Back to Journals » Veterinary Medicine: Research and Reports » Volume 12
Genogrouping of Infectious Bursal Disease Viruses Circulating in Ethiopian Chickens: Proposal for Assigning Very Virulent Strains in the Country into New Sub Genogroup 3d
Authors Bari FD
Received 23 December 2020
Accepted for publication 10 February 2021
Published 26 February 2021 Volume 2021:12 Pages 43—52
DOI https://doi.org/10.2147/VMRR.S296367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Young Lyoo
Fufa Dawo Bari
Department of Microbiology, Immunology and Veterinary Public Health, College of Veterinary Medicine and Agriculture, Addis Ababa University, Bishoftu, Ethiopia
Correspondence: Fufa Dawo Bari
Department of Microbiology, Immunology and Veterinary Public Health, College of Veterinary Medicine and Agriculture, Addis Ababa University, P O Box 34, Bishoftu, Ethiopia
Tel +251 929190312
Fax +251 114339933
Email [email protected]
Introduction: In 2017 infectious bursal disease viruses (IBDVs) were reclassified into genogroups based on nature of clustering on a phylogenetic tree constructed using VP2 gene sequence data rather than according to their pathotype and/or antigenic types. Ethiopian IBD viruses were not reclassified according to the proposed genogrouping.
Methods: In order to genogroup the Ethiopian IBDVs, available VP2 gene sequences data together with reference strain sequences were retrieved from GenBank and genogrouped as recently recommended based on evolutionary tree reconstruction and determination of their clustering on the phylogenetic tree.
Results: The Ethiopian IBDVs were grouped into genogroups 1 and 3 that antigenically represent classically virulent and very virulent IBDVs, respectively. The genogroup 1 IBDVs were clustered with the vaccine strain while the genogroup 3 viruses were clustered with four known viruses belonging to sub-genogroup 3a and sub-genogroup 3b. Almost half of the Ethiopian IBDVs reported did not cluster with the specific sub-groups of genogroup 3; rather, the isolates were clustered differently suggesting they deserve a different sub-genogroup tentatively proposed as 3d. The two genogroups observed based on clustering on a phylogenetic tree were supported by corresponding deduced amino acid changes in similar positions in VP2 sequences. In addition, virulence marker amino acid genes coupled with second major hydrophilic region (amino acid positions 314– 325) were predicted in these sequences that could be responsible for the occurrence of IBD outbreaks.
Conclusion: A new sub-genogroup of IBDVs, 3d, were observed in the sequences that could be one of the reasons for the frequent occurrence of IBD outbreaks and questions the protective potential of the existing vaccine. To institute disease control in the country, the effectiveness of the vaccine in use needs to be assessed in vivo against both genogroups 1 and 3 viruses and all three sub-genogroup 3 viruses circulating in the country.
Keywords: infectious bursal disease virus, VP2 gene sequence, genogroups, sub-genogroups, virulent strains
Introduction
Infectious bursal disease (IBD), also known as Gumboro,1 is a contagious and immunosuppressive disease of chickens circulating globally. The disease was detected for the first time in Ethiopian domestic chickens in 2002 in which 45–50% mortality was reported.2 Currently, the disease is the second priority chicken disease in the country next to Newcastle disease (NCD). The etiological agent of the disease is IBD virus (IBDV) that belongs to the genus Avibirnavirus of the family Birnaviridae.3 The virus family has no envelope and is icosahedral in shape, with a double-stranded and bi-segmented (segments A and B) RNA genome.3,4 Segment A contains viral protein (VP) 2, VP3, and VP4 genes and has the key region required for pathogenicity, antigenicity, and virulence. In addition, segment A has protective antigens while segment B contains VP1, which is the RNA-dependent RNA polymerase responsible for viral genome replication.
Historically, since 2004, highly pathogenic IBDV strains emerged globally via mutation and genetic re-assortment5 and during the same time Ethiopia reported the first outbreak of the disease.2,6 Unless the chickens get protective vaccines during their early life (2 to 6 weeks of age), it leads to considerable economic loss. Therefore, vaccination is considered an important means of protecting domestic chickens during their first weeks of life.7 As a result, in Ethiopia, commercial poultry farms vaccinate their chickens against IBD regularly using IBDV LC-75 live vaccine and they have improved biosecurity measures. The immune response to the attenuated IBDV LC-75 vaccine prepared from classically virulent IBDVs against very virulent virus is variable, but it is still in use in the country. Despite that, IBD outbreaks due to very virulent and classically virulent IBDVs were frequently reported from different parts of Ethiopia.8–10 The use of live vaccine for IBD control could lead to the emergence of very virulent strains via genome re-assortment and recombination.11 The economic consequence of disease caused by such virulent strains on poultry health, production, and the overall economy is substantial. Hence, any IBD outbreak needs to be assessed by IBDV full gene sequencing of both segments A and B to have clear evidence of any genetic changes leading to emergence of new strains like gene recombination and gene re-assortment coupled with occurrence of mutations.
The possible means of emergence of the IBDV strains are mutation, natural gene recombination,11,12 and genome re-assortment with vaccine strains.11,13 The IBDV genetic changes contributing to these outbreaks and detailed evolutionary analysis for the emergence of the viruses were assessed via molecular evolutionary analysis and deduced amino acid comparisons of the VP2 region of the IBDV genome. The VP2 contains exposed projection (P) domain that encompasses the hypervariable region (hVP2).14 The P domain is composed of four loop structures designated PBC, PDE, PFG, and PHI that are exposed on the surface of the virion, and this domain is responsible for inducing a protective immune response in chickens.15,16 Experimental evidence indicated that the amino acids in the P domain of VP2 play a critical role for the binding of neutralizing monoclonal antibodies.16 The partial VP2 region that comprises the hVP2 region (amino acid positions 220–345), in general, is widely used for phylogenetic analysis to compare the molecular evolution of the IBDVs.16, 17 The widely used nomenclature of the virus had been based on antigenicity and pathogenicity that is confusing most of the time. As a result, recently Jackwood et al.18 revised the nomenclature based on phylogenetic analysis and used the term genogroups and sub-groups based on the nature of clustering on the evolutionary tree. The Ethiopian IBDVs were not genogrouped and sub-genogrouped according to the proposed nomenclature. Therefore, in this study evidence of genogrouping of Ethiopian isolates in known genogroups, new sub-genogroups, and key mutations contributing to virulence and classification of the virulent viruses are presented. In addition, the difficulty of sub-grouping the genogroup 3a and 3b viruses is assessed and a new sub-genogrouping (3d?) is proposed for Ethiopian vvIBDVs.
Materials and Methods
Data Source for Reference and Ethiopian IBDV Sequences Search and Retrieval
Freely available and/or published VP2 nucleotide sequence data of IBDVs reported from Ethiopia and other reference sequences available in GenBank were retrieved (Table 1) and used for this study. A total of 101 sequences of which 23 were Ethiopian IBDV virus sequences were downloaded. In addition, representative reference sequences of the seven known genotypes18,19 were used as a base to compare and contrast the analysis, including phylogenetic tree reconstruction, genogrouping, and sub-groupings, and for comparison of critical amino acid changes/variability per position.
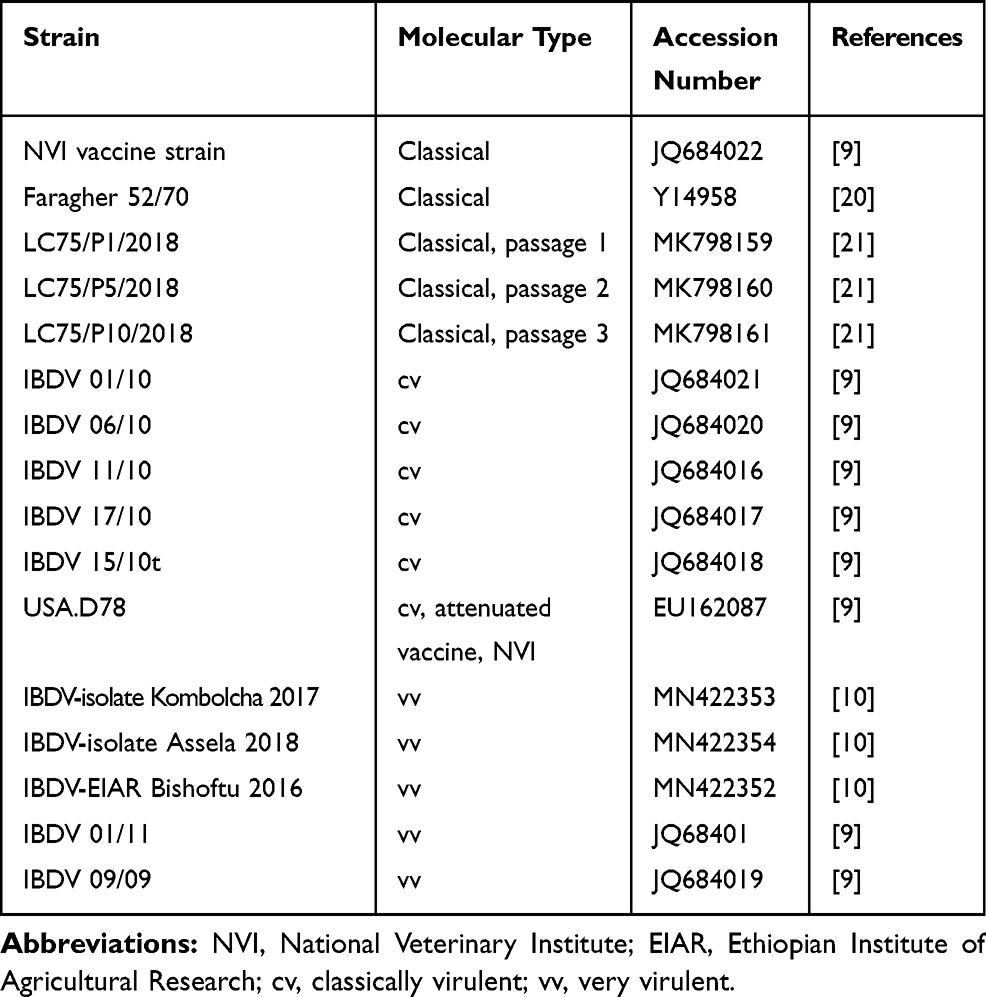 |
Table 1 Virulence-based Classification of Some IBDV Sequences Retrieved from GenBank and Used in the Analysis with Their Accession Numbers |
Sequence Data Analysis
The collected IBDV VP2 gene sequences were aligned using the Clustal W multiple sequence alignment program embedded in MEGA 7 software.22 The maximum likelihood statistical method was used for best fit model selection in MEGA 7. The selected model was used to reconstruct molecular evolution of the viruses using the same software. In addition, the nucleotide sequences were used to deduce their amino acid readings using BioEdit23 software to compare amino acid changes per site at critical amino acid positions. The amino acid positions undergoing changes were inferred with reference to the vaccine strain sequence data used in the country (reference accession number JQ684022). Both phylogenetic tree reconstruction and genetic distance calculations were used for subdivision of genogroup 3 viruses in MEGA 7 software.22 Hydrophilic regions as an indicator of neutralising antigenic sites were predicted from deduced amino acid sequence data using the method of the Hopp and Woods24 program that is embedded in BioEdit software.23 Recombinant gene detection was carried out using Recombination Detection Program software (RDP).25
Results and Discussion
Recombination Detection
IBDVs are believed to emerge via homologous recombination and re-assortant phenomena, besides mutations, that occur naturally among different IBDV strains including vaccine strains. These sequences used for phylogenetic tree reconstruction were initially screened for evidence of recombination using RDP25 before being used for further analysis. The sequences showed no evidence of recombination. The other method by which recombinants are visualized is by difference in nature of clustering on a phylogenetic tree upon repeated tree reconstruction, which was also not observed in this analysis.
The absence of recombination in Ethiopian IBDV isolates could be due to use of short nucleotide sequence data in the analysis that could not show evidence of change in the nature of clustering on a phylogenetic tree. Further, investigation using full genome sequence data, at least for segment A (and possibly also segment B), and reanalysis of the sequences for evidence of genetic changes like recombination, re-assortment, and mutations in general are very important to arrive at conclusive evidence. Such study could give insight into the emergence of new strains, the efficacy of existing vaccines against these strains, and the need for further research for vaccine development.
Model Selection
The molecular evolutionary relationship was assessed via phylogenetic tree reconstruction of VP2 sequences of IBDV using both known genogroup and ungrouped sequences. The ungrouped Ethiopian IBDV sequences and reference sequences used for genogrouping were included to have the overall phylogenetic tree and demonstrate their evolutionary relationship. For this, model selection was first carried out in MEGA721 that resulted in selection of a K2 + G (Kimura 2-parameter with gamma distribution) model according to Bayesian Information Criterion (Supplementary Material 1). The best fit model, K2 + G model, was subsequently used for phylogenetic tree reconstruction. Model selection is widely applied in other viruses, like the Newcastle disease virus.26
Genogrouping Based on Partial VP2 Gene Sequence
The Ethiopian IBDVs were genogrouped into two genogroups, namely, genogroup-1 and genogroup-3 (Figure 1). These genogroups 1 and 3 represent classically virulent and very virulent IBDVs according to their virulence/pathotype, respectively.9 Interestingly, the genogroup 1 IBDVs were always clustered together with a bootstrap value of > 50% up on repeated phylogenetic tree reconstruction in MEGA 7.21 On the phylogenetic tree, the genogroup 3 viruses were supported by a very high bootstrap value of 96% and the Ethiopian genogroup 3 IBDVs were found on the tip of the tree. In addition, they did not cluster with any of the proposed and/or sub-genogroups of genogroup 3 IBDVs, namely, sub-groups 3a, 3b, and 3c, suggesting that they deserve different sub-group/s of their own.
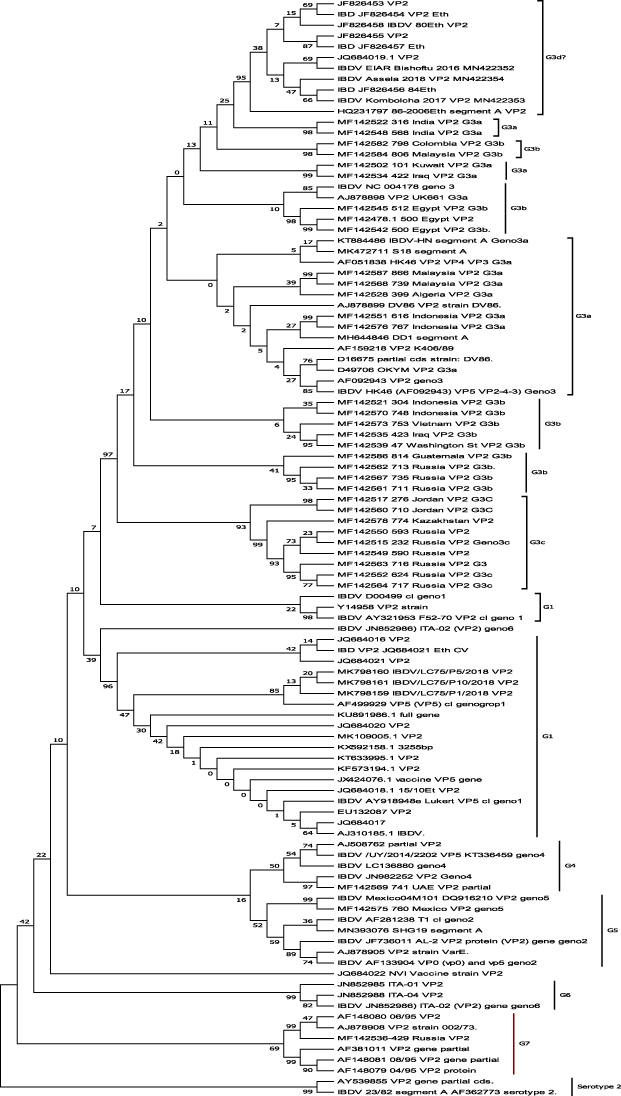 |
Figure 1 Evolutionary relationships of IBDVs. The evolutionary history was inferred using the neighbour-joining method.27 The optimal tree with the sum of branch length = 1.47232579 is shown. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches.28 The evolutionary distances were computed using the Kimura 2-parameter method29 and are in the units of the number of base substitutions per site. The rate variation among sites was modelled with a gamma distribution (shape parameter = 1). The analysis involved 100 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All ambiguous positions were removed for each sequence pair. There were a total of 413 positions in the final dataset. Evolutionary analyses were conducted in MEGA 7.22Note: IBDVs = infectious bursal disease viruses; G = genogroup. |
Revisiting Previous Sub-grouping of Genogroup 3 IBDVs
The subgrouping of genogroup 3 IBDVs reported by Jackwood et al.18 used 45 nucleotide sequences of 560 positions that resulted in forming three sub-groups, namely, 3a, 3b, and 3c. References used for the demarcation of sub-groupings like genetic distance, other than nature of clustering, were not indicated in the publication. According to the report, sub-group 3a contains viruses mainly from all over the world, sub-group 3b contains viruses from the Americas, and sub-group 3c contains viruses from Asian and Russian countries. Except sub-group 3c, the sub-groups 3a and 3b of genogroup 3 viruses were mixed up or scattered here and there on a phylogenetic tree. In the current analysis, these sub-groups were not fully repeated as reported before.18 The unrepeatability of the nature of clustering for sub-grouping of genogroup 3 viruses could be due to either use of relatively shorter gene sequences data or unrepeatability of the clustering when new sub-groups were formed, as observed in this analysis.
Sub-grouping of Genogroup 3 Ethiopian Isolates Based on Their Molecular Evolutionary Analysis
A total of 410 nt sequence data of 51 viruses that included sequence data used by Jackwood et al.18 and Michaela and Jackwood19 for sub genogrouping were used for phylogenetic tree reconstruction using the same software and program except use of the K2 + G model rather than the maximum composite likelihood method. Molecular evolutionary analysis of genogroup 3 viruses are depicted in Figure 2. Unfortunately, there is no clear demarcation between the sub-groups reported by Jackwood et al.18,19 with or without inclusion of Ethiopian isolates when the tree was reconstructed using 410 nucleotide sequences by both the neighbour-joining method (Figure 2) and minimum evolution analysis (figure not shown) in MEGA 7.21 From the phylogenetic tree, it is clear that sub-genogroup 3c always clustered together and the majority, but not all, of the reported sub-group 3a were also clustered together. Sub-group 3b of genogroup 3 viruses are the most diverse of the proposed sub-groups of genogroup 3 because they clustered here and there on the tree. In this analysis, there is no clear demarcation between sub-groups of genogroup 3b IBDVs other than for sub-genogroup 3c and almost all sub-genogroup 3a viruses that might be determined based on longer and/or full gene sequence analysis of segment A (possibly together with full or some parts of the segment B genome). Interestingly, the Ethiopian genogroup 3 viruses always cluster together and deserve a new sub-genogroup proposed to be sub-genogroup 3d (Figure 2).
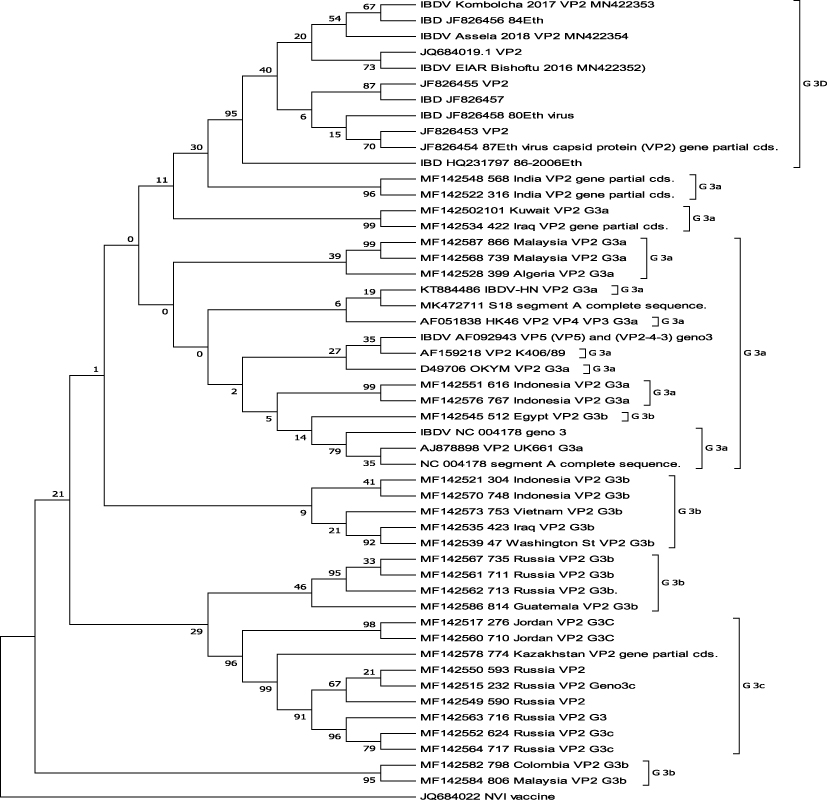 |
Figure 2 Evolutionary relationships of genogroup 3 IBDVs. The evolutionary history was inferred using the neighbour-joining method.27 The optimal tree with the sum of branch length = 0.66910707 is shown. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches.28 The evolutionary distances were computed using the selected Kimura 2-parameter method29 and are in the units of the number of base substitutions per site. The rate variation among sites was modelled with a gamma distribution (shape parameter = 1). The analysis involved 51 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All ambiguous positions were removed for each sequence pair. There were a total of 410 positions in the final dataset. Evolutionary analyses were conducted in MEGA 7.21Note: G = genogroup. |
Demarcation of Sub-genogroups Based on Genetic Distance
Phylogeny-based sub-grouping of genogroup 3 viruses illustrated that the Ethiopian IBDV isolates did not cluster with other virus sequences compared, including the sequence used for genogrouping and/or sub-grouping,18,19 suggesting they emerged differently. The local genogroup 3 IBDVs were clustered together with bootstrap values of 96% (Figure 2), while it is 97% when all genogroups are included (Figure 1). The nature of clustering of Ethiopian isolates was repeatable upon repeated phylogenetic tree reconstruction using two frequently used methods, namely, neighbour-joining28 and minimum evolution analysis (figure not shown). As can be evidenced from the phylogenetic tree, sub-groups 3a and 3b of genogroup 3 are mixed up on the tree (Figure 2) but almost all sub-genogroup 3a viruses cluster together except four viruses.
To gain further insight into the evolutionary relationship of the genogroup 3 viruses, the genetic distances of the viral sequences that could possibly differentiate the known genogroup 3 sub-groups and the Ethiopian isolates were calculated. A genetic distance of 4.7% was the minimum distance that separated sub-genogroups 3a and 3b of the IBD virus sequences used by Jackwood et al.18 In this analysis, the sub-genogroup 3d of the Ethiopian isolates had a minimum genetic distance of 4.9%, which is slightly higher than the cut-off point (i.e. 4.7%) estimated. This finding further supports the separate nature of clustering on a phylogenetic tree (Figure 2) and the need to assign into new sub genogroup 3d. Therefore, considering the nature of clustering reported by Jackwood et al.,18 a 4.7% genetic distance could be considered as the minimum genetic distance that could separate the sub-genogroups. The limitation of this report is that the genetic distance was estimated based on the nature of clustering reported by Jackwood et al.,18 with the exception that the Ethiopian isolates genogroup 3 was taken as it is because they always cluster together. The use of genetic distance for sub-grouping viruses is widely used in other viruses like Newcastle disease virus. Interestingly, a closer value of 5% was used as a cut-off value for demarcation of sub-groups in the case of Newcastle disease virus.25
Critical Mutation Contributing to Antigenicity and Evolution of IBDVs
In the sequences analyzed, the second major hydrophilic region was predicted using multiple amino acid sequence data (Figure 3) that represents region 314–325 in peak B15 of the PHI loop.16 The PHI loop, together with loop PBC, of the IBDV VP2 gene contains neutralizing epitopes that strongly influence the neutralization reactivity.16 The hypervariable region of IBDV is within the two major hydrophilic regions that stretch from 206–35014 and is responsible for virulence and antigenicity.30 Surprisingly, the predicted hydrophilic region is more conserved in the sequence aligned except amino acid position 330, a closely located amino acid on a linear sequence, for which R to S amino acid substitution was observed between genogroup 1 and genogroup 3 viruses, respectively. Details of the amino acid changes in partial VP2 sequence per position are provided in Supplementary Material 2.
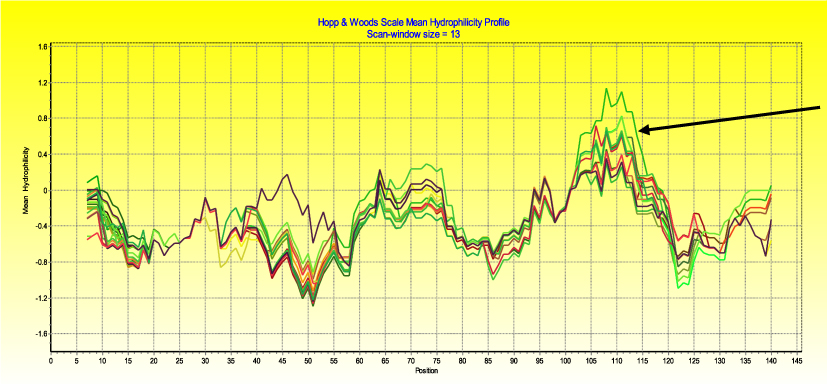 |
Figure 3 Predicted hydrophilic region from deduced amino acid sequences of IBDVs. The arrow on the right-hand side of the figure indicates region 105–113, which represents major hydrophilic region 314–325 of peak B of the PHI loop.30 |
Amino acid changes in the VP2 sequence further confirmed the circulation of two groups of IBDVs in the country: those which are true field outbreaks and those most likely derived from the vaccine strain used in the country. The two groups and the vaccine strain have different amino acid changes in the VP2 region of the virus (Table 2). Of these positions, known virulence marker amino acids per positions A222, Q253, I256, I294, and S299 were observed as reported before30 (Table 2).
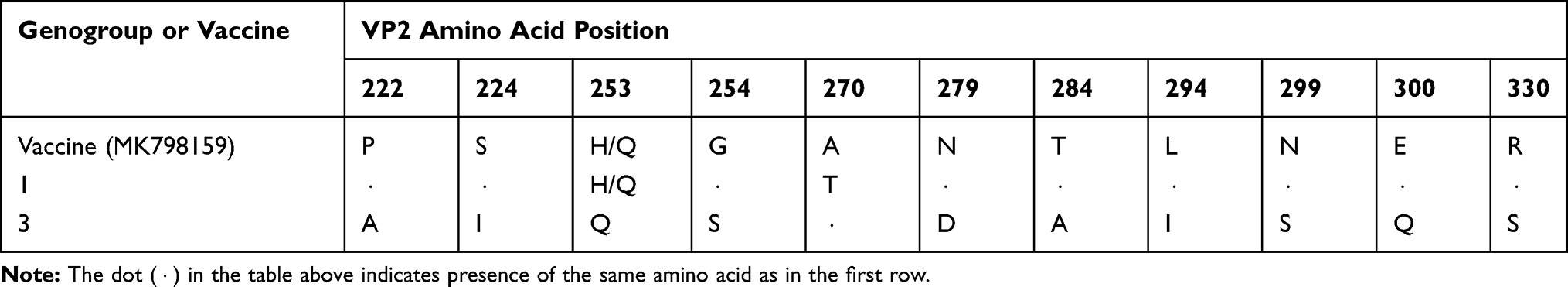 |
Table 2 Deduced Amino Acid Differences in VP2 Regions Among Genogroup 1, Genogroup 3 and Current Vaccine Strain in Ethiopia |
An amino acid position 222 is located in the first major hydrophilic region and the change from a P to an A amino acid as observed in genogroup 3 viruses at this position could lead to significant antigenic differences.30
In addition, H amino acid was observed in genogroup 1 viruses at position 253 while it is Q/N in genogroup 3 viruses. It was already reported that H253Q/N substitution, as observed in Ethiopian isolates, increased virulence of attenuated IBDV vaccine experimentally. At amino acid position 254 (loop PDE), there is S amino acid in genogroup 3 viruses while it is G in most genogroup 1 viruses including the vaccine strain except in three viruses (Table 2). At this amino acid position, when Q is substituted by S amino acid, as observed in very virulent field isolates, it leads to vaccination failure.31 In addition, the presence of S mutation at this position (254) in vaccine strain was reported to lead to severe bursal lesions in vaccinated chickens.32
The sequence of virulent IBDV strains analyzed had amino acids Q253, D279, and A284 in the VP2 gene. Experimental evidence indicated that these amino acids are involved in the cell tropism, tissue culture infectivity, and pathogenicity of virulent strains.33 Among the virulence marker genes, the virulence of some amino acid positions was verified experimentally; for example, a V256I substitution in attenuated vaccine strains increased virulence.34
A unique amino acid and position that differentiates the vaccine strain from both the field isolates (genogroup 1 and genogroup 3 viruses) is the presence of M amino acid at position 290 (Figure 2). Previously, Pikuła et al.11 reported that M294, a closely located amino acid on the linear amino acid sequence, was observed in re-assortant viruses that could not be reckoned out from the small VP2 sequence analyzed.
Some genogroup 1 viruses like JQ684022, a former vaccine strain used in Ethiopia, had Q amino acid at position 253 and it could have contributed to some outbreaks in the country because two virus sequences (JQ684016 and JQ684021) have almost 100% amino acid sequence similarity except at amino acid position 330. Some amino acid changes observed in the vaccine strains and in genogroup 1 viruses in VP2 were reported to be due to cell culture adaptation like N279 and T28435,36 that was observed in these sequences. At both 279 and 284 amino acid positions, the genogroup 3 viruses had a D and an I amino acids reported in vvIBDV, respectively. This suggests that these viruses did not emerge from the vaccine used to control the disease in domestic chickens. In Nigeria, Adamu et al.37 reported that N279T substitution was observed in attenuated viruses; in Ethiopia, however, N amino acid is available in vaccine strains and all genogroup 1 viruses, while it is D in genogroup 3 viruses. Experimentally, D to N substitution at amino acid position 279 of the VP2 gene resulted in loss of virulence,36 suggesting that the amino acid position is relevant to virulence.
An R330S mutation was observed in the Ethiopian sequences analyzed. This amino acid 330 is located at the interface between sub-units in the trimeric spike, away from the main antigenic site formed by loops PBC and PHI.38 Experimental evidence indicates that amino acid position 330 is an epitope in the PHI loop of the VP2 gene that was recognized by its reactivity with a monoclonal antibody.16 An R330S mutation, for example, leads to subtle alterations in the conformation of loop PBC16 thereby affecting its antigenicity.
In general, the presence of reported virulence gene markers30,39,40 like Q222, Q253, I294, S299, S330, and A28434 in Ethiopian IBDV sequences provided strong evidence regarding the circulation of virulent (both genogroups 1 and 3) strains in domestic chickens. In general, the amino acid substitution that clearly differentiates genogroup 1 and genogroup 3 is observed at amino acid positions P222A, V242I, L254S, V256I, T284A, L294I, N299S, E300Q, and R330S except in the old vaccine strain JQ684022.
S amino acid at position 254 of VP2 was observed in some sequences, the presence of which was reported to be suggestive of vaccine failure and was also accompanied by severe bursal lesions in infected chickens.32 In addition, at amino acid position 249 both field virus and vaccine strains in Ethiopia have Q while two field strains have R amino acid. At this position, a change from R to Q in VP2 markedly increased the virulence of an attenuated IBDV vaccine strain.31
Conclusion
In Ethiopia, two genogroups of IBDVs were circulating so far that were clustered separately on a phylogenetic tree and supported by amino acid changes at identical positions, including reported virulence marker amino acid per positions. The former classification of the viruses based on pathotype as classically virulent and very virulent corresponded to genogroup 1 and 3 viruses, respectively. A minimum genetic distance of 4.7% demarcated the reference sub-groups, while the Ethiopian genogroup 3 viruses had a genetic distance of 4.9%. As a result, the Ethiopian genogroup 3 viruses deserve another new sub-group tentatively proposed as sub-genogroup 3d. The limitations in this sub-grouping were use of relatively short sequence for genetic distance analysis and use of sequences reported based on the nature of clustering on a phylogenetic tree proposed by Jackwood et al.18 together with the Ethiopian sequences. Further refining of the sub-genogrouping is needed as it is not reliable for sub-genogroup 3b viruses.
Typical virulence marker genes reported previously were observed in Ethiopian sequences. As the vaccine strain clustered with genogroup 1 viruses was linked with outbreak cases, any IBD vaccine used to control the disease needs to be checked for side effects, efficacy, and disease prevention and/or causation in vivo before being used for mass vaccination to control the disease.
Ethical Approval
Approval for this article was obtained from the College of Veterinary Medicine and Agriculture (Reference number VM/ERC/16/03/2020) and followed all ethical standards for research without direct contact with animal subjects.
Acknowledgments
I thank all researchers who uploaded the IBDV sequence data to the NCBI website for access to, use and comparison of their research.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Cosgrove AS. An apparently new disease of chickens: avian nephrosis. Avian Dis. 1962;6(3):385–389. doi:10.2307/1587909
2. Zeleke A, Yami M, Kebede F, et al. An emerging disease threat to poultry Debre Zeit. Ethio Vet J. 2002;6:1–7.
3. Delmas B, Attoui H, Ghosh S, et al. ICTV virus taxonomy profile: Birnaviridae. J Gen Virol. 2019;100(1):5–6. doi:10.1099/jgv.0.001185
4. Muller H, Scholtissek C, Becht H. The genome of infectious bursal disease virus consists of two segments of double-stranded RNA. J Virol. 1979;31(3):584–589. doi:10.1128/JVI.31.3.584-589.1979
5. Mardassi H, Khabouchi N, Ghram A, et al. A very virulent genotypes of infectious bursal disease virus predominantly associated with recurrent infectious bursal disease outbreaks in Tunisian vaccinated flocks. Avian Dis. 2004;48(4):829–840. doi:10.1637/7210-052004R
6. Zeleke A, Gelaye E, Sori T, et al. Investigation on infectious bursal disease outbreak in Debre Zeit. Int J Poult Sci. 2005;7:504–506.
7. Camilotti E, Moraes LB, Furian TQ, et al. Infectious bursal disease: pathogenicity and immunogenicity of vaccines. Braz J Poult Sci. 2016;18(2):303–308. doi:10.1590/1806-9061-2015-0148
8. Negash T, Gelaye E, Petersen H, et al. Molecular evidence of very virulent infectious bursal disease viruses in chickens in Ethiopia. Avian Dis. 2012;56(3):605–610. doi:10.1637/10086-022012-ResNote.1
9. Jenbere S, Lynch SE, Kebede F, et al. Genetic characterization of infectious bursal disease virus isolates in Ethiopia. Acta Trop. 2014;130:39–43. doi:10.1016/j.actatropica.2013.09.025
10. Shegu D, Sori T, Tesfaye A, et al. Sequence‑based comparison of field and vaccine strains of infectious bursal disease virus in Ethiopia reveals an amino acid mismatch in the immunodominant VP2 protein. Arch Virol. 2020;165(6):1367–1375. doi:10.1007/s00705-020-04622-6
11. Pikuła A, Lisowska A, Jasik A, et al. Identification and assessment of virulence of a natural reassortant of infectious bursal disease virus. Vet Res. 2017;48(1):49–89. doi:10.1186/s13567-017-0453-2
12. Jackwood DJ. Molecular epidemiologic evidence of homologous recombination in infectious bursal disease viruses. Avian Dis. 2012;56(3):574–577. doi:10.1637/10053-010912-ResNote.1
13. Wei Y, Li J, Zheng J, et al. Genetic reassortment of infectious bursal disease virus in nature. Bioch Bioph Res Commun. 2006;350(2):277–287. doi:10.1016/j.bbrc.2006.09.040
14. Wei Y, Yu X, Zheng J, et al. Reassortant infectious bursal disease virus isolated in China. Virus Res. 2008;131(2):279–282. doi:10.1016/j.virusres.2007.08.013
15. Coulibaly F, Chevalier C, Gutsche I, et al. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell. 2005;120(6):761–772. doi:10.1016/j.cell.2005.01.009
16. Schnitzler D, Bernstein F, Miiller H. The genetic basis for the antigenicity of the VP2 protein of the infectious bursal disease virus. J Gen Virol. 1993;74(8):1563–1571. doi:10.1099/0022-1317-74-8-1563
17. Letzel T, Coulibaly F, Rey FA. Molecular and structural bases for the antigenicity of VP2 of infectious bursal disease virus. J Virol. 2007;81(23):12827–12835. doi:10.1128/JVI.01501-07
18. Jackwood DJ, Schat KA, Michel LO. A proposed nomenclature for infectious bursal disease virus isolates. Avian Pathol. 2018;47(6):576–584. doi:10.1080/03079457.2018.1506092
19. Michael LO, Jackwood DJ. Classification of infectious bursal disease virus into genogroups. Arch Virol. 2017;162(12):3661–3670. doi:10.1007/s00705-017-3500-4
20. Lupini C, Giovanardi D, Pesente P. A molecular epidemiology study based on VP2 gene sequences reveals that a new genotype of infectious bursal disease virus is dominantly prevalent in Italy. Avian Pathol. 2016;45(4):458–464. doi:10.1080/03079457.2016.1165792
21. Kebede W, Bitew M, Dawo F Sequence analysis of infectious bursal disease virus vaccine strain adapted on Vero cell culture, Ethiopia; 2019. Available from: https://www.ncbi.nlm.nih.gov/nuccore/MK798159,MK798160,MK798161.
22. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi:10.1093/molbev/msw054
23. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98.
24. Hopps TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. PNAS. 1981;78(6):3824–3828. doi:10.1073/pnas.78.6.3824
25. Martin DP, Murrell B, Golden M, et al. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):vev003. doi:10.1093/ve/vev003
26. Dimitrove KM, Abolnik C, Afonso CL, et al. Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infect Gen Evol. 2019;74:103917. doi:10.1016/j.meegid.2019.103917
27. Saitou N, Nei M. The neighbour-joining method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406. doi:10.1093/oxfordjournals.molbev.a040454
28. Felsentein J. Confidence limits on phylogenetic: an approach using the bootstrap. Evolution. 1985;39(4):783–791. doi:10.1111/j.1558-5646.1985.tb00420.x
29. Kimura M. A simple method for estimating evolutionary rate of base substitution through nucleotides sequences. J Mol Evol. 1980;16(2):111–120. doi:10.1007/BF01731581
30. Brown MD, Green P, Skinner MA. VP2 sequences of recent European ‘very virulent’ isolates of infectious bursal disease virus are closely related to each other but are distinct from those of ‘ classical’ strains. J Gen Virol. 1994;75(3):675–680. doi:10.1099/0022-1317-75-3-675
31. Jackwood DJ, Sreedevi B, LeFever LJ, et al. Studies on naturally occurring infectious bursal disease viruses suggest that a single amino acid substitution at position 253 in VP2 increases pathogenicity. Virology. 2008;377:110–116. doi:10.1016/j.virol.2008.04.018
32. Jackwood DJ, Sommer-Wagner SE. Amino acids contributing to antigenic drift in the infectious bursal disease Birnavirus (IBDV). Virology. 2011;409(1):33–37. doi:10.1016/j.virol.2010.09.030
33. Brandt M, Yao K, Liu M, et al. Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. J Virol. 2001;75(24):11974–11982. doi:10.1128/JVI.75.24.11974-11982.2001
34. Qi X, Zhang L, Chen Y, et al. Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious bursal disease virus. PLoS One. 2013;8(7):e70982. doi:10.1371/journal.pone.0070982
35. Lim BL, Cao YC, Yu T, et al. Adaption of very virulent infectious bursal disease virus to chicken embryonic fibroblasts by site-directed mutagenesis of residues 279 and 284 of viral coat protein VP2. J Virol. 1999;73(4):2854–2862. doi:10.1128/JVI.73.4.2854-2862.1999
36. Mundt E. Tissue culture infectivity of different strains of infectious bursal disease virus is determined by distinct amino acids in VP2. J Gen Virol. 1999;80(8):2067–2076. doi:10.1099/0022-1317-80-8-2067
37. Adamu J, Owoade AA, Abdu PA, et al. Characterization of field and vaccine infectious bursal disease viruses from Nigeria revealing possible virulence and regional markers in the VP2 minor hydrophilic peaks. Avian Pathol. 2013;42(5):420–433. doi:10.1080/03079457.2013.822055
38. Coulibaly F, Chevalier C, Delmas B. Crystal structure of an Aquabirnavirus particle: insights into antigenic diversity and virulence determinism. J Virol. 2010;84(4):1792–1799. doi:10.1128/JVI.01536-09
39. Yamaguchi T, Ogawa M, Miyoshi M, et al. Sequence and phylogenetic analyses of highly virulent infectious bursal disease virus. Arch Virol. 1997;142(7):1441–1458. doi:10.1007/s007050050171
40. Xia RX, Wang HY, Huang GM, et al. Sequence and phylogenetic analysis of a Chinese very virulent infectious bursal disease virus. Arch Virol. 2008;153(9):1725–1729. doi:10.1007/s00705-008-0140-8
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.