Back to Journals » Cancer Management and Research » Volume 11
Genome-wide DNA methylation and RNA expression profiles identified RIPK3 as a differentially methylated gene in Chlamydia pneumoniae infection lung carcinoma patients in China
Authors Xiong WM, Xu QP, Xiao RD, Hu ZJ, Cai L ![]() , He F
, He F ![]()
Received 2 September 2018
Accepted for publication 5 May 2019
Published 28 June 2019 Volume 2019:11 Pages 5785—5797
DOI https://doi.org/10.2147/CMAR.S186217
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Beicheng Sun
Wei-Min Xiong,1 Qiu-Ping Xu,1 Ren-Dong Xiao,2 Zhi-Jian Hu,2 Lin Cai,1,3–5 Fei He1,3–5
1Department of Epidemiology and Health Statistics, School of Public Health, Fujian Medical University, Fuzhou 350108, People’s Republic of China; 2Department of Thoracic Surgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou 350001, People’s Republic of China; 3Fujian Provincial Key Laboratory of Tumor Microbiology, Fujian Medical University, Fuzhou 350108, People’s Republic of China; 4Key Laboratory of Ministry of Education for Gastrointestinal Cancer, Fujian Medical University, Fuzhou 350108, People’s Republic of China; 5Fujian Provincial Key Laboratory of Environment Factors and Cancer, Fujian Medical University, Fuzhou 350108, People’s Republic of China
Aim: To explore the relationship between Chlamydia pneumonia (Cpn) infection and lung cancer using integrative methylome and transcriptome analyses.
Methods: Twelve primary lung cancer patients who were positive for Cpn and twelve patients who were negative were selected for demographic, clinicopathological, and lifestyle matching. Genomic DNA and RNA were extracted and DNA methylation and mRNA levels were detected using the Infinium Human Methylation 450 Beadchip array and mRNA + lncRNA Human Gene Expression Microarray. We identified differentially expressed methylation and genes profiles.
Results: Integrative analysis revealed an inverse correlation between differentially expressed genes and DNA methylation. Cpn-related lung cancer methylated genes (target genes) were introduced into the gene ontology and KEGG, PID, BioCarta, Reactome, BioCyc and PANTHER enrichment analyses using a q-value cutoff of 0.05 to identify potentially functional methylation of abnormal genes associated with Cpn infection. Gene sets enrichment analysis was evaluated according to MsigDB. Levels of differentially expressed methylated sites were quantitatively verified. The promoter methylation sites of 62 genes were inversely related to expression levels. According to the quantitative analysis of DNA methylation, the methylation level of the RIPK3 promoter region was significantly different between Cpn-positive cancerous and adjacent tissues, but not between Cpn-negative cancerous and adjacent tissues.
Conclusion: Hypomethylation of the RIPK3 promoter region increases RIPK3 expression, leading to regulated programmed necrosis and activation of NF-κB transcription factors, which may contribute to the development and progression of Cpn-related lung cancer.
Keywords: lung neoplasms, chlamydia pneumonia, DNA methylation, gene expression
Introduction
According to GLOBOCAN,1 lung cancer accounted for the majority of global cancer incidence and deaths in 2012. Pathogenic factors for lung cancer include both genetic and environmental factors.2,3 In recent years, the role of infection in the etiology of cancer has gained scientific interest.
Chlamydia pneumoniae (Cpn) is a common cause of acute respiratory infections, and the inflammation caused by chronic infection may lead to carcinogenesis. Epidemiological evidence suggests that Cpn infection may be associated with the development and progression of lung cancer.4 A recent meta-analysis of 2,549 patients with lung cancer and 2,764 controls showed that Cpn infection significantly increased the risk of lung cancer (odds ratio =2.07, 95% confidence interval: 1.43–2.99).5 Cell experiments found that Cpn infection can induce human mesothelial cell transformation, thereby increasing the risk of lung cancer.6 Studies using integrated computer methods found that Cpn triggered lung cancer growth by changing host cell replication, transcription, and DNA damage repair mechanisms7
The underlying molecular mechanism of Cpn chronic infection-mediated lung cancer is not yet clear. Studies have shown that inflammation-induced lung cancer differs from genetic- and epigenetic-mediated cancer development.8 DNA methylation is an important epigenetic mechanism that regulates gene transcription and cell function.9 It regulates gene expression without changing gene sequences. DNA methylation usually inhibits gene expression, while demethylation induces gene activation and expression. Early carcinogenesis can cause abnormal changes in methylation, and methylation can be detected in bodily fluids and early lesions of biopsied tissues. Methylation can be reversed by demethylating drugs, suggesting that methylation-mediated gene abnormalities may serve as a biological target for early detection of lung cancer and treatment interventions.
To explore the pathogenic effect of Cpn on lung cancer and its underlying epigenetic mechanism, we used the high-throughput Illumina methylation 450 k array to determine the methylation levels of the whole genome in lung cancer tissues and para-cancer controls. We revealed methylation characteristics of serologic Cpn positive lung cancer and found that the combination of methylation and expression profiles identified 62 potentially functional methylated abnormal genes associated with Cpn. Furthermore, we identified the role of abnormal methylation in the development and progression of lung cancer at the molecular level.
Materials and methods
Subjects
Twelve newly diagnosed primary lung cancer cases that tested positive for Cpn serological antibodies (IgA and IgG) and 12 cases that were negative following thoracic surgery in the First Affiliated Hospital of Fujian Medical University were selected and matched for demographic characteristics, clinicopathological features, and lifestyles (Table 1). Twelve pairs of patients were tested using methylation and expression microarrays, and methylation levels of the promoter region in the target genes were verified using the Sequenom methylation method (see Table 1, No. 1–6); the remaining twelve pairs were validated using the MethylTarget method (see Table 1, No. 7–12). No patient received adjuvant therapy, such as radiotherapy and chemotherapy, before surgery. Lung cancer tissues and corresponding normal tissues were resected and stored at −80 °C within 10 min after surgery. Histopathological diagnosis of each specimen was performed independently by two pathologists (Figure 1). The study protocol was approved by the institutional review board of Fujian Medical University, and written informed consent was obtained from all patients. This study was conducted in accordance with the Declaration of Helsinki.
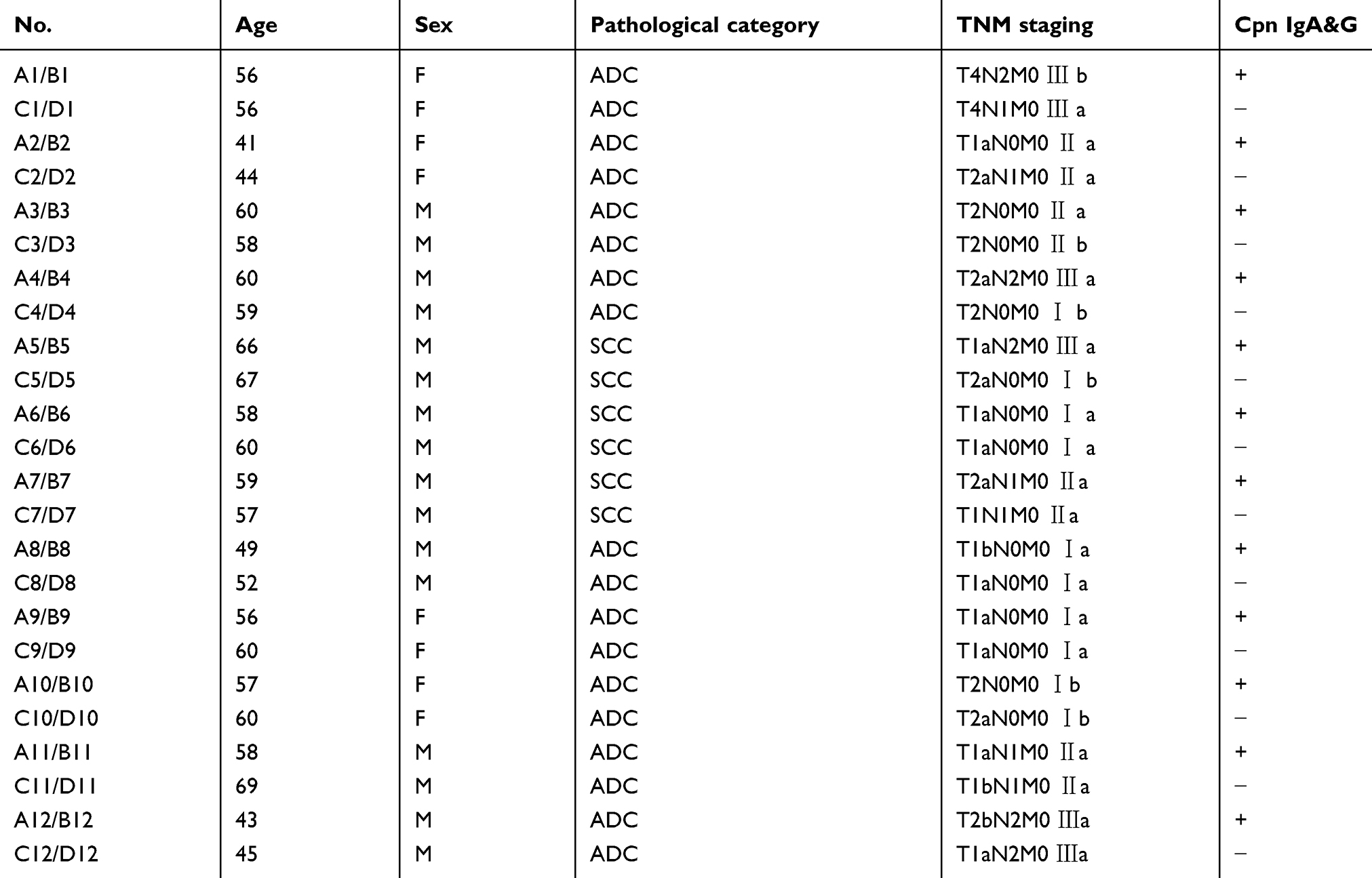 |
Table 1 Demographic and clinical characteristics of the lung cancer patients |
 |
Figure 1 Pathological diagnosis of (A) lung cancer and (B) adjacent normal tissues (×400 times, hematoxylin and eosin staining). |
Serological tests, DNA and total RNA extraction
We used Cpn IgA SeroFIATM and IgG SeroFIATM kits (Savyon, Israel) and the themicro-indirect immunofluorescence (MIF) method to detect serum Cpn IgG and IgA antibodies in patients with lung cancer. DNA was extracted using the Qiagen® Genomic DNA Mini Kit, and total RNA was extracted using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacture’s protocol. Purity and concentration were determined using the NanoDrop® ND-2000 spectrophotometer. DNA and total RNA integrity were determined using 2% agarose gel electrophoresis.
DNA methylation and gene expression profiling
After bisulfite conversion of DNA, the methylation profile of genomic DNA was assessed using the Infinium Human Methylation 450 Beadchip Kit (Illumina, San Diego, CA, USA), which can detect more than 450,000 methylation sites located within and outside CpG islands.10 Relative total RNA levels were also determined using the mRNA + lncRNA Human Gene Expression Microarray V4.0 (4×180 K format, CapitalBio Corp, Beijing, China), which is more specific for DNA-DNA hybridization and achieves low abundance of RNA amplification and detection efficiency. All samples were processed at the same time to avoid chip-to-chip variation. The quality of hybridization and overall chip performance were monitored using visual inspection of both the internal quality control checks and the raw scanned data.
Quantitative methylation analysis
The Sequenom MassARRAY platform (CapitalBio Corp, Beijing, China) was used to perform quantitative methylation analysis of the target gene promoter.11 This system uses matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry in combination with RNA base-specific cleavage (MassCLEAVE). PCR primers were designed with Methprimer (http://epidesigner.com). Each forward primer was tagged with a 10-mer to balance the PCR, and each reverse primer had an additional T7 promoter tag for in vitro transcription. The target regions were amplified using the primer pairs shown in Table 2. The MethylTarget platform (GeneskyBio Corp, Shanghai, China) was also used to quantitatively analyze DNA methylation levels. The targeted DNA sequence was amplified by net-PCR and designed DNA fragments were sequenced using Illumina Hiseq 2000. PCR primers were designed with primer3 (http://primer3.ut.ee/).
 |
Table 2 Primer design for validating the methylation level of gene fragments |
Accurate experimental methods and uniform judgment standards were adopted. The reagents were kept consistent during the experiment, and the consumables were autoclaved before each experiment. All samples were processed simultaneously to avoid heterogeneity between chips. Hybridization quality and overall chip performance were monitored using internal quality control checks and raw scan data.
Data processing and analysis
Infinium methylation data were processed using the GenomeStudio Methylation Module (v. 2011.1) in which methylation values (expressed as β-values, ranging from 0 to 1) and detection P-values were calculated. For quality control, methylation measures with a detection P-value >0.05 were removed. The paired empirical Bayes moderated t-test in the limma package was used to obtain differential methylation positions (DMPs) (adjusted P-values (q-values) ≤0.05 and | β-Difference | ≥0.2) between cancer and adjacent normal tissues. The q-values were corrected using the Benjamini Hochberg method for multiple hypotheses, known as the false discovery rate method.12 Hierarchical cluster analysis of DMPs was carried out using complete linkage and Euclidean distance as a measure of similarity. Heatmaps were generated using the gplots package in R. We also used the hypergeometric test to investigate whether the CpG islets were enriched at a particular genomic location using 450 k as the background.
The 450 K methylation probes can be annotated into a gene’s transcription start sites (TSS) 1500, TSS200, the 5ʹ untranslated region (UTR), the 1st exon, body, and the 3ʹUTR. Taking the TSS1500 region as an example, the TSS1500 region of one gene may contain multiple CpG sites. The mean value of the Beta values using multiple CpG sites on the gene TSS1500 region indicates the degree of methylation in this region. Therefore, we were interested in the impact of differential methylation regions (DMRs) on gene expression. The paired empirical Bayes moderated t test in the limma package was used to obtain DMRs (q-values ≤0.05) between cancerous and adjacent normal tissues.
Human Gene Expression raw signal data were processed using the Agilent Feature Extraction V12.0 and array normalization using the quantile method with limma packages. The genes detected in more than eight samples were used for differential expression analysis, and the paired empirical Bayes moderated t test in the limma package was used to identify differentially expressed genes (DEGs) (q-values ≤0.05 and | Fold-Change (FC) | >2.0) between cancerous and adjacent normal tissues. The q-values were corrected using the Benjamini Hochberg method for multiple hypotheses. DEGs were further hierarchical clustered and filtered using Heatmaps and Volcano plots in R, respectively. Volcano plots were constructed using | FC | > 2.0 and q-value ≤0.05 as cut-offs.
Figure 2 shows a schematic diagram of the steps followed to identify the target genes. DMRs of the methylome and DEGs of the transcriptome were identified for Cpn positive lung cancer vs Cpn positive adjacent normal tissues (Figure 2A vs B) and Cpn negative lung cancer vs Cpn negative adjacent normal tissues (Figure 2C vs D). Integrative analysis using the Pearson’s correlation was used to screen significantly different genes (DGs) with a negative correlation between DNA methylation of up-stream gene regions and expression levels (Pearson Correlation Coefficient < −0.7 and P-value <0.05). After removing the overlap of Cpn+ DGs and Cpn- DGs, the remaining set included Cpn-related lung cancer methylation genes (target genes). The target genes were entered into KOBAS (http://kobas.cbi.pku.edu.cn/) for Gene Ontology (GO) and pathway (KEGG, PID, BioCarta, Reactome, BioCyc and PANTHER) enrichment analysis with a q-value cutoff of 0.05. For all genes, gene sets enrichment analysis (GSEA) was evaluated according to MsigDB. In the validation analysis, the average methylation levels of each DNA fragment and CpG site were compared using the Wilcoxon Rank Sum test with a P-value cutoff of 0.05.
 |
Figure 2 Schematic diagram of integrative methylome and transcriptome analyses. |
Results
Genome-wide DNA methylation distribution in Cpn-related NSCLC
We identified 5,309 DMPs including 3,619 hypermethylated and 1,690 hypomethylated probes in A vs B, and 6,213 DMPs including 4,032 hypermethylated and 2,181 hypomethylated probes in C vs D (Figure S1). Hierarchical clustering of DMPs showed separate clustering of non-small cell lung carcinoma (NSCLC) and adjacent normal tissues in both A vs B (Figure S2) and C vs D (Figure S3) indicating distinct epigenetic regulation.
Further we classified these DMPs based on their enrichment at CpG islands (CGIs), flanking CGIs (shores and shelves, 2–4 kb from CGIs), and open sea (non-related to CGIs). Our data showed that the majority of the hypermethylated probes (52%, 43%) were enriched in the CGIs (Figure S4A–C), whereas hypomethylated probes (68%, 70%) were mostly enriched in the open sea (Figure S4B–D). Hypermethylation in NSCLC was primarily enriched in the CpG-rich regions, while hypomethylation was primarily enriched in the open sea. When comparing the distribution of hypermethylated probes between A vs B (Figure S4A) and C vs D (Figure S4C), we found both were enriched in the CGIs. However, for all DMPs, we observed that A vs B was more enriched in the CGIs compared to C vs D (Table S1).
Gene expression profiles in Cpn-related NSCLC
When subjected to principal component analysis (PCA) analysis, the contribution rate of the top 2 principal components was 51.90%. The gene expression profiles of the lung cancer and normal pairs in the two different Cpn status normal tissues (B, D) were relatively close, while that of Cpn positive and negative NSCLC cancer tissues (A, C) were markedly different, which tended to be disperse (Figures S5 and S6).
There was a total of 32,676 probe sets (transcripts) representing 20,753 genes on the Gene Chip. We observed significant differences in DEGs in A vs B and C vs D. Among these 2,765 genes 1,454 genes were up-regulated and 1,311 were down-regulated in A vs B, and 50 genes were up-regulated and 792 were down-regulated in C vs D (Figure S1 and Table S2). Hierarchical clustering was applied to the expression profiles of the DEGs for Cpn positive NSCLC vs Cpn positive adjacent normal tissues (A vs B) and Cpn negative NSCLC vs Cpn negative adjacent normal tissues (C vs D). These data are represented by heatmaps (Figure S7 for A vs B; Figure S8 for C vs D) and volcano plots (Figure S9 for A vs B and Figure S10 for C vs D). The results indicate that DEGs could be used to distinguish cancerous tissues from normal tissues.
Transcriptional effects of aberrant DNA methylation
Changes of methylation in the upstream region of genes were inversely related to differential gene expression, so we screened a set of locus genes that were linked to the TSS1500, TSS200, 5ʹUTR and 1st Exon regions (all in the up-stream region) with an identified methylation site. For Cpn positive patients, 179 methylation probes were identified in A vs B; for Cpn negative patients, 174 methylation probes were identified in C vs D. For A vs B, we removed the data of the probes of C vs D. We found 124 methylation probes that were inversely related to gene expression, which indicated that we found 42 genes (Table S3).
We also observed a negative correlation between gene upstream region methylation and gene expression (Figure 3A for A vs B; Figure 3B for C vs D). We used a combined analysis of the DEGs and DMRs. We found that gene expression profiles of 62 aberrantly methylated genes that existed in A vs B but not in C vs D were inversely correlated with promoter methylation (Table 3).
 |
Figure 3 The inverse relationship between gene upstream region methylation and gene expression. (A) Heatmaps of negative correlation between DMRs and DEGs for Cpn positive NSCLC vs Cpn positive adjacent normal tissues (A vs B), and (B) Cpn negative NSCLC vs Cpn negative adjacent normal tissues (C vs D). In DNA methylation, “Red” indicates relative hypermethylation, and “Blue” indicates relative hypomethylation. In gene expression, “Red” indicates up-regulated genes, and “Blue” indicates down-regulated genes. |
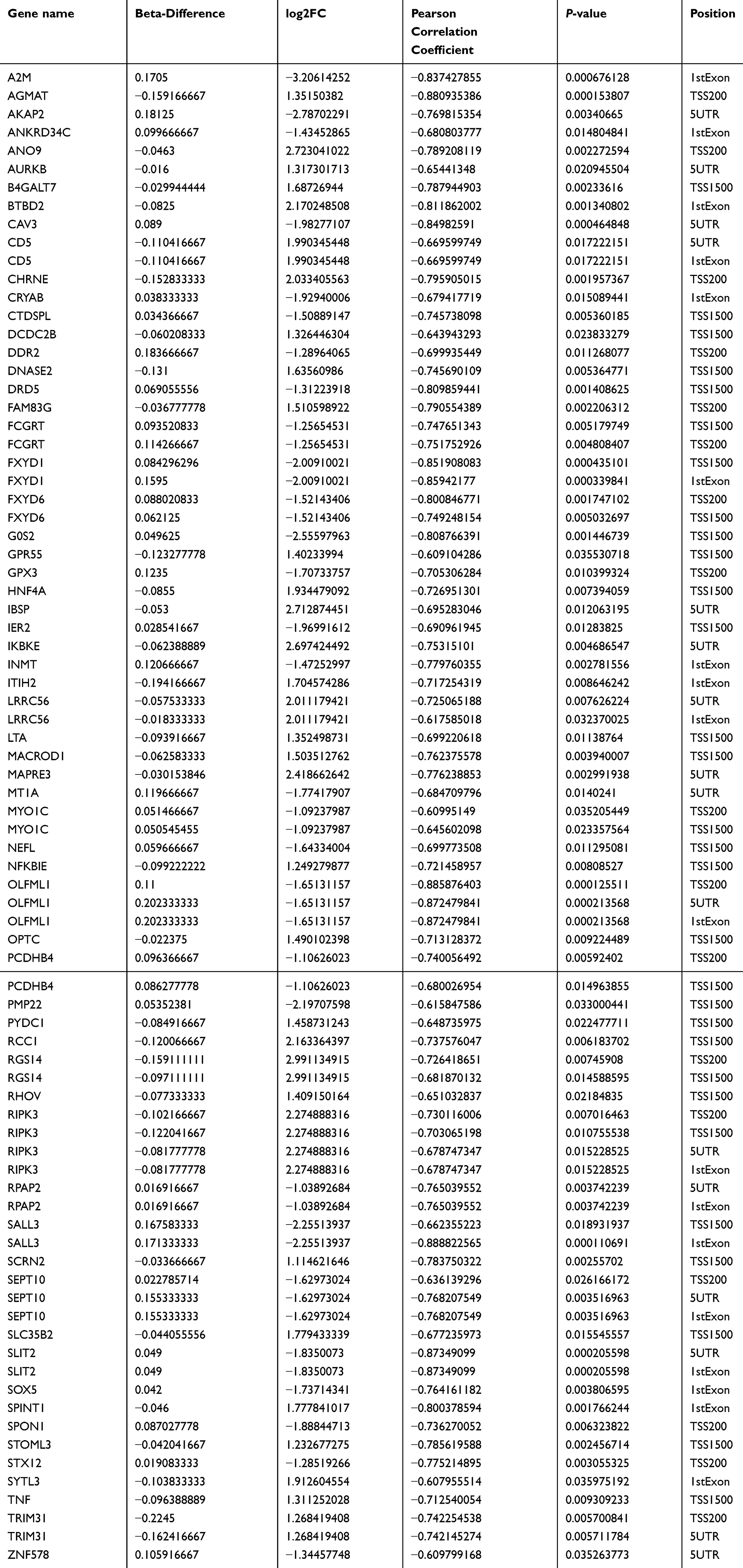 |
Table 3 Combined analysis of differentially expressed genes and regional differential methylation |
Cpn-related lung cancer genes (target genes) and potential biological function analysis
In total, 62 target genes were submitted for GO and pathway (KEGG, PID, BioCarta, Reactome, BioCyc and PANTHER) enrichment analysis. For biological processes, positive regulation of chronic inflammatory response to antigenic stimulus (GO: 0002876) was the most represented GO term, followed by negative regulation of growth (GO: 0045926) and regulation of chronic inflammatory response to antigenic stimulus (GO: 0002874). For cellular components, the major represented categories were membrane raft (GO: 0045121). Genes involved in important molecular functions were also identified, such as nuclear factor-kappa B-inducing kinase activity (GO: 0004704) (Figure 4). Using the KEGG pathway database, we found that tumor necrosis factor (TNF) and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) overexpression by hypomethylation of the promoter region may activate the TNF signaling pathway (hsa04668, Figure S11). RIPK3 and inhibitor of nuclear factor-kappa B kinase subunit epsilon (IKBKE) may play a role in responding to bacterial invasion in the cytosolic DNA-sensing pathway (hsa04623, Figure S12). Furthermore, we also found that the first ranked pathway in the Reactome database was Methylation of MeSeH for excretion (R-HSA-2408552). In PANRHER, we found a number of target genes that were involved in the adrenaline and noradrenaline biosynthesis pathway (P00001) and Toll receptor signaling pathway (P00054). Details are shown in Figure 5. We also conduced GSEA for all 20,198 genes. The Heat map and gene list correlation profile in the dataset for the comparison of A vs B (Figure S13) and C vs D (Figure S14) are shown. Our data indicate that DNA methylation can affect the binding of transcription factors to genes.
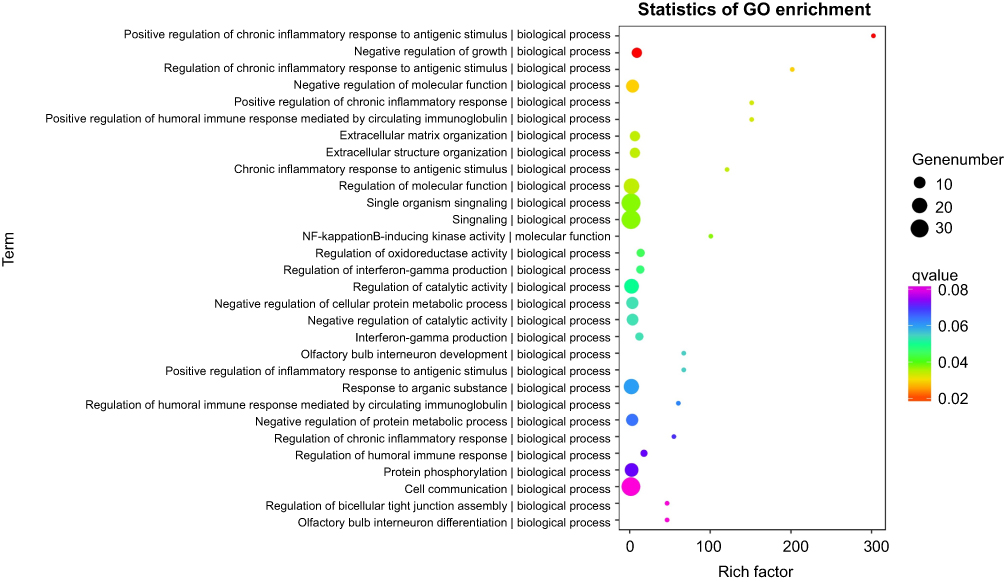 |
Figure 4 Cpn-related lung cancer genes (target genes) and potential biological function analysis. Gene ontology for the top 30 biological processes, cellular components, and molecular functions annotation. |
 |
Figure 5 KEGG, PID, BioCarta, Reactome, BioCyc and PANTHER pathway enrichment analysis for all 62 target genes. |
Quantitative methylation analysis and validation
Among the 62 target genes that were the most representative aberrantly methylated genes of Cpn-associated lung cancer, RIPK3 was verified using 24 microarray samples as the most methylated in all promoter regions (TSS1500, TSS200, 5ʹUTR and 1stExon regions) and was enriched in innate immune responses for foreign DNA from invading microbes in pathway analysis. Furthermore, methylation levels were quantified against the DMRs in the promoter regions of RIPK3. The Wilcoxon Rank Sum test showed that, in the Cpn positive group, the average methylation level of RIPK3 in lung cancer samples was significantly lower than that in the para-cancer control samples (7.25% vs 11.67%, P=0.005), but the difference was not significant in the Cpn negative group (8.96% vs 12.54%, P=0.093). The lowest methylation level of RIPK3 was found in Cpn-positive lung cancer samples, and in the 2nd, 4th, and 5th CpG sites there was a significant difference in methylation levels between lung cancer and para-cancer control samples in the Cpn-positive group, but not in the Cpn negative group (Figure 6). For the differentially expressed methylation sites in the RIPK3 promoter regions screened using chip and first validation, DNA methylation levels were verified on samples numbered ABDC 7–12. Only the 5th CpG site of RIPK3 was statistically significant in A vs B (P=0.041), and not in C vs D (P=0.310).
 |
Figure 6 Methylation levels of the CpG sites in the RIPK3 promoter. (A) Schematic diagram of the RPIK3 promoter. The sequence represents a 276-base pair fragment (−43 bp to +232 bp) in RPIK3. The start of the 1st exon was considered as +1 of the sequence. Numbers 1–6 refer to locations of the CpG sites within the RIPK3 elements tested, and underlining shows the number of multiple CpG sites that were tested simultaneously. (B) Comparison of the methylation levels of CpG sites in RIPK3 promoter regions. Data are expressed as Median (P25, P75). * Wilcoxon Rank Sum test was performed: *P<0.05(relative to the respective control: B or D). ncase =6, ncontrol =6. |
Discussion
Lung cancer is the most prevalent and deadly cancer in the world. In addition to an imbalance in proto- and anti-oncogenes, aberrant promoter hypermethylation is now recognized as an essential component in lung cancer progression.13 Infection has been found to be closely related to abnormal DNA methylation and can further induced cancer.14,15 The development of chip techniques has led to novel approaches to lung cancer classification and diagnosis. We investigated the genome-wide DNA methylation and gene expression profiling of 12 NSCLC (6 Cpn positive and 6 Cpn negative) and 12 paired adjacent lung tissues. The target genes of Cpn infection-associated lung cancer were enriched in several representative pathways, including positive regulation of chronic inflammatory response to antigenic stimulus (GO: 0002876), negative regulation of growth (GO: 0045926), regulation of chronic inflammatory response to antigenic stimulus (GO: 0002874), membrane raft (GO: 0045121) and nuclear factor-kappa B-inducing kinase activity (GO: 0004704). RIPK3 and IKBKE were enriched in the TNF signaling pathway (hsa04668) and the cytosolic DNA-sensing pathway (hsa04623). Chip and validation screening demonstrated that the abnormal methylation sites in the promoter regions of RIPK3 were associated with Cpn-related lung cancer.
A recent study16 on Chlamydia trachomatis (Ct) and ovarian cancer suggested that Chlamydia infection promotes host DNA damage, causing malignant cell proliferation, which permanently affects the host at the genomic and epigenetic levels, particularly through altering host chromatin structure by DNA methylation and post-translational histone modifications. Cpn can induce histone H3 and H4 modifications, which have a major effect on cytokine production.16 Ct infection was associated with increased expression of two mesenchymal cell markers: fibronectin and α-smooth muscle actin (α-SMA). The DNA methylation status of selected regions of E-cadherin, fibronectin, and α-SMA genes revealed that Ct infection was accompanied by changes in DNA methylation of the E-cadherin promoter.17 A whole genome sequencing study18 of Cpn showed that there are many enzymes involved in the synthesis and metabolism of aromatic compounds, such as synthetase, hydroxylase, decarboxylase, and methylase. However, there have been no Cpn-related methylation studies. We speculated that Cpn may lead to abnormal methylation of human genomic DNA, resulting in abnormal activation of oncogenes and transcriptional silencing of tumor suppressor genes, causing disordered cell growth and differentiation.
It is well known that DNA methylation of the promoter region strongly correlates with transcriptional repression, and that DNA methylation downstream of the TSS, in particular of the 1st exon, is critical for transcriptional silencing, independent of the cell type. In the current study, we found an inverse relationship between Cpn-related DMPs and DEGs. We identified Cpn-related DMRs for 62 significant target genes. These genes were enriched in several representative pathways, including positive regulation of chronic inflammatory response to antigenic stimulus, regulation of chronic inflammatory response to antigenic stimulus, and nuclear factor-kappa B-inducing kinase activity, among others. The biological function of most of these genes was related to chronic infection, which indicates that Cpn might be involved in the progression of lung cancer through DNA methylation changes.
Validation experiments showed that RIPK3 was enriched in the TNF signaling pathway and cytosolic DNA-sensing pathway, and was hypomethylated in the corresponding promoter regions. We also found that RIPK3 was a unique aberrant methylated gene in Cpn-positive lung cancer tissues. There was an inverse relationship between methylation of the promoter region and transcriptional activity.19,20 Hypomethylation of CpG islands in the promoter can result in gene overexpression. The start of the 1st exon was considered as +1 of the sequence, and RIPK3 was located in the promoter region from −43 bp to +232 bp (276 bp total). A previous study showed that reduced methylation of the RIPK3 promoter triggered expression of RIPK3 in RIPK3-null cancer cells21 and treatment with hypomethylating agents restored RIPK3 expression.22 RIPK3 was a component of the tumor necrosis factor receptor-I signaling complex that activated NF-κB transcription factors23,24 and further initiated cell proliferation, apoptosis resistance, and malignant transformation. However, recent studies showed that RIPK3, RIPK1, and mixed-lineage kinase domain-like protein (MLKL) formed a multi-protein complex, called a necrotic body,23,25,26 which regulated programmed cell necrosis.27 This was considered a new type of necrosis that is dependent on genetic programming and regulation.28 Programmed necrosis does not depend on the traditional caspase apoptotic pathway and has been defined as RIPK3-dependent necrotizing cell death.29 In contrast, programmed necrosis provided the host with an autonomous defense, and cell rupture caused by programmed necrosis induced inflammation by releasing the damage-related molecules. Increasing evidence suggests that programmed necrosis is related to clinical diseases.29 Necrosis played not only a protective role but also a detrimental role in the pathogenesis of pulmonary diseases, such as chronic obstructive pulmonary disease and lung cancer,30 and is therefore an important potential therapeutic target.28 Contrary to the result of our study, recent data suggest that RIPK3 expression is often silenced in cancer cells due to genomic methylation near its transcriptional start site.22,31 Fukasawa et al32 compared the methylation levels of 288 cancer-associated gene promoters between lung cancer cells and normal lung cells. In the majority of lung cancer cell lines, RIPK3 was hypermethylated and down-regulated. The methylated frequency of RIPK3 was highest in small cell carcinoma. Outcome differences might be due to the different pathological types of lung cancer studied. It would be interesting to discuss the role of RIPK3 in development of lung cancer and its association with methylation by histological subtype.
In the present study, we also identified other Cpn-related target genes. Neurofilament (NF) is an intermediate filament found in the cytoplasm of central and peripheral nervous system neurons. It is a type IV intermediate filament heterodimer composed of light (NEFL), middle, and heavy chains,33 which provide structural support of axons and adjusts axon diameter to control the rate of electrical signals traveling along the axon.34 A recent study reported that abnormal methylation and expression of NF genes were detected in esophageal squamous cell carcinoma,35 breast cancer,36 renal cell carcinoma,37 hepatocellular carcinoma,38 and Ewing’s sarcoma.39 We found that hypermethylation of NEFL TSS1500 regions in Cpn-positive lung cancer tissues inhibited the expression of NEFL, which might contribute to the development of Cpn-associated lung cancer. Another study showed that NEFL expression regulated by promoter hypomethylation, which inhibits the NF-κB pathway, was associated with suppressed invasion and migration of NSCLC cell lines and patients.40
More recently dopamine and its receptors have been indicated as regulatory factors for immune cells and malignant cell proliferation.41 The bioavailability of dopamine is regulated by dopamine receptors (DRs),42 which are G protein-coupled receptors. DRs are divided into different classes - D1 (D1 and D5) and D2 (D2, D3, and D4) – and are expressed in alveolar epithelial cells. The expression of DRs might be affected by corresponding DNA methylation levels. Liao et al43 used methylation-sensitive representational difference analysis to screen gastric cancer and normal gastric mucosa, and found the promoter and exon regions of DRD5 were differentially methylated DNA sequences. DRD5 promoted adenylate cyclase to synthesize cAMP by activating the Gαs/olf family of G proteins.44 cAMP is an important second messenger in many biological processes and cAMP pathway dysregulation is related to the development of cancer. Our results showed that, in Cpn-related lung cancer, down-regulated DRD5 expression mediated by changes in corresponding promoter methylation status inhibited adenylate cyclase, thereby decreasing intracellular cAMP accumulation. This promoted the mitotic proliferation of cells, which may ultimately lead to development of lung cancer. The function of DRD5 in lung cancer requires further investigation.
There is currently no gold standard for detecting Cpn antigen levels, and the detection rate of existing methods is relatively low, increasing the difficulty of determining if patients have an active infection. Thus, in this study, serum Cpn IgG and IgA levels were detected using MIF, which is the standard for serologic detection of Chlamydia infection. Limited by the design of our case-control study, we only explored the relationship between chlamydia and lung cancer. Thus, Cpn infection may be involved in lung cancer development, and well-designed cohort studies and randomized controlled trials are needed to minimize the effect of disease on antibody titers, reduce selection bias, and better adjust for potential confounders.
This study explored the epigenetic mechanism of Cpn infection in lung cancer. Our integrative methylome and transcriptome analyses revealed that Cpn infection contributed to methylation-mediated changes in RIPK3. Activation of the NF-κB pathway, TNF signaling pathway, and cytosolic DNA-sensing pathway might be involved in the development and progression of lung cancer. Case-control studies have inherent limitations, such as selection bias and recall bias, that are difficult to avoid, and thus the same methylation sites may have distinctly different regulatory roles depending on factors such as race, tissue sample, and time of expression. New study designs and sample sizes may provide better insight into the association between Cpn infection, gene methylation, and lung cancer. Therefore, the results of this study need to be further validated in a multi-center, rigorously designed large sample study, and the specific mechanism needs to be further explored using relevant functional studies.
Abbreviation list
Cpn, Chlamydia pneumonia; GO, gene ontology; MIF, micro-indirect immunofluorescence; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; MassCLEAVE, mass spectrometry in combination with RNA base-specific cleavage; DMPs, differentially methylated positions; DMRs, differentially methylated regions; DEGs, differentially expressed genes; DGs, differentially genes; FC, Fold-Change; TSS, transcription start sites; GSEA, gene sets enrichment analysis;NSCLC, non-small cell lung cancer; CGIs, CpG islands; PCA, principal component analysis; TGFBR, transforming growth factor beta receptor; IKBKE, inhibitor of nuclear factor-kappa B kinase subunit epsilon; RIPK3, receptor-interacting serine/threonine-protein kinase 3; DRD5, dopamine receptor D5; NEFM, neurofilament medium; NEFL, neurofilament light; DR, dopamine receptors; MS-RDA, methylation-sensitive representational difference analysis; cAMP, cyclic adenosine monophosphate.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant number 81402738), Fujian Provincial Natural Science Foundation Project (grant number 2016J01355), Fujian Program for Outstanding Young Researchers in University awarded by Education Department of Fujian (grant number 2017B019), and the National Key Research and Development Program of China (grant number 2017YFC0907100).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Broaddus VC, Mason RC, Ernst JD, et al. Murray & Nadel’s Textbook of Respiratory Medicine E-Book. Philadelphia: Elsevier Health Sciences; 2015:6.
3. Pallis AG, Syrigos KN. Lung cancer in never smokers: disease characteristics and risk factors. Crit Rev Oncol Hematol. 2013;88(3):494–503. doi:10.1016/j.critrevonc.2013.06.011
4. Littman AJ, Jackson LA, Vaughan TL. Chlamydia pneumoniae and lung cancer: epidemiologic evidence. Cancer Epidemiol Biomarkers Prev. 2005;14(4):773–778. doi:10.1158/1055-9965.EPI-04-0599
5. Hua‑Feng X, Yue‑Ming W, Hong L, et al. A meta‑analysis of the association between Chlamydia pneumoniae infection and lung cancer risk. Indian J Cancer. 2015;52(Suppl 2):e112–e115. doi:10.4103/0019-509X.172506
6. Rizzo A, Carratelli CR, De Filippis A, et al. Transforming activities of Chlamydia pneumoniae in human mesothelial cells. Int Microbiol. 2014;17:185–193. doi:10.2436/20.1501.01.221
7. Khan S, Imran A, Khan AA, et al. Systems biology approaches for the prediction of possible role of Chlamydia pneumoniae proteins in the etiology of lung cancer. PLoS One. 2016;11(2):e0148530. doi:10.1371/journal.pone.0148530
8. Blanco D, Vicent S, Fraga MF, et al. Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16, activation of the DNA damage response pathway. Neoplasia. 2007;9(10):840–852.
9. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. doi:10.1038/nbt.1685
10. Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–295. doi:10.1016/j.ygeno.2011.07.007
11. Ehrich M, Correll D, van Den Boom D. Introduction to EpiTYPER for quantitative DNA methylation analysis using the MassARRAY system. Sequenom Application Note. 2006;1–7.
12. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
13. Haugen A. Molecular biology in the diagnosis of lung cancer. Tidsskrift for Den Norske Laegeforening. 2005;125(23):3283–3285.
14. Yeung CLA, Tsang TY, Yau PL, et al. Human papillomavirus type 16 E6 suppresses microRNA-23b expression in human cervical cancer cells through DNA methylation of the host gene C9orf3. Oncotarget. 2017;8(7):12158–12173. doi:10.18632/oncotarget.14555
15. Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer. 2017;20(1):8–15. doi:10.1007/s10120-016-0650-0
16. Chumduri C, Gurumurthy RK, Zadora PK, et al. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage tesponse. Cell Host Microbe. 2013;13(6):746–758. doi:10.1016/j.chom.2013.05.010
17. Schmeck B, Beermann W, N’Guessan PD, et al. Simvastatin reduces Chlamydophila pneumoniae-mediated histone modifications and gene expression in cultured human endothelial cells. Circ Res. 2008;102(8):888–895. doi:10.1161/CIRCRESAHA.107.161307
18. Jovana R, Aleksandra I-K, Elisabeth S, et al. Infection is associated with E-cadherin promoter methylation, downregulation of E-cadherin expression, and increased expression of fibronectin and α-SMA-implications for epithelial-mesenchymal transition. Front Cell Infect Microbiol. 2017;7:253. doi:10.3389/fcimb.2017.00517
19. Schubeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–326. doi:10.1038/nature14192
20. Zemach A, McDaniel IE, Silva P, et al. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328(5980):916–919. doi:10.1126/science.1186366
21. Yang C, Li J, Yu L, et al. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell Death Dis. 2017;8:e3084. doi:10.1038/cddis.2017.518
22. Koo G-B, Morgan MJ, Lee D-G, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–725. doi:10.1038/cr.2015.56
23. Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
24. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16(7):689–697. doi:10.1038/ni.3206
25. Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1):213–227. doi:10.1016/j.cell.2011.11.031
26. Li J, McQuade T, Siemer AB, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339–350. doi:10.1016/j.cell.2012.06.019
27. Newton K, Dugger DL, Wickliffe KE, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343(6177):1357–1360. doi:10.1126/science.1249361
28. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311. doi:10.1038/nature14191
29. Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi:10.1056/NEJMra1310050
30. Mizumura K, Maruoka S, Gon Y, et al. The role of necroptosis in pulmonary diseases. Respir Investig. 2016;54(6):407–412. doi:10.1016/j.resinv.2016.03.008
31. Morgan MJ, You-Sun K. The serine threonine kinase RIP3: lost and found. BMB Rep. 2015;48:303–312.
32. Fukasawa M, Kimura M, Morita S, et al. Microarray analysis of promoter methylation in lung cancers. J Hum Genet. 2006;51(4):368–374. doi:10.1007/s10038-005-0355-4
33. Tu PH, Elder G, Lazzarini RA, et al. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol. 1995;129(6):1629–1640. doi:10.1083/jcb.129.6.1629
34. Alberts B. Molecular Biology of the Cell.
35. Kim MS, Chang X, LeBron C, et al. Neurofilament heavy polypeptide regulates the Akt-β-catenin pathway in human esophageal squamous cell carcinoma. PLoS One. 2010;5(2):e9003. doi:10.1371/journal.pone.0009003
36. Jeschke J, Van Neste L, Glöckner SC, et al. Biomarkers for detection and prognosis of breast cancer identified by a functional hypermethylome screen. Epigenetics. 2012;7(7):701–709. doi:10.4161/epi.20445
37. Dubrowinskaja N, Gebauer K, Peters I, et al. Neurofilament heavy polypeptide CpG island methylation associates with prognosis of renal cell carcinoma and prediction of antivascular endothelial growth factor therapy response. Cancer Med. 2014;3(2):300–309. doi:10.1002/cam4.181
38. Revill K, Wang T, Lachenmayer A, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145(6):
39. Alholle A, Brini AT, Gharanei S, et al. Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics. 2013;8(11):1198–1204. doi:10.4161/epi.26266
40. Shen Z, Chen B, Gan X, et al. Methylation of neurofilament light polypeptide promoter is associated with cell invasion and metastasis in NSCLC. Biochem Biophys Res Commun. 2016;470(3):627–634. doi:10.1016/j.bbrc.2016.01.094
41. Pornour M, Ahangari G, Hejazi S, et al. Dopamine receptor gene (DRD1-DRD5) expression changes as stress factors associated with breast cancer. Asian Pac J Cancer Prev. 2013;15:10339–10343. doi:10.7314/APJCP.2014.15.23.10339
42. Campa D, Zienolddiny S, Lind H, et al. Polymorphisms of dopamine receptor/transporter genes and risk of non-small cell lung cancer. Lung Cancer. 2007;56(1):17–23. doi:10.1016/j.lungcan.2006.11.007
43. Liao AJ, Su Q, Wang X, et al. Isolation and bioinformatics analysis of differentially methylated genomic fragments in human gastric cancer. World J Gastroenterol. 2008;14(9):1333. doi:10.3748/wjg.14.1333
44. Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217. doi:10.1124/pr.110.002642
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.