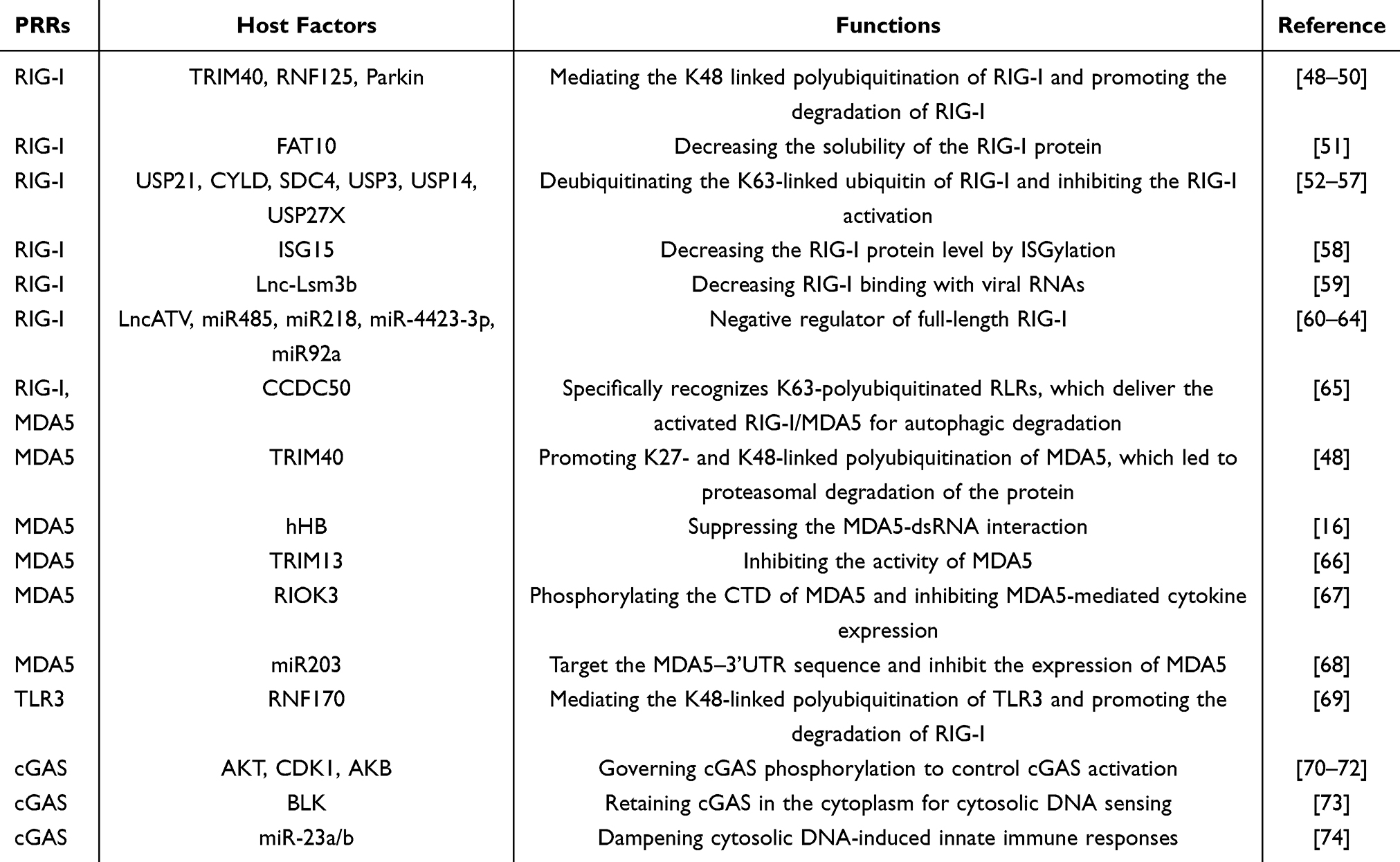 |
Table 2 Negative Regulatory Factors on PRRs |
RIG-I-Like Pattern Recognition Receptors (RLRs)
RIG-I-like pattern recognition receptors include RIG-I, MDA5, and laboratory of genetics and physiology 2 (LGP2), which are expressed in most cells and exhibit strong immune responses in bone marrow cells, epithelial cells, and central nervous system cells. RIG-I and MDA5 contain two N-terminal caspase activation and recruitment domains (CARDs), which activate the innate immune signaling pathway. A central DExD/H RNA helicase domain promotes ATP hydrolysis and RNA binding, and a C-terminal RNA binding helicase domain (CTD) assists in the recognition and specific binding of RNA ligands. RIG-I and MDA5 act as pattern recognition receptors to recognize exogenous RNA viruses and induce a common signaling pathway to produce interferon. However, whether LGP2 serves as a pattern recognition receptor is controversial due to the lack of CARD domains that are required for signal transmission. RIG-I and MDA5 CARD domains interact with other proteins that have CARD domains,75,76 such as mitochondrial antiviral protein (MAVS, also known as IPS-1/VISA/Cardif). Binding of the CARD-CARD domain activates MAVS,77–79 recruits, phosphorylates and activates TBK1 and IKK-ε kinases, which phosphorylate downstream transcription factors, such as IRF3. These transcription factors form dimers and translocate from the cytoplasm to the nucleus to induce interferon expression via binding to interferon promoters. Activated MAVS also activate the downstream transcription factor NF-κB, after the phosphorylation and activation of IKKα/IKKβ, and transport it from the cytoplasm to the nucleus to induce the expression of inflammatory factors.80–82 The expression and activation of RLRs play fundamental roles in eliminating invading RNA viruses and maintaining immune homeostasis. Among the many types of posttranslational modifications, ubiquitination and phosphorylation directly regulate the activation of RLRs and CARD-mediated downstream signaling. SUMOylation and ISGylation are newly discovered posttranslational modification modes of RLR regulation.83–86
Retinoic Acid Inducible Gene-I (RIG-I)
RIG-I is involved in the identification of paramyxovirus families, such as Newcastle disease virus (NDV), Sendai virus (SeV), respiratory syncytial virus (RSV), rhabdoviruses, herpes stomatitis virus (VSV), and rabies virus. The Orthomyxoviridae family includes influenza A, influenza B viruses (IAV, IBV), flaviviruses, hepatitis C virus (HCV), Japanese encephalitis virus (JEV), filovirus, and Ebola virus.87–90 RIG-I was initially used as a dsRNA mimic poly (I:C) ligand to induce interferon.91 Later, it was discovered that RIG-I was also a pattern recognition receptor for innate immune signaling pathways.92,93 RIG-I typically recognizes the 5’ triphosphate terminus of RNA sequences,94 which distinguishes host RNA from viral RNA. The 5’ppp domain of mature host tRNA and rRNA is covered by ribosomal proteins, and the host RNA is not recognized by RIG-I.95,96 The DNA analog poly(dA:dT) also induces interferon production via RIG-I-dependent signaling because intracellular RNA polymerase III, which transcribes DNA into RNA, is recognized by RIG-I. Some DNA viruses, such as herpesvirus-1, adenovirus, Epstein‒Barr virus, and vaccinia virus (VV), also induce interferon production via RIG-I-dependent signaling pathways. Notably, the intracellular Gram-negative bacterium Legionella pneumophila also activates the RIG-I signaling pathway.97,98 The CARD and helicase domains of RIG-I envelop the RNA binding site in cell resting states, which causes RIG-I to be in an inhibitory state. As a virus PAMP, RIG-I undergoes conformational changes by hydrolyzing ATP, which exposes the RNA binding domain. RIG-I binds to viral RNA via closer interactions and is activated to interact with MAVS.99
Positive Regulatory Factors on RIG-I
Posttranslational modification is crucial for controlling RIG-I-mediated signaling. Numerous host factors, including ubiquitin ligases and protein kinases, regulate RIG-I (Figure 1). The SPRY domain of TRIM25 at the carboxy terminus interacts with the N-terminal CARD domain of RIG-I, which promotes K63-linked ubiquitination and significantly enhances RIG-I-induced signaling pathways.11,12 TRIM4 also plays a crucial role in controlling virus-triggered IFN induction by facilitating the K63-linked ubiquitination of RIG-I.13 RNF135 is another ubiquitin ligase that preferentially binds to the RIG-I CTD and facilitates the polyubiquitination of K63 at K788, K849, K851, K888, K907, and K909. Elimination of RNF135 leads to significant impairments in the body’s natural antiviral immune reaction, including reduced production of type I IFN and decreased survival rates in mice infected with the virus. RNF135-driven RIG-I ubiquitination plays a vital role in natural antiviral immune responses.14 RNF135 enhances the interaction of TRIM25 with RIG-I, which increases the activation of RIG-I via TRIM25.100,101 NDR2 enhances the antiviral immune reaction by promoting the activation of RIG-I in macrophages via TRIM25.102 MEX3C is an E3 ubiquitin ligase that aligns with RIG-I within the stress granules of cells infected by viruses, and its increased expression triggers the lysine-63-linked ubiquitination of RIG-I, which activates the IFN-β promoter.15 Human hemoglobin subunit beta (hHB) amplifies the RIG-I-mediated antiviral reactions via the promotion of RIG-I ubiquitination, which is contingent on hHB-induced reactive oxygen species (ROS).16 In addition to the Lys63-linked ubiquitin of RIG-I, several deubiquitinating enzymes participate in the regulation of RIG-I activity. USP4 and ovarian tumor-domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) help stabilize RIG-I proteins by removing the K48-linked ubiquitin chains attached to RIG-I.18,19 SUMOylation inhibits the K48-linked degradation of RLRs. TRIM38 actively controls RIG-I and MDA5 via SUMOylation at K43/K865 and K96/K888, respectively.20
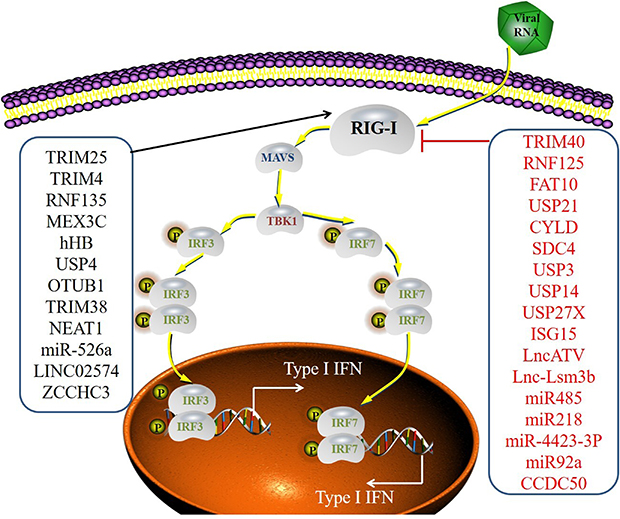 |
Figure 1 The regulator of the RIG-I signaling pathway. RIG-I recognizes RNA derived from actively replicating RNA viruses. RIG-I contains CARD-like structures that mediate interactions with the adaptor MAVS. MAVS initiates signaling pathways leading to IRF3 and IRF7 via TBK1. Activated IRF3 and IRF7 form dimers and translocate to the nucleus to induce the production of type I IFN. RIG-I is regulated by various mechanisms, such as PTMs (phosphorylation, ubiquitylation, SUMOylation and ISGylation) and small non-coding RNAs. |
LncRNA also enhances the function of RIG-I in a positive manner. For example, the lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) stimulates the RIG-I-IRF7 signaling pathway by enhancing RIG-I expression, which suppresses Hantan virus (HTNV) infection.21 LINC02574 inhibits IAV replication by increasing the expression of RIG-I, MDA5 and TLR3 and promoting the phosphorylation of IRF3, which beneficially modulates the innate immune reaction.22
Recent research suggests that miRNAs help control RLR signaling. MiR-136 is an immune agonist for RIG-I that leads to the buildup of IL-6 and IFN-β in A549 cells, and ultimately to the inhibition of IAV infection.23 RNA virus infection upregulates the production of miR-526a via IRF-dependent pathways. Therefore, miR-526a positively regulates the production of virus-induced type I IFN and prevents virus replication, primarily via the amplification of RIG-I K63 ubiquitination by miR-526a, which is achieved by inhibition of CYLD expression.17
Negative Regulatory Factors on RIG-I
K48-linked polyubiquitination controls the expression of RIG-I, which is a widely recognized process for protein degradation that is reliant on proteasomes. A number of E3 ligases that inhibit RIG-I have been reported (Figure 1). For example, TRIM40 attaches to RIG-I, which facilitates its polyubiquitination linked to K27 and K48 via its E3 ligase function and promotes the degradation of RIG-I by the proteasome to ultimately suppress the production of IFN.48 Parkin is an E3 ubiquitin-protein ligase that facilitates mitophagy, which inhibits the activation of RIG-I and MDA5 via the direct interaction and catalysis of K48-linked polyubiquitination and leads to the degradation of RIG-I and MDA5.50 RNF125 is an ubiquitin ligase that facilitates the K48-linked polyubiquitination of RIG-I, which leads to the degradation of RIG-I via proteasomes and a reduction of RIG-I-triggered IFN-β synthesis.49 During the advanced phase of infection, SUMO-specific protease 2 (SENP2) promotes the de-SUMOylation of RIG-I and MDA5, which lead to their K48-linked polyubiquitination and degradation.20 The process of ubiquitination is reversible. Multiple additional mechanisms that block the activation of RIG-I via inhibition of the K63-linked polyubiquitin chains on RIG-I have been demonstrated. USP21 is an enzyme responsible for deubiquitination, and it eliminates the K63-linked polyubiquitin chain from RIG-I, which suppresses innate immune reactions.52 CYLD is a different enzyme involved in deubiquitination, and it eliminates the K63-linked polyubiquitin chain from RIG-I to act as a negative regulatory factor.53 Syndecan-4 (SDC4) is a transmembrane (TM) protein that forms complexes with RIG-I and CYLD and facilitates the CYLD-driven deubiquitination of RIG-I, which inhibits the IFN signaling pathway.54 USP3, USP14, and USP27X remove K63-linked ubiquitin from RIG-I, which hinders RIG-I function.55–57 In addition to deubiquitinating enzymes and ubiquitin ligases, the regulatory mechanism of RIG-I via modifications similar to ubiquitin has been reported. Preliminary research indicated that conjugation of the IFN-inducible ubiquitin-like protein ISG15 (ISGylation) to RIG-I suppressed the expression of RIG-I.58 Earlier research indicated that the non-covalent attachment of FAT10 to RIG-I reduced the solubility of the RIG-I protein, which decreased RIG-I-mediated IFN expression.51
Long noncoding RNAs (lncRNAs) influence innate immune reactions by controlling RIG-I. The IFN-triggered lncRNA Lnc-Lsm3b competes with viral RNAs for binding to RIG-I monomers, which leads to the deactivation of RIG-I innate function in the advanced phase of the innate immune response.59 Analogous to lnc-Lsm3b, the lncRNA inhibitor of antiviral signaling (lncATV) is a strong inhibitor of RIG-I, which suppresses the synthesis of IFN-I and IFN-stimulated genes (ISGs) to avoid excessive activation of signaling pathways.60
The host generates miR-485, which targets and eliminates RIG-I mRNA to slow the antiviral reaction and increase viral multiplication.61 A correlational study pinpointed miR-218 as an innovative viral-triggered miRNA triggered that disrupted RIG-I expression and hindered interferon production to aids in viral immune evasion.62 MiR-4423-3p significantly enhances HCV infection by inhibiting the RIG-I/IFN pathway via direct attachment to RIG-I mRNA.63 MiR-92a specifically targets RIG-I and diminishing its expression to decrease the VSV-induced activation of TBK1 and IRF3, which are vital for initiating the transcription of type-I IFN genes.64
Melanoma Differentiation-Associated Gene 5 (MDA5)
MDA5 exhibits a similar sequence and structural resemblance to RIG-I. In contrast to RIG-I, MDA5 shows amino acid similarities of 23% and 35% in the N-terminal tandem CARD and C-terminal helicase domains, respectively. MDA5 is primarily identifies members of the small RNA virus family. However, there is growing evidence that the types of viruses identified by MDA5 are not specific. MDA5 also identifies Dengue, West Nile, respiratory enteroviruses and murine noroviruses. The viruses identified by MDA5 are not limited to RNA viruses. Recent studies have shown that MDA5 also identifies HDV. Various processes govern the function of MDA5, including posttranslational modifications (PTMs) and immunomodulatory noncoding RNAs99 (Figure 2).
 |
Figure 2 The regulator of the MDA5 signaling pathway. MDA5 recognizes RNA derived from actively replicating RNA viruses. MDA5 contains CARD-like structures that mediate interactions with the adaptor MAVS. MAVS initiates signaling pathways leading to IRF3 and IRF7 via TBK1. Activated IRF3 and IRF7 form dimers and translocate to the nucleus to induce the production of type I IFN. RIG-I is regulated by various mechanisms, such as PTMs (phosphorylation, ubiquitylation, SUMOylation and ISGylation) and small non-coding RNAs. |
Positive Regulatory Factors on MDA5
Earlier research demonstrated that multiple types of PTMs regulated MDA5 activity, including phosphorylation, ubiquitylation, SUMOylation and ISGylation. Phosphorylation of the 2 CARDs and the CTD of MDA5 inhibit abnormal activation in resting cells.24,67 Once a viral RNA is identified by MDA5, phosphatase 1 (PP1) removes phosphate groups from MDA5, which triggers the activation of its subsequent signaling pathways.24,25 Excessive activation of MDA5 leads to systemic lupus erythematosus-like autoimmune diseases. Therefore, the phosphorylation of MDA5 is likely to be crucial for the prevention of harmful MDA5 activation in cells without infection.
The MDA5 protein exhibits polyubiquitination. Similar to the RIG-I protein, K63-linked polyubiquitin chains bind to the CARD domain of MDA5.26 The K174 residue of the MDA5 CARD domain is vital for this binding. A ubiquitination-defective K174A mutant of MDA5 did not trigger type I IFN expression. TRIM65 facilitates ubiquitination linked to K63 at Lys743 of MDA5, which increases the oligomerization of MDA5 on dsRNAs.27 The ISGylation of MDA5 is similar to K63-linked ubiquitination, and it is controlled by phosphorylation, which facilitates the oligomerization of CARDs and the assembly of MDA5 filaments.103 Current research identified multiple proteins that interact with MDA5 and modulate its function. Pulldown and coimmunoprecipitation assays of glutathione S-transferase revealed that porcine 20–50-oligoadenylate synthetase-like protein (pOASL) binds to MDA5, which amplified MDA5-driven type I IFN signaling.30 The chaperone protein 14-3-3η enhances antiviral innate immunity by aiding in the oligomerization of MDA5 and its redistribution within cells.29 The zinc-finger protein ZCCHC3, which acts as a co-receptor for RIG-I and MDA5, attaches to dsRNA and increases the attachment of RIG-I and MDA5 to dsRNA. ZCCHC3 also recruits the E3 ubiquitin ligase TRIM25 to the RIG-I and MDA5 complexes, which aids in its K63-associated polyubiquitination and activation.28
LncRNAs play a role in controlling MDA5 activation, and lncITPRIP-1 amplifies the natural immune reaction to viral infections by encouraging oligomerization and activating MDA5.31 LINC02574 suppresses the replication of IAV by increasing the expression of RIG-I, TLR3 and MDA5 and the phosphorylation of IRF3 and the production of IFN.22 LINC1392 enhanced the expression of several interferon-stimulated genes (ISGs), such as IFIT1, IFIT2, and IFITM3, by stimulating MDA5, which hindered the replication of coxsackievirus B5 (CVB5) in vitro.32
Negative Regulatory Factors on MDA5
TRIM40 attaches to the CARD domain of MDA5 and accelerates the K27- and K48-linked polyubiquitination of MDA5, which results in the proteasomal degradation of MDA5.48 Viral infection amplifies the expression of CCDC50, and CCDC50 specifically recognizes RLRs that are K63-polyubiquitinated, which delivers the activated RIG-I/MDA5 for degradation by autophagy and inhibits the type I interferon (IFN) signaling pathway.65 The protein kinase RIOK3 phosphorylates the CTD domain of MDA5 and inhibits IFN expression mediated by MDA5. However, RIOK3 does not phosphorylate the CARD domain of MDA5, and a different protein kinase likely phosphorylates the CARDs of MDA5.67 The E3 ubiquitin ligase TRIM13 in immune cells shows increased expression in macrophages derived from bone marrow after stimulation with type I IFN inducers. TRIM13 interacts with MDA5 to inhibit the MDA5-mediated production of type I IFN in vitro during EMCV infection.66 Human hemoglobin subunit beta (hHB) impedes MDA5-driven antiviral signals by inhibiting the MDA5–dsRNA interaction.16 The dual-luciferase reporter assay results showed that miR-203 targeted the MDA5–3’UTR sequence, and miRNA-203 may act as an inhibitor of MDA5 in miiuy croaker.68
Toll-Like Pattern Recognition Receptors (TLRs)
TLRs are highly expressed in many types of cells, such as macrophages, dendritic cells, neutrophils, natural killer cells, and fibroblasts.104 TLRs are a class of transmembrane proteins that include an extracellular region, transmembrane region, and the Toll interleukin-1 receptor region (TIR). The extracellular region is a leucine-rich repeat sequence that recognizes PAMPs of pathogens.105 Transmembrane and TIR regions bind to adaptor proteins to activate downstream signaling pathways. Thirteen toll-like receptors have been reported. TLR1-9 is conserved in humans and mice. TLR10 lost function after retroviral insertion in mic, but TLR11-13 is only expressed in mice.106–108
The localization of TLRs differs. TLR2, TLR4 and TLR5 are located on the outer membrane of the cell, and TLR3, TLR7, TLR8 and TLR9 are located on the endosome. Different TLRs recognize different ligands, which are primary membrane structures of microorganisms, such as lipids, lipoproteins, and proteins. TLR4 recognizes the lipopolysaccharides of Gram-positive bacteria.109,110 TLR2 combines with TLR1 or TLR6 to bind various bacterial components, such as peptidoglycan from Gram-positive bacteria, lipopeptides, lipoprotein bacteria, and mycoplasma lipopeptides.111–114 TLR3 primarily recognizes double-stranded RNA (dsRNA) during viral replication, such as hepatitis C virus (HCV), Japanese encephalitis virus (JEV), dengue virus (DENV), and enterovirus 71 (EV71).115 TLR5 primarily recognizes the flagella of bacteria.116 TLR7 recognizes synthetic imidazoline-like quinoline molecules, guanosine analogs, or single-stranded RNA (ssRNA) from viruses, such as human immunodeficiency virus type I (HIV-1), vesicular stomatitis virus (VSV), and influenza viruses, as well as certain small interfering RNAs (siRNAs).117,118 TLR9 recognizes CpG-DNA sequences in bacteria and viruses.119,120 Human TLR8 recognizes single-stranded RNA (ssRNA), but mouse TLR8 is nonfunctional. All TLRs mediate the production of inflammatory cytokines. The activation of TLR3, TLR4, TLR7, TLR8, and TLR9 leads to the production of IFNs, which are important for antiviral immune responses. This review focuses on Toll-like pattern recognition receptor 3, which recognizes viruses (Figure 3).
 |
Figure 3 The regulator of the TLR3 signaling pathway. TLR3 recognizes viral dsRNA. TLR3 mediates the interaction with the adaptor TRIF. TRIF initiates signaling pathways leading to IRF3 via TBK1. Activated IRF3 dimerizes and translocates to the nucleus to induce the production of type I IFN. TLR3 is regulated by various mechanisms, such as PTMs. |
Toll-Like Pattern Recognition Receptor 3 (TLR3)
Toll-like receptor 3 (TLR3) facilitates the transcriptional activation of type I interferons (IFNs), proinflammatory cytokines and chemokines, which together form an antiviral response in hosts. In contrast to other TLR family proteins, TLR3 entirely relies on the Toll/interleukin-1 receptor (TIR) domain-containing adaptor inducing IFN-β (TRIF) for RNA sensing.120 TLR3 and TRIF interact to recruit TNF receptor-associated factor (TRAF3) and activate two kinases, TANK-binding kinase 1 (TBK) and inhibitor-κB kinase ε (IKKε). Ultimately, the translation factor IRF3 is activated by phosphorylation and dimerizes. The IRF3 dimer translocates from the cytoplasm to the nucleus where it induces the production of type I IFN.
Positive Regulatory Factors on TLR3
TLR3 activation is linked to K63-linked polyubiquitination. The E3 ubiquitin ligase TRIM3 primarily resides in the Golgi apparatus and is transported to early endosomes when stimulated by the dsRNA analog poly (I:C). TRIM3 facilitates the K63-associated polyubiquitination of TLR3 at K831, a process that intensifies after poly (I:C) stimulation.33 Bruton’s tyrosine kinase (BTK) in macrophages increases the production of inflammatory cytokines and IFN-β via TLR3, which suppresses intracellular dengue virus infection.35 Mex3B is an RNA-binding protein that enhances TLR3-mediated signaling in two distinct manners. The attachment of Mex3B to dsRNA enhances the ability of TLR3 to bind to dsRNA. Mex3B facilitates the proteolytic processing of TLR3, which is essential for its activation.36 Mex3B enhances the innate immune response by facilitating the K63-linked ubiquitination of TLR3.34 The FYVE domain of ZFYVE1 increases TLR3-driven innate immune and inflammatory responses by facilitating the binding of ligands to TLR3.37
Negative Regulatory Factors on TLR3
E3 ubiquitin ligases and deubiquitinase act as master regulators of TLR signaling by co-regulating the dynamic and reversible ubiquitination process.121 The TLR3-binding E3 ligase RNF170 mediates the K48-linked poly-ubiquitination of K766 in the TIR domain of TLR3. RNF170 selectively inhibits the TLR3-mediated pathway by promoting TLR3 degradation via the proteasome pathway.69
Intracellular DNA Sensors
RNA viruses induce immune responses by activating TLR or RLR signaling pathways, and DNA viruses are recognized by other types of pattern recognition receptors, such as DDX41, IFI16, and cGAS. Compared to the regulation of RLRs by PTMs, we know little about PTMs that directly regulate cGAS or IFI16 activity. Recent studies have shown that phosphorylation, acetylation, and glutamylation play important roles in controlling these sensors. However, the mechanism by which host factors regulate the production of IFN via PTMs on DDX41 and IFI16 has rarely been reported. The following section focuses on the host regulators of the pattern recognition receptor cyclic adenylate synthetase (cGAS).
Cyclic Adenylate Synthetase (cGAS)
cGAS contains one nucleotide transferase domain and two DNA-binding domains. cGAS is self-inhibited in the resting state. cGAS can bind to DNA in the cytoplasm to form a 2:2 complex, which is activated by conformational changes and catalyzes the cyclization of adenosine triphosphate (ATP) and guanosine (GTP) to form the second messenger cyclic guanosine (cGAMP).120,122 cGAMP binds to the adaptor protein STING, located in the ER, to cause a conformational change in STING, which may be the cause of STING activation. Activated STING migrates from the endoplasmic reticulum to the Golgi apparatus.123 During the transfer process, the carboxyl terminus of STING recruits and activates TBK1, which forms dimers after phosphorylation and activation of the transcription factor IRF3. IRF3 migrates from the cytoplasm to the nucleus to induce IFN-β production. STING also activates the IKK complex, and the IKK complex can phosphorylate and ubiquitinate the NF-κB transcription inhibitor IkB-α to separate IkB-α from NF-κB, which promotes its activation. Translocation to the nucleus induces the production of inflammatory factors, such as TNF, IL-β, and IL-6.124,125
cGAS is activated by double-stranded DNA, such as HSV-1. Its ability to recognize DNA is not sequence specific. The crystal structure of the cGAS and DNA complex indicates that cGAS primarily binds to the sugar-phosphate chain of double-stranded DNA. Single-stranded DNA is also recognized by cGAS via the formation of internal double-stranded structures. The cGAS-STING signaling pathway produces an immune response against the invasion of DNA-containing pathogens and generates an anti-tumor immune response. However, overactivation of the cGAS-STING signaling pathway by DNA may also lead to autoimmune and inflammatory diseases, such as systemic lupus erythematosus. Therefore, proper regulation of the cGAS-STING signaling pathway is crucial for protection against foreign pathogens.126 Posttranslational modifications, such as phosphorylation, ubiquitination, SUMOylation, and acetylation, play key roles in cGAS signaling regulation127 (Figure 4).
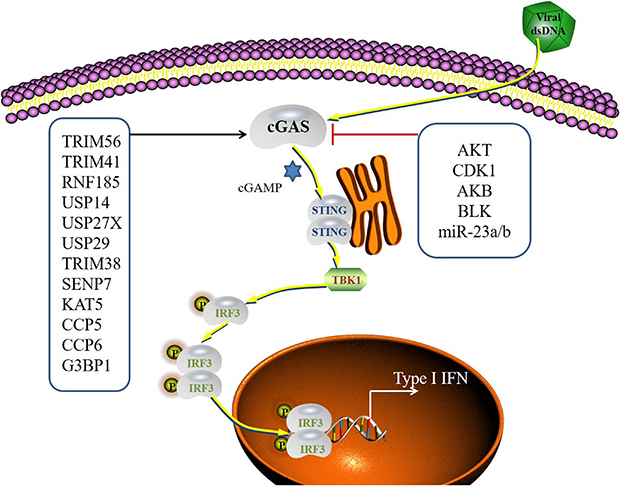 |
Figure 4 The regulator of the cGAS signaling pathway. cGAS recognizes viral dsRNA and activates a STING-dependent signaling pathway. STING initiates signaling pathways leading to IRF3 via TBK1. Activated IRF3 dimerizes and translocates to the nucleus to induce the production of type I IFN. cGAS is regulated by various mechanisms, such as PTMs (phosphorylation, ubiquitylation, SUMOylation, glutamylation and acetylation) and small non-coding RNAs. |
Positive Regulatory Factors on cGAS
Ubiquitination
Several ubiquitin ligases have been identified as regulators of cGAS activity. TRIM56 is an E3 ligase that monoubiquitinates cGAS at Lys335 to significantly enhance its dimerization, DNA-binding ability and cGAMP generation.38 Like TRIM56, TRIM41 also increases the production of IFN by promoting cGAS monoubiquitination.39 RNF185 is an ER ubiquitin ligase that specifically facilitates the K27-linked polyubiquitination of cGAS at K-384 to enhance its enzymatic function and IFN production.40 Patients with systemic lupus erythematosus (SLE) exhibit continuous activation of cGAMP-STING signaling128 and increased RNF185 mRNA levels. Whether the increase in RNF185 mRNA plays a role in enhancing cGAS-STING signaling is not certain. Reports indicate that during DNA virus infection, TRIM14 enlists USP14 to eliminate K48-linked ubiquitin chains at the Lys414 site in h-cGAS, which results in cGAS stabilization and the enhancement of antiviral innate immunity.41 Similarly, USP27X42 and USP2943 contribute to the stabilization of cGAS proteins via the removal of K48-linked polyubiquitin chains from cGAS, and both of these factors act as enhancers of innate immunity to combat DNA viral infections.
SUMOylation
Similar to ubiquitination, TRIM38 preserves the SUMOylation of m-cGAS at the Lys217 and Lys464 residues (equivalent to Lys231 and Lys479 in hcGAS), which inhibit the K48-linked polyubiquitination and degradation of cGAS.42 Another study revealed that SENP7, a protease specific to SUMO, played a positive role in controlling cGAS-mediated signaling. cGAS undergoes SUMOylation at K335, K372, and K382. Because the SUMOylation sites are located on the DNA-binding surface or dimerization interface of cGAS, the SUMOylation of cGAS at these locations hinders its capacity for DNA binding and self-association. Upon HSV-1 infection, SENP7 actively engages with cGAS to deSUMOylate cGAS and consequently activating cGAS.44
Acetylation
The acetylation process of cGAS can control its activity in a positive or negative manner depending on the specific acetylation location. Protein acetylation of lysine residues is facilitated by acetyltransferases and deacetylases.129 In the resting state, deacetylation of hcGAS at the Lys384, Lys394, and Lys414 residues enhances cGAS activity.130 Recently, the lysine acetyltransferase KAT5 was shown to acetylate the N-terminal region of human cGAS at the Lys47/Lys56/Lys62/Lys83 residues. Acetylation enhances the binding of cGAS to DNA and its activity in response to DNA challenge.45 Additional functional verification indicated that acetylation at Lys198 enhanced the activation of h-cGAS. Notably, quantitative proteomic analysis revealed a reduction in h-cGAS-Lys198 acetylation following infection by HSV-1 or HCMV (human cytomegalovirus), which suggests that these DNA viruses bypass innate immune monitoring by commandeering the acetylation of cGAS at these locations.131
Glutamylation
Glutamylation is a posttranslational alteration that is facilitated by tubulin tyrosine ligase-like (TTLLs) glutamylases and is counteracted by enzymes belonging to the cytosolic carboxypeptidase (CCP) family.132 Glutamylation stringently controls cGAS. Mice lacking CCP5 or CCP6 are sensitive to DNA virus infections due to the unsuccessful activation of type I interferons.46 This study revealed that polyglutamatergic TTLL6 at Glu272 reduces the binding capacity of cGAS for DNA, and monoglutamatergic TTLL4 at Glu302 inhibits the enzymatic function of cGAS. Conversely, CCP6 and CCP5, which are analogous to TTLL6 and TTLL4, respectively, facilitate the deglutamylation of cGAS at glu272 and glu302, which alleviates the inhibitory impact of glutamylation. TTLL4, TTLL6, CCP5, and CCP6 collaboratively influence cGAS-driven immune reactions by dynamically controlling cGAS glutamylation. However, the mechanism by which these enzymes are activated or controlled to regulate the glutamylation, and deglutamylation of cGAS is ambiguous. Additional research is required to clarify this question and enhance our understanding of cGAS regulation.
In addition to the posttranslational modifications of cGAS, some host factors promote IFN synthesis by directly increasing the formation of cGAS complexes. For example, the GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) plays a pivotal role in DNA detection and the effective activation of cGAS, and G3BP1 enhances the binding of cGAS to DNA by facilitating the formation of cGAS complexes.47
Negative Regulatory Factors on cGAS
Phosphorylation
The phosphorylation of cGAS by serine kinases, including serine/threonine protein kinase (AKT), cyclin-dependent kinase-1 (CDK1), and Aurora kinase B (AKB), inhibits cGAS function, which affects the immune response.70–72 Infection by HSV-1 in 293T cells triggers AKT activation, which subsequently phosphorylates cGAS at positions S291 (m-cGAS) and S305 (h-cGAS).71 The phosphorylation of cGAS at positions S291/S301 hindered the production of cGAMP, which affected the generation of type I IFNs.71 Another study revealed that HSV-1 infection in human primary fibroblasts and HEK293T cells led to the phosphorylation of cGAS at various serine residues (Ser37, Ser116, Ser201, Ser221, and Ser263), which inhibited cGAS function during their posttranslational alteration. However, the specific signaling mechanisms involved in this phosphorylation are not clear.131 Zhong et al demonstrated a comparable reduction in cGAS activity following phosphorylation at the S305 site by the CDK1-cyclin B kinase complex in cells undergoing mitosis.72 Recent findings by Li et al revealed that AKB hyperphosphorylation of the cGAS N-terminus at specific serine and threonine sites (Ser13, Ser37, Ser64, Thr69, Thr91, Ser116, Ser129, and Ser143) inhibited chromatin DNA detection in cells undergoing mitosis.70 Therefore, it seems likely that AKT, CDK1, and AKB regulate cGAS phosphorylation to manage cGAS activation in vital cell cycle stages, such as DNA replication and mitotic division. However, the phosphorylation of cGAS by these kinases was counteracted by protein phosphatase 1 (PP1)72 or PP6,133 which restored the capacity of cGAS to sense DNA. Notably, cGAS encompasses multiple tyrosine residues whose functions are not known. Research revealed that the phosphorylation of Tyr215 in BLK (B lymphocyte kinase) in hcGAS resulted in the retention of cGAS within the cytoplasm, which is essential for the detection of cytosolic DNA.73
cGAS activity is regulated by miRNAs. Overexpression of miR-23a/b markedly suppresses cytosolic DNA-induced innate immune responses.74
Conclusion
Innate immunity is the first line of defense of host cells against the invasion of foreign microorganisms and resistance to the entry of viruses. It plays an important role in the invasion process. There are many host factors that regulate innate immune responses. Host factors promote the immune response and enhance the body’s clearance of invading viruses. However, an excessive immune response leads to tissue damage and induces autoimmune diseases. Host factors suppress the immune response, so that the body cannot clear the virus quickly, which results in chronic infection. To balance the immune response induced by viral infection, the immune response was modified, and a complete regulatory mechanism was established. In this review, we focused on the host factors that regulate the IFN signaling pathway induced by virus infection via pattern recognition receptors. Pattern recognition receptors (RIG-I, MDA5, TLR3, and cGAS) are regulated by various mechanisms, such as PTMs (phosphorylation, ubiquitylation, SUMOylation and ISGylation) and small non-coding RNAs. Posttranslational modification is one of the key factors in host antiviral immune defense and viral immune escape, which strictly regulates the activation of the virus-induced innate immune response signaling pathway. Viral infection promotes the expression of host factors, which inhibit interferon production by targeting PRRs and results in immune escape. We found that some host factors regulate the production of IFNs via different PRRs. For example, ZCCHC3 positively regulates the IFN signaling pathway via MDA5 and RIG-I, which suggests crosstalk between different PRR-mediated IFN signaling pathways. Exploring the regulatory role of host factors on PRRs will help us better understand the mechanism of antiviral immune defense in the body and provide targets and a theoretical basis for accurate clinical diagnosis and treatment of virus-related diseases caused by IFNs. Increasing focus has been placed on the functions of PRRs in autoimmune disorders and cancer-fighting immunity, but the regulatory mechanisms of PRRs by environmental, pathogen and host factors are largely unknown. Delving deeper into these inquiries will undoubtedly elucidate our comprehension of PRR-driven PAMP detection pathways and help formulate approaches to avert and manage associated diseases.
Author Contributions
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all of these areas, took part in drafting, and revising or critically reviewing the article. All authors gave final approval of the manuscript version to be published, agreed on the journal for submission, and agreed to be accountable for all aspects of the work.
Funding
This study was financially supported by the Doctoral Research Start-up Fund of the Henan University of Science and Technology (JJW), Fundamental Research Funds for Henan University of Science and Technology (QNY, grant No. 13510001) and National College Students’ innovation and entrepreneurship training program (YRD, grant No. 202310464088).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hoffmann HH, Schneider WM, Rice CM. Interferons and viruses: an evolutionary arms race of molecular interactions. Trend Immunol. 2015;36(3):124–138. doi:10.1016/j.it.2015.01.004
2. Liu J, Cao X. Cellular and molecular regulation of innate inflammatory responses. Cell Mol Immunol. 2016;13(6):711–721. doi:10.1038/cmi.2016.58
3. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Ann Rev Immunol. 2014;32:461–488. doi:10.1146/annurev-immunol-032713-120156
4. Hornung V, Hartmann R, Ablasser A, Hopfner KP. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol. 2014;14(8):521–528. doi:10.1038/nri3719
5. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87–103. doi:10.1038/nri3787
6. Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16(1):35–50. doi:10.1038/nri.2015.8
7. Hu MM, Shu HB. Cytoplasmic mechanisms of recognition and defense of microbial nucleic acids. Annu Rev Cell Dev Biol. 2018;34:357–379. doi:10.1146/annurev-cellbio-100617-062903
8. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trend Immunol. 2014;35(2):88–93. doi:10.1016/j.it.2013.10.010
9. Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3(6):920–940. doi:10.3390/v3060920
10. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
11. Gack MU, Shin YC, Joo CH, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446(7138):916–920. doi:10.1038/nature05732
12. Okamoto M, Kouwaki T, Fukushima Y, Oshiumi H. Regulation of RIG-I Activation by K63-Linked Polyubiquitination. Front Immunol. 2017;8:1942. doi:10.3389/fimmu.2017.01942
13. Yan J, Li Q, Mao AP, Hu MM, Shu HB. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J Molecul Cell Biol. 2014;6(2):154–163. doi:10.1093/jmcb/mju005
14. Lai Y, Liang M, Hu L, et al. RNF135 is a positive regulator of IFN expression and involved in RIG-I signaling pathway by targeting RIG-I. Fish Shellfish Immunol. 2019;86:474–479. doi:10.1016/j.fsi.2018.11.070
15. Kuniyoshi K, Takeuchi O, Pandey S, et al. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc Natl Acad Sci USA. 2014;111(15):5646–5651. doi:10.1073/pnas.1401674111
16. Yang Q, Bai SY, Li LF, et al. Human hemoglobin subunit beta functions as a pleiotropic regulator of RIG-I/MDA5-mediated antiviral innate immune responses. J Virol. 2019;93(16). doi:10.1128/JVI.00718-19
17. Xu C, He X, Zheng Z, et al. Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J Virol. 2014;88(19):11356–11368. doi:10.1128/JVI.01400-14
18. Wang L, Zhao W, Zhang M, et al. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J Virol. 2013;87(8):4507–4515. doi:10.1128/JVI.00031-13
19. Jahan AS, Biquand E, Muñoz-Moreno R, et al. OTUB1 is a key regulator of RIG-I-dependent immune signaling and is targeted for proteasomal degradation by influenza A NS1. Cell Rep. 2020;30(5):1570–1584.e1576. doi:10.1016/j.celrep.2020.01.015
20. Hu MM, Liao CY, Yang Q, Xie XQ, Shu HB. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J Exp Med. 2017;214(4):973–989. doi:10.1084/jem.20161015
21. Ma H, Han P, Ye W, et al. The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J Virol. 2017;91(9). doi:10.1128/JVI.02250-16
22. Zhang Y, Chi X, Hu J, et al. LncRNA LINC02574 inhibits influenza A virus replication by positively regulating the innate immune response. Int J Mol Sci. 2023;24(8):1.
23. Zhao L, Zhu J, Zhou H, et al. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci Rep. 2015;5:14991. doi:10.1038/srep14991
24. Wies E, Wang MK, Maharaj NP, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38(3):437–449. doi:10.1016/j.immuni.2012.11.018
25. Davis ME, Wang MK, Rennick LJ, et al. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe. 2014;16(1):19–30. doi:10.1016/j.chom.2014.06.007
26. Wallach D, Kovalenko A. Phosphorylation and dephosphorylation of the RIG-I-like receptors: a safety latch on a fateful pathway. Immunity. 2013;38(3):402–403. doi:10.1016/j.immuni.2013.02.014
27. Lang X, Tang T, Jin T, Ding C, Zhou R, Jiang W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J Exp Med. 2017;214(2):459–473. doi:10.1084/jem.20160592
28. Lian H, Zang R, Wei J, et al. The zinc-finger protein ZCCHC3 Binds RNA and Facilitates Viral RNA sensing and activation of the RIG-I-like Receptors. Immunity. 2018;49(3):438–448.e435. doi:10.1016/j.immuni.2018.08.014
29. Lin JP, Fan YK, Liu HM. The 14-3-3η chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathogens. 2019;15(2):e1007582. doi:10.1371/journal.ppat.1007582
30. Li LF, Yu J, Zhang Y, et al. Interferon-inducible oligoadenylate synthetase-like protein acts as an antiviral effector against classical swine fever virus via the MDA5-mediated type I interferon-signaling pathway. J Virol. 2017;91(11). doi:10.1128/JVI.01514-16
31. Xie Q, Chen S, Tian R, et al. Long noncoding RNA ITPRIP-1 positively regulates the innate immune response through promotion of oligomerization and activation of MDA5. J Virol. 2018;92(17). doi:10.1128/JVI.00507-18
32. Li J, Li J, Teng P, et al. Long noncoding RNA 1392 regulates MDA5 by interaction with ELAVL1 to inhibit coxsackievirus B5 infection. Virologica Sin. 2023;38(5):699–708. doi:10.1016/j.virs.2023.08.001
33. Li WW, Nie Y, Yang Y, et al. Ubiquitination of TLR3 by TRIM3 signals its ESCRT-mediated trafficking to the endolysosomes for innate antiviral response. Proc Natl Acad Sci USA. 2020;117(38):23707–23716. doi:10.1073/pnas.2002472117
34. Hu J, Xiang Y, Zhu X, et al. Grass carp (Ctenopharyngodon idella) Mex3B positively regulates innate immunity by promoting the K63-linked ubiquitination of TLR3. Fish Shellfish Immunol. 2023;141:109023. doi:10.1016/j.fsi.2023.109023
35. Lee KG, Xu S, Kang ZH, et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci USA. 2012;109(15):5791–5796. doi:10.1073/pnas.1119238109
36. Yang Y, Wang SY, Huang ZF, et al. The RNA-binding protein Mex3B is a coreceptor of Toll-like receptor 3 in innate antiviral response. Cell Res. 2016;26(3):288–303. doi:10.1038/cr.2016.16
37. Zhong X, Feng L, Xu WH, et al. The zinc-finger protein ZFYVE1 modulates TLR3-mediated signaling by facilitating TLR3 ligand binding. Cell Mol Immunol. 2020;17(7):741–752. doi:10.1038/s41423-019-0265-6
38. Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun. 2018;9(1):613. doi:10.1038/s41467-018-02936-3
39. Liu ZS, Zhang ZY, Cai H, et al. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018;8:35. doi:10.1186/s13578-018-0233-3
40. Wang Q, Huang L, Hong Z, et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLOS Pathogens. 2017;13(3):e1006264. doi:10.1371/journal.ppat.1006264
41. Chen M, Meng Q, Qin Y, et al. TRIM14 Inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Molecular Cell. 2016;64(1):105–119. doi:10.1016/j.molcel.2016.08.025
42. Hu MM, Yang Q, Xie XQ, et al. Sumoylation promotes the stability of the DNA Sensor cGAS and the Adaptor STING to regulate the kinetics of response to DNA virus. Immunity. 2016;45(3):555–569. doi:10.1016/j.immuni.2016.08.014
43. Zhang Q, Tang Z, An R, Ye L, Zhong B. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 2020;30(10):914–927. doi:10.1038/s41422-020-0341-6
44. Cui Y, Yu H, Zheng X, et al. SENP7 potentiates cGAS activation by relieving SUMO-mediated inhibition of cytosolic DNA sensing. PLoS Pathogens. 2017;13(1):e1006156. doi:10.1371/journal.ppat.1006156
45. Song ZM, Lin H, Yi XM, Guo W, Hu MM, Shu HB. KAT5 acetylates cGAS to promote innate immune response to DNA virus. Proc Natl Acad Sci USA. 2020;117(35):21568–21575. doi:10.1073/pnas.1922330117
46. Xia P, Ye B, Wang S, et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol. 2016;17(4):369–378. doi:10.1038/ni.3356
47. Liu ZS, Cai H, Xue W, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. 2019;20(1):18–28. doi:10.1038/s41590-018-0262-4
48. Zhao C, Jia M, Song H, et al. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. 2017;21(6):1613–1623. doi:10.1016/j.celrep.2017.10.020
49. Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. 2007;104(18):7500–7505. doi:10.1073/pnas.0611551104
50. Bu L, Wang H, Hou P, et al. The Ubiquitin E3 ligase parkin inhibits innate antiviral immunity through K48-linked polyubiquitination of RIG-I and MDA5. Front Immunol. 2020;11:1926. doi:10.3389/fimmu.2020.01926
51. Nguyen NT, Now H, Kim WJ, Kim N, Yoo JY. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci Rep. 2016;6:23377. doi:10.1038/srep23377
52. Fan Y, Mao R, Yu Y, et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J Exp Med. 2014;211(2):313–328. doi:10.1084/jem.20122844
53. Friedman CS, O’Donnell MA, Legarda-Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9(9):930–936. doi:10.1038/embor.2008.136
54. Lin W, Zhang J, Lin H, et al. Syndecan-4 negatively regulates antiviral signalling by mediating RIG-I deubiquitination via CYLD. Nat Commun. 2016;7:11848. doi:10.1038/ncomms11848
55. Cui J, Song Y, Li Y, et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014;24(4):400–416. doi:10.1038/cr.2013.170
56. Li H, Zhao Z, Ling J, et al. USP14 promotes K63-linked RIG-I deubiquitination and suppresses antiviral immune responses. Europ J Immunol. 2019;49(1):42–53. doi:10.1002/eji.201847603
57. Tao X, Chu B, Xin D, Li L, Sun Q. USP27X negatively regulates antiviral signaling by deubiquitinating RIG-I. PLoS Pathogens. 2020;16(2):e1008293. doi:10.1371/journal.ppat.1008293
58. Kim MJ, Hwang SY, Imaizumi T, Yoo JY. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008;82(3):1474–1483. doi:10.1128/JVI.01650-07
59. Jiang M, Zhang S, Yang Z, et al. Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response. Cell. 2018;173(4):906–919.e913. doi:10.1016/j.cell.2018.03.064
60. Fan J, Cheng M, Chi X, Liu X, Yang W. A human long non-coding RNA LncATV promotes virus replication through restricting RIG-I-mediated innate immunity. Front Immunol. 2019;10:1711. doi:10.3389/fimmu.2019.01711
61. Ingle H, Kumar S, Raut AA, et al. The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci Signaling. 2015;8(406). doi:10.1126/scisignal.aab3183
62. Qiu Y, Geng X, Ban J, Liu Y. MicroRNA-218 inhibits type I interferon production and facilitates virus immune evasion via targeting RIG-I. Biotechnol Appl Biochem. 2020;67(3):396–403. doi:10.1002/bab.1882
63. Qian X, Wu B, Xu C, Qi Z. Hepatitis C virus infection cycle-specific microRNA profiling reveals stage-specific miR-4423-3p Targets RIG-I to facilitate infection. Front Cell Infect Microbiol. 2022;12:851917. doi:10.3389/fcimb.2022.851917
64. Sheng Y, Wang Y, Lu W, et al. MicroRNA-92a inhibits macrophage antiviral response by targeting retinoic acid inducible gene-I. Microbiol Immunol. 2018;62(9):585–593. doi:10.1111/1348-0421.12640
65. Hou P, Yang K, Jia P, et al. A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. 2021;31(1):62–79. doi:10.1038/s41422-020-0362-1
66. Narayan K, Waggoner L, Pham ST, et al. TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J Virol. 2014;88(18):10748–10757. doi:10.1128/JVI.02593-13
67. Takashima K, Oshiumi H, Takaki H, Matsumoto M, Seya T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015;11(2):192–200. doi:10.1016/j.celrep.2015.03.027
68. Chu Q, Han J, Sun L, Cui J, Xu T. Characterization of MDA5 and microRNA-203 negatively regulates the RLR signaling pathway via targeting MDA5 in miiuy croaker. Dev Comp Immunol. 2022;126:104259. doi:10.1016/j.dci.2021.104259
69. Song X, Liu S, Wang W, Ma Z, Cao X, Jiang M. E3 ubiquitin ligase RNF170 inhibits innate immune responses by targeting and degrading TLR3 in murine cells. Cell Mol Immunol. 2020;17(8):865–874. doi:10.1038/s41423-019-0236-y
70. Li T, Huang T, Du M, et al. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science. 2021;371(6535). doi:10.1126/science.abc5386
71. Seo GJ, Yang A, Tan B, et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 2015;13(2):440–449. doi:10.1016/j.celrep.2015.09.007
72. Zhong L, Hu MM, Bian LJ, Liu Y, Chen Q, Shu HB. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discovery. 2020;6:26. doi:10.1038/s41421-020-0162-2
73. Zhang F, Yuan Y, Ma F. Function and Regulation of Nuclear DNA Sensors During Viral Infection and Tumorigenesis. Front Immunol. 2020;11:624556. doi:10.3389/fimmu.2020.624556
74. Yu Q, Chu L, Li Y, et al. miR-23a/b suppress cGAS-mediated innate and autoimmunity. Cell Mol Immunol. 2021;18(5):1235–1248. doi:10.1038/s41423-021-00668-x
75. Jiang F, Ramanathan A, Miller MT, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479(7373):423–427. doi:10.1038/nature10537
76. Kowalinski E, Lunardi T, McCarthy AA, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147(2):423–435. doi:10.1016/j.cell.2011.09.039
77. Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988. doi:10.1038/ni1243
78. Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi:10.1038/nature04193
79. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Molecular Cell. 2005;19(6):727–740. doi:10.1016/j.molcel.2005.08.014
80. Benhaim M, Lee KK, Guttman M. Tracking higher order protein structure by hydrogen-deuterium exchange mass spectrometry. Protein Pept Lett. 2019;26(1):16–26. doi:10.2174/0929866526666181212165037
81. Cadena C, Hur S. Filament-like assemblies of intracellular nucleic acid sensors: commonalities and differences. Molecular Cell. 2019;76(2):243–254. doi:10.1016/j.molcel.2019.09.023
82. Peisley A, Wu B, Yao H, Walz T, Hur S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Molecular Cell. 2013;51(5):573–583. doi:10.1016/j.molcel.2013.07.024
83. Jiang X, Kinch LN, Brautigam CA, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36(6):959–973. doi:10.1016/j.immuni.2012.03.022
84. Kato H, Takahasi K, Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev. 2011;243(1):91–98. doi:10.1111/j.1600-065X.2011.01052.x
85. Oshiumi H, Matsumoto M, Seya T. Ubiquitin-mediated modulation of the cytoplasmic viral RNA sensor RIG-I. J Biochem. 2012;151(1):5–11. doi:10.1093/jb/mvr111
86. Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020;20(9):537–551. doi:10.1038/s41577-020-0288-3
87. Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi:10.1038/nature04734
88. Faul EJ, Wanjalla CN, Suthar MS, Gale M, Wirblich C, Schnell MJ. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathogens. 2010;6(7):e1001016. doi:10.1371/journal.ppat.1001016
89. Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82(1):335–345. doi:10.1128/JVI.01080-07
90. Cárdenas WB, Loo YM, Gale M, et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80(11):5168–5178. doi:10.1128/JVI.02199-05
91. Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–737. doi:10.1038/ni1087
92. Schlee M, Hartmann E, Coch C, et al. Approaching the RNA ligand for RIG-I? Immunol Rev. 2009;227(1):66–74. doi:10.1111/j.1600-065X.2008.00724.x
93. Schlee M, Hartmann G. The chase for the RIG-I ligand--recent advances. Molecular Therapy. 2010;18(7):1254–1262. doi:10.1038/mt.2010.90
94. Hornung V, Ellegast J, Kim S, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi:10.1126/science.1132505
95. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10(10):1065–1072. doi:10.1038/ni.1779
96. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138(3):576–591. doi:10.1016/j.cell.2009.06.015
97. Melchjorsen J, Rintahaka J, Søby S, et al. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J Virol. 2010;84(21):11350–11358. doi:10.1128/JVI.01106-10
98. Valentine R, Smith GL. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J Gen Virol. 2010;91(Pt 9):2221–2229. doi:10.1099/vir.0.021998-0
99. Zheng J, Shi W, Yang Z, et al. RIG-I-like receptors: molecular mechanism of activation and signaling. Advan Immunol. 2023;158:1–74. doi:10.1016/bs.ai.2023.03.001
100. Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 2009;284(2):807–817. doi:10.1074/jbc.M804259200
101. Oshiumi H, Miyashita M, Matsumoto M, Seya T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathogens. 2013;9(8):e1003533. doi:10.1371/journal.ppat.1003533
102. Liu Z, Wu C, Pan Y, et al. NDR2 promotes the antiviral immune response via facilitating TRIM25-mediated RIG-I activation in macrophages. Sci Adv. 2019;5(2):1.
103. Liu G, Lee JH, Parker ZM, et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nature Microbiol. 2021;6(4):467–478. doi:10.1038/s41564-021-00884-1
104. Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. doi:10.1038/41131
105. Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164(11):5564–5574. doi:10.4049/jimmunol.164.11.5564
106. Kawai T, Akira S. TLR signaling. Semin Immunopathol. 2007;19(1):24–32. doi:10.1016/j.smim.2006.12.004
107. Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388(4):621–625. doi:10.1016/j.bbrc.2009.08.062
108. Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420(1):1–16. doi:10.1042/BJ20090272
109. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi:10.1126/science.282.5396.2085
110. Hoshino K, Takeuchi O, Kawai T, et al. Pillars article: cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the lps gene product. J Immunol. 1999;162:3749–3752. doi:10.4049/jimmunol.162.7.3749
111. Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. doi:10.1016/S1074-7613(00)80119-3
112. Takeuchi O, Kaufmann A, Grote K, et al. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol. 2000;164(2):554–557. doi:10.4049/jimmunol.164.2.554
113. Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169(1):10–14. doi:10.4049/jimmunol.169.1.10
114. Takeuchi O, Kawai T, Mühlradt PF, et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Internat Immunol. 2001;13(7):933–940. doi:10.1093/intimm/13.7.933
115. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi:10.1038/35099560
116. Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410(6832):1099–1103. doi:10.1038/35074106
117. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3(2):196–200. doi:10.1038/ni758
118. Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101(15):5598–5603. doi:10.1073/pnas.0400937101
119. Krug A, French AR, Barchet W, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21(1):107–119. doi:10.1016/j.immuni.2004.06.007
120. Coban C, Ishii KJ, Kawai T, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med. 2005;201(1):19–25. doi:10.1084/jem.20041836
121. Zheng Y, Gao C. Fine-tuning of antiviral innate immunity by ubiquitination. Advan Immunol. 2020;145:95–128.
122. Li X, Shu C, Yi G, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39(6):1019–1031. doi:10.1016/j.immuni.2013.10.019
123. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:7213):674–678. doi:10.1038/nature07317
124. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING activation by translocation from the ER Is associated with infection and autoinflammatory disease. Cell Host Microbe. 2015;18(2):157–168. doi:10.1016/j.chom.2015.07.001
125. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signaling. 2012;5(214):ra20. doi:10.1126/scisignal.2002521
126. Herzner AM, Hagmann CA, Goldeck M, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. 2015;16(10):1025–1033. doi:10.1038/ni.3267
127. Joshi B, Joshi JC, Mehta D. Regulation of cGAS activity and downstream signaling. Cells. 2022;11(18):2812. doi:10.3390/cells11182812
128. Kato Y, Park J, Takamatsu H, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheumatic Dis. 2018;77(10):1507–1515. doi:10.1136/annrheumdis-2018-212988
129. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20(3):156–174. doi:10.1038/s41580-018-0081-3
130. Dai J, Huang YJ, He X, et al. Acetylation Blocks cGAS activity and inhibits Self-DNA-induced autoimmunity. Cell. 2019;176(6):1447–1460.e1414. doi:10.1016/j.cell.2019.01.016
131. Song B, Greco TM, Lum KK, Taber CE, Cristea IM. The DNA Sensor cGAS is Decorated by Acetylation and Phosphorylation Modifications in the Context of Immune Signaling. Molecul Cellul Proteom. 2020;19(7):1193–1208. doi:10.1074/mcp.RA120.001981
132. Rogowski K, van Dijk J, Magiera MM, et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143(4):564–578. doi:10.1016/j.cell.2010.10.014
133. Li M, Shu HB. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein and Cell. 2020;11(8):584–599. doi:10.1007/s13238-020-00729-3
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.