Back to Journals » Journal of Inflammation Research » Volume 16
HDAC1 is Involved in Neuroinflammation and Blood-Brain Barrier Damage in Stroke Pathogenesis
Authors Wang HK ![]() , Su YT, Ho YC, Lee YK
, Su YT, Ho YC, Lee YK ![]() , Chu TH, Chen KT, Wu CC
, Chu TH, Chen KT, Wu CC ![]()
Received 2 May 2023
Accepted for publication 12 September 2023
Published 18 September 2023 Volume 2023:16 Pages 4103—4116
DOI https://doi.org/10.2147/JIR.S416239
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Hao-Kuang Wang,1– 3 Yu-Ting Su,4 Yu-Cheng Ho,3,5 Yung-Kuo Lee,6 Tian-Huei Chu,6 Kuang-Ti Chen,7 Cheng-Chun Wu3,5
1Department of Neurosurgery, E-DA Hospital, I-Shou University, Kaohsiung City, Taiwan; 2School of Medicine for International Students, College of Medicine, I-Shou University, Kaohsiung City, Taiwan; 3Graduate Institute of Medicine, College of Medicine, I-Shou University, Kaohsiung City, Taiwan; 4Department of Obstetrics and Gynecology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung City, Taiwan; 5School of Medicine, College of Medicine, I-Shou University, Kaohsiung City, Taiwan; 6Medical Laboratory, Medical Education and Research Center, Kaohsiung Armed Forces General Hospital, Kaohsiung City, Taiwan; 7Department of Veterinary Medicine, Nation Chung-Hsing University, Taichung City, Taiwan
Correspondence: Cheng-Chun Wu, School of Medicine, College of Medicine, I-Shou University, Kaohsiung City, Taiwan, Tel +886-7-6151100-7961, Fax +886-7-6155150, Email [email protected]
Background: Stroke is a common cause of disability and mortality worldwide; however, effective therapy remains limited. In stroke pathogenesis, ischemia/reperfusion injury triggers gliosis and neuroinflammation that further activates matrix metalloproteinases (MMPs), thereby damaging the blood–brain barrier (BBB). Increased BBB permeability promotes macrophage infiltration and brain edema, thereby worsening behavioral outcomes and prognosis. Histone deacetylase 1 (HDAC1) is a repressor of epigenomic gene transcription and participates in DNA damage and cell cycle regulation. Although HDAC1 is deregulated after stroke and is involved in neuronal loss and DNA repair, its role in neuroinflammation and BBB damage remains unknown.
Methods: The rats with cerebral ischemia were evaluated in behavioral outcomes, levels of inflammation in gliosis and cytokines, and BBB damage by using an endothelin-1-induced rat model with cerebral ischemia/reperfusion injury.
Results: The results revealed that HDAC1 dysfunction could promote BBB damage through the destruction of tight junction proteins, such as ZO-1 and occludin, after stroke in rats. HDAC1 inhibition also increased the levels of astrocyte and microglial gliosis, tumor necrosis factor-alpha, interleukin-1 beta, lactate dehydrogenase, and reactive oxygen species, further triggering MMP-2 and MMP-9 activity. Moreover, modified neurological severity scores for the cylinder test revealed that HDAC1 inhibition deteriorated behavioral outcomes in rats with cerebral ischemia.
Discussion: HDAC1 plays a crucial role in ischemia/reperfusion-induced neuroinflammation and BBB damage, thus indicating its potential as a therapeutic target.
Keywords: HDAC1, stroke, blood-brain barrier, TNF-α, IL-1β, ROS, mNSS, cylinder test
Introduction
Stroke is a common cause of disability and death worldwide. Ischemic stroke, the most common type of stroke, results from the interruption of adequate blood and oxygen supply to the brain, leading to neuronal death and brain functional impairment.1,2 Approximately 14 million individuals have stroke per year; among them, 5.5 million patients die and 5 million are permanently disabled.3,4 Currently, effective therapy for stroke remains limited.
The blood–brain barrier (BBB) is a critical interface between the brain parenchyma and vasculature and regulates the transcellular transport of nutrients and essential components.5 The BBB mainly regulates ion homeostasis,6 hormone7 and transmitter movement,8 blood flow, angiogenesis, neuronal development, and synaptic activity.9 The BBB, a dynamic structure that acts as a physical and metabolic barrier, is composed of cells sealed by tight junction proteins.10 The BBB protects the brain from intrusive elements and is involved in the bidirectional transport of materials.11
In stroke, ischemia causes immediate neuronal death due to decreased ATP synthesis. This is followed by reactive oxygen species (ROS)-induced DNA and cell membrane damage, which leads to cell death or degeneration hours or days after ischemic stroke. Furthermore, ATP dysfunction causes excitotoxicity that triggers cell apoptosis and ROS accumulation.12,13 In addition, neuronal damage elicits a neuroinflammatory response from reactive microglia and astrocytes. The gliosis mediates cytokine release, thereby contributing to neuronal loss and ischemia/reperfusion injury or secondary injuries.14,15 Moreover, neuroinflammation during stroke pathogenesis can cause BBB damage, leading to cytotoxic edema and increased permeability. Ischemia/reperfusion injury is associated with the restoration of blood and oxygen supply and may progress over hours to weeks after artery occlusion.16 The reactive microglia and astrocytes secrete inflammatory cytokines and cytotoxic compounds, such as interleukin (IL)-1β, IL-1α, tumor necrosis factor (TNF)-α, IL-6, and nitric oxide,17,18 that disrupt the BBB and increase its permeability by intruding tight junction proteins, such as claudin-5, occludin, and zonula occludens (ZO)-1.19 Furthermore, TNF-α can trigger apoptotic cascades and matrix metalloproteinases (MMPs) in neurons and glial cells.20 The IL-1-induced endothelial cell reactivation and immune cell infiltration after BBB disruption additionally contribute to increases in cytokine, chemokine, and MMP-9 levels.21 The loss of BBB tight junction integrity disturbs paracellular permeability, exacerbating immune cell infiltration, brain edema, and hemorrhagic transformation, resulting in worsened neurological outcomes and mortality.22 Therefore, mechanisms underlying BBB disruption should be elucidated for the development of novel stroke therapeutics.
Gene transcription is controlled by histone acetylation, which is antagonistically modulated by histone acetyltransferases and deacetylases (HDACs).23 HDAC1, a member of the HDAC family, modulates DNA damage and neuronal survival in the brain under neuronal damage and degeneration conditions.24–27 HDAC1 plays a critical role in stroke pathogenesis. Ischemia/reperfusion injury induces HDAC1 dysfunction, which exacerbates neuronal loss and DNA damage after stroke. This explains the poor behavioral outcomes of rats with ischemia and HDAC1 inhibition in some studies.28,29 However, the role of HDAC1 in ischemia/reperfusion-induced neuroinflammation and BBB damage remains unknown, and the effect of HDAC1 on the activity of glial cells after stroke remains controversial. Therefore, we examined the interplays of HDAC1 in this pathologic progress.
Materials and Methods
Animal Experiments and Drug Administration
In this study, the Institute of Animal Care and Use Committee of I-Shou University and E-Da Hospital approved the procedures of animal experiments (IACUC-EDAH-108017, 6 August 2019; IACUC-EDAH-108037, 21 February 2021; IACUC-ISU-108015, 8 October 2019), and we followed the Guidelines for the Care and Use of Laboratory Animals, Council of Agriculture, Taiwan to care the animal and conduct experiments. We purchased adult male SD rats from the Lasco biotechnology company (Taipei, Taiwan), a total of 102 male rats were used in this study. We used a rat model of ischemia/reperfusion injury established through the stereotactic intracerebral injection of endothelin-1, a vessel constrictor. An HDAC1 selective inhibitor, N-(2-aminophenyl)-4-[N-(pyridine-3yl-methoxy-carbonyl) aminomethyl] benzamide (MS-275), was stereotactically injected to inhibit HDAC1 function in 8 weeks old of rats weighed 250–300 g.27–29 We randomly allocated the rats for the experimental groups as sham control, endothelin-1 cerebral microinjection, and endothelin-1 combined MS-275 microinjection. In the cerebral ischemia/reperfusion model creation, we stereotaxic injected 3 µL of 100 pM endothelin-1 (Sigma, E7764; St. Lois, MO, USA) into the brain followed the brain atlas30 by three coordinates: AP 0, ML + 2.5, DV − 2.3; AP + 2.3, ML + 2.5, DV − 2.3; AP + 0.7, ML + 3.8, DV − 7.0. HBSS was stereotaxically injected as a sham control. MS-275 was stereotaxic injected following pre-mixed with endothelin-1; a total of 3 μL of mixed volume in a final concentration of 100μM of MS-275 and 100 pM of endothelin-1 was synchronized and injected into the brain. Twenty-four hours after the surgery, the experimental rats were sacrificed for further examinations by Western blot and immunostainings.
Evaluation of HDACs Enzymatic Activity
To detect in vivo HDAC1 activity, we employed a nuclear extraction kit (Active Motif; Carlsbad, CA) to isolate nuclear protein fractions, following the manufacturer’s guidelines. In summary, dissected brain samples were homogenized in hypotonic buffer on ice for 15 minutes, followed by centrifugation at 850 g for 10 minutes. The resulting pellet was reconstituted in a hypotonic buffer containing 0.5% detergent and kept on ice for 15 minutes, after which centrifugation at 14,000 g for 1 minute occurred. The nuclear protein was solubilized by adding a complete lysis buffer. For nuclear HDAC1 analysis, 800 μg of fresh nuclear protein was extracted from each sample and subjected to immunoprecipitation (IP) at 4°C overnight. The IP products were utilized to measure HDACs activity using the activity assay kits (Enzo Life Sciences, Farmingdale, NY; BML-AK500-0001, BML-AK512, BML-AK531, BML-AK518), following the provided instructions.
Evans Blue Assay
We conducted Evan`s blue (EB) assay to evaluate the BBB permeability 24 h after stroke, following the previously reported procedures. After anesthesia, 2% of EB dissolved in saline was injected for 4 mL/kg by the tail vein. The rats were trans-cardially perfused and removed from the brain after waiting for 30 min for complete circulation.31 The brain was sliced by a rat brain slicer (World Precision Instruments, Sarasota, FL, USA). A total of 8 brain sections were prepared from one brain, and the EB extravasation was quantified by Image J. Values from the contralateral region were used as control.
Immunofluorescent Staining
In the work of Immunofluorescent staining, we followed the procedure from our previous study.29 First, we anesthetized the rats and performed trans-cardio perfusion with PBS and 4% paraformaldehyde. Then we removed the brain for post-fixation and dehydration and embedded the tissue with an optical coherence tomography compound. The brain region within bregma +2 to −4 mm was separated and prepared for brain sections. We collected the brain sections at ten µm per section; one of three sections was ordered and adhered to the slide to accumulate the brain samples. We performed antibody hybridization on brain slides using the primary antibodies: Zo-1 (Genetex GTX108627; Hsinchu City, Taiwan), Iba-1 (Genetex GTX635363), GFAP (Abclonal A0237; Xinbei City). We further adopt AlexaFluor-conjugated secondary antibodies (Thermo; Waltham, MA) to recognize the primary antibodies. The nucleus of the tissue was stained with DAPI and mounted by a mounting medium (Dako; Glostrup, Denmark). In quantification of the immunoreactivity signals, we conducted a microscopic platform of high throughput screening (ImageXpress® Automated Imaging System, Molecular Device; San Jose, CA) to acquire the high-content images from the penumbra area of injured brains and analyzed the data by its accompanied software (MetaMorph®, Molecular Device).
Western Blots, ELISA, and Assays for ROS, LDH, and MMP Activity
Brain protein extractions were prepared from the brain tissue from the regions within bregma: +3 to −1 mm), and we used a brain slicer to separate the brain regions. The detailed procedure of Western blots was described in our previous study.32 The ROS and LDH assay kits were purchased from BioVision K936-100-250 and K726 (Milpitas, CA, USA) and followed the manual’s instructions. ELISA kits for detecting IL-1β and TNF-α were acquired from R&D RLB00 and RTA00 (Minneapolis, MN). In the MMP9 activity assay, we purchased the kit from Abcam AB234057 (Cambridge, UK). We experimented and followed the manufacturer’s directions using fresh samples of brain lysates.
Behavioral Tests
We evaluated the neurological outcomes of experimental rats on PSDs 1, 3, and 7 by performing the modified neurological severity score (mNSS). The experimental procedures and detail can reference our previous work.29 The evaluations include gait, climbing, body symmetry, forelimb flexion, turning ability, compulsory circling, and sensory response. Three grades evaluated each task, and total scores were summarized as the performance assessment. In addition, we experimented with the cylinder test following our previous work.29 The rats were put in a transparent cylinder and allowed to move freely. We measured the number of using forepaws from the contralateral and ipsilateral sides to place on the cylinder wall. The ratio of forepaw use was quantified as R/(L + R)*100%. Two days before the surgery, we conducted a pre-preconditioning that allowed rats to stay in the cylinder for 5 min, two times, to help the rats acclimate to the experimental condition.
Statistics
All data were confirmed with normal distribution. They were presented as the mean ± SEM and were analyzed by one-way ANOVA with post hoc Tukey’s test for multiple-group comparison or by Student’s t-test for 2-group comparison. Differences with p < 0.05 were considered statistically significant.
Results
HDAC1 Inhibition Exacerbated BBB Damage in vivo
We employed Evans blue staining to assess the permeability of the blood-brain barrier (BBB) 24 hours after ischemia/reperfusion injury induced by endothelin-1. To inhibit HDAC1 function, we utilized a neutralization approach involving microinjection of MS-275 into the brain concurrently with endothelin-1. Previous studies have characterized the specificity of MS-275 for HDAC1.27,28 However, partial studies also indicated that MS-275 may affect other class 1 HDAC members such as HDAC 2, 3, and 8,33 in addition to the brain tissue. Therefore, we further investigated and confirmed the specificity of MS-275 for HDAC1 in the brain at the current concentration, as shown in Figure S1.
At 24 hours post-surgery, the rats were euthanized, and brain slices were prepared. The level of BBB disruption was determined by analyzing the area of the brain stained with Evans blue. The findings revealed that rats experiencing cerebral ischemia and HDAC1 inhibition had higher BBB permeability (Figure 1A), which was demonstrated by a greater stained brain area. Additionally, HDAC1 inhibition exacerbated the disruption of the BBB (Figure 1B), and a statistically significant difference was observed in the stained area between rats injected with the vehicle and those injected with MS-275, both of which had cerebral ischemia. Thus, severe extravasation of Evans blue was observed in rats with cerebral ischemia and HDAC1 inhibition following stroke, suggesting that HDAC1 is involved in maintaining the integrity of the BBB after stroke.
 |
Figure 1 HDAC1 dysfunction promoted BBB damage in rats 24 h after stroke. (A) The representative data of Evan`s blue staining from the cerebral ischemia rats 24 h after surgery. (B) The quantified data for Evan`s blue staining from cerebral ischemia rats. Sham n=6, Stroke+Vehicle n=8, Stroke+MS275 n=8. Data was evaluated by one-way ANOVA, *p < 0.05, **p <0.01, ***p <0.001. |
To further confirm the role of HDAC1 in BBB damage, we employed HDAC1 siRNA through stereotactic microinjection of antisense oligomers simultaneously with endothelin-1. The data revealed an increase in BBB disruption at 24 h after stroke when HDAC1 was knocked down (Figure S2A), supporting the crucial function of HDAC1 in BBB damage. Subsequently, we investigated the effect of neuroinflammation in BBB damage at the sub-acute stage by conducting a delayed MS-275 stereotactic injection two weeks after the stroke. The data demonstrated that stroke-induced BBB damage remained, but no difference was observed between cerebral ischemia rats with delayed MS-275 injection and cerebral ischemia rats only (Figure S2B). This suggests that HDAC1 plays a vital role in the acute stage of neuroinflammation. Finally, to determine whether MS-275 can directly induce BBB damage, we injected it into the rat brain. Compared to the sham control, there was no significant difference in BBB damage in the Evans blue assay (Figure S2C). This indicates that BBB damage is primarily mediated by ischemia/reperfusion injury in our model.
HDAC1 Inhibition Worsened Disruption of Tight Junction Structure
To further determine the effect of HDAC1 in BBB disruption undergoing cerebral ischemia. We performed immunofluorescence staining for ZO-1 (Figure 2A and C), an essential component of BBB tight junctions. Rats with cerebral ischemia exhibited decreased ZO-1 immunofluorescence activity, indicating that ischemia/reperfusion injury disrupted BBB tight junctions (Figure 2B). Notably, the decline in ZO-1 expression was more prominent in rats with cerebral ischemia and HDAC1 inhibition (Figure 2B), suggesting that HDAC1 is essential for the maintenance of the BBB after stroke.
 |
Figure 2 HDAC1 dysfunction decreased the expression of tight junction associated protein- ZO-1 24 h after stroke. (A) The representative figures of immunofluorescent staining for ZO-1 in cerebral ischemia rats 24 h after stroke. The white square denotes an amplified view from the merged figure. Bar: 100 μm; Bar: 400 μm in amplify (B) The quantified data of immunofluorescent staining. (C) The red square denotes where the general view of immunostainings was captured in the brain sections. Sham n=6, Stroke+Vehicle n=8, Stroke+MS275 n=8. Data was evaluated by one-way ANOVA, *p < 0.05, **p <0.01, ***p <0.001. |
Next, we performed Western blot analysis to evaluate the levels of ZO-1 and occludin, proteins associated with the structural maintenance of tight junctions. We collected the brain tissue from the rats with cerebral ischemia and prepared protein lysates 24 h after stroke. Decreased levels of ZO-1 and occludin were observed, indicating that ischemia/reperfusion considerably damaged the BBB (Figure 3A). Furthermore, HDAC1 inhibition promoted the disruption of BBB structural proteins in rats with cerebral ischemia, leading to a further reduction in the protein levels (Figure 3A and B). Therefore, HDAC1 inhibition exacerbates the destruction of tight junction proteins, further increasing BBB permeability.
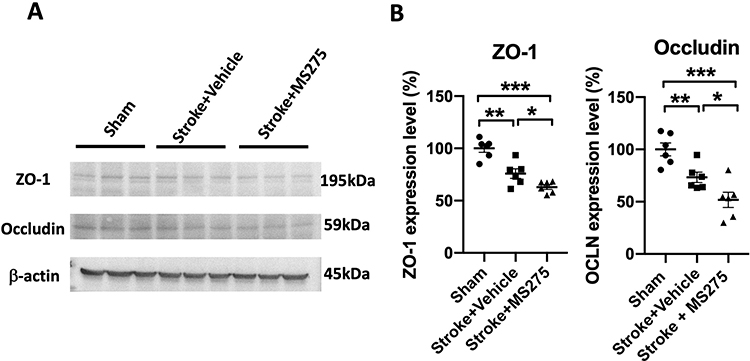 |
Figure 3 HDAC1 dysfunction reduced the tight junction-associated proteins in ZO-1 and occluding 24 h after stroke. (A) The representative Western blotting data for ZO-1 and occludin 24 h after stroke. β-actin served as an internal control. (B) The quantified levels of ZO-1 and occluding normalized to internal control. N=6 per group. Data was evaluated by one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001. |
HDAC1 Inhibition Promoted Gliosis
Neuronal damage induced by ischemia/reperfusion injury can trigger the reactivation of astrocytes and microglia, leading to the accumulation of inflammatory cytokines and the promotion of BBB disruption in stroke pathogenesis. Therefore, we investigated the levels of gliosis in astrocytes and microglia. The rats with cerebral ischemia and with or without HDAC1 inhibition were sacrificed and perfused to remove the brain for section preparation. Sham-operated rats were used as control. To evaluate astrocyte gliosis, we performed immunofluorescence staining for glial fibrillary acidic protein (GFAP) and determined the cell number in the ischemic core and penumbra (Figure 4A and C). Tissue quantification revealed an increased number of cells in the ischemic core and penumbra, indicating that HDAC1 inhibition worsened astrocyte gliosis (Figure 4B). In addition, we performed immunofluorescence staining for ionized calcium-binding adapter molecule 1 (Iba-1) to evaluate microglial gliosis (Figure 5A and C). An increased number of round cells with extended protrusions was observed in rats with cerebral ischemia and HDAC1 inhibition (Figure 5B); this finding indicated that HDAC1 inhibition increased the number of reactive microglial cells. Therefore, HDAC1 inhibition increased the reactive cell numbers of astrocytes and microglia, suggesting the essential role of HDAC1 in BBB disruption through gliosis.
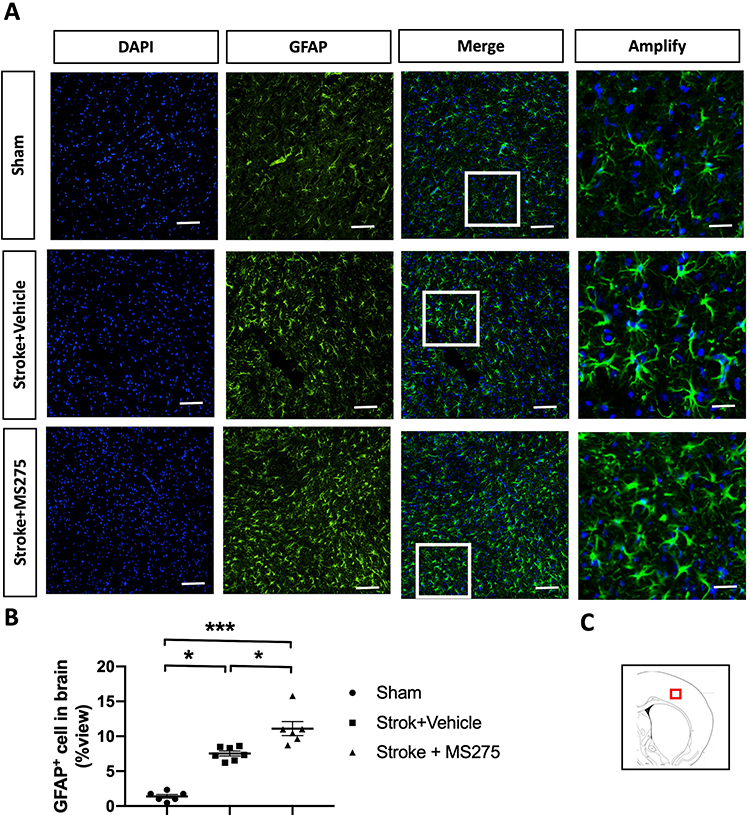 |
Figure 4 HDAC1 dysfunction increased the astrocytic gliosis 24 h after stroke. (A) The representative figures of immunofluorescent staining for GFAP in cerebral ischemia rats. The white square denotes an amplified view from the merged figure. Bar: 50 μm; Bar: 200 μm in amplify (B) The quantified data of immunofluorescent staining for GFAP. (C) The red square denotes where the general view of immunostainings was captured in the brain sections. Sham n=6, Stroke+Vehicle n=8, Stroke+MS275 n=8. Data was evaluated by one-way ANOVA, *p < 0.05, ***p <0.001. |
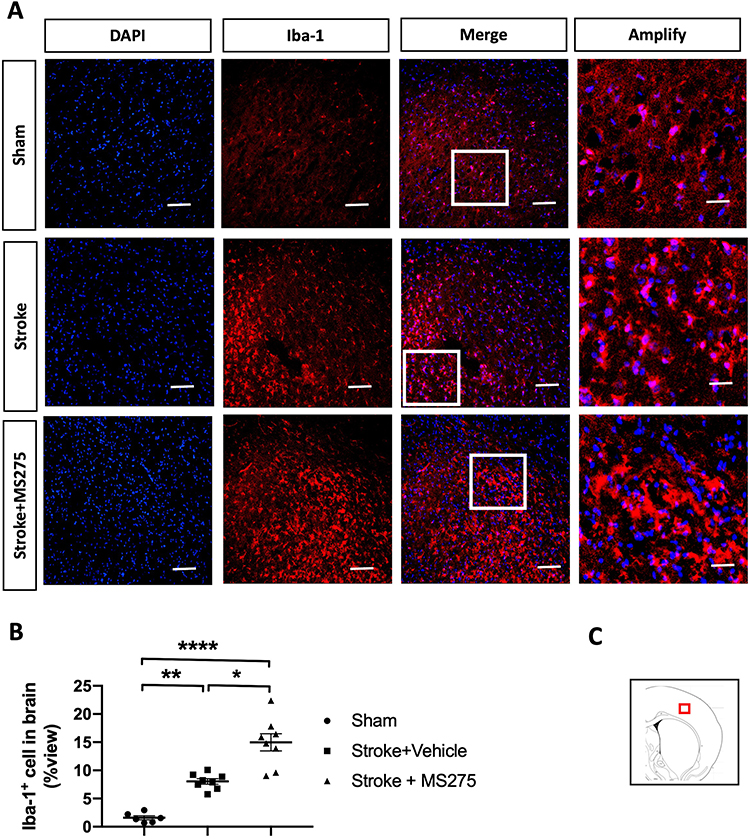 |
Figure 5 HDAC1 dysfunction increased the microglial gliosis 24 h after stroke. (A) The representative figures of immunofluorescent staining for GFAP in cerebral ischemia rats. The white square denotes an amplified view from the merged figure. Bar: 50 μm; Bar: 200 μm in amplify (B) The quantified data of immunofluorescent staining for GFAP. (C) The red square denotes where the general view of immunostainings was captured in the brain sections. Sham n=6, Stroke+Vehicle n=8, Stroke+MS275 n=8. Data was evaluated by one-way ANOVA, *p < 0.05, **p <0.01, ****p <0.0001. |
HDAC1 Inhibition Aggravated Neuroinflammation
BBB disruption is highly associated with inflammatory cytokine levels following ischemia/reperfusion-induced gliosis. Therefore, we examined the levels of the inflammatory cytokines IL-1β and TNF-α 24 h after stroke. HDAC1 inhibition increased the cytokine levels and gliosis in rats with cerebral ischemia (Figure 6A and B). In addition, we determined the levels of lactate dehydrogenase (LDH) and ROS in the brain lysates 24 h after stroke. The results indicated that HDAC1 inhibition caused severe damage to the brain, resulting in increased levels of LDH and ROS (Figure 6C and D). Moreover, TNF-α and IL-1β promoted BBB damage through MMP-9. Therefore, we determined the enzymatic activity of MMP-2 and MMP-9 24 h after stroke by using the gelatinase activity assay. The results indicated that ischemia/reperfusion injury significantly increased the activity of MMP-2 and MMP-9. Compared with vehicle-injected rats, MS-275-injected rats exhibited increased MMP-2 and MMP-9 activity (Figure 6E), suggesting that HDAC1 dysfunction exacerbates BBB damage through gliosis-induced inflammatory cytokines and MMPs.
 |
Figure 6 HDAC1 dysfunction worsened the levels of inflammation cytokines, ROS, and LDH 24h after stroke. (A and B) ELISA was conducted for inflammation cytokines in IL-1b and TNF-a. (C and D) ROS and LDH were detected 24 h after stroke from the brain lysates of cerebral ischemia rats. (E) MMPs activity assay was conducted to evaluate the enzymatic activity of MMP2 and MMP9 24 h after stroke. Sham n=6, Stroke+Vehicle n=8, Stroke+MS275 n=8. Data was evaluated by one-way ANOVA, *p < 0.05, **p <0.01, ***p <0.001. |
HDAC1 Inhibition Deteriorated Behavioral Outcomes After Stroke
BBB disruption considerably affects neurological outcomes and mortality. To investigate the effect of HDAC1-induced BBB damage on the behavioral outcomes of rats with cerebral ischemia, we evaluated the modified neurological severity score (mNSS) for neuromuscular function and cylinder tests. The results indicated that ischemia/reperfusion significantly reduced rats’ neuromuscular ability. HDAC1 inhibition increased brain damage (Figure 7A), further impairing the neuromuscular response. The cylinder test results revealed that HDAC1 inhibition reduced the ability of rats with cerebral ischemia to use their contralateral forepaw (Figure 7B). Therefore, HDAC1 dysfunction negatively affects behavioral outcomes and increases neuroinflammation and BBB disruption.
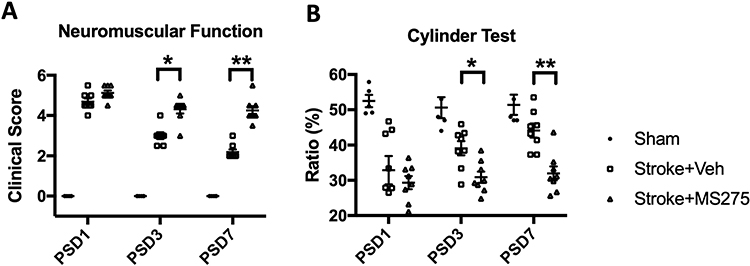 |
Figure 7 HDAC1 dysfunction elicited worsened behavioral outcomes in the mNSS and cylinder tests at post-stroke days (PSD) 1, 3, 7. (A) Evaluations for neuromuscular function by modified neural severity scores (mNSS) in cerebral ischemia rats at PSD 1, 3, 7. (B) Evaluation for forepaw using ability in cerebral ischemia rats at PSD 1, 3, 7. Sham n=6, Stroke+Vehicle n=8, Stroke+MS275 n=8. Data was evaluated by one-way ANOVA, *p < 0.05, **p <0.01. |
Discussion
This study evaluated the role of HDAC1 in stroke-induced BBB damage. The results indicated that BBB disruption is more severe when HDAC1 activity is inhibited. HDAC1 inhibition increased neuroinflammation by increasing the levels of gliosis, TNF-α, IL-1β, ROS, and LDH in the brain after stroke. Cerebral ischemia and HDAC1 inhibition deteriorated behavioral outcomes in rats after stroke. Our study provides novel insights into the role of HDAC1 in the pathogenesis of ischemia/reperfusion injury and its involvement in neuroinflammation and BBB damage after stroke. These results may aid in the development of therapeutic strategies for ischemia/reperfusion injury.
Although promising HDAC-based therapeutic approaches for stroke have been reported, the role of a specific HDAC in stroke pathogenesis remains controversial. Most studies have focused on nonselective HDAC inhibitors, such as suberoylanilide hydroxamic acid (a selective inhibitor of HDAC1, 2, 3, 4, 5, 6, 7, and 9), sodium 4-phenylbutyrate (a nonspecific HDAC inhibitor), valproic acid (VPA, a selective inhibitor of HDAC1, 2, 3, and 8), and trichostatin A (TSA, a selective inhibitor of HDAC1, 4, 6, 10, and 11), that exert neuroprotective effects to ameliorate inflammation, endoplasmic reticulum stress, excitotoxicity, oxidative stress, apoptosis, and BBB damage after stroke.34 VPA protects against the disruption of BBB tight junction proteins, such as claudin-5 and ZO-1, and attenuates NF-κB and MMP-9 levels in the brain.35 In addition, the class IIA HDAC inhibitor TMP269 maintains tight junction proteins, such as ZO-1, occludin, and claudin-5, and protects the BBB after ischemia/reperfusion injury.36
The exact role of a specific HDAC in stroke pathogenesis remains controversial, with few studies mentioning the involvement of a specific HDAC in the pathological progression of stroke. A study reported that HDAC4 levels increased following stroke, thereby protecting against BBB damage through the stabilization of tight junction proteins and reduction of nicotinamide adenine dinucleotide phosphate oxidase and MMP-9 levels.37 Sodium butyrate (a selective HDAC4 inhibitor) may exert a neuroprotective effect against BBB damage.38,39 Furthermore, oxygen–glucose deprivation (OGD) can upregulate HDAC9 in the endothelial cells of the brain, resulting in decreased levels of tight junction proteins and impairment of BBB permeability.40 Furthermore, OGD can upregulate HDAC3 and peroxisome proliferator-activated receptor gamma (PPARγ) in human microvascular endothelial cells, resulting in elevated BBB permeability and decreased tight junction protein expression. Therefore, RGFP966 (an HDAC3 inhibitor) treatment can recover paracellular permeability and promote claudin-5 expression through the PPARγ receptor.41 This study provides insights into the role of HDAC1 in stroke pathogenesis. In addition to protecting against DNA and neuronal damage, HDAC1 participated in the pathological mechanism of neuroinflammation and BBB disruption. HDAC1 dysfunction can deteriorate pathological outcomes; therefore, HDAC1 modulation should be studied for the development of therapeutic strategies.
Most studies developing therapeutic approaches for neuroinflammation have focused on nonspecific HDAC inhibitors. For example, VPA can ameliorate microglial reactivation in LPS-induced neuroinflammation,42 and TSA can reduce the expression of GFAP in astrocytes, thus inhibiting astrocyte reactivation.43 Additional studies on TSA, VPA, sodium butyrate, and ginger-rhizome-derived compounds have indicated that nonspecific HDAC inhibition reduced the levels of inflammatory cytokines, such as IL-6, inducible nitric oxide synthase (iNOS), cyclooxygenase-2, IL-1β, heat shock protein 70, and TNF-α both in vitro and in vivo.42,44–46 In addition, belinostat and TSA, nonspecific HDAC inhibitors, ameliorated experimental autoimmune encephalitis–induced neuroinflammation by reducing the levels of M1 microglia, TNF-α, and IL-1β.47,48 However, the role of a specific HDAC in neuroinflammation and BBB damage remains unexplored.
An HDAC2-based study reported that HDAC2 overexpression activated gliosis and increased the mRNA levels of GFAP, Iba1, and iNOS in a mouse model of retina ischemia; however, HDAC1 overexpression was not involved in the pathological progression of retina ischemia.49 Another study reported that HDAC3 exacerbates neuroinflammation by activating microglia through the cGAS-STING pathway.50,51 Therefore, the selective inhibition of HDAC3 through RGF966 or knockdown ameliorated neuroinflammation levels in the mouse models of demyelination, middle cerebral artery occlusion, and LPS-induced neuroinflammation.52–54 Similar results have been reported in studies on HDAC6 and HDAC8 using the model of LPS-induced microglial activation.55,56 Therefore, the inhibition of specific HDACs is crucial for the function of cells and tissues in some diseases. Moreover, it can reduce disease severity and can be used as a potential therapeutic approach.
In stroke pathogenesis, ischemia/reperfusion injury induces a considerable inflammatory response in the brain to control the injured microenvironment, remove the damaged neural tissue, and repair the damaged brain. However, the severe neuroinflammation promotes MMP activity, thereby degrading tight junction proteins, triggering BBB damage, and worsening neurological outcomes in animal models and patients.57 In this study, the selected dose of MS-275 was determined based on findings from our prior study, in which we confirmed its impact on behavioral outcomes.28 Thus, we have maintained the same dosage in the current study to further assess BBB damage post-stroke. In addition, given the limitations imposed by intracerebral injection volume, we lack information regarding whether higher doses of MS-275 could yield more significant effects on HDAC1. However, our earlier research has already established that HDAC1 dysregulation exacerbates brain damage and impairs motor function after stroke, evident in terms of infarction volume, neuronal loss, DNA damage, and elevated inflammation cytokines.28 Remarkably, by delayed MS-275 stereotactic injection at two weeks after stroke as the sub-acute stage, HDAC1 dysfunction-induced BBB damage was not found (Figure S2B). This data further highlights that inflammation levels could be essential in stroke-associated BBB damage.
In our data, MS275 seems to trigger a more intense BBB damage locally as seen by increased Evan’s blue staining intensity. In our investigation, the stroke model was induced through the stereotaxic injection of endothelin-1. This design establishes the affected region based on the diffusion pattern of endothelin-1 post intracerebral injection. As a result, both the extent of infarct volume and the region of BBB damage are profoundly linked to this diffusion process. Moreover, the infiltration of monocytes can potentially escalate inflammation subsequent to BBB damage, thereby intensifying the brain injury within the context of stroke pathogenesis.58,59 Hence, based on our observations, we posit that this insight provides a plausible explanation for the occurrence of localized BBB damage.
In a study, HDAC1 overexpression reduced microglia viability in an OGD model in vitro and promoted M1-type microglial polarization and TNF-α synthesis.60 HDAC1 modulates M1-type microglial polarization by targeting the acetylation levels of kinesin family member 4A.61 In addition, HDAC1 knockdown may reduce LPS-induced cytokine expression in microglia in vitro.62 Unlike previous studies, we adopted a neutralization approach and inhibited HDAC1. Our data indicated that HDAC1 dysfunction could further worsen neuroinflammation and BBB damage. Moreover, in our study, HDAC1 inhibition triggered microglia and astrocyte reactivation, leading to a further increase in the levels of inflammatory cytokines and MMP-9; a result contradictory to those of previous studies. The varying results may be attributed to the difference in the study model. The results of previous studies were obtained from LPS-induced neuroinflammation in microglia; however, we used a rat model of ischemia/reperfusion injury. In addition, the time point of the experimental assessment may have resulted in varying results. We focused on the acute stage and evaluated the experiments 24 h after stroke, which is difficult to reproduce in the macrophage cell line by using LPS stimulation.
This study is an extension of our previous research on stroke.28 In this study, HDAC1 inhibition enhanced gliosis and IL-1β and TNF-α levels 24 h after stroke. We believe this is crucial in the pathological progression of stroke because IL-1β amplifies the inflammatory response and promotes neutrophil infiltration after ischemia/reperfusion injury.63 Infiltration may further activate ROS production, causing oxidative stress.64,65 In addition, the elevated TNF-α level can trigger ROS production and exacerbate the infarct area after stroke.66 Furthermore, IL-1β, TNF-α, and ROS can promote BBB disruption.19 TNF-α can induce MMP reactivation and promote BBB disruption,20 and IL-1β can cause endothelial reactivation and increase BBB permeability.21 Therefore, HDAC1 plays a crucial role in stroke-induced BBB damage, and HDAC1 dysfunction can increase BBB damage through elevated neuroinflammation after stroke.
Although our data support HDAC1 is essential in stroke-associated BBB damage, due to the experimental limitation, it cannot be ruled out that the changes in neuroinflammation seen in this study are derived from a worsening of neuronal damage or glial cell reactivation. Future studies should determine the exact role of microglia and astrocytes in neuroinflammation-induced BBB damage. Selective HDAC1 inhibition/activation/RNA interference approaches may be useful in elucidating mechanisms underlying BBB damage. The mechanisms include HDAC1-mediated M1/M2 microglia polarization, astrocyte activation, endothelial cell activation, and pericyte responses. Such studies would extend insights into neuroinflammation and assist the development of specific HDAC-based therapeutic strategies.
Conclusion
HDAC1 plays an essential role in the modulation of BBB damage. HDAC1 inhibition promotes microglial and astrocyte gliosis, increases inflammatory cytokine levels, and triggers MMP activity. Therefore, HDAC1 inhibition promotes the damage to tight junction proteins, which led to worsened behavioral outcomes in rats with cerebral ischemia in this study. These results indicate the role of HDAC1 in neuroinflammation, indicating the potential of selective-HDAC1-based therapeutic strategies for ischemia/reperfusion injury.
Data Sharing Statement
The data used to support the findings of this study are included within the article, containing Figures 1–7. Other data that might be useful for the findings of this study will be supplied by the corresponding author (C.C. Wu) upon request.
Acknowledgments
We want to acknowledge the technical support of the Basic Medical Core Laboratory, I-Shou University College of Medicine.
Funding
This study was supported by E-DA Hospital and I-Shou University, Kaohsiung, Taiwan (EDAHP110001, EDAHP110032, EDAHP111001, EDAHP111006, EDAHP111044, EDAHS109003, ISU-110-IUC-07, ISU 110-S-01, ISU-111-IUC-06). It was also partly supported by grants from the Ministry of Science and Technology, Taiwan (MOST-109-2320-B-214-001, MOST 110-2320-B-214 −004 -MY2, MOST 111-2628-B-214 −001 -MY3, MOST 110-2314-B-182A-158, MOST 111-2314-B-182A-102, MOST 111-2314-B-214-009, NSTC 112-2314-B-182A-069).
Disclosure
The authors declare there is no conflict of interest in this study.
References
1. Kuriakose D, Xiao Z. Pathophysiology and treatment of stroke: present status and future perspectives. Int J Mol Sci. 2020;21(20):7609. doi:10.3390/ijms21207609
2. Feigin VL, Stark BA, Johnson CO, et al. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet Neurol. 2021;20(10):795–820. doi:10.1016/S1474-4422(21)00252-0
3. Donkor ES. Stroke in the 21(st) century: a snapshot of the burden, epidemiology, and quality of life. Stroke Res Treat. 2018;2018:3238165. doi:10.1155/2018/3238165
4. Katan M, Luft A. Global burden of stroke. Semin Neurol. 2018;38(2):208–211. doi:10.1055/s-0038-1649503
5. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: important unanswered questions. J Exp Med. 2020;217(4). doi:10.1084/jem.20190062
6. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412. doi:10.1101/cshperspect.a020412
7. Hampl R, Bicikova M, Sosvorova L. Hormones and the blood-brain barrier. Horm Mol Biol Clin Investig. 2015;21(3):159–164. doi:10.1515/hmbci-2014-0042
8. Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. 2020;17(1):69. doi:10.1186/s12987-020-00230-3
9. Cabezas R, Avila M, Gonzalez J, et al. Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci. 2014;8:211. doi:10.3389/fncel.2014.00211
10. Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC. The blood-brain barrier: an engineering perspective. Front Neuroeng. 2013;6(7). doi:10.3389/fneng.2013.00007
11. Keaney J, Campbell M. The dynamic blood-brain barrier. FEBS J. 2015;282(21):4067–4079. doi:10.1111/febs.13412
12. Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29(3):705–718. doi:10.1161/01.str.29.3.705
13. Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6(1):11. doi:10.1186/1750-1326-6-11
14. Macrez R, Ali C, Toutirais O, et al. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 2011;10(5):471–480. doi:10.1016/S1474-4422(11)70066-7
15. Chen S, Shao L, Ma L. Cerebral edema formation after stroke: emphasis on blood-brain barrier and the lymphatic drainage system of the brain. Front Cell Neurosci. 2021;15:716825. doi:10.3389/fncel.2021.716825
16. Sorby-Adams AJ, Marcoionni AM, Dempsey ER, Woenig JA, Turner RJ. The role of neurogenic inflammation in blood-brain barrier disruption and development of cerebral oedema following acute central nervous system (CNS) injury. Int J Mol Sci. 2017;18(8):1788. doi:10.3390/ijms18081788
17. Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3(10):136. doi:10.3978/j.issn.2305-5839.2015.03.49
18. Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:214–228. doi:10.1111/j.1749-6632.2003.tb07478.x
19. Han X, Zhang E, Shi Y, Song B, Du H, Cao Z. Biomaterial-tight junction interaction and potential impacts. J Mater Chem B. 2019;7(41):6310–6320. doi:10.1039/c9tb01081e
20. Kimura-Ohba S, Yang Y. Oxidative DNA damage mediated by intranuclear MMP activity is associated with neuronal apoptosis in ischemic stroke. Oxid Med Cell Longev. 2016;2016:6927328. doi:10.1155/2016/6927328
21. Qiu YM, Zhang CL, Chen AQ, et al. Immune cells in the BBB disruption after acute ischemic stroke: targets for immune therapy? Front Immunol. 2021;12:678744. doi:10.3389/fimmu.2021.678744
22. Yang C, Hawkins KE, Dore S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2019;316(2):C135–C153. doi:10.1152/ajpcell.00136.2018
23. Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011;2011:371832. doi:10.1155/2011/371832
24. Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem. 2012;287(42):35444–35453. doi:10.1074/jbc.M112.394544
25. Demyanenko SV, Dzreyan VA, Neginskaya MA, Uzdensky AB. Expression of histone deacetylases HDAC1 and HDAC2 and their role in apoptosis in the penumbra induced by photothrombotic stroke. Mol Neurobiol. 2020;57(1):226–238. doi:10.1007/s12035-019-01772-w
26. Kim D, Frank CL, Dobbin MM, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60(5):803–817. doi:10.1016/j.neuron.2008.10.015
27. Kim JY, Shen S, Dietz K, et al. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci. 2010;13(2):180–189. doi:10.1038/nn.2471
28. Chen JS, Wang HK, Hsu CY, et al. HDAC1 deregulation promotes neuronal loss and deficit of motor function in stroke pathogenesis. Sci Rep. 2021;11(1):16354. doi:10.1038/s41598-021-95837-3
29. Chen JS, Wang HK, Su YT, et al. Restoration of HDAC1 enzymatic activity after stroke protects neurons from ischemia/reperfusion damage and attenuates behavioral deficits in rats. Int J Mol Sci. 2021;22(19). doi:10.3390/ijms221910654
30. Horie N, Maag AL, Hamilton SA, Shichinohe H, Bliss TM, Steinberg GK. Mouse model of focal cerebral ischemia using endothelin-1. J Neurosci Methods. 2008;173(2):286–290. doi:10.1016/j.jneumeth.2008.06.013
31. Nakazaki M, Sasaki M, Kataoka-Sasaki Y, et al. Intravenous infusion of mesenchymal stem cells inhibits intracranial hemorrhage after recombinant tissue plasminogen activator therapy for transient middle cerebral artery occlusion in rats. J Neurosurg. 2017;127(4):917–926. doi:10.3171/2016.8.JNS16240
32. Wu CC, Lien CC, Hou WH, Chiang PM, Tsai KJ. Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for alzheimer’s disease. Sci Rep. 2016;6:27358. doi:10.1038/srep27358
33. Du L, Wang D, Wei X, et al. MS275 as Class I HDAC inhibitor displayed therapeutic potential on malignant ascites by iTRAQ-based quantitative proteomic analysis. BMC Gastroenterol. 2022;22(1):29. doi:10.1186/s12876-022-02101-7
34. Ihezie SA, Mathew IE, McBride DW, Dienel A, Blackburn SL, Thankamani Pandit PK. Epigenetics in blood-brain barrier disruption. Fluids Barriers CNS. 2021;18(1):17. doi:10.1186/s12987-021-00250-7
35. Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J Cereb Blood Flow Metab. 2011;31(1):52–57. doi:10.1038/jcbfm.2010.195
36. Su L, Liang D, Kuang SY, Dong Q, Han X, Wang Z. Neuroprotective mechanism of TMP269, a selective class IIA histone deacetylase inhibitor, after cerebral ischemia/reperfusion injury. Neural Regen Res. 2020;15(2):277–284. doi:10.4103/1673-5374.265562
37. Zhang Y, Hu DN, Zhu Y, et al. Regulation of matrix metalloproteinase-2 secretion from scleral fibroblasts and retinal pigment epithelial cells by miR-29a. Biomed Res Int. 2017;2017:2647879. doi:10.1155/2017/2647879
38. Li H, Sun J, Wang F, et al. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 2016;1642:70–78. doi:10.1016/j.brainres.2016.03.031
39. Park MJ, Sohrabji F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J Neuroinflammation. 2016;13(1):300. doi:10.1186/s12974-016-0765-6
40. Shi W, Wei X, Wang Z, et al. HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2016;20(6):1139–1149. doi:10.1111/jcmm.12803
41. Zhao Q, Yu Z, Zhang F, et al. HDAC3 inhibition prevents oxygen glucose deprivation/reoxygenation-induced transendothelial permeability by elevating PPARgamma activity in vitro. J Neurochem. 2019;149(2):298–310. doi:10.1111/jnc.14619
42. Chen PS, Wang CC, Bortner CD, et al. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. 2007;149(1):203–212. doi:10.1016/j.neuroscience.2007.06.053
43. Kanski R, Sneeboer MA, van Bodegraven EJ, et al. Histone acetylation in astrocytes suppresses GFAP and stimulates a reorganization of the intermediate filament network. J Cell Sci. 2014;127(Pt 20):4368–4380. doi:10.1242/jcs.145912
44. Kannan V, Brouwer N, Hanisch UK, Regen T, Eggen BJ, Boddeke HW. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J Neurosci Res. 2013;91(9):1133–1142. doi:10.1002/jnr.23221
45. Shim S, Kim S, Choi DS, Kwon YB, Kwon J. Anti-inflammatory effects of [6]-shogaol: potential roles of HDAC inhibition and HSP70 induction. Food Chem Toxicol. 2011;49(11):2734–2740. doi:10.1016/j.fct.2011.08.012
46. Suh HS, Choi S, Khattar P, Choi N, Lee SC. Histone deacetylase inhibitors suppress the expression of inflammatory and innate immune response genes in human microglia and astrocytes. J Neuroimmune Pharmacol. 2010;5(4):521–532. doi:10.1007/s11481-010-9192-0
47. Shen Y, Yang R, Zhao J, et al. The histone deacetylase inhibitor belinostat ameliorates experimental autoimmune encephalomyelitis in mice by inhibiting TLR2/MyD88 and HDAC3/ NF-kappaB p65-mediated neuroinflammation. Pharmacol Res. 2022;176:105969. doi:10.1016/j.phrs.2021.105969
48. Gong Y, Liu YC, Ding XL, Fu Y, Cui LJ, Yan YP. Tanshinone IIA ameliorates CNS autoimmunity by promoting the differentiation of regulatory T cells. Neurotherapeutics. 2020;17(2):690–703. doi:10.1007/s13311-019-00789-2
49. Sung MS, Heo H, Eom GH, et al. HDAC2 regulates glial cell activation in ischemic mouse retina. Int J Mol Sci. 2019;20(20):5159. doi:10.3390/ijms20205159
50. Zhao Y, Mu H, Huang Y, et al. Microglia-specific deletion of histone deacetylase 3 promotes inflammation resolution, white matter integrity, and functional recovery in a mouse model of traumatic brain injury. J Neuroinflammation. 2022;19(1):201. doi:10.1186/s12974-022-02563-2
51. Liao Y, Cheng J, Kong X, et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. 2020;10(21):9644–9662. doi:10.7150/thno.47651
52. Sun W, Zhang N, Liu B, et al. HDAC3 Inhibitor RGFP966 ameliorated neuroinflammation in the cuprizone-induced demyelinating mouse model and LPS-Stimulated BV2 cells by downregulating the P2X7R/STAT3/NF-kappaB65/NLRP3 activation. ACS Chem Neurosci. 2022;13:2579–2598. doi:10.1021/acschemneuro.1c00826
53. Bian HT, Xiao L, Liang L, Xie YP, Wang HL, Wang GH. RGFP966 is protective against lipopolysaccharide-induced depressive-like behaviors in mice by inhibiting neuroinflammation and microglial activation. Int Immunopharmacol. 2021;101(Pt B):108259. doi:10.1016/j.intimp.2021.108259
54. Matheson R, Chida K, Lu H, et al. Neuroprotective effects of selective inhibition of histone deacetylase 3 in experimental stroke. Transl Stroke Res. 2020;11(5):1052–1063. doi:10.1007/s12975-020-00783-3
55. Lin FL, Yen JL, Kuo YC, et al. HADC8 Inhibitor WK2-16 therapeutically targets lipopolysaccharide-induced mouse model of neuroinflammation and microglial activation. Int J Mol Sci. 2019;20(2). doi:10.3390/ijms20020410
56. Song Y, Qin L, Yang R, et al. Inhibition of HDAC6 alleviating lipopolysaccharide-induced p38MAPK phosphorylation and neuroinflammation in mice. Pharm Biol. 2019;57(1):263–268. doi:10.1080/13880209.2018.1563620
57. Takata F, Nakagawa S, Matsumoto J, Dohgu S. Blood-brain barrier dysfunction amplifies the development of neuroinflammation: understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front Cell Neurosci. 2021;15:661838. doi:10.3389/fncel.2021.661838
58. Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci Ther. 2021;27(1):36–47. doi:10.1111/cns.13569
59. Varvel NH, Neher JJ, Bosch A, et al. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A. 2016;113(38):E5665–74. doi:10.1073/pnas.1604263113
60. Wang J, Zhao H, Fan Z, et al. Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization. Stroke. 2017;48(8):2211–2221. doi:10.1161/STROKEAHA.117.017387
61. Ji J, Wang J, Yang J, et al. The Intra-nuclear SphK2-S1P axis facilitates M1-to-M2 shift of microglia via suppressing HDAC1-Mediated KLF4 deacetylation. Front Immunol. 2019;10:1241. doi:10.3389/fimmu.2019.01241
62. Durham BS, Grigg R, Wood IC. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J Neurochem. 2017;143(2):214–224. doi:10.1111/jnc.14144
63. McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27(16):4403–4412. doi:10.1523/JNEUROSCI.5376-06.2007
64. Aizawa H, Makita Y, Sumitomo K, et al. Edaravone diminishes free radicals from circulating neutrophils in patients with ischemic brain attack. Intern Med. 2006;45(1):1–4. doi:10.2169/internalmedicine.45.1491
65. Simi A, Tsakiri N, Wang P, Rothwell NJ. Interleukin-1 and inflammatory neurodegeneration. Biochem Soc Trans. 2007;35(Pt 5):1122–1126. doi:10.1042/BST0351122
66. Pawluk H, Wozniak A, Grzesk G, et al. The role of selected pro-inflammatory cytokines in pathogenesis of ischemic stroke. Clin Interv Aging. 2020;15:469–484. doi:10.2147/CIA.S233909
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.