Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Ginsenoside Rb1 Promotes Hepatic Glycogen Synthesis to Ameliorate T2DM Through 15-PGDH/PGE2/EP4 Signaling Pathway
Authors Liang M, Zhan W, Wang L, Bei W, Wang W
Received 18 August 2023
Accepted for publication 11 October 2023
Published 17 October 2023 Volume 2023:16 Pages 3223—3234
DOI https://doi.org/10.2147/DMSO.S431423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Mingjie Liang,1,2,* Wenjing Zhan,1,2,* Lexun Wang,1,2 Weijian Bei,1,2 Weixuan Wang1,2
1Traditional Chinese Medicine Research Institute, Guangdong Pharmaceutical University, Guangzhou, Guangdong Province, People’s Republic of China; 2Guangdong Provincial Research Center of Integration of Traditional Chinese Medicine and Western Medicine in Metabolic Diseases, Guangdong Pharmaceutical University, Guangzhou, Guangdong Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weixuan Wang, Guangdong Pharmaceutical University, No. 280, Waihuan East Road, University Town, Guangzhou, Guangdong Province, 510006, People’s Republic of China, Tel +86-020-39352607, Email [email protected]
Purpose: Ginsenoside Rb1 (Rb1), one of the crucial bioactive constituents in Panax ginseng C. A. Mey., possesses anti-type 2 diabetes mellitus (T2DM) property. Nevertheless, the precise mechanism, particularly the impact of Rb1 on hepatic glycogen production, a crucial process in the advancement of T2DM, remains poorly understood. 15-hydroxyprostaglandin dehydrogenase (15-PGDH) is responsible for prostaglandin E2 (PGE2) inactivation. A recent study has reported that inhibition of 15-PGDH promoted hepatic glycogen synthesis and improved T2DM. Therefore, herein, we aimed to investigate whether Rb1 ameliorated T2DM through 15-PGDH/PGE2-regulated hepatic glycogen synthesis.
Methods: By combining streptozotocin with a high-fat diet, we successfully established a mouse model for T2DM. Afterward, these mice were administered Rb1 or metformin for 8 weeks. An insulin-resistant cell model was established by incubating LO2 cells with palmitic acid. Liver glycogen and PGE2 levels, the expression levels of 15-PGDH, serine/threonine kinase AKT (AKT), and glycogen synthase kinase 3 beta (GSK3β) were measured. Molecular docking was used to predict the binding affinity between 15-PGDH and Rb1.
Results: Rb1 administration increased the phosphorylation levels of AKT and GSK3β to enhance glycogen synthesis in the liver of T2DM mice. Molecular docking indicated that Rb1 had a high affinity for 15-PGDH. Moreover, Rb1 treatment resulted in the suppression of elevated 15-PGDH levels and the elevation of decreased PGE2 levels in the liver of T2DM mice. Furthermore, in vitro experiments showed that Rb1 administration might enhance glycogen production by modulating the 15-PGDH/PGE2/PGE2 receptor EP4 pathway.
Conclusion: Our findings indicate that Rb1 may enhance liver glycogen production through a 15-PGDH-dependent pathway to ameliorate T2DM, thereby offering a new explanation for the positive impact of Rb1 on T2DM and supporting its potential as an effective therapeutic approach for T2DM.
Keywords: ginsenoside Rb1, type 2 diabetes mellitus, 15-hydroxyprostaglandin dehydrogenase, prostaglandin E2, hepatic glycogen synthesis
Introduction
By the year 2021, the global number of diabetes patients reached 537 million,1 with type 2 diabetes mellitus (T2DM) constituting 90% of all diabetes instances.2 As a pervasive global epidemic, T2DM not only exerts pressure on public healthcare infrastructure but also imposes a growing economic burden on families. Since conventional treatment strategies for T2DM usually have side effects,3 active ingredients from medicinal plants may be promising treatment strategies and deserve further study.
The development of T2DM is greatly influenced by insulin resistance.4 The liver is a vital organ responsible for coordinating metabolic homeostasis.5 In insulin resistance states, insulin fails to maintain hepatic glucose homeostasis, leading to hyperglycemia,5 therefore, improving hepatic insulin resistance may be critical in treating T2DM. Serine/threonine kinase AKT (AKT) plays a vital role in regulating the insulin signaling pathway and governing hepatic glucose metabolism.5 AKT enhances the synthesis of glycogen by phosphorylating and suppressing the activity of glycogen synthase kinase 3 beta (GSK3β).5,6 In the occurrence of hepatic insulin resistance, the insulin-induced activation of AKT is hindered, leading to the suppression of glycogen synthesis in the liver and consequently causing an increase in blood glucose levels.6 Previous research has reported that the decreased hepatic glycogen synthesis contributed to T2DM progression,7 making the AKT/GSK3β signaling pathway a promising target for treating T2DM.
Prostaglandin E2 (PGE2) is a vital lipid mediator that modulates various biological processes. Although PGE2 has been shown to promote inflammation, mounting studies have demonstrated that PGE2 played positive roles in numerous pathophysiological processes.8,9 PGE2 has been shown to promote glycogen synthesis, thereby maintaining glucose homeostasis.10 PGE2 also reduced the activity of lipogenic enzymes and the level of tumor necrosis factor-α to inhibit hepatic steatosis and inflammation.11,12 The levels of PGE2 are tightly controlled. The degradation of PGE2 occurs through the action of 15-hydroxyprostaglandin dehydrogenase (15-PGDH).13 Although previous studies have shown that the suppression of 15-PGDH activity promoted tissue regeneration and protected against tissue damage,13,14 its influence on T2DM is still not well clarified. In our recent investigation, we initially demonstrated that inhibition of 15-PGDH activity promoted glycogen synthesis in mice with T2DM,15 indicating that targeting 15-PGDH could potentially improve glucose metabolism abnormalities in T2DM.
Ginsenosides are the main bioactive ingredients of Panax ginseng C. A. Mey. (P. ginseng), and among the numerous ginsenosides, ginsenoside Rb1 (Rb1) is considered the predominant component.16,17 Previous studies have demonstrated notable anti-obesity and anti-diabetes properties of Rb1.3,18,19 Rb1 can decrease body weight and fasting blood glucose levels, ameliorate impaired glucose tolerance and insulin resistance, and attenuate liver steatosis.3,20 However, the effect and mechanism of Rb1 on hepatic glycogen synthesis is still unclear. Hence, the objective of this research was to investigate whether Rb1 promoted hepatic glycogen synthesis and improved T2DM by reducing the expression of 15-PGDH in vivo (based on a T2DM mouse model induced by a high-fat diet/streptozotocin injection) and in vitro (employing an insulin-resistant cell model induced by palmitic acid treatment).
Materials and Methods
Resources
Sigma-Aldrich (St. Louis, MO, USA) provided streptozotocin (S0130) and palmitic acid (P5585). Prostaglandin E2 receptor EP4 (EP4) inhibitor CJ-42794 (C4037) was acquired from APExBIO (Houston, TX, USA). Cell Signaling Technology (Danvers, MA, USA) provided the following antibodies: anti-AKT (9272, 1:1000), anti-phosphorylated (p)-AKTSer473 (9271, 1:1000), anti-β-actin (3700, 1:2000), anti-rabbit secondary antibody (7074, 1:3000), and anti-mouse secondary antibody (7076, 1:3000). The anti-15-PGDH antibody (ab187161, Abcam, Cambridge, MA, USA) was diluted at a ratio of 1:1000. The anti-p-GSK3βSer9 antibody (AF2016, 1:1000) was obtained from Affinity Biosciences (Cincinnati, USA). The anti-GSK3β antibody (22104-1-AP, 1:1000) was obtained from Proteintech (Rosemont, IL, USA). Rb1 was obtained from PUSH BIO-TECHNOLOGY (Chengdu, China).
Animal Experiments
Male C57BL/6J mice were kept in a specific pathogen-free environment at the animal center of Guangdong Pharmaceutical University. Approval for all animal studies was obtained from the Animal Experimentation Ethics Committee of Guangdong Pharmaceutical University (Approval number: gdpulacspf2017586). Animal experiments followed the NIH guidelines for the care and use of laboratory animals (8th edition). Mice were kept in a room with a controlled temperature of 25 ± 2 °C, with a light/dark cycle of 12 h/12 h. Mice were given unrestricted access to food and water during the entire study.
All mice underwent a one-week acclimatization period before the commencement of the experiments. The control group (Ctrl, n = 6) mice were given a chow diet, whereas the remaining mice (n = 24) were induced to develop T2DM by combining a high-fat diet (HF60, Dyets, Bethlehem, PA, USA) with streptozotocin injection. Mice were given a high-fat diet for 4 consecutive weeks.21 Subsequently, the mice received intraperitoneal injections of streptozotocin (30 mg/kg of body weight) for 3 continuous days.21,22 Two weeks after injection of streptozotocin, mice with fasting blood glucose exceeding 11.1 mmol/L21 were randomly assigned to 4 groups: T2DM group (T2DM, n = 6), 20 mg/kg Rb1-treated group (L-Rb1, n = 6), 50 mg/kg Rb1-treated group (H-Rb1, n = 6), and 250 mg/kg metformin-treated group (MET, n = 6). Rb1 or metformin was given orally for 8 consecutive weeks, with the doses of Rb1 or metformin being determined based on prior studies.23–25 After 8 weeks of drug treatment, the blood and liver specimens were gathered subsequent to a 12 h fasting duration. After anesthesia with 3% isoflurane, blood samples were obtained through retro-orbital bleeding, and euthanasia was performed by cervical dislocation.26–28 The schematic diagram of animal experiment process is presented in Figure S1.
Biochemical Analysis
An ELISA kit (E-EL-0034c) brought from Elabscience (Wuhan, China) was utilized to measure the levels of PGE2 in the liver. Liver glycogen levels were measured using a glycogen assay kit (A043-1-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Periodic Acid-Schiff (PAS) Staining
The PAS staining kit was purchased from Servicebio (Wuhan, China). The liver specimens were fixed using 4% paraformaldehyde, embedded with paraffin, and cut into 4 μm-thick slices. The paraffin-embedded slices underwent dewaxing using xylene, followed by rehydration with ethanol and rinsing with distilled water. After being oxidized with 0.5% periodic acid for 15 min, each section was stained with Schiff reagent for 10 min, washed with distilled water for 5 min, and then counterstained with hematoxylin for 30 sec. Sequentially, the slices underwent washing, dehydration, and sealing. The microscope (Olympus, Tokyo, Japan) was used to obtain pictures of the stained sections.
Molecular Docking
The structure of Rb1 was provided by the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The Protein Data Bank (PDB) (https://www.pdb.org/) provided the crystal structure of 15-PGDH. Rb1 was identified as the ligand, and the 15-PGDH protein was designated as the receptor. The original ligand of 15-PGDH protein and water molecules were eliminated using PyMOL (version 4.3.0). Tasks such as hydrogenation, constructing a docking grid box for the protein, and converting the PDB format to PDBQT were carried out using AutoDockTools (version 4.2). Then, AutoDock Vina (version 1.1.2) was employed for molecular docking analysis. The docking result was displayed as binding free energy, which serves as an indicator of binding affinity. A lower binding energy represents a higher binding affinity between the protein and the ligand.
Western Blot Analysis
Liver specimens and LO2 cells were obtained and then lysed in RIPA buffer (Solarbio, Beijing, China), which included inhibitors for proteases and phosphatases (Thermo Fisher Scientific, Waltham, MA, USA). The BCA protein assay kit (Solarbio) was used to measure the concentration of protein. Following separation using SDS-PAGE, the proteins were transferred onto a PVDF transfer membrane (IPVH00010, Millipore, Burlington, MA, USA). The Western blot assays were conducted following a standard procedure as previously described.15 The Western blot images were quantified using the Image Lab software (Bio-Rad, Hercules, CA, USA).
Cell Experiments
LO2 hepatocytes were brought from the Cell Bank of The Chinese Academy of Sciences (Shanghai, China). LO2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, Waltham, MA, USA), which contained 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Wisent, Montreal, QC, Canada). The dose and incubation time of palmitic acid,29 Rb1,30 and CJ-4279415 were chosen based on previous studies. To induce insulin resistance in LO2 cells, palmitic acid was added at a concentration of 250 μM for 24 h. Insulin-resistant LO2 cells were then treated with Rb1 (20 μM) or a combination of Rb1 (20 μM) and CJ-42794 (40 nM) for 24 h.
The measurement of glucose consumption was conducted using a glucose assay kit (F006-1-1, Nanjing Jiancheng Bioengineering Institute) as previously described.15 An ELISA kit (E-EL-0034c, Elabscience) was utilized to measure the PGE2 levels in the supernatant of the cell culture medium.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8.0 (La Jolla, CA, USA). To compare the average values among different groups, one-way ANOVA was employed, followed by the application of Tukey’s test. The data were presented in the form of mean ± standard deviation (SD). A significance level of P < 0.05 was deemed to be statistically significant.
Results
Rb1 Treatment Increases Hepatic Glycogen Levels in T2DM Mice
Previously, we have reported that Rb1 treatment reduces fasting blood glucose levels in T2DM mice.20 Nevertheless, the precise mechanism is still not well clarified. The liver plays a crucial role in regulating glucose metabolism.6 In response to insulin stimulation, the liver can store glucose as glycogen.31 Given that reduced hepatic glycogen synthesis is associated with increased blood glucose levels and contributes to the advancement of T2DM, the liver was collected for further examination.6,7 The glycogen levels in different groups were visualized by PAS staining (Figure 1A). Under the same area, compared to the control group, a significant decrease in the amount of PAS staining was observed in T2DM mice, however, this decrease was markedly increased after administration of Rb1 or metformin (Figure 1A), which is consistent with the result of a glycogen assay kit (Figure 1B).
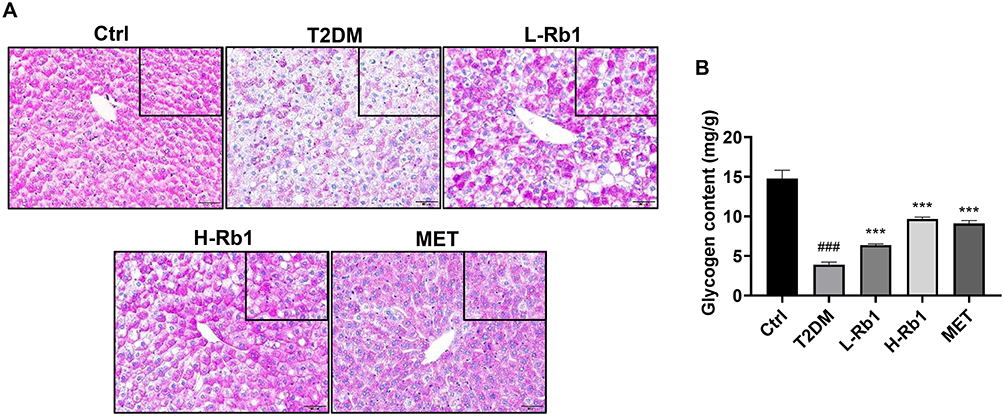 |
Figure 1 Rb1 treatment enhances hepatic glycogen levels in type 2 diabetes mellitus (T2DM) mice. (A) Liver samples were obtained from control mice (Ctrl), T2DM mice, mice treated with 20 mg/kg of Rb1 (L-Rb1), mice treated with 50 mg/kg of Rb1 (H-Rb1), and mice treated with 250 mg/kg of metformin (MET) and conducted periodic acid-Schiff staining. Scale bar = 50 μm. The upper right corner box refers to the coloring area of PAS under the same area. (B) Liver glycogen levels; n = 6. ###P < 0.001 vs the Ctrl group; ***P < 0.001 vs the T2DM group. |
Rb1 Promotes Hepatic Glycogen Synthesis in T2DM Mice by Modulating the AKT/GSK3β Signaling Pathway
AKT is a key kinase in the insulin signaling pathway and serves as a pivotal regulator in the maintenance of glucose homeostasis.5 AKT can phosphorylate and inactivate GSK3β, therefore promoting glycogen synthesis.5 AKT phosphorylation is decreased in the state of insulin resistance, leading to the activation of GSK3β and a decrease in glycogen synthesis.5 The Western blot analysis demonstrated a decrease in the hepatic phosphorylation levels of AKTSer473 and GSK3βSer9 in the T2DM group, however, these phosphorylation levels were up-regulated following the administration of Rb1 or metformin (Figure 2A–C). These results suggest that Rb1 might promote hepatic glycogen synthesis to prevent T2DM progression through the AKT/GSK3β signaling pathway.
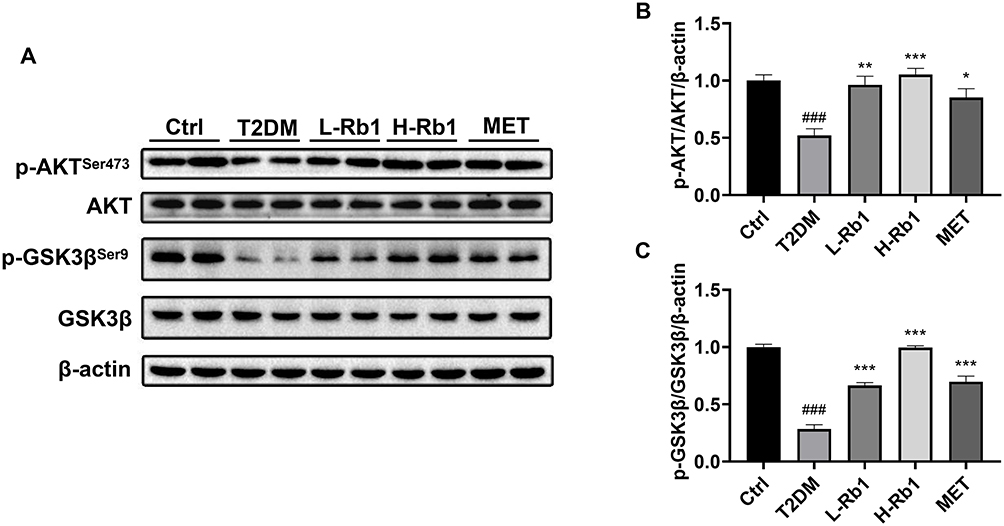 |
Figure 2 Rb1 treatment enhances hepatic glycogen production by regulating the AKT/GSK3β signaling pathway in T2DM mice. (A) Levels of AKT, p-AKTSer473, GSK3β, and p-GSK3βSer9 were analyzed using Western blot analysis. β-actin was used as the control for loading. (B) The bar chart displayed the quantitative data of p-AKTSer473/AKT/β-actin obtained from Western blot analysis. (C) The bar chart displayed the quantitative data of p-GSK3βSer9/GSK3β/β-actin obtained from Western blot analysis. n = 4. ###P < 0.001 vs the Ctrl group; *P < 0.05, **P < 0.01, ***P < 0.001 vs the T2DM group. |
Rb1 Treatment Decreases the 15-PGDH Expression Level in the Liver of T2DM Mice
15-PGDH is a dehydrogenase that is responsible for PGE2 degradation. In a recent study, we demonstrated for the first time that administration of 15-PGDH inhibitor resulted in an increase in hepatic glycogen synthesis in T2DM mice.15 Given that 15-PGDH may be a core regulator in Rb1-ameliorated T2DM, molecular docking analysis was performed to investigate the potential interaction between Rb1 and 15-PGDH (Figure 3A). The molecular docking result suggested that Rb1 may interact with 15-PGDH, as indicated by binding free energy of −6.2 kcal/mol. Moreover, as shown in Figure 3A, Rb1 may interact with several amino acids of 15-PGDH protein, such as Val159, Ser164, and Tyr263.
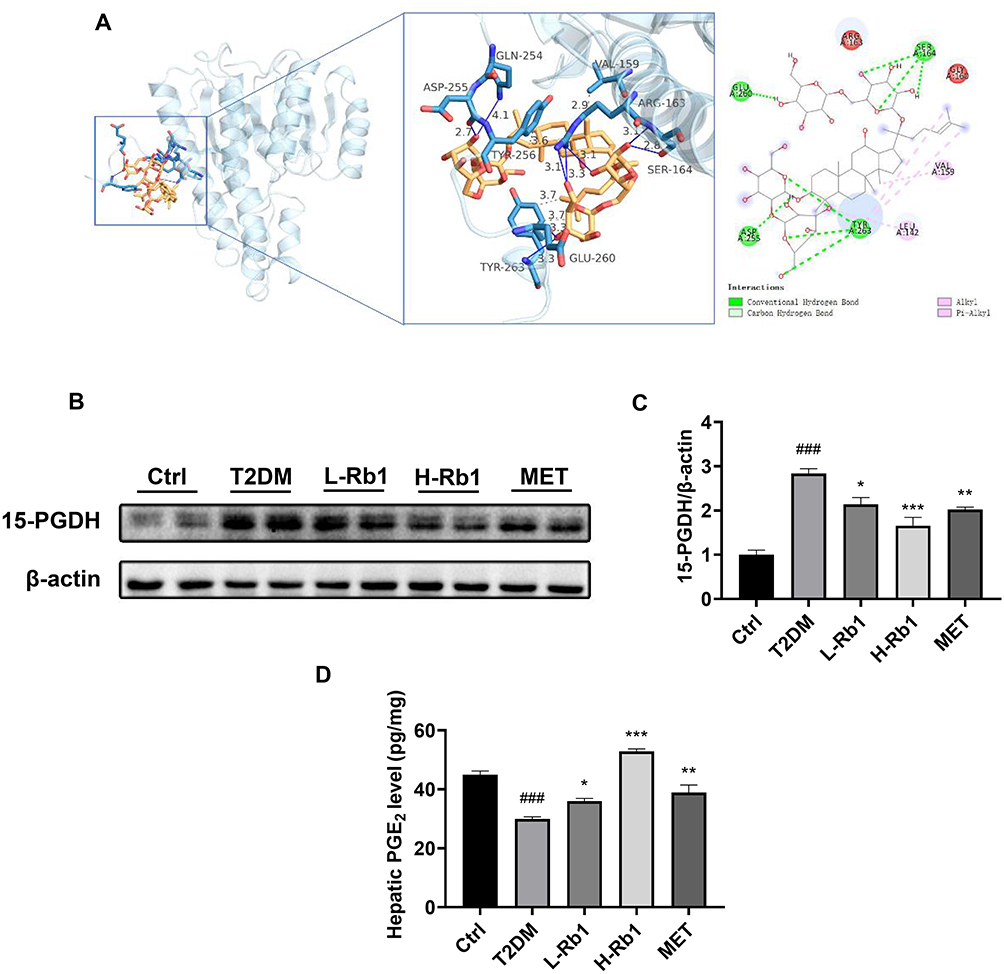 |
Figure 3 15-hydroxyprostaglandin dehydrogenase (15-PGDH) is a vital regulator of Rb1 in preventing T2DM. (A) Molecular docking analysis of the interaction between Rb1 and 15-PGDH. (B) The hepatic expression level of 15-PGDH was analyzed using Western blotting analysis. β-actin was used as the control for loading. (C) The bar chart displayed the quantitative data of 15-PGDH/β-actin obtained from Western blot analysis; n = 4. (D) Hepatic prostaglandin E2 (PGE2) levels; n = 6. ###P < 0.001 vs the Ctrl group; *P < 0.05, **P < 0.01, ***P < 0.001 vs the T2DM group. |
Western blot analysis found that Rb1 or metformin treatment decreased the up-regulated 15-PGDH expression level in the liver of T2DM mice (Figure 3B and C). Moreover, the levels of PGE2 in the liver were assessed in different groups. In the T2DM group, the hepatic PGE2 levels were lower compared to the control group, and Rb1 or metformin treatment significantly enhanced the PGE2 levels (Figure 3D). These findings indicate that 15-PGDH may be a crucial regulator in Rb1-ameliorated T2DM.
Rb1 Enhances Glycogen Synthesis by Regulating the 15-PGDH/PGE2/EP4 Pathway
Insulin resistance is a key feature of T2DM.4 Therefore, palmitic acid-induced insulin-resistant LO2 hepatocytes were used in the following experiments. The cells treated with palmitic acid showed a decrease in glucose consumption, indicating the successful creation of an insulin-resistant cell model (Figure 4A). Furthermore, the administration of Rb1 resulted in an enhancement of glucose consumption in LO2 cells, suggesting the beneficial impact of Rb1 on insulin resistance in vitro (Figure 4A). Moreover, we found that Rb1 reduced the elevated protein expression level of 15-PGDH caused by palmitic acid treatment (Figure 4B and C). Accordingly, Rb1 enhanced the decreased PGE2 levels caused by palmitic acid induction (Figure 4D). These results suggest that Rb1 might promote glycogen production through the regulation of the 15-PGDH/PGE2 signaling pathway.
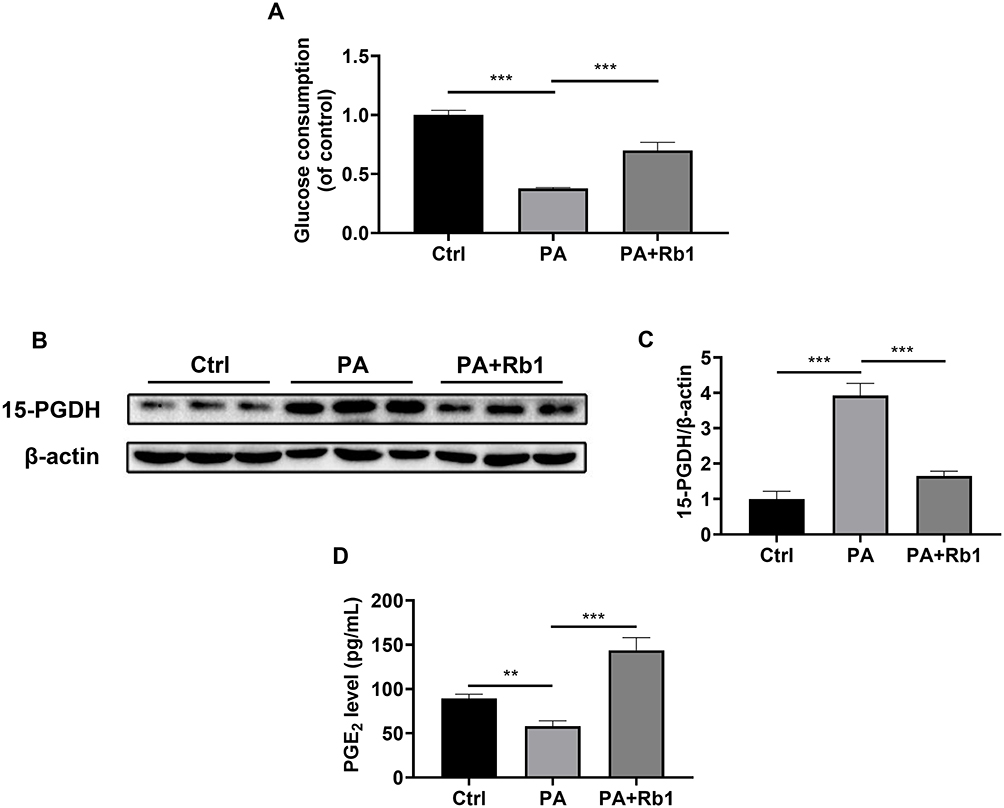 |
Figure 4 Rb1 decreases the 15-PGDH level and increases the PGE2 level in palmitic acid-treated LO2 cells. (A) Glucose consumption was measured in control (Ctrl), palmitic acid-treated (PA), and palmitic acid+ Rb1-treated (PA+Rb1) LO2 cells. (B) The expression level of 15-PGDH was analyzed using Western blotting analysis. β-actin was used as the control for loading. (C) The bar chart displayed the quantitative data of 15-PGDH/β-actin obtained from Western blot analysis. (D) Levels of PGE2 in the LO2 cell culture medium. n = 3. **P < 0.01, ***P < 0.001. |
In our prior investigation, it was demonstrated that the direct application of PGE2 increased the reduced levels of phosphorylation in AKT (Ser473) and GSK3β (Ser9) in cells treated with palmitic acid, suggesting the role of PGE2 in the regulation of the AKT/GSK3β signaling pathway.15 PGE2 activates downstream signaling pathways through four trans-membrane receptors, prostaglandin E2 receptor EP1-4 (EP1-4). EP4 is abundantly expressed in the liver, and multiple studies have indicated that activation of EP4 improved T2DM.32–34 Thus, we next examined whether Rb1 promoted glycogen synthesis through EP4. Following the administration of palmitic acid, the levels of phosphorylation for AKT (Ser473) and GSK3β (Ser9) experienced a notable decrease, however, co-treatment of Rb1 and palmitic acid up-regulated the phosphorylation levels of AKT and GSK3β (Figure 5A–C). Furthermore, palmitic acid-treated cells were subjected to the administration of Rb1 and CJ-42794, an EP4 inhibitor, and observed a reduction in AKT and GSK3β phosphorylation levels compared to cells treated with palmitic acid and Rb1 (Figure 5A–C). These results indicate that Rb1 might promote glycogen synthesis by modulating the EP4/AKT/GSK3β signaling pathway.
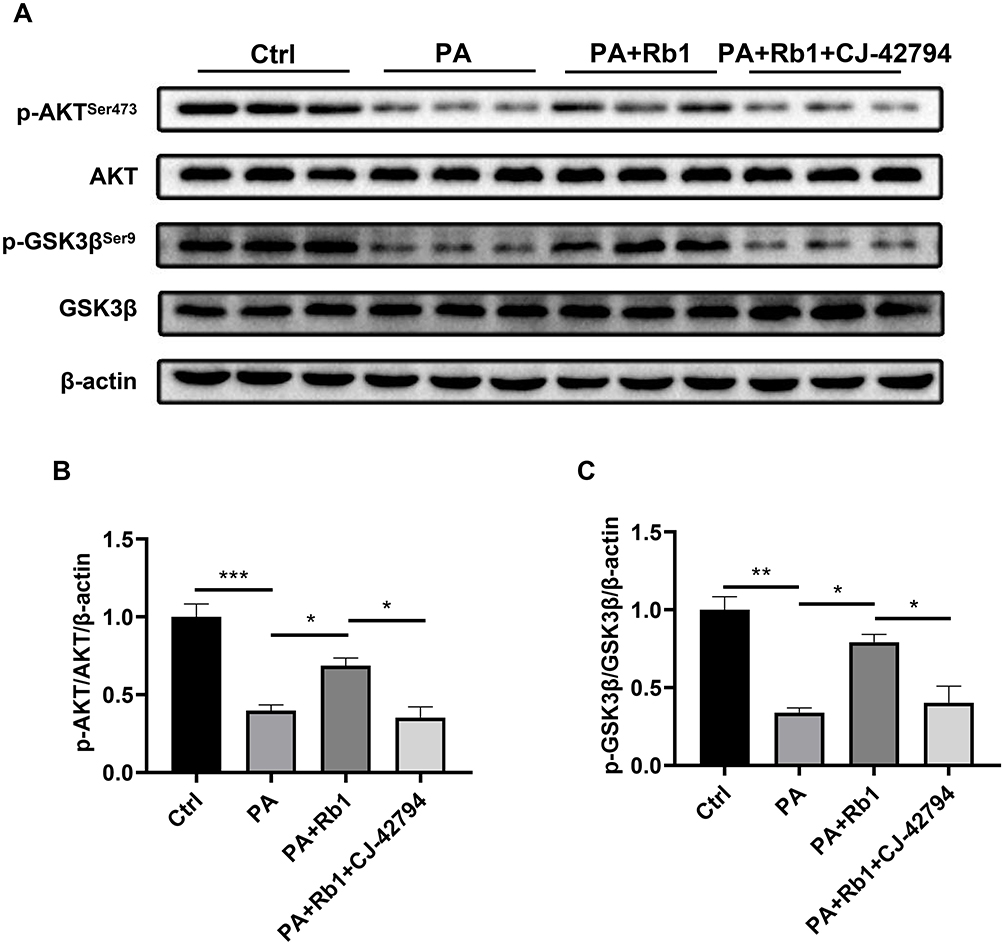 |
Figure 5 EP4 is involved in Rb1-promoted glycogen synthesis. (A) Levels of AKT, p-AKTSer473, GSK3β, and p-GSK3βSer9 in control (Ctrl), palmitic acid-treated (PA), palmitic acid+Rb1-treated (PA+Rb1), and palmitic acid+Rb1+EP4 inhibitor CJ-42794-treated (PA+Rb1+CJ-42794) LO2 cells were analyzed using Western blot analysis. β-actin was used as the control for loading. (B) The bar chart displayed the quantitative data of p-AKTSer473/AKT/β-actin obtained from Western blot analysis. (C) The bar chart displayed the quantitative data of p-GSK3βSer9/GSK3β/β-actin obtained from Western blot analysis. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001. |
Discussion
Previous therapeutic options in treating T2DM, such as sulfonylureas, biguanides, α-glucosidase inhibitors, thiazolidinedione, glucagon-like peptide 1 receptor agonists, and sodium/glucose cotransporter 2 inhibitors, often have various side effects, including hypoglycemia, gain of weight, gastrointestinal events, hypotension, and drug resistance.3,35,36 Rb1 can be extracted from P. ginseng and has been shown to exert anti-diabetic effects through numerous targets and mechanisms, thus, Rb1 administration may be a promising strategy in T2DM treatment.3 Herein, we examined the molecular mechanism of Rb1 on hepatic glycogen synthesis in a T2DM mouse model induced by a high-fat diet combined with streptozotocin, as well as in an insulin-resistant cell model induced by palmitic acid.
T2DM is characterized by impaired glucose homeostasis, which is mainly owing to insulin resistance in target organs and tissues.5 As a core insulin target organ, the liver exerts vital roles in controlling blood glucose homeostasis.5,37 The liver is a main organ for glycogen storage. In the postprandial condition, additional glucose in the blood is stored as glycogen in hepatocytes.31 Under physiological conditions, glycogen synthesis promotes glucose uptake and storage and maintains glucose homeostasis.38 However, in metabolic diseases, such as T2DM, hepatic insulin resistance occurs, liver glycogen synthesis decreases, and subsequently leads to the elevation of blood glucose levels.39 AKT plays a vital role in the insulin signaling pathway.5 AKT can be activated by phosphorylation at Ser473.5 The activated AKT alleviates T2DM by phosphorylating and inhibiting GSK3β activity, which activates glycogen synthase to promote glycogen synthesis.5,6 In this current investigation, we observed that T2DM mice exhibited reduced hepatic glycogen content. Furthermore, the hepatic AKTSer473 and GSK3βSer9 phosphorylation levels were significantly decreased in T2DM mice, indicating the occurrence of insulin resistance in the liver.
According to reports, Rb1 has shown positive effects on T2DM by reducing fasting blood glucose levels, improving impaired glucose tolerance and insulin sensitivity, and decreasing body weight gain and fat accumulation.18–20 As for maintaining glucose homeostasis in the liver, Rb1 has been demonstrated to enhance the rate of liver glycogen synthesis and suppress hepatic gluconeogenesis, thereby improving liver glucose metabolism.24,40 Mechanistically, Rb1 inhibited the cAMP/CREB/MPC1 signaling to down-regulate pyruvate carboxylase and up-regulate pyruvate dehydrogenase, thus inhibiting gluconeogenesis.24 However, the effects and mechanisms of Rb1 on hepatic glycogen synthesis need further investigation. Herein, we discovered that Rb1 administration enhanced hepatic glycogen content in T2DM mice. Further investigations found that Rb1 administration significantly increased hepatic AKTSer473 and GSK3βSer9 phosphorylation levels in T2DM mice, indicating that Rb1 may promote glycogen synthesis by regulating AKT/GSK3β signaling pathway.
15-PGDH catalyzes the dehydrogenation of PGE2 and leads to PGE2 inactivation.41,42 15-PGDH is widely expressed in mammalian tissues, especially with higher expression in the liver, lung, bladder, small intestine, and colon.42 Accumulating studies indicate that 15-PGDH has negative impacts on various pathophysiological processes and inhibiting 15-PGDH is regarded as a promising therapeutic strategy in promoting tissue repair and regeneration, preventing organ damage, and resisting aging.41,42 15-PGDH inhibition or knockdown promoted liver, bone marrow, and colon regeneration and repair.13 The expression level of 15-PGDH was elevated in acute kidney injury caused by lipopolysaccharide induction, whereas 15-PGDH inhibition enhanced PGE2 levels and improved kidney injury.43 Similarly, 15-PGDH expression was elevated among individuals diagnosed with idiopathic pulmonary fibrosis, and inhibition of 15-PGDH improved pulmonary performance, reduced apoptosis of alveolar epithelial cells, and inhibited fibroblasts proliferation.44 A recent study found that in aged mice, the activity and expression of 15-PGDH were significantly increased, and led to decreased PGE2 levels, while inhibiting 15-PGDH raised PGE2 levels, bolstered mitochondrial function, and enhanced both muscle mass and strength.41 However, tumor development may be inhibited by 15-PGDH. The presence of 15-PGDH was universally lacking in human colon cancer and 15-PGDH knockout mice had increased colon tumorigenesis and were more sensitive to the carcinogen azoxymethane.45 Nevertheless, the effects and mechanisms of 15-PGDH on T2DM remain poorly understood. Our latest study indicated that inhibition of 15-PGDH promoted glycogen synthesis in T2DM mice, implying a potential link between 15-PGDH and T2DM progression.15 In the current investigation, molecular docking analysis first indicated that Rb1 exhibited a prospective binding affinity with 15-PGDH, specifically binding to the C-terminal region of this enzyme. Interestingly, previous studies have shown a high correlation between the catalytic effectiveness of 15-PGDH and its C-terminal region.46,47 Mechanistically, the efficiency of 15-PGDH in catalyzing prostaglandin oxidation can be influenced by the C-terminal region.46,47 Subsequently, Western blot analysis was conducted and determined that in T2DM mice or insulin-resistant cells, the 15-PGDH expression levels were increased, while administration of Rb1 reduced the levels of 15-PGDH both in vivo and in vitro. These results suggest that 15-PGDH may be a core regulator in Rb1-ameliorated T2DM.
PGE2 is a vital lipid mediator in numerous biological processes. PGE2 was once regarded as a pro-inflammatory factor with detrimental effects; nevertheless, an increasing number of studies have indicated its beneficial roles in metabolic processes.8,9,48 PGE2 can alleviate insulin resistance, protect hepatocytes, and promote anti-inflammatory M2 macrophage polarization, suggesting its protective roles in T2DM.8,9,49 PGE2 can bind to four trans-membrane receptors, namely EP1-4, and EP4 has a high expression in the liver.32 EP4 activation improved insulin sensitivity and glucose tolerance and reduced the levels of pro-inflammatory cytokines.33 By binding to EP4, PGE2 inhibited the NF-κB heterodimer p50/p65 formation and prevented M1 macrophage activation.8 Furthermore, in hepatocytes, EP4 inhibition hampered the increased glycogen synthesis and reduced gluconeogenesis caused by PGE2 administration,15 suggesting the vital role of EP4 in regulating hepatic glucose homeostasis. In the present study, we found that the administration of Rb1 increased the reduced PGE2 levels in mice with T2DM or insulin-resistant cells, indicating the involvement of PGE2 in Rb1-ameliorated T2DM. Furthermore, in vitro studies showed that EP4 inhibition blocked the increased phosphorylation levels of AKTSer473 and GSK3βSer9 caused by Rb1 treatment. Therefore, we speculate that Rb1 might promote glycogen synthesis and prevent T2DM progression by regulating the 15-PGDH/PGE2/EP4/AKT/GSK3β signaling pathway.
Our study has several limitations that should be considered. Firstly, our experiment focused on administering Rb1 after T2DM modeling and investigated its therapeutic effect on treating T2DM. However, whether Rb1 can prevent the occurrence of T2DM deserves further exploration. Additionally, our investigation only examined the effect of Rb1 on high-fat diet/streptozotocin-induced T2DM mice. It would be valuable to investigate whether Rb1 can inhibit disease progression in db/db mice, a spontaneous T2DM mice model, and explore the underlying mechanisms. Lastly, our current study focused on the effect of Rb1 on liver glycogen synthesis. It would be worthwhile to investigate the effects and mechanisms of Rb1 on the pancreas and other insulin-target tissues, such as adipose tissue and skeletal muscle.
Conclusion
In conclusion, this study suggests that Rb1 may prevent T2DM progression via the promotion of hepatic glycogen synthesis, at least partially through regulation of the 15-PGDH/PGE2/EP4 pathway (Figure 6). These results indicate that Rb1, a compound that improves hepatic glucose metabolism, may offer a promising treatment strategy for T2DM. However, additional experiments investigating the effects of Rb1 on T2DM mice, apart from hepatic glycogen synthesis, are necessary. In the future, it would be worthwhile to investigate whether the combined use of Rb1 and other conventional drugs could be more effective in T2DM treatment. Furthermore, it would be valuable to explore the downstream effects of the 15-PGDH/PGE2/EP4 pathway, other than influencing glycogen synthesis, which could enhance our understanding of the pathological mechanisms of T2DM and might provide new targets for the clinical treatment of T2DM.
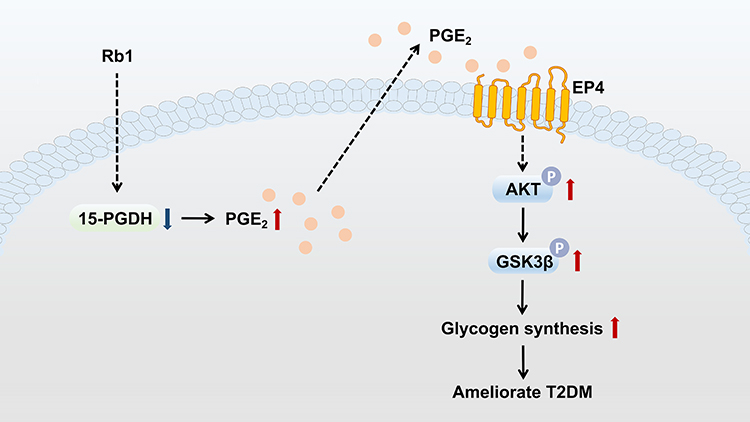 |
Figure 6 Graph illustrating how Rb1 administration prevents T2DM. Hepatic 15-PGDH expression level is decreased following Rb1 treatment, resulting in increased PGE2 levels in the liver. PGE2 binds to the prostaglandin E2 receptor EP4, leading to the phosphorylation and activation of AKT, which results in an enhanced phosphorylation level of GSK3β. The phosphorylated GSK3β promotes glycogen synthesis and ameliorates T2DM. |
Abbreviations
15-PGDH, 15-hydroxyprostaglandin dehydrogenase; AKT, serine/threonine kinase AKT; EP4, prostaglandin E2 receptor EP4; GSK3β, glycogen synthase kinase 3 beta; MET, metformin; PA, palmitic acid; PAS, periodic acid-Schiff; PDB, protein data bank; PGE2, prostaglandin E2; P. ginseng, Panax ginseng C. A. Mey.; Rb1, ginsenoside Rb1; T2DM, type 2 diabetes mellitus.
Funding
This work was supported by Guangdong Basic and Applied Basic Research Foundation [grant numbers 2021A1515012553, 2019A1515110123] and National Natural Science Foundation of China [grant number 82300927].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sun H, Saeedi P, Karuranga S, et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119. doi:10.1016/j.diabres.2021.109119
2. DeFronzo RA, Ferrannini E, Groop L, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. 2015;1:15019. doi:10.1038/nrdp.2015.19
3. Zhou P, Xie W, He S, et al. Ginsenoside Rb1 as an Anti-Diabetic Agent and Its Underlying Mechanism Analysis. Cells. 2019;8(3):204. doi:10.3390/cells8030204
4. Flamment M, Hajduch E, Ferré P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23(8):381–390. doi:10.1016/j.tem.2012.06.003
5. Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol Metab. 2017;28(7):497–505. doi:10.1016/j.tem.2017.03.003
6. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177–197. doi:10.1002/cphy.c130024
7. Liu TY, Shi CX, Gao R, et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin Sci (Lond). 2015;129(10):839–850. doi:10.1042/cs20150009
8. Wang W, Liang M, Wang L, et al. Role of prostaglandin E2 in macrophage polarization: insights into atherosclerosis. Biochem Pharmacol. 2023;207:115357. doi:10.1016/j.bcp.2022.115357
9. Wang W, Zhong X, Guo J. Role of 2‑series prostaglandins in the pathogenesis of type 2 diabetes mellitus and non‑alcoholic fatty liver disease (Review). Int J Mol Med. 2021;47(6):114. doi:10.3892/ijmm.2021.4947
10. Okumura T, Kanemaki T, Kitade H. Stimulation of glucose incorporation into glycogen by E-series prostaglandins in cultured rat hepatocytes. Biochim Biophys Acta. 1993;1176(1–2):137–142. doi:10.1016/0167-4889(93)90188-u
11. Henkel J, Coleman CD, Schraplau A, et al. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci Rep. 2018;8(1):16127. doi:10.1038/s41598-018-34633-y
12. Henkel J, Frede K, Schanze N, et al. Stimulation of fat accumulation in hepatocytes by PGE2-dependent repression of hepatic lipolysis, β-oxidation and VLDL-synthesis. Lab Invest. 2012;92(11):1597–1606. doi:10.1038/labinvest.2012.128
13. Zhang Y, Desai A, Yang SY, et al. TISSUE REGENERATION. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science. 2015;348(6240):aaa2340. doi:10.1126/science.aaa2340
14. Yao L, Chen W, Song K, et al. 15-hydroxyprostaglandin dehydrogenase (15-PGDH) prevents lipopolysaccharide (LPS)-induced acute liver injury. PLoS One. 2017;12(4):e0176106. doi:10.1371/journal.pone.0176106
15. Liang M, Wang L, Wang W. The 15-hydroxyprostaglandin dehydrogenase inhibitor SW033291 ameliorates abnormal hepatic glucose metabolism through PGE2–EP4 receptor–AKT signaling in a type 2 diabetes mellitus mouse model. Cell Signal. 2023;108:110707. doi:10.1016/j.cellsig.2023.110707
16. Mohanan P, Subramaniyam S, Mathiyalagan R, Yang DC. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J Ginseng Res. 2018;42(2):123–132. doi:10.1016/j.jgr.2017.01.008
17. Ratan ZA, Haidere MF, Hong YH, et al. Pharmacological potential of ginseng and its major component ginsenosides. J Ginseng Res. 2021;45(2):199–210. doi:10.1016/j.jgr.2020.02.004
18. Wu Y, Yu Y, Szabo A, Han M, Huang XF. Central inflammation and leptin resistance are attenuated by ginsenoside Rb1 treatment in obese mice fed a high-fat diet. PLoS One. 2014;9(3):e92618. doi:10.1371/journal.pone.0092618
19. Yu X, Ye L, Zhang H, et al. Ginsenoside Rb1 ameliorates liver fat accumulation by upregulating perilipin expression in adipose tissue of db/db obese mice. J Ginseng Res. 2015;39(3):199–205. doi:10.1016/j.jgr.2014.11.004
20. Wang W, Zhan W, Liang M, et al. Ginsenoside Rb1 ameliorates the abnormal hepatic glucose metabolism by activating STAT3 in T2DM mice. J Funct Foods. 2023;104:105534. doi:10.1016/j.jff.2023.105534
21. Dai S, Liu S, Li C, Zhou Z, Wu Z. Site-selective modification of exendin 4 with variable molecular weight dextrans by oxime-ligation chemistry for improving type 2 diabetic treatment. Carbohydr Polym. 2020;249:116864. doi:10.1016/j.carbpol.2020.116864
22. Parilla JH, Willard JR, Barrow BM, Zraika S. A Mouse Model of Beta-Cell Dysfunction as Seen in Human Type 2 Diabetes. J Diabetes Res. 2018;2018:6106051. doi:10.1155/2018/6106051
23. Dong JY, Xia KJ, Liang W, et al. Ginsenoside Rb1 alleviates colitis in mice via activation of endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 signaling pathway. Acta Pharmacol Sin. 2021;42(9):1461–1471. doi:10.1038/s41401-020-00561-9
24. Lou MD, Li J, Cheng Y, et al. Glucagon up-regulates hepatic mitochondrial pyruvate carrier 1 through cAMP-responsive element-binding protein; inhibition of hepatic gluconeogenesis by ginsenoside Rb1. Br J Pharmacol. 2019;176(16):2962–2976. doi:10.1111/bph.14758
25. Zhang C, Han M, Zhang X, Tong H, Sun X, Sun G. Ginsenoside Rb1 Protects Against Diabetic Cardiomyopathy by Regulating the Adipocytokine Pathway. J Inflamm Res. 2022;15:71–83. doi:10.2147/jir.S348866
26. Terasaki M, Hiromura M, Mori Y, et al. Amelioration of Hyperglycemia with a Sodium-Glucose Cotransporter 2 Inhibitor Prevents Macrophage-Driven Atherosclerosis through Macrophage Foam Cell Formation Suppression in Type 1 and Type 2 Diabetic Mice. PLoS One. 2015;10(11):e0143396. doi:10.1371/journal.pone.0143396
27. Wang X, Chen S, Lu R, et al. Adipose-derived stem cell-secreted exosomes enhance angiogenesis by promoting macrophage M2 polarization in type 2 diabetic mice with limb ischemia via the JAK/STAT6 pathway. Heliyon. 2022;8(11):e11495. doi:10.1016/j.heliyon.2022.e11495
28. Biggs EK, Liang L, Naylor J, et al. Development and characterisation of a novel glucagon like peptide-1 receptor antibody. Diabetologia. 2018;61(3):711–721. doi:10.1007/s00125-017-4491-0
29. Li L, Xue J, Wan J, et al. LRP6 Knockdown Ameliorates Insulin Resistance via Modulation of Autophagy by Regulating GSK3β Signaling in Human LO2 Hepatocytes. Front Endocrinol (Lausanne). 2019;10:73. doi:10.3389/fendo.2019.00073
30. Li Y, Zhang S, Zhu Z, et al. Upregulation of adiponectin by Ginsenoside Rb1 contributes to amelioration of hepatic steatosis induced by high fat diet. J Ginseng Res. 2022;46(4):561–571. doi:10.1016/j.jgr.2021.10.005
31. Gursan A, Prompers JJ. Magnetic Resonance Imaging and Spectroscopy Methods to Study Hepatic Glucose Metabolism and Their Applications in the Healthy and Diabetic Liver. Metabolites. 2022;12(12):1223. doi:10.3390/metabo12121223
32. Nakanishi T, Nakamura Y, Umeno J. Recent advances in studies of SLCO2A1 as a key regulator of the delivery of prostaglandins to their sites of action. Pharmacol Ther. 2021;223:107803. doi:10.1016/j.pharmthera.2021.107803
33. Yasui M, Tamura Y, Minami M, et al. The Prostaglandin E2 Receptor EP4 Regulates Obesity-Related Inflammation and Insulin Sensitivity. PLoS One. 2015;10(8):e0136304. doi:10.1371/journal.pone.0136304
34. Yasui-Kato M, Patlada S, Yokode M, Kamei K, Minami M. EP4 signalling is essential for controlling islet inflammation by causing a shift in macrophage polarization in obesity/type 2 diabetes. Diab Vasc Dis Res. 2020;17(4):1479164120945675. doi:10.1177/1479164120945675
35. Gourdy P, Darmon P, Dievart F, Halimi JM, Guerci B. Combining glucagon-like peptide-1 receptor agonists (GLP-1RAs) and sodium-glucose cotransporter-2 inhibitors (SGLT2is) in patients with type 2 diabetes mellitus (T2DM). Cardiovasc Diabetol. 2023;22(1):79. doi:10.1186/s12933-023-01798-4
36. Ishii H, Hayashino Y, Akai Y, Yabuta M, Tsujii S. Dipeptidyl peptidase-4 inhibitors as preferable oral hypoglycemic agents in terms of treatment satisfaction: results from a multicenter, 12-week, open label, randomized controlled study in Japan (PREFERENCE 4 study). J Diabetes Investig. 2018;9(1):137–145. doi:10.1111/jdi.12659
37. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572–587. doi:10.1038/nrendo.2017.80
38. Ren T, Ma A, Zhuo R, et al. Oleoylethanolamide Increases Glycogen Synthesis and Inhibits Hepatic Gluconeogenesis via the LKB1/AMPK Pathway in Type 2 Diabetic Model. J Pharmacol Exp Ther. 2020;373(1):81–91. doi:10.1124/jpet.119.262675
39. Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol. 2007;47(1):142–156. doi:10.1016/j.jhep.2007.04.002
40. Shen L, Haas M, Wang DQ, et al. Ginsenoside Rb1 increases insulin sensitivity by activating AMP-activated protein kinase in male rats. Physiol Rep. 2015;3(9):e12543. doi:10.14814/phy2.12543
41. Palla AR, Ravichandran M, Wang YX, et al. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science. 2021;371(6528):eabc8059. doi:10.1126/science.abc8059
42. Sun CC, Zhou ZQ, Yang D, et al. Recent advances in studies of 15-PGDH as a key enzyme for the degradation of prostaglandins. Int Immunopharmacol. 2021;101(Pt B):108176. doi:10.1016/j.intimp.2021.108176
43. Miao S, Lv C, Liu Y, et al. Pharmacologic Blockade of 15-PGDH Protects Against Acute Renal Injury Induced by LPS in Mice. Front Physiol. 2020;11:138. doi:10.3389/fphys.2020.00138
44. Bärnthaler T, Theiler A, Zabini D, et al. Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J Allergy Clin Immunol. 2020;145(3):818–833. doi:10.1016/j.jaci.2019.11.032
45. Myung SJ, Rerko RM, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(32):12098–12102. doi:10.1073/pnas.0603235103
46. Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12(8):955–962. doi:10.2174/138161206776055958
47. Zhou H, Yan F, Tai HH. C-Terminal region of human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase is involved in the interaction with prostaglandin substrates. Eur J Biochem. 2001;268(12):3368–3374. doi:10.1046/j.1432-1327.2001.02218.x
48. Wang W, Hu Y, Wang X, Wang Q, Deng H. ROS-Mediated 15-Hydroxyprostaglandin Dehydrogenase Degradation via Cysteine Oxidation Promotes NAD(+)-Mediated Epithelial-Mesenchymal Transition. Cell Chem Biol. 2018;25(3):255–261. doi:10.1016/j.chembiol.2017.12.008
49. Cheng H, Huang H, Guo Z, Chang Y, Li Z. Role of prostaglandin E2 in tissue repair and regeneration. Theranostics. 2021;11(18):8836–8854. doi:10.7150/thno.63396
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.