Back to Journals » International Journal of Nanomedicine » Volume 18
Glycosylation-Engineered Platelet Membrane-Coated Interleukin 10 Nanoparticles for Targeted Inhibition of Vascular Restenosis
Authors Li F, Rong Z, Chen T ![]() , Wang P, Di X, Ni L, Liu C
, Wang P, Di X, Ni L, Liu C
Received 27 May 2023
Accepted for publication 25 August 2023
Published 5 September 2023 Volume 2023:18 Pages 5011—5030
DOI https://doi.org/10.2147/IJN.S423186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Fengshi Li,1,* Zhihua Rong,1,* Tianqi Chen,2 Peng Wang,1 Xiao Di,1 Leng Ni,1 Changwei Liu1
1Department of Vascular Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100730, People’s Republic of China; 2Department of Medical Research Center, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100730, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Leng Ni; Changwei Liu, Tel +86 010 69152500 ; +86 010 69152501, Email [email protected]; [email protected]
Purpose: The purpose of this study was to improve the immune compatibility and targeting abilities of IL10 nanoparticles coated with platelet membrane (IL10-PNPs) by glycosylation engineering in order to effectively reduce restenosis after vascular injury.
Materials and Methods: In this study, we removed sialic acids and added α (1,2)-fucose and α (1,3)-fucose to platelet membrane glycoprotein, thus engineering the glycosylation of IL10-PNPs (IL10-GE-PNPs). In vitro and in vivo experiments were conducted to evaluate the targeting and regulatory effects of IL10-GE-PNPs on macrophage polarization, as well as the influence of IL10-GE-PNPs on the phenotypic transformation, proliferation, and migration of smooth muscle cells, and its potential in promoting the repair function of endothelial cells within an inflammatory environment. In order to assess the distribution of IL10-GE-PNP in different organs, in vivo imaging experiments were conducted.
Results: IL10-GE-PNPs were successfully constructed and demonstrated to effectively target and regulate macrophage polarization in both in vitro and in vivo settings. This regulation resulted in reduced proliferation and migration of smooth muscle cells and promoted the repair of endothelial cells in an inflammatory environment. Consequently, restenosis after vascular injury was reduced. Furthermore, the deposition of IL10-GE-PNPs in the liver and spleen was significantly reduced compared to IL10-PNPs.
Conclusion: IL10-GE-PNPs emerged as a promising candidate for targeting vascular injury and exhibited potential as an innovative drug delivery system for suppressing vascular restenosis. The engineered glycosylation of IL10-PNPs improved their immune compatibility and targeting abilities, making them an excellent therapeutic option.
Keywords: vascular restenosis, platelet membrane, glycosylation, interleukin 10, targeting delivery
Introduction
Atherosclerosis-related cardiovascular diseases remain the leading cause of death and disability worldwide.1 Interventional angioplasty is a common treatment for these diseases to reconstruct vascular patency. Angioplasty has been shown to lead to vascular injury, resulting in vascular restenosis, which has a high reintervention rate and has a negative effect on the patient’s prognosis.2,3 Local de-endothelialization damage causes immune cells and platelets to be recruited into the injured vessel wall. Smooth muscle cells (SMCs) proliferate and migrate as a consequence of inflammatory cytokines secreted by immune cells and platelets, leading to vascular restenosis.4 At the site of restenosis, macrophages play a critical role in inflammatory reactions. During the formation of vascular restenosis, macrophages are active, polarize into the M1 phenotype and secrete cytokines such as IL-1β, TNF-α, and IL-6, which transform SMCs into the secretory cell type, contributing to the proliferation and migration of the cells.5 Alternatively activated macrophages, called the M2 phenotype, result in a significant decrease in vascular restenosis, inhibiting smooth muscle cell phenotypic transformation and promoting endothelial cell repair by different cytokine secretion profiles. Consequently, targeted stimulation of macrophage polarization into the M2 phenotype is an effective method for preventing vascular restenosis.6
Interleukin 10 (IL10) is an all-encompassing anti-inflammatory cytokine with immunoregulatory roles.7 IL10 is recognized for its various mechanisms in governing M2 macrophage polarization.8 Nevertheless, the systemic administration of IL10 leads to adverse reactions, including anemia and thrombocytopenia.9 In preceding investigations, nanoparticles containing IL10 were formulated with the aim of suppressing the localized inflammatory response within atherosclerotic lesions.10,11 The hemostasis function of platelets has long been known. In recent years, platelets were shown to play a critical role in inflammatory reactions.12 After vascular injury, platelets accumulate at the site of vascular injury and bind to endothelial cells (ECs), injury-exposed SMCs, and immune cells to promote the local inflammatory response. Accordingly, a previous study prepared platelet membrane-coated paclitaxel nanoparticles to target the site of vascular injury and inhibit restenosis.13 However, there are still some problems, such as the large number of platelet cells needed and the long-term side effects of local deposition of paclitaxel. In our previous study, we demonstrated that IL10 nanoparticles coated with platelet membrane (IL10-PNPs) could target the regulation of M2 macrophage polarization at the site of vascular injury, thereby creating a local “immune cascade”. As a result, SMC proliferation and migration are inhibited, and endothelial repair is promoted, thereby significantly reducing vascular restenosis.14 Notably, however, IL10-PNPs still accumulated in the liver and spleen in significant amounts. Consequently, it is imperative that the current PNPs be further improved in terms of their immune-compatibility and targeting abilities.
Glycosylation is one of the most important post-translational modifications of proteins in vivo and plays a crucial role in protein function.15 Recent research indicates that platelet surface glycosylation is significantly associated with a wide range of diseases. In vivo, platelets with deficient sialylation are more likely to be removed in thrombocytopenia.16 Desialylation may be the major cause of increased thrombosis in patients with congenital disorders of N-glycosylation.17 There was a decrease in sialic acid, β-galactose (β-Gal) and α-mannose on the surface of platelets during coronary heart disease, while an increase in α1,6-fucose and β-N-acetylglucosamine (β-GlcNAc) was observed, increasing the adhesion between platelets and other cells.18,19 Thus, platelet function is closely related to surface glycosylation. Moreover, platelets are a rich source of glycosyltransferases and donor sugar substrates, which can be released into the extracellular space when activated.20 Desialylated platelets have stronger cell adhesion and will expose β-GlcNAc and β-Gal, which make them more easily phagocytosed by the liver and spleen.21 Fucose can mediate intercellular adhesion and bind to β-GlcNAc and β-Gal.22 It is therefore feasible to improve the performance of PNPs by utilizing the effect of glycosylation on platelet membrane glycoprotein function.
Herein, we prepared a glycosylation-engineered platelet membrane with surface sialic acid removed and fucose conjugated. Glycosylation-engineered platelet membrane-coated IL10 nanoparticles (IL10-GE-PNPs) showed good biocompatibility and targeting. This treatment showed a significant inhibitory effect on restenosis after vascular injury.
Methods and Materials
Glycosylation Engineering (GE) of Platelet Membrane Glycoprotein
To collect platelets from platelet-rich plasma (PRP), we used the same procedure described previously.13 An adenosine 50-diphosphate solution (ADP; Sigma-Aldrich, USA) was used to activate platelets, followed by a 1/4 volume of an acid citrate dextrose solution (ACD; Sigma-Aldrich, USA) to prevent aggregation as previously described. For removal of sialic acids on both N- and O-glycans, isolated platelets were treated for 1 hour with neuraminidase (NEU; Sigma-Aldrich, USA) at room temperature.18 Fucose engineering was performed by adding GDP-fucose as a donor of fucose, α (1,2)-fucose as an adjunct to galactose by adding fucosyltransferase I (FUT I), and α (1,3)-fucose as an adjunct to GlcNAc by adding fucosyltransferase VII (FUT VII).22 In brief, platelets were treated with 1 mM GDP-fucose (EMD Biosciences, USA) and 20 mU/mL FUT I/VII (EMD Biosciences, USA) for 30 minutes at 37 °C as previously described.23 Thus, glycosylation-engineered platelets with sialic acid removal and α (1,2)-fucose and α (1,3)-fucose adjunction were prepared.
Fluorescein isothiocyanate (FITC)-conjugated lectins (Vector Laboratories, USA) were used to bind to specific sugars, including Ricinus communis Agglutinin I (RCA I), which binds to β-galactose to identify the removal of sialic acids; Ulex europaeus agglutinin-I (UEA-I), which binds to α (1,2)-fucose; and Lotus tetragonolobus lectin (LTL), which binds to α (1,3)-fucose.24,25 Platelets were incubated with FITC-conjugated lectins for 20 min at room temperature. Glycosylation engineering indicated by the levels of FITC-conjugated lectin binding was evaluated by flow cytometry (LSRFortessaTM, Biosciences, USA).
Preparation and Characterization of IL10-NPs, IL10-PNPs and IL10-GE-PNPs
In the laboratory experiment, platelets were obtained from healthy student volunteers who abstained from taking anti-platelet medication for at least 14 days and provided informed consent. Platelet/glycosylation engineer platelet membranes were extracted by freeze‒thaw cycles as previously described.13 Platelet aggregation was measured using a BioTek Synergy H1 plate reader. Platelets dispersed in calcium-free Tyrode’s buffer had an OD650 value of 0% aggregation. After activating with ADP and adding ACD at different volume ratios, the sample’s OD650 value was measured 30 minutes later. A platelet aggregation rate is calculated as the ratio between the sample’s OD650 value and 0%. ACD significantly reduced platelet aggregation when 1/4 volume was added, and glycosylation engineering did not affect platelet aggregation (Figure S1). The IL10 nanoparticles (IL10-NPs) were prepared by a modified double solvent evaporation method, which has been described previously and modified slightly.11,26 In summary, 1 mL of PLGA (Sigma-Aldrich, USA) was dissolved 60 mg/mL in ethyl acetate and mixed with recombinant rat IL10 (Abcam, UK) and D- (+)-glucosamine (4 and 6 w/w % of total PLGA mass) dissolved in distilled water that was ultrapure and free of DNases and RNases. With a sonicator for 1 minute, the solution was emulsified to form an initial W1/O (water/oil) emulsion. The solution was then blended with 5 mL of 1% PVA (Sigma-Aldrich, USA) aqueous solution. For the final emulsion of W1/O/W2, two minutes of sonication were performed. The final emulsion was injected into a 0.3% (w/v) PVA solution and stirred overnight to eliminate ethyl acetate from the solution. After IL10-NPs were synthesized, the resulting particles were washed by centrifugation at 13,000 rpm for 5 minutes 5 times in ultrapure DNase/RNase-free distilled water and resuspended in PBS. To prepare IL10-PNP/IL10-GE-PNP, we took IL10-NPs with a loading of 1 mg of IL10 and mixed them with 1.5 mL of platelet membrane/GE- platelet membrane (containing approximately 3×108 platelets) and then sonicated the sample at a power of 100 W for 2 minutes to coat IL10-NPs with platelet membranes.13 For analysis of the targeting effect, nanoparticles were loaded with 1,10-dioctadecyl-3,3,30,30-tetramethylindotri-carbocyanine iodide (DiR, Invitrogen). Preparation of DiR-NPs and DiR-PNPs/DiR-GE-PNPs was performed in the same way. To conduct the in vitro experiment, we collected blood samples from healthy individuals (healthy university students with informed consent obtained) for at least 14 days without antiplatelet drugs to obtain platelets. Platelets were glycosylation engineered in the same way, and IL10-NPs, IL10-PNPs and IL10-GE-PNPs were made using human IL10 recombinant protein (Abcam, USA). We used transmission electron microscopy (TEM; JEOL JEM2100F, JEOL, Ltd., Japan) to examine the morphology of IL10-NPs, IL10-PNPs, and IL10-GE-PNPs stained with phosphotungstic acid. In accordance with previous reports, the encapsulation efficacy and loading capacity were evaluated.27 The IL10-NPs/IL10-PNPs/IL10-GE-PNPs should be dispersed in a solution of 2.5% SDS/0.04 M NaOH in a volume of 5 mL. The nanoparticles were hydrolyzed at 37 °C and 100 rpm in an orbital shaker. A rat IL10 enzyme-linked immunosorbent assay (ELISA) kit (Solarbio, China, SEKR-0006) was used to measure IL10 concentrations. The IL10 encapsulation efficiency was calculated by the ratio of loaded IL10 to initial IL10, and the loading capacity was calculated by the ratio of loaded IL10 to the total quantity of IL10-NPs/IL10-PNPs/IL10-GE-PNPs. DLS measurements were carried out using a Zetasizer Nano S (Malvern Instruments, UK) to determine the size and zeta potential of the IL10-NPs, IL10-PNPs, and IL10-GE-PNPs. We evaluated the release profiles of IL10 from IL10-NP, IL10-PNP, and IL10-GE-PNP by incubating them at 37 °C in PBS and then measuring the protein released using a rat IL10 ELISA kit (Solarbio, China, SEKR-0006) at various intervals for up to 240 h.
Pharmacokinetics Study
To evaluate the circulation half-life of different IL10 formulations, male SD rat were injected with IL10, IL10-NP, IL10-PNP and IL10-GE-PNP at a dosage of 25μg IL10/kg via the tail vein (n = 3). A total of 30 μL of blood was collected at different time points (0, 5, 15, and 30 min, and 1, 2, 4, 8, 24, 48, and 72 h). The collected blood samples were centrifuged to separate the plasma. The amount of IL10 in plasma was analyzed by a rat IL10 ELISA kit (Solarbio, China, SEKR-0006). Pharmacokinetic parameters were calculated to fit a two-compartment model and a one-way nonlinear model.
Cell Culture
According to previous results, macrophages were derived from THP-1 cell lines (ATCC, USA, TIB-202).28 We collected platelets from healthy volunteers. As a means of simulating the inflammatory environment after vascular injury, we used a coculture system (platelets:macrophage = 100:1) in which macrophages were polarized in response to platelets.
Flow Cytometry
To assess glycosylation engineering of the platelet membrane on macrophage phagocytosis of PNP, we labeled NPs with FITC, and macrophages were cultured with NPs/PNPs/GE-PNPs for 24 h. Then, cells were harvested and washed by ethylene diamine tetra-acetic acid (EDTA) and incubated with anti-CD61 antibody (Biolegend, USA) and analyzed by flow cytometry on an LSRFortessaTM (BD Biosciences, USA).
To assess the binding effect of IL10-GE-PNP on endothelial cells (ECs), smooth muscle cells (SMCs) and macrophages, we cultured IL10-PNPs/IL10-GE-PNPs with ECs/SMCs/macrophages. After culture for 2/6 h, cells were harvested and incubated with anti-CD61 antibody (Biolegend, USA) and analyzed by flow cytometry on an LSRFortessaTM (BD Biosciences, USA).
To assess the regulatory role of IL10-GE-PNP on macrophage polarization, we added IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs (containing 0.1 μg IL10) to the coculture system. Following 24 hours of coculture, we digested and stained the cells with anti-CD86 antibody (Biolegend, USA) to identify M1 macrophages and anti-CD163 antibody (Biolegend, USA) to identify M2 macrophages. After incubation for 20 minutes at room temperature, the samples were analyzed by flow cytometry on an LSRFortessaTM (BD Biosciences, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
The chemokines in the supernatant, including TNF-a, IL-1b, and IL-6, secreted by M1 macrophages were measured using the corresponding ELISA kits (Abcam, USA) according to the manufacturer’s instructions.
Real-Time Quantitative Polymerase Chain Reaction (RT‒qPCR)
The effect of IL10-GE-PNP application on the phenotypic transformation-related gene expression of SMCs was detected by RT‒qPCR. TRIzol Reagent (Ambion, USA) was used to isolate RNA from SMCs after 24 hours of culture in the coculture system with PBS/IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs. With a reverse transcription kit (TaKaRa, Japan), 1 mg of RNA was reverse transcribed. An internal control was conducted using β-actin. On the QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific, USA), RT‒qPCR was performed using SYBR Green mix (Thermo Fisher Scientific, USA). The forward primer (F) and reverse primer (R) were as follows (5’ −3’): OPN-F: CAGCCGTGGGAAGGACAGTT-ATG; OPN-R: TCACATCGGAATGCTCATTGCTCTC; SM22-F: GCAGTCCAAAATCGA- GAAGAAG; SM22-R: CAGAATCACGCCATTCTTCAG; β-actin-F: TGACGTGGACATCCG- CAAAG; β-actin-R: CTGGAAGGTGGACAGCGAGG. By using 2-ΔΔCt, we calculated relative gene expression. The expression of genes was normalized to the expression of β-actin.
Western Blot
We identified glycoproteins on the surface of platelets using sodium dodecyl sulfate‒polyacrylamide gel electrophoresis (SDS‒PAGE) and Western blotting. Radioimmunoprecipitation assay (RIPA) lysis buffer containing protease inhibitor (1:100) was used to lyse platelet membrane vesicles and empty PNPs/GE-PNPs. To determine total protein contents, we used a BCA Protein Assay Kit (Thermo Fisher Scientific, USA, 23225), followed by heating samples for 10 minutes at 100 °C. Using equal amounts of protein (30 mg/well), we ran the samples for two hours at 120 V on 10% SDS‒PAGE gels. Coomassie Blue staining was applied to the gel for two hours, and the gel was washed overnight. Furthermore, polyvinylidene difluoride membranes were used for gel transfer. By Western blotting, key proteins were identified by using primary antibodies, including anti-CD42b antibody (Proteintech, China, 12860-1-AP), anti-P-selectin antibody (Proteintech, China, 60322-1-Ig), anti-CD61 antibody (Proteintech, China, 18309-1-AP), anti-CD41 antibody (Proteintech, China, 24552-1-AP), anti-GP VI antibody (ABclonal, China, A14999) and anti-CD40 L antibody (Proteintech, China, 16668-1-AP), at 4 °C overnight and incubated with the appropriate secondary antibodies.
As described above, Western blot analysis was also performed to detect contractile phenotype-related proteins in SMCs after culture in a coculture system with PBS/IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs, which included anti-SMMHC (Proteintech, China, 18569-1-AP), anti-SMTN (Proteintech, China, 23567-1-AP), anti-a-SMA (Proteintech, China, 14395-1-AP), anti-SM-22 (Abcam, USA, ab14106), anti-β-actin (Proteintech, China, 66009-1-Ig), and anti-calponin1 (CST, USA, 17819).
Cell Viability Assay
We evaluated the effect of IL10-GE-PNPs on the proliferation of SMCs. Human SMCs (ScienCell, 6110) were cultured in the lower chamber of Transwells (Corning Incorporated, USA) (106 cells/well), while the coculture system, including platelets and macrophages, was seeded in the upper chamber with IL10-GE-PNPs. As a control, we substituted IL10-GE-PNPs with IL10-PNPs, IL10-NPs or IL10, or PBS. As a blank group, SMCs were cultured alone. We assessed the proliferation of SMCs using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Japan) in accordance with the manufacturer’s instructions. SMCs were harvested after 1–5 days and dispensed into a 96-well plate. Then, the cells were incubated at 37 °C for 2 h. After that, the absorbance was measured using a microplate spectrophotometer at 450 nm (Varioskan; Thermo Fisher, USA).
Transwell Assay
Transwell assays were used to assess the migration of SMCs. As described above, the coculture system with PBS/IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs was seeded in the lower Transwell chamber, with SMCs seeded in the upper Transwell chamber. The cells were incubated at 37 °C and 5% CO2 for 24 hours, and then, the filter was removed using a cotton tip to remove the cells from the upper chamber. Four percent paraformaldehyde was used to fix SMCs in the lower chamber, and 1% crystal violet in 2% ethanol was used for staining.
Wound Healing Assay
We performed wound healing assays to assess the effectiveness of IL10-GE-PNPs in promoting EC repair. In a 24-well plate, human umbilical vein endothelial cells (HUVECs) were plated and cultured for 24 hours to achieve an 80% fusion rate. Afterward, the ECs were scratched with a 200-mL sterile pipette tip, and the coculture systems mentioned above with PBS/IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs were seeded into Transwell chambers and placed in 24-well plates. HUVECs were cultured alone as a blank group. Images were taken at 0 and 24 hours after culture for 24 hours.
Tube Formation Assay
After 24 hours of culture, harvested HUVECs were placed in 96-well plates coated with Matrigel (Corning Incorporated, USA), and the medium from the blank group as well as the coculture system (with PBS/IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs added) was added. Following an incubation period of 6 h, imaging was conducted.
PNP Circulation Retention Property
Blood clearance of PNPs was studied using DiR-labeled PNPs. IVIS Lumina XRMS Series III (PerkinElmer, USA) was used to detect the fluorescence of 100 mL of blood collected in 96-well plates at various time intervals. Based on the 2-minute value, values were normalized.
Experimental Model of Angioplasty-Induced Vascular Injury
All animal studies were conducted in accordance with the Chinese Ministry of Health’s Animal Management Regulations and were reviewed and approved by the Peking Union Medical College’s Animal Ethics Committee (No. XHDW-2019-001). Previously described balloon-induced carotid artery injuries were performed on all rats. Anesthesia was achieved with pentobarbital sodium (40 mg/kg, i.p.), and a neck incision was made at the midline. Blunt dissection exposed the left common carotid artery and carotid bifurcation. The left external carotid artery, the inner carotid artery, and the left common carotid artery were clamped temporarily. A 2F Fogarty arterial embolectomy catheter (Edwards Lifesciences, USA) was slowly inducted into the common carotid artery via an arteriotomy made on the external carotid artery, inflated to a pressure of 2 atm and withdrawn three times with rotation. After that, 4–0 sutures were used to close the wound.
Fluorescence Imaging ex vivo
Following wound closure, rats were randomly divided into four groups to determine whether PNPs targeted vascular injury. For vascular localization studies, saline was injected intravenously into the control group, and FITC-labeled NPs, FITC-labeled PNPs, and FITC-labeled GE-PNPs were injected intravenously into the corresponding group. Left carotid arteries were harvested after 24 h for ex vivo fluorescence analysis. After that, the samples were fixed in 10% formalin, embedded in paraffin, and cut into 5 μm sections. The sections were stained with DAPI for 20 min, and fluorescence images of the sections were obtained by a confocal laser scanning microscope (Nikon, Japan).
DiR saline solution was injected intravenously into the control group, and DiR-labeled NPs, DiR-labeled PNPs and DiR-labeled GE-PNPs were injected intravenously into the corresponding group. For injection of DiR into the tail vein of all rats, 5 μg was administered. After 24 hours of the procedure, the rats were sacrificed with fluorescence intensities measured on the bilateral carotid arteries, the heart, the liver, the spleen, the lung, and the kidney by IVIS Lumina XRMS Series III (PerkinElmer, USA).
In vivo Release of IL10-GE-PNPs
The level of IL10 in different organs was measured to examine the release function of IL10-GE-PNPs in vivo. We homogenized frozen tissues (carotid artery, heart, liver, spleen, lung, kidney) 24 hours after treatment. The BCA Protein Assay Kit (Thermo Fisher Scientific, USA, 23225) was used to adjust the supernatants to the same protein concentration, and then, an ELISA kit (Solarbio, China, SEKR-0006) for rat IL10 was used to measure the IL10 concentration.
Histology and Immunohistology
After wound closure, rats were randomly divided into five groups. Saline was injected intravenously into the saline group. IL10/IL10-NPs/IL10-PNPs/IL10-GE-PNPs containing 1 μg IL10 were injected intravenously into the rats of the corresponding group on Day 0 and Day 5 after vascular injury. Ketamine and xylazine were used to euthanize the rats on Day 14, followed by a perfusion of PBS and 4% paraformaldehyde (PFA) at 120–140 mmHg. Left and right carotid arteries were fixed with 10% formalin, embedded in paraffin, and sectioned at 5 μm. Each slide was stained with hematoxylin and eosin (H&E) to determine the areas of intima and media. Immunofluorescence was performed by incubating sections with antibodies against F4/80 (Abcam, USA, ab16911) for identifying macrophages, CD86 (Abcam, USA, ab238468) for locating M1 macrophages, and CD163 (Abcam, USA, 182422) for locating M2 macrophages. Immunohistochemistry was performed by incubating sections with CD31 for identification of endothelial cells and with a-SMA for identification of SMCs with contractile phenotypes. Investigators performing quantitative analysis of histology were blinded to the experimental group until all data had been collected.
Proteins from the frozen left carotid arteries were extracted and adjusted in concentration as described above. For detection of the contractile phenotype-related proteins of SMCs, Western blotting was performed as described above. Key proteins were identified by using primary antibodies, including anti-α-SMA (Proteintech, China, 14395-1-AP), anti-SM-22 (Abcam, USA, ab14106), anti-β-actin (Proteintech, China, 66009-1-Ig), and anti-calponin1 (CST, USA, 17819).
Biocompatibility Evaluation
The in vitro cytotoxicity of IL10-GE-PNPs, IL10-PNPs, IL10-NPs or IL10 against macrophages, SMCs and HUVECs was investigated by the CCK-8 assay. After 12 h of culture, cells were treated with different IL10-loaded formulations for 48 h and the cytotoxicity was determined by the CCK-8 assay. As a control, we substituted IL10-loaded formulations with PBS. As a blank group, Macrophages, SMCs or HUVECs were cultured alone.
For in vivo biocompatibility evaluation, the complete blood count, biochemical parameters, and coagulation parameters of the blood were measured 24 hours and 28 days following treatment. In addition, 28 days following treatment, major organs (heart, liver, spleen, lung, kidney) were harvested and stained with H&E.
Statistical Analysis
Data was displayed as mean values with corresponding standard deviations. Statistical analysis was carried out using GraphPad Prism version 7.0 (GraphPad Software, USA). The Student’s t-test or a non-parametric test was used to evaluate the differences between two groups, and a one-way analysis of variance (ANOVA) with Tukey’s post-hoc test was employed to compare multiple groups. Results were considered statistically significant if p < 0.05. Statistical power was kept at 80%.
Results
Identification of Glycosylation Engineering of Platelet Membrane Glycoproteins
A schematic diagram of glycosylation engineering is shown in Figure 1A. Platelets in the NEU group were treated with NEU alone and those in the NEU+fucose engineered group underwent a complete glycosylation engineering process. After treatment with NEU for 1 hour, β-galactose was significantly exposed in the NEU group, as demonstrated by RCA I binding, and β-galactose was affected after fucose engineering (Figure 1B and C). As demonstrated by UEA-I binding, α (1,2)-fucose was effectively bound after fucose engineering (Figure 1B and C). As demonstrated by LTL binding, α (1,3)-fucose was effectively bound after fucose engineering (Figure 1B and C).
 |
Figure 1 Schematic diagram and identification of glycosylation engineering of platelet membrane glycoproteins and the characterization of GE-PNP. (A) Schematic diagram of glycosylation engineering. (B and C) Flow cytometry detected lectins binding form in neuraminidase group (NEU) and neuraminidase + fucose engineered(FE) group. (D) Transmission electron microscopy of empty PLGA nanoparticles (NPs), IL10-NPs, IL10-PNPs, and IL10-GE-PNPs. Scale bar = 100μm. (E) Coomassie stain to visualize platelet vesicles and empty PNPs/GE-PNPs on SDS-PAGE at equal protein levels. (F) Western blot to identify platelet membrane glycoprotein in platelet vesicles and PNPs/GE-PNPs. (G) Size and (H) Zeta potential of the empty PLGA core, IL10-NP, empty PNP, IL10-PNP, empty GE-PNP, and IL10-GE-PNP are evaluated. (I) Monitoring the diameter of GE-PNP in deionized water on day 0 and day 7. (J) Determining the encapsulation efficiency and (K) loading capacity of IL10 in IL10-NP, IL10-PNP, and IL10-GE-PNP. (L) Measuring the in vitro cumulative release of IL10-NP, IL10-PNP, and IL10-GE-PNP after incubation at 37°C in PBS Data are presented as mean ± SD, ***p < 0.001, n=3. Abbreviation: ns not significant. |
Production and Characterization of IL10-NPs, IL10-PNPs and IL10-GE-PNPs
We carried out transmission electron microscopy (TEM) and dynamic light scattering (DLS) to characterize the structure of the nanoparticles and their fabrication efficiency. According to TEM results, IL10-PNPs and IL10-GE-PNPs possess a similar core-shell structure and are adequately coated with platelet membranes compared to IL10-NPs and empty PLGA nanoparticles (Figure 1D). According to Coomassie Blue staining, platelet membrane vesicles, PNPs and GE-PNPs retain and enrich membrane proteins at similar amounts (Figure 1E). Western blotting revealed similar levels of membrane protein expression between groups (Figure 1F). The surface zeta potential and size of IL10-PNPs and IL10-GE-PNPs were slightly higher than those of IL10-NPs (Figure 1G and H). A 7-day storage in deionized water resulted in no significant changes in the size of IL10-GE-PNPs, suggesting that IL10-GE-PNPs are stable (Figure 1I). As measured under experimental conditions, the encapsulation efficiency was 81.30 ± 1.00%, 80.43 ± 0.68% and 80.63 ± 0.73% for IL10-NPs, IL10-PNPs and IL10-GE-PNPs, respectively (Figure 1J). The loading capacity was 2.52 ± 0.09%, 2.50 ± 0.07% and 2.52 ± 0.06% for IL10-NPs, IL10-PNPs and IL10-GE-PNPs, respectively (Figure 1K). The controlled-release profiles of IL10 were observed in IL10-NPs, IL10-PNPs, and IL10-GE-PNPs. In the first 40 h, IL10-NPs released IL10 faster than IL10-PNPs and IL10-GE-PNPs, with a peak at 100 h, reaching 85.60 ± 0.70%, 84.77 ± 0.64% and 86.50 ± 0.90% of the total encapsulated IL10, respectively (Figure 1L). According to the release profile, glycosylation engineering did not affect the release capacity of PNPs.
Pharmacokinetic and Pharmacodynamic Characteristics
A pharmacokinetic study showed that IL10-NP, IL10-PNP, and IL10-GE-PNP had a longer systemic retention time than IL10 (Figure S2 and Table S1).
In vitro Binding Function of IL10-GE-PNPs
The binding function of IL10-GE-PNPs to ECs/SMCs/macrophages was assessed by detecting the expression of the platelet membrane glycoprotein CD61 on these cells. The ratio of CD61+ ECs/SMCs/macrophages was significantly higher at 2 h and 6 h in the GE-PNP group than in the PNP group (Figure S3). The binding rate and ratio suggest that the binding function of IL10-GE-PNPs to target cells in vitro was higher than that of PNPs.
Effect of Glycosylation Engineering on Macrophage Phagocytosis of PNP
Effect of glycosylation engineering of platelet membrane protein on macrophage phagocytosis of PNP was accessed by detecting FITC-labeled NPs in macrophages after 24 h co-culture. Macrophages with FITC high signal indicated phagocytosis of NPs/PNPs/GE-PNPs, with FITC and CD61 high signal indicated adherent with PNPs/GE-PNPs. The adherent between macrophages and PNPs/ GE-PNPs was detached after EDTA wash (Figure 2A). And glycosylation engineering significantly reduced macrophage phagocytosis of PNP (Figure 2A and B).
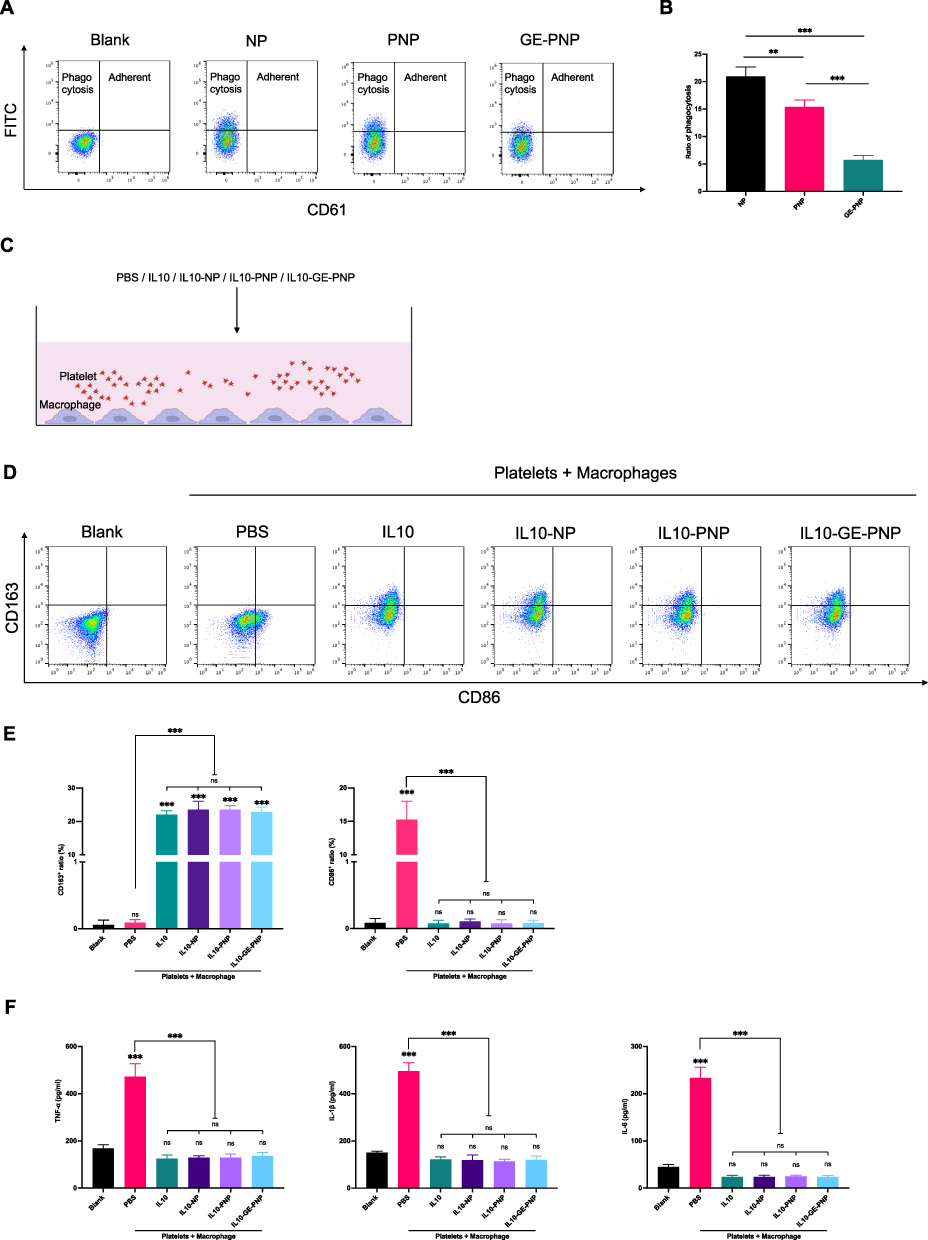 |
Figure 2 Macrophage phagocytosis of GE-PNP and the impact of IL10-GE-PNP on macrophage polarization in an in vitro modeled inflammatory environment is investigated. (A and B) Flow cytometry detected the ratio of phagocytosis of NPs, PNPs and GE-PNPs. (C) A diagram of the co-culture system is displayed. (D) Flow cytometry is utilized to analyze the polarization of macrophages that are cultured alone or co-cultured with platelets + PBS, platelets + IL10, platelets + IL10-NPs, platelets + IL10-PNPs, and platelets + IL10-GE-PNPs for 24 hours. (E) The ratio of M1 to M2 macrophages in different groups is determined. (F) The levels of TNF-α, IL-1β, and IL-6 in the cell supernatant are quantified using ELISA. Data are presented as mean ± SD, **p<0.01, ***p < 0.001, n=3. Abbreviation: ns not significant. |
Regulatory Effect of IL10-PNPs on Macrophage Polarization in vitro
To assess the effect of IL10-GE-PNPs on macrophage polarization at the vascular injury site, we added platelets to the media to mimic the local inflammatory responses (Figure 2C). Flow cytometry was used to detect the M1 macrophage marker CD86 and the M2 macrophage marker CD163. The ratio of M1 macrophages increased significantly in the PBS group, suggesting that platelets contribute to macrophage inflammation (Figure 2D and E). A significant increase in the number of M2 macrophages was observed in all four groups after 24 h of coculture: IL10, IL10-NPs, IL10-PNPs, and IL10-GE-PNPs (Figure 2D and E). M1 macrophage-released cytokines (TNF-a, IL-1b, and IL-6) in the PBS group were significantly increased and inhibited by IL10, IL10-NPs, IL10-PNPs, and IL10-GE-PNPs (Figure 2F). The preparation of nanoparticles and glycosylation-engineered platelet membrane coating had no significant impact on the release and regulatory function of IL10 in vitro.
The Effects of IL10-GE-PNPs on Smooth Muscle Cell Phenotypic Transformation and Function
For in vitro simulation of the inflammatory environment, platelets and macrophages were cocultured with SMCs or ECs (Figure 3A). Hyperplastic intima is formed when SMCs undergo phenotypic transformation into secretory cells. In the PBS group, there was a significant increase in the expression of a gene associated with the secretory phenotype, OPN, and a decrease in the expression of a gene associated with the contractile phenotype, SM22. IL10, IL10-NP, IL10-PNP, and IL10-GE-PNP application significantly decreased the expression of OPN and increased the expression of SM22 (Figure 3B and C). In the PBS group, SMCs’ contractile phenotype-related proteins were significantly decreased, while IL10, IL10-NP, IL10-PNPs, and IL10-GE-PNPs inhibited their phenotypic transformation (Figure 3D). A CCK-8 assay and an EdU assay were used to assess SMC proliferation. Compared with those in the blank group, SMCs in the PBS group showed significantly increased proliferation, whereas SMCs in the IL10, IL10-NP, IL10-PNP, and IL10-GE-PNP groups showed significantly decreased proliferation (Figure 3E and F). Transwell assays were used to assess the migration of SMCs. SMCs in the PBS group were significantly more likely to migrate than those in the blank group, while SMCs in the IL10, IL10-NP, IL10-PNP, and IL10-GE-PNP groups were significantly less likely to migrate (Figure 3G).
 |
Figure 3 The role of IL10-GE-PNP in regulating SMCs in an inflammatory environment simulated by the platelets + macrophages co-culture system in vitro is explored. (A) A diagram of the co-culture system is shown. (B) RT-qPCR is used to assess the relative expression of SM22 and (C) osteopontin (OPN) in SMCs, with n=3. (D) The expression of a contractile phenotype related protein in SMCs is analyzed after 24 hours of co-culture, with n=3. (E) The impact of IL10-GE-PNP on SMC cell viability is determined using the CCK-8 assay, with n=5. The results are normalized based on the blank group value. (F) The effect of IL10-GE-PNP on SMC proliferative ability is evaluated using the Edu assay, with n=3. (G) The impact of IL10-PNP on the migration ability of SMCs is determined through the transwell assay. n=3. Data are presented as mean ± SD, * p<0.05, ** p<0.01, *** p <0.001. Scale bar = 100μm. Abbreviation: ns not significant. |
EC Repair Function in Response to IL10-GE-PNPs
By repairing ECs as soon as possible after vascular injury, immune cell infiltration and SMC proliferation could be decreased. Tube formation assays and wound healing assays were used to assess the repair function of ECs. The tube formation assay revealed significantly decreased EC function in the PBS group and significantly increased EC function in the IL10, IL10-NP, IL10-PNP, and IL10-GE-PNP groups (Figure 4A and B). ECs in the IL10, IL10-NP, IL10-PNP, and IL10-GE-PNP groups healed significantly faster than those in the PBS group during the endothelial cell wound healing assay. PNPs, NPs, and GE-PNPs released IL10, which suppressed macrophage inflammatory responses and enhanced EC repair, while platelet membrane coating did not significantly affect EC repair (Figure 4C and D).
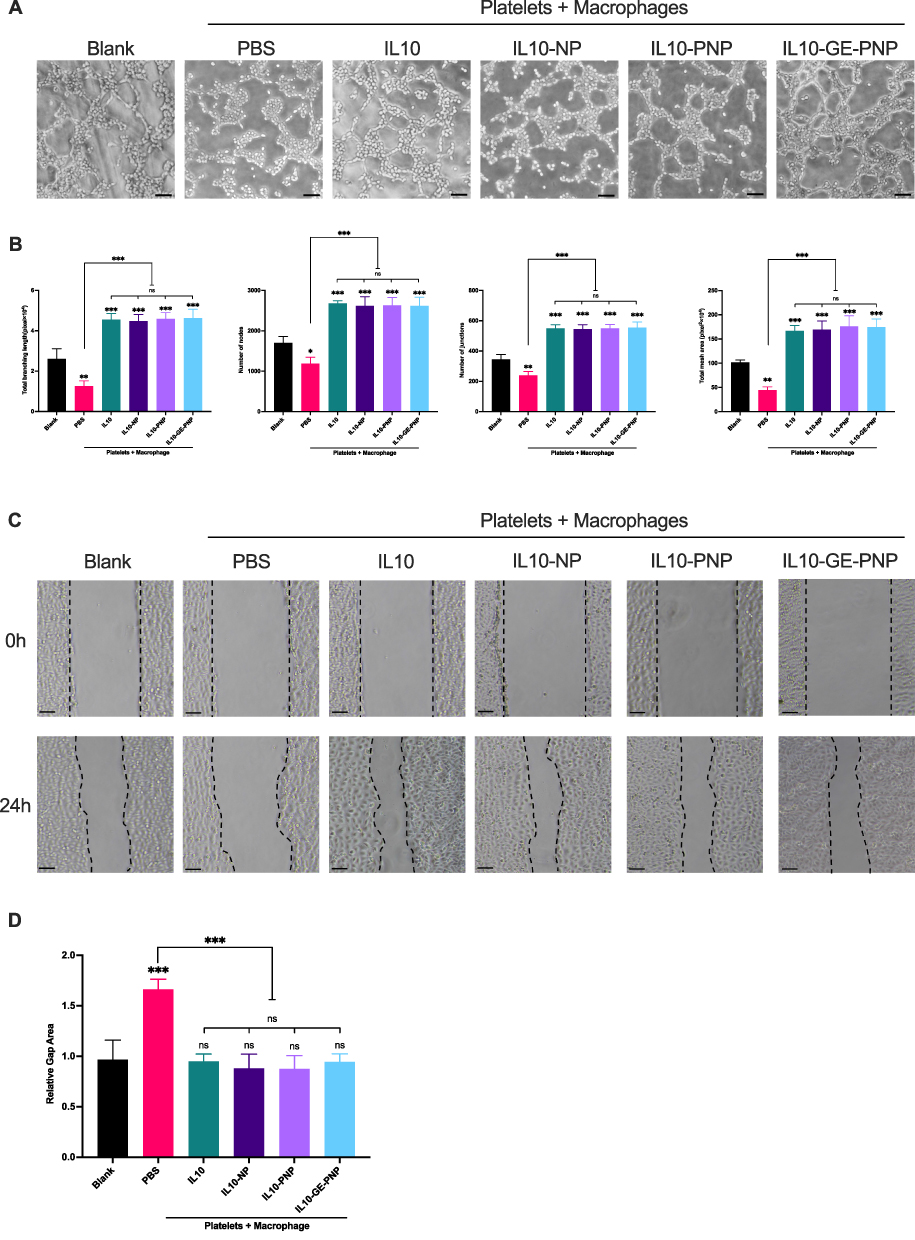 |
Figure 4 Regulation role of IL10-GE-PNP on ECs functions in an inflammatory environment simulated by platelets + macrophages coculture system in vitro. (A) The impact of IL10-GE-PNP on HUVECs function by tube formation assay. (B) Quantitative analysis of tube formation assay in different groups. (C) The impact of IL10-GE-PNP on HUVECs repair function by wound healing assay. (D) The relative gap area of wound healing assay in different groups, values were normalized based on the value of the blank group. Data are presented as mean ± SD, *p<0.05, **p<0.01, ***p <0.001. n=3. Scale bar = 50μm. Abbreviation: ns not significant. |
Vascular Injury Targeting and Release Function of Platelet Membrane-Coated Nanoparticles
Histological examination demonstrated that enhanced accumulation was found in the vascular injury in GE-PNP group, while little NP was detected in the vascular injury (Figure 5A). An intravenous injection of DiR-labeled NPs, PNPs and GE-PNPs was performed after vascular injury to evaluate whether circulating GE-PNPs accumulated there. Fluorescence was examined 24 hours after injection in the bilateral carotid arteries and major organs, including the heart, liver, spleen, lungs, and kidneys, ex vivo due to the thick tissue structure of rats, which prevented fluorescence from being observed in vivo. In the GE-PNP group, fluorescence intensity at the vascular injury site was significantly increased, suggesting that GE-PNP can accumulate more easily at sites of vascular injury (Figure 5B and C). Twenty-four hours after IL10-GE-PNP treatment, IL10 concentrations were measured in various organs to evaluate its release function. In comparison with the IL10-PNP and IL10-NP groups, the IL10-GE-PNP group had significantly higher IL10 concentrations in the left carotid artery (Figure 5D).
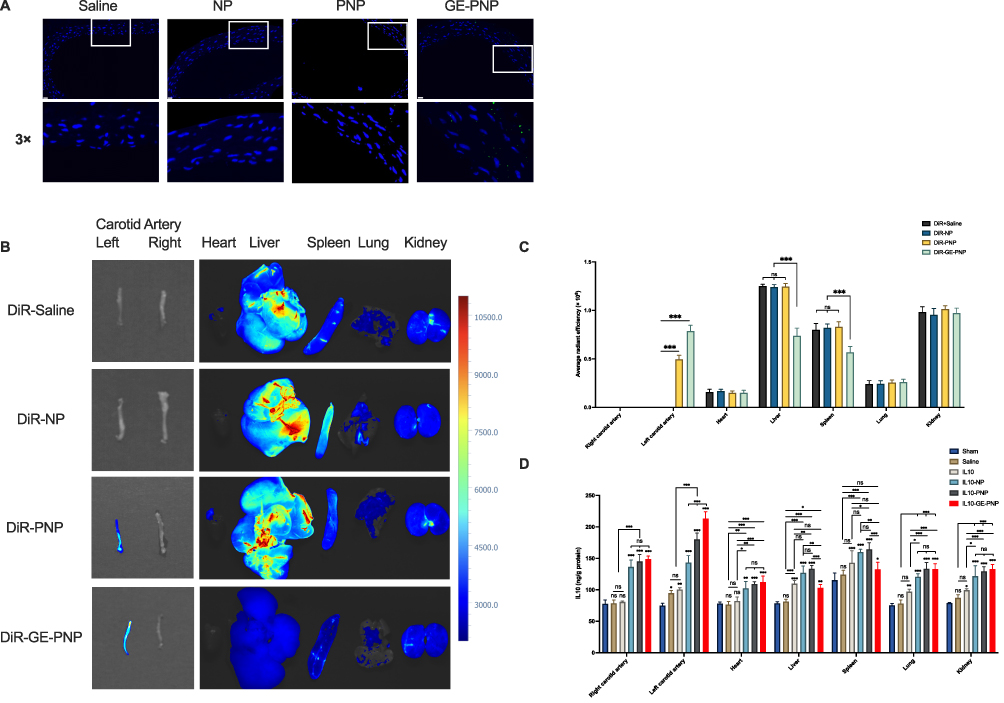 |
Figure 5 The targeting binding and release effects of GE-PNPs on vascular injury are studied. (A) Fluorescent microscopic images show the binding of NPs, PNPs, and GE-PNPs to the site of vascular injury. (B) Near-infrared fluorescence images of carotid arteries and main organs are obtained from the DiR solution group, DiR-NP group, DiR-PNP group, and DiR-GE-PNP group. (C) The average radiant efficiency of the carotid arteries and main organs is calculated for each group. (D) The concentration of IL10 in different organs is measured 24 hours after treatment. Data are presented as mean ± SD, *p<0.05, **p<0.01, ***p < 0.001, n=3. Scale bar = 20μm. Abbreviation: ns not significant. |
Targeted Polarization of Macrophages by IL10-GE-PNPs in vivo
We examined the expression of M1 and M2 macrophage-associated markers in response to IL10-GE-PNPs to determine its effects on macrophage phenotype polarization. For histological analysis, the injured left common carotid artery was obtained on Day 14. We stained tissue sections with F4/80 and CD86 or with F4/80 and CD163 to determine how many macrophages (F4/80+), M1 macrophages (F4/80+/CD86+) and M2 macrophages (F4/80+/CD163+) were present at the site of vascular injury (Figure 6A and B). The proportion of macrophages in the saline group, IL10 group, IL10-NP group, IL10-PNP group and IL10-GE-PNP group was significantly higher than that in the sham group (Figure 6C). The evidence indicates that vascular injury may result in macrophage infiltration in the local area. The proportion of macrophages significantly decreased after the application of IL10-PNPs and IL10-GE-PNPs. Additionally, in the IL10-NP, IL10-PNP, and IL10-GE-PNP groups, M1 macrophages were significantly decreased compared with those in the saline and IL10 groups (Figure 6D). These results suggest that following vascular injury, macrophages undergo a phenotypic change toward the M1 phenotype, which is likely to increase local inflammation. One milligram of IL10 administered systemically is ineffective at the site of vascular injury due to its low dose. There was a significant increase in M2 macrophages in the IL10-GE-PNP group compared to the other groups (Figure 6E). By promoting the polarization of local macrophages to the M2 phenotype, IL10-GE-PNPs suppress inflammation and promote tissue regeneration.
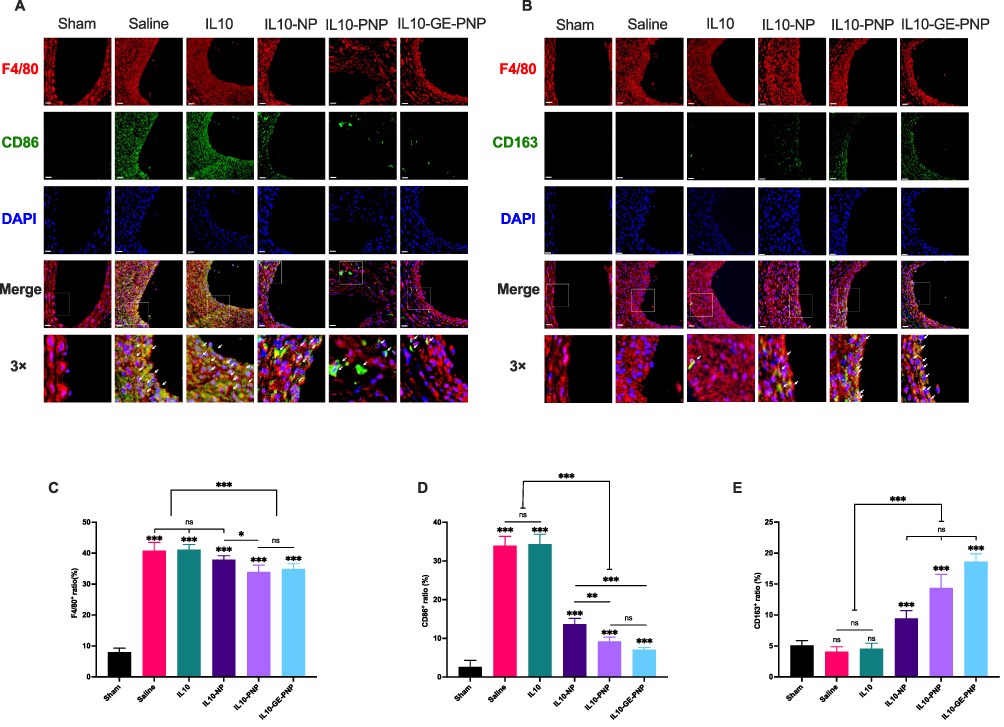 |
Figure 6 The role of IL10-GE-PNP in the regulation of macrophage polarization in vivo. (A) Dual immunofluorescence images showing F4/80 (red) and CD86 (green) in the sham group and in carotid arteries with different treatments, with nuclei stained by DAPI (blue). White arrows indicate F4/80+CD86+cells. (B) Dual immunofluorescence images showing F4/80 (red) and CD163 (green) in the sham group and in carotid arteries with different treatments, with nuclei stained by DAPI (blue). White arrows indicate F4/80+CD163+cells. (C) The percentage of macrophages among all cells. (D) The percentage of CD86-positive macrophages among all macrophages. (E) The percentage of CD163-positive macrophages among all macrophages. Data are presented as mean ± SD, *p<0.05, **p<0.01, ***p < 0.001, n=6. Scale bar = 20μm. Abbreviation: ns not significant. |
The Role of IL10-GE-PNPs in the Prevention of Restenosis
Rat left common carotid arteries were harvested 14 days after balloon injury and stained with H&E to analyze restenosis. The intimal to medial ratio (I/M) was measured as an index. At 14 days, IL10-GE-PNPs significantly reduced the neointimal areas and I/M compared to those of the IL10-PNP, IL10-NP, IL10, and saline groups (Figure 7A-C). To examine SMC proliferation and EC repair, we used immunohistochemistry. There was a significant increase in CD31 positivity in the endothelium of the blood vessels in the IL10-GE-PNP group compared to the other groups, suggesting that IL10-GE-PNPs could promote endothelial cell regeneration (Figure 7A, B and D). IL10-GE-PNPs significantly increased α-SMA expression, which is consistent with the more contractile phenotypes of SMCs (Figure 7A, B and E). According to Western blotting results, contractile phenotype-related proteins were significantly reduced after vascular injury, and IL10-GE-PNPs significantly reversed SMC phenotypic transformation (Figure 7F and G). The application of equal doses of IL10 systemically did not produce effective plasma concentrations and therefore did not inhibit restenosis.
 |
Figure 7 Anti-restenosis effect of IL10-GE-PNP in vivo. (A) H&E staining images, immunohistochemistry images of CD31 and α-SMA in the sham group and injured carotid artery with different treatments. (B) Quantitative analysis of intima area and (C) ratio of intima to media in different groups. (D) The ratio of CD31+ cells on the intimal side in different groups. (E) Quantitative analysis of mean integrated option density (IOD) in intima and media of different groups. (F and G) Relative expression of a contractile phenotype-related protein in left carotid artery 14 days after treatment. Data are presented as mean ± SD, *p<0.05, **p<0.01, ***p < 0.001, n=6. Scale bar = 100μm. Abbreviation: ns not significant. |
Evaluation of IL10-GE-PNP Biocompatibility in vivo
The in vitro cytotoxicity assay showed no significant difference in cell viability between different IL10 formulations and blank group. H&E-stained sections showed no evidence of integrity damage or distinguishable injuries in major organs. In addition, the treatment group did not differ significantly from the sham group in terms of complete blood count, biochemical and coagulation parameters, suggesting the safety of IL10-GE-PNPs (Figure S4).
Discussion
As the basic unit of life, cells have extremely precise and complex functional network systems, and their engineering can provide an effective method for the development of high-tech bioengineering technologies. Generally, cell engineering refers to the application of theories and methods of cell biology and molecular biology to carry out genetic manipulation and large-scale cell and tissue culture at the cellular level according to artificial design blueprints.29 The well-known CAR-T-cell therapy is a very typical application of cell engineering in disease treatment.30 Unlike traditional molecular engineering, the focus of cell engineering is not to optimize individual genes and proteins as therapeutic agents but to reprogram cells using molecular assemblies as modules to achieve regulation of in vivo functions.29
The development of cell membrane-coated nanoparticles has attracted increased attention because of their unique capabilities for biosimulation and biointerfaces. If other functions can be integrated into these nanoparticles, their applications could be greatly broadened as technology advances.31 In our previous study, IL10-loaded platelet membrane-coated nanoparticles were identified as an excellent candidate for targeted suppression of vascular restenosis based on immune regulation.14 However, it was found that a large quantity of PNPs were damaged, degraded and deposited in the liver and spleen. Therefore, it is necessary to further improve its targeting performance and antiphagocytic ability. Desialylated platelets are more capable of forming platelet-leukocyte aggregates since sialic acid is negatively charged and gives platelets a high electronegative charge. Upon the removal of sialic acid from platelets, the electronegative charge of the platelets is reduced, which in turn leads to more intercellular interactions between platelets and leukocytes and the formation of platelet-leukocyte aggregates. However, the desialylation of platelet glycoproteins can result in an increased number of binding sites for other cells. Thus, desialylation of platelet glycoproteins will likely enhance their ability for targeted adhesion to the site of vascular injury. Glycan modifications of platelet surface glycoproteins are also critical for platelet destruction and clearance. The terminal residues on N- and O-glycans are typically sialic acid with an adjacent penultimate sugar (β-gal). Therefore, as platelets are desalylated, they are exposed to more β-gal. This change is recognized by Ashwell-Morell receptors (AMRs), which are multimeric endocytic receptor complexes, also known as asialoglycoprotein receptors, located on the surface of hepatocytes and/or liver macrophages, causing platelets to be eliminated from circulation.32–34 In the present study, we removed sialic acid by NEU and successfully enhanced the targeting ability of PNPs (Figure 8).
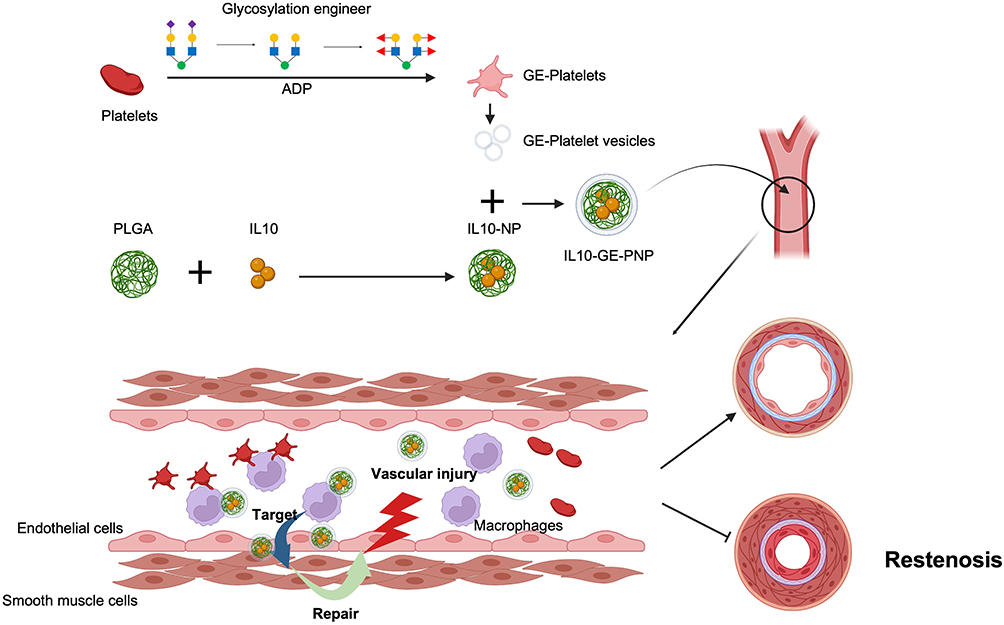 |
Figure 8 Schematic of this study. Created with BioRender.com. |
Glycoproteins with fucosylated moieties participate in many physiological and pathological processes, such as cell adhesion, tissue formation, angiogenesis, and fertilization.35 In previous studies, fucosylation modification was shown to enhance the homing of stem cells.23,36 Fucosylation biology has been studied extensively using genetic engineering, glycan-specific inhibitors of fucosylation, and competing inhibitors of fucose binding. Moreover, shielding of exposed β-GlcNac can effectively reduce the phagocytosis of platelets in the liver.37 In a previous study, FUT1 linked α (1,2)-fucose to β-Gal, and FUT7 linked α (1,3)-fucose to β-GlcNAc, which successfully shielded the exposed β-Gal and β-GlcNAc.22 In this study, we applied GDP-fucose as a donor and successfully linked α (1,2)-fucose and α (1,3)-fucose by corresponding fucosyltransferases, which effectively reduced the phagocytosis of PNP, resulted in a shorter preparation cycle and resulted in a wider clinical translation and application value. In the context of biosafety assessment, IL10-GE-PNP exhibited negligible cytotoxicity and exerted no substantial impact on the microstructure or functionality of vital organs in vivo. Collectively, these experimental findings underscore the robust biosafety profile of GE-PNP, signifying great potential for its prospective applications.
As an anti-inflammatory cytokine with immunoregulatory properties, IL10 has a broad spectrum of activity.7 Human recombinant IL10 is currently being studied for various inflammatory diseases, including inflammatory bowel disease, chronic hepatitis C, psoriasis, and rheumatoid arthritis. Further trials are underway.9 Vascular injury induces macrophage polarization to the M1 phenotype, resulting in SMC transformation to the secretory phenotype, which promotes proliferative growth and migration and ultimately leads to restenosis.5 However, the M2 phenotype reduces vascular restenosis by inhibiting inflammatory cytokine secretion, thus inhibiting the secretory phenotype transformation of SMCs and promoting the repair of ECs to reduce inflammatory cell infiltration.6,38 IL10 can also inhibit vascular SMC activation in vitro and in vivo. Vascular injury triggers the release of inflammatory factors, causing SMCs to switch from an inactive contractile phenotype to an active secretory phenotype, resulting in restenosis. Through I-κB degradation and NF-κB activation, IL10 inhibits SMC proliferation and migration, resulting in a reduction in intimal hyperplasia.39 IL10 can also recruit endothelial progenitor cells, which promote endothelial sprouting and network formation, leading to faster re-epithelialization.40 Notably, systemic application of IL10 can result in side effects such as anemia and thrombocytopenia, which are serious health risks.9,41 Interleukin-based nanoparticles have shown good targeting effects and stability.11,26,42,43 IL10-NP could effectively regulate and target advanced atherosclerosis, which is also a vascular disease based on inflammation.11 Nevertheless, its regulatory effects on local immune cells have not been fully explored, and its targeted delivery capabilities may further be improved. Our previous study showed that platelet membrane-coated IL10-NPs have a stronger targeting effect and a stronger ability to regulate the local immune environment at the lesion site.14 However, there are still many IL10-PNP deposits in the liver and spleen. In our study, the immunomodulatory function of IL10 and its effects on SMCs and ECs were also verified. Additionally, the preparation of nanoparticles and platelet membrane coatings with or without glycosylation engineering did not significantly influence the release and regulatory efficacy of IL10 in vitro regardless of the method used to prepare them. Based on the findings outlined above, it is foreseeable that IL10-GE-PNP holds the potential to emerge as a potent and targeted inhibitor for combating vascular restenosis in forthcoming approaches. This glycosylation editing technique exhibits wide-ranging applicability in enhancing the targeting efficacy of cell-based therapeutic interventions. This study, however, presents several limitations. The validation of glycosylation outcomes relied on flow cytometry rather than employing the more precise glycosylation mass spectrometry. For potential upcoming preclinical investigations, additional comprehensive and standardized verification is required for cytotoxicity assessments and the evaluation of biocompatibility.
Conclusions
Glycosylation engineering has been proven to be an effective way to reduce PNP phagocytosis and enhance target performance. In conclusion, IL10-GE-PNPs is an excellent candidate for targeting vascular injury and shows promise as an innovative drug delivery system for suppressing vascular restenosis.
Data Sharing Statement
The data used to support the findings of this study have not been made available online because some of them are essential data for further study. The data are available from the corresponding authors ([email protected] Leng Ni; [email protected] Changwei Liu) upon reasonable request.
Ethics Approval and Consent to Participate
For the in vitro experiment, platelets were collected from healthy volunteers (healthy university students) without any anti-platelet drugs taken for at least 14 days, and informed consent was obtained, and approved by Peking Union Medical College Hospital ethics committee (KS2022314). All animal studies were conducted in accordance with the Animal Management Rules of the Chinese Ministry of Health, and the Animal Ethics Committee of Peking Union Medical College reviewed and approved the procedures of the studies (No. XHDW-2019-001).
Consent for Publication
All authors gave their consent for publication.
Acknowledgments
We thank the public laboratory platform, cell biology center facility and animal research laboratory platform of the National Science and Technology Key Infrastructure on Translational Medicine in Peking Union Medical College Hospital. This work was supported by the National Natural Science Foundation of China (No. 81970417).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81970417) and National High Level Hospital Clinical Research Funding (No. 2022-PUMCH-A-079).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Cannon B. Cardiovascular disease: biochemistry to behaviour. Nature. 2013;493(7434):S2–3. doi:10.1038/493S2a
2. Bønaa KH, Mannsverk J, Wiseth R, et al. Drug-Eluting or Bare-Metal Stents for Coronary Artery Disease. N Engl J Med. 2016;375(13):1242–1252. doi:10.1056/NEJMoa1607991
3. Jukema JW, Verschuren JJW, Ahmed TAN, Quax PHA. Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat Rev Cardiol. 2011;9(1):53–62. doi:10.1038/nrcardio.2011.132
4. Clare J, Ganly J, Bursill CA, Sumer H, Kingshott P, de Haan JB. The Mechanisms of Restenosis and Relevance to Next Generation Stent Design. Biomolecules. 2022;12(3):430. doi:10.3390/biom12030430
5. Xing Y, Pan S, Zhu L, et al. Advanced Glycation End Products Induce Atherosclerosis via RAGE/TLR4 Signaling Mediated-M1 Macrophage Polarization-Dependent Vascular Smooth Muscle Cell Phenotypic Conversion. Oxid Med Cell Longev. 2022;2022:9763377. doi:10.1155/2022/9763377
6. Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19(6):526–537. doi:10.1038/s41590-018-0113-3
7. de Vries JE. Immunosuppressive and anti-inflammatory properties of interleukin 10. Ann Med. 1995;27(5):537–541. doi:10.3109/07853899509002465
8. Ip WE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–519. doi:10.1126/science.aal3535
9. Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy--review of a new approach. Pharmacol Rev. 2003;55(2):241–269. doi:10.1124/pr.55.2.4
10. Kim M, Sahu A, Hwang Y, et al. Targeted delivery of anti-inflammatory cytokine by nanocarrier reduces atherosclerosis in Apo E-/- mice. Biomaterials. 2020;226:119550. doi:10.1016/j.biomaterials.2019.119550
11. Kamaly N, Fredman G, Fojas JJR, et al. Targeted Interleukin-10 Nanotherapeutics Developed with a Microfluidic Chip Enhance Resolution of Inflammation in Advanced Atherosclerosis. ACS Nano. 2016;10(5):5280–5292. doi:10.1021/acsnano.6b01114
12. Margraf A, Zarbock A. Platelets in Inflammation and Resolution. J Immunol. 2019;203(9):2357–2367. doi:10.4049/jimmunol.1900899
13. Hu CM, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526(7571):118–121. doi:10.1038/nature15373
14. Li F, Rong Z, Zhang R, et al. Vascular restenosis reduction with platelet membrane coated nanoparticle directed M2 macrophage polarization. iScience. 2022;25(10):105147. doi:10.1016/j.isci.2022.105147
15. Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21(12):729–749. doi:10.1038/s41580-020-00294-x
16. Jones C, Denecke J, Sträter R, et al. A novel type of macrothrombocytopenia associated with a defect in α2,3-sialylation. Am J Pathol. 2011;179(4):1969–1977. doi:10.1016/j.ajpath.2011.06.012
17. de la Morena-Barrio ME, Di Michele M, Lozano ML, et al. Proteomic analysis of platelet N-glycoproteins in PMM2-CDG patients. Thromb Res. 2014;133(3):412–417. doi:10.1016/j.thromres.2013.12.024
18. Li L, Qu C, Lu Y, et al. The platelet surface glycosylation caused by glycosidase has a strong impact on platelet function. Blood Coagul Fibrinolysis. 2019;30(5):217–223. doi:10.1097/MBC.0000000000000826
19. Li L, Qu C, Wu X, et al. Patterns and levels of platelet glycosylation in patients with coronary heart disease and type 2 diabetes mellitus. J Thromb Thrombolysis. 2018;45(1):56–65. doi:10.1007/s11239-017-1573-2
20. Wandall HH, Rumjantseva V, Sørensen ALT, et al. The origin and function of platelet glycosyltransferases. Blood. 2012;120(3):626–635. doi:10.1182/blood-2012-02-409235
21. Quach ME, Chen W, Li R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood. 2018;131(14):1512–1521. doi:10.1182/blood-2017-08-743229
22. Li J, Hsu HC, Mountz JD, Allen JG. Unmasking Fucosylation: from Cell Adhesion to Immune System Regulation and Diseases. Cell Chem Biol. 2018;25(5):499–512. doi:10.1016/j.chembiol.2018.02.005
23. Xia L, McDaniel JM, Yago T, Doeden A, McEver RP. Surface fucosylation of human cord blood cells augments binding to P-selectin and E-selectin and enhances engraftment in bone marrow. Blood. 2004;104(10):3091–3096. doi:10.1182/blood-2004-02-0650
24. Niu S, Fei M, Cheng C, et al. Altered beta-1,4-galactosyltransferase I expression during early inflammation after spinal cord contusion injury. J Chem Neuroanat. 2008;35(3):245–256. doi:10.1016/j.jchemneu.2008.01.002
25. Lee J, Yeo I, Kim Y, et al. Comparison of Fucose-Specific Lectins to Improve Quantitative AFP-L3 Assay for Diagnosing Hepatocellular Carcinoma Using Mass Spectrometry. J Proteome Res. 2022;21(6):1548–1557. doi:10.1021/acs.jproteome.2c00196
26. Zeng L, Ma W, Shi L, et al. Poly(lactic-co-glycolic acid) nanoparticle-mediated interleukin-12 delivery for the treatment of diabetic retinopathy. Int J Nanomedicine. 2019;14:6357–6369. doi:10.2147/IJN.S214727
27. Chen H, Gao J, Lu Y, et al. Preparation and characterization of PE38KDEL-loaded anti-HER2 nanoparticles for targeted cancer therapy. J Control Release. 2008;128(3):209–216. doi:10.1016/j.jconrel.2008.03.010
28. Baxter EW, Graham AE, Re NA, et al. Standardized protocols for differentiation of THP-1 cells to macrophages with distinct M(IFNγ+LPS), M(IL-4) and M(IL-10) phenotypes. J Immunol Methods. 2020;478:112721. doi:10.1016/j.jim.2019.112721
29. Lim WA. The emerging era of cell engineering: harnessing the modularity of cells to program complex biological function. Science. 2022;378(6622):848–852. doi:10.1126/science.add9665
30. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379(1):64–73. doi:10.1056/NEJMra1706169
31. Ai X, Wang S, Duan Y, et al. Emerging Approaches to Functionalizing Cell Membrane-Coated Nanoparticles. Biochemistry. 2021;60(13):941–955. doi:10.1021/acs.biochem.0c00343
32. Li Y, Fu J, Ling Y, et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc Natl Acad Sci U S A. 2017;114(31):8360–8365. doi:10.1073/pnas.1707662114
33. Sørensen AL, Rumjantseva V, Nayeb-Hashemi S, et al. Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood. 2009;114(8):1645–1654. doi:10.1182/blood-2009-01-199414
34. Grewal PK. The Ashwell-Morell receptor. Methods Enzymol. 2010;479:223–241. doi:10.1016/S0076-6879(10)79013-3
35. Miyoshi J, Yajima T, Okamoto S, et al. Ectopic expression of blood type antigens in inflamed mucosa with higher incidence of FUT2 secretor status in colonic Crohn’s disease. J Gastroenterol. 2011;46(9):1056–1063. doi:10.1007/s00535-011-0425-7
36. Chen L, Luo W, Wang Y, et al. Directional homing of glycosylation-modified bone marrow mesenchymal stem cells for bone defect repair. J Nanobiotechnology. 2021;19(1):228. doi:10.1186/s12951-021-00969-3
37. Hoffmeister KM, Josefsson EC, Isaac NA, Clausen H, Hartwig JH, Stossel TP. Glycosylation restores survival of chilled blood platelets. Science. 2003;301(5639):1531–1534. doi:10.1126/science.1085322
38. Yan W, Li T, Yin T, et al. M2 macrophage-derived exosomes promote the c-KIT phenotype of vascular smooth muscle cells during vascular tissue repair after intravascular stent implantation. Theranostics. 2020;10(23):10712–10728. doi:10.7150/thno.46143
39. Mazighi M, Pellé A, Gonzalez W, et al. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am J Physiol Heart Circ Physiol. 2004;287(2):H866–871. doi:10.1152/ajpheart.00918.2003
40. Short WD, Steen E, Kaul A, et al. IL-10 promotes endothelial progenitor cell infiltration and wound healing via STAT3. FASEB J. 2022;36(7):e22298. doi:10.1096/fj.201901024RR
41. Fredman G, Kamaly N, Spolitu S, et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7(275):275ra20. doi:10.1126/scitranslmed.aaa1065
42. Raimondo TM, Mooney DJ. Functional muscle recovery with nanoparticle-directed M2 macrophage polarization in mice. Proc Natl Acad Sci U S A. 2018;115(42):10648–10653. doi:10.1073/pnas.1806908115
43. Cappellano G, Woldetsadik AD, Orilieri E, et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. 2014;32(43):5681–5689. doi:10.1016/j.vaccine.2014.08.016
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.