Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Genetic Architecture of Comorbidity Between Chronic Obstructive Pulmonary Disease and Cardiovascular Diseases: Exploring Shared Mechanisms and Potential Therapeutic Targets
Received 7 October 2025
Accepted for publication 18 December 2025
Published 8 January 2026 Volume 2026:21 572586
DOI https://doi.org/10.2147/COPD.S572586
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
Shiyu Chen,1,* Xiaojian Li,1,* Rongfang Xie1,2
1Clinical Medical College,Jiangxi University of Chinese Medicine, Nanchang, 330004, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Affiliated Hospital of Jiangxi University of Chinese Medicine, Nanchang, 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Rongfang Xie, Clinical Medical College, Jiangxi University of Chinese Medicine, Nanchang, 330004, People’s Republic of China, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) and cardiovascular diseases (CVDs), including hypertension (HTN), coronary heart disease (CHD), and heart failure (HF), are major global health burdens. The shared genetic mechanisms underlying the high comorbidity between COPD and CVDs remain unclear.
Methods: We integrated large-scale GWAS summary statistics for COPD and three major CVDs (HTN, CHD, HF). Several analytic approaches were applied, including linkage disequilibrium score regression (LDSC), high-definition likelihood (HDL), multi-marker analysis of genomic annotation (MAGMA), pleiotropic analysis under composite null hypothesis (PLACO), and summary-data-based Mendelian randomization (SMR). These methods were used to evaluate genetic correlations, identify shared risk loci, and prioritize potential therapeutic targets.
Results: LDSC and HDL analyses revealed significant positive genetic correlations between COPD and the three CVDs (rg = 0.23– 0.38, P < 0.05). MAGMA enrichment analysis identified 277 unique pleiotropic genes enriched in pathways such as Notch signaling and nicotinic acetylcholine receptor signaling. Tissue-specific analyses indicated that shared genetic signals were enriched not only in the lung and heart but also in neuroendocrine-related tissues such as the cerebellum and pituitary, suggesting the involvement of a potential “lung–heart–brain” multi-organ axis. PLACO identified 94 pleiotropic SNPs, with consistent colocalization signals at 15q25.1 (CHRNA3/5, IREB2) and 4q22 (SOX7). SMR analysis further prioritized 626 candidate genes, including ZNF652, XRCC3, SLC22A5, and SOX7, which may serve as potential therapeutic targets.
Conclusion: This study provides genetic evidence for shared mechanisms linking COPD with HTN, CHD, and HF. It highlights the roles of neurotransmitter receptors, iron metabolism, vascular development, and energy metabolism in COPD–CVD comorbidity. These findings offer insights into precision prevention and therapeutic strategies targeting COPD–CVD comorbidity.
Keywords: chronic obstructive pulmonary disease, hypertension, coronary heart disease, heart failure, GWAS, pleiotropy, drug targets
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation and chronic airway inflammation, and has become a major global public health burden. According to projections by the World Health Organization (WHO), COPD is expected to rank as the third leading cause of death worldwide by 2030, following cardiovascular diseases (CVDs) and cancer.1
Emerging evidence suggests that COPD is not merely a pulmonary disorder, but rather a complex multisystem syndrome. Among its comorbidities, cardiovascular diseases—including hypertension (HTN), coronary heart disease (CHD), and heart failure (HF)—are among the most common.2,3 Epidemiological studies indicate that patients with COPD have a substantially elevated risk of cardiovascular events, and CVDs represent a leading cause of mortality in this population.4,5 The high frequency of comorbidity suggests that COPD and cardiovascular diseases may share underlying pathological mechanisms, such as oxidative stress and endothelial dysfunction, rather than being merely the result of an additive effect of risk factors like smoking, chronic inflammation, or lifestyle factors.6
With the advent of large-scale genome-wide association studies (GWAS), extensive genetic data have provided new opportunities to explore the shared genetic basis of complex diseases. Previous studies have identified multiple genetic loci—such as the 15q25 region associated with nicotine dependence/nicotinic receptor signaling and the 4q22 region encompassing the HHIP gene—that are jointly implicated in COPD and CVD risk.7,8 However, systematic genetic investigations into the mechanisms underlying COPD–CVD comorbidity remain limited.
This study integrates GWAS summary statistics and employs a range of genetic analysis methods, including linkage disequilibrium score regression (LDSC),9 high-resolution likelihood (HDL),10 generalized gene set analysis (MAGMA),11 and composite null hypothesis pleiotropy analysis (PLACO),12 to systematically evaluate the genetic correlation between COPD and cardiovascular diseases, identify shared risk loci, and elucidate their common mechanisms at the genetic, tissue, and pathway levels. By combining multiple genetic approaches, this study aims to provide genetic evidence for understanding the comorbid mechanisms of COPD and cardiovascular diseases and offer new perspectives for the development of precision prevention and treatment strategies (Figure 1).
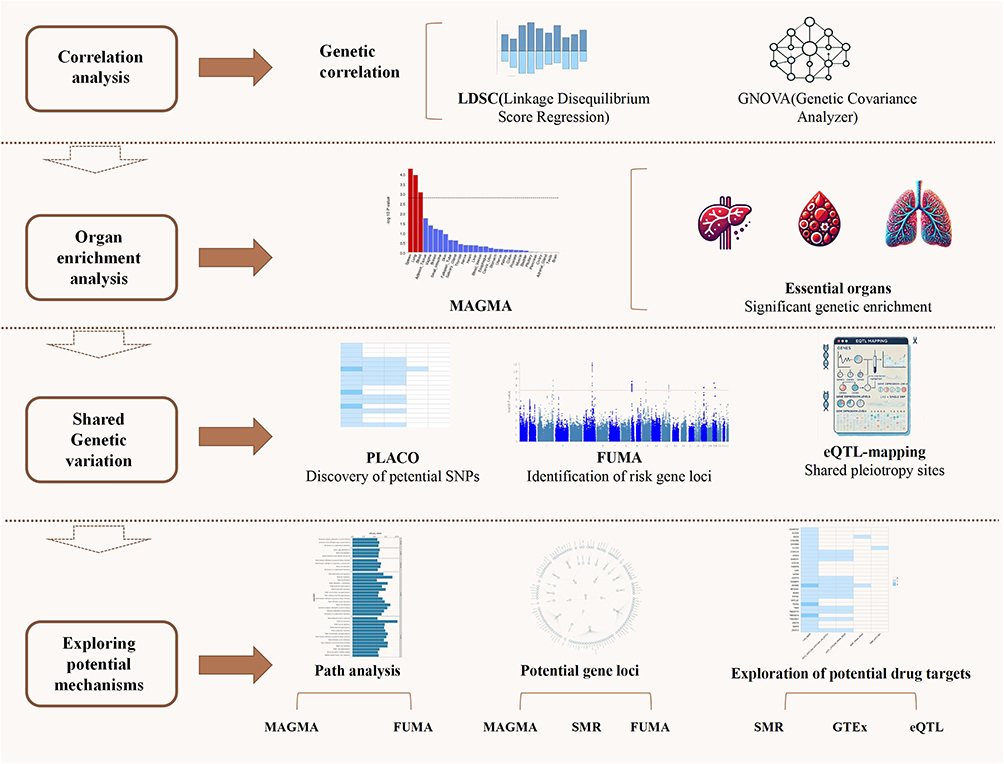 |
Figure 1 Flowchart. Abbreviations: LDSC, linkage disequilibrium score regression; HDL, high-definition likelihood; MAGMA, multi-marker analysis of genomic annotation; PLACO, pleiotropic analysis under composite null hypothesis; SMR, summary-data-based Mendelian randomization. |
Materials and Methods
GWAS Datasets
The GWAS summary statistics used in this study for COPD and major cardiovascular diseases were obtained from publicly available resources from large cohorts, including UK Biobank and FinnGen, with a primary focus on participants of European ancestry. The original GWAS studies have undergone standard quality control at the individual level, and this study is based on the publicly released summary statistics for subsequent analysis.For essential hypertension (HTN, GWAS ID: ukb-b-12493) in the UK Biobank, cases were defined using ICD-10 code I10 for hospital discharge and death registry records, including 54,358 cases and 408,652 controls. Controls were participants in the same cohort who did not have an ICD-10 I10 or related hypertension diagnosis during the follow-up period.13 Major coronary heart disease events (CHD, GWAS ID: ukb-d-I9_CHD) were defined by ICD-10 codes for myocardial infarction and other major coronary artery disease events, with 10,157 cases having corresponding records, and 351,037 controls without any CHD diagnosis.14 Heart failure (HF, GWAS ID: ukb-d-I50) was defined using ICD-10 code I50 as the main diagnosis in hospital records, with 1,088 cases and 360,106 controls who did not have ICD-10 I50 or related diagnoses.15 COPD data were obtained from FinnGen R11 (endpoint J10_COPD, GWAS ID: finngen_R11_J10_COPD), comprising 6,408 COPD cases defined by ICD-10 J44 and related codes and 397,564 controls without any COPD diagnosis. Each study followed the same quality control procedures, and logistic regression was used to assess SNP–phenotype associations in each study, with genetic principal components included as covariates in the association analysis.
Quality Control
In this study, stringent quality control measures were implemented to ensure the accuracy and reliability of the GWAS data. First, SNPs located within the major histocompatibility complex (MHC) region on chromosome 6 (25Mb–35Mb) were excluded to avoid false positives due to the complex gene structure and linkage disequilibrium in this region. Second, only common variants with a minor allele frequency (MAF) greater than 0.01 were retained to improve statistical power and reduce the likelihood of false positives. Additionally, we excluded all non-biallelic SNPs, ambiguous strand SNPs (A/T or C/G), duplicate SNPs, indels, poorly imputed variants (INFO < 0.8), and SNPs inconsistent with the 1000 Genomes Phase 3 reference. To minimize genotype missingness bias, SNPs with a call rate of less than 95% were also removed. Furthermore, SNPs in the control group that significantly deviated from Hardy-Weinberg equilibrium (HWE, P < 1×10−6) were excluded, as such deviations may indicate genotyping errors or potential population stratification.
Genome-Wide Genetic Correlation Analysis
To assess the shared genetic architecture between COPD and cardiovascular traits, we employed the linkage disequilibrium score regression (LDSC) method.9 The LD scores used in the LDSC analysis were calculated based on common SNP genotypes from European-ancestry samples in the 1000 Genomes Project.16 Standard errors (SE) were estimated using the leave-one-out method in LDSC and were further used to correct for attenuation bias. Additionally, the LDSC intercept was used to assess potential population overlap between the different studies. Importantly, it should be emphasized that no significant population overlap was observed between the trait combinations analyzed, which enhances the reliability of our findings. To further validate the results from the LDSC analysis, we introduced the High-definition likelihood (HDL) method, a recent methodological extension of LDSC, designed to estimate genetic correlations after stratifying variants by minor allele frequency (MAF).17 Compared to LDSC, HDL has a stronger capability for multidimensional data processing of phenotypes, enabling the integration of various data sources such as genomic, transcriptomic, and epigenomic data, thereby providing more accurate genetic association analysis results. The validation using HDL ensures the robustness of the genome-wide genetic overlap analysis.
Gene-Level Analysis
To explore the shared mechanisms of the identified loci, we mapped the nearby genes based on the lead SNP for each locus. We performed multi-marker effect analysis on the GWAS data using the Generalized Gene Set Analysis (MAGMA) method to clarify the biological functions of these pleiotropic loci. Specifically, we conducted MAGMA gene analysis, incorporating linkage disequilibrium (LD) between markers to identify pleiotropic genes and detect multi-marker effects (Pfdr < 0.05).11 We ultimately tested 10,678 gene sets from the Molecular Signatures Database (MSigDB), including curated gene sets (c2.all) and Gene Ontology (GO) terms (c5.bp, c5.cc, c5.mf)18 to analyze the biological functions of the lead SNPs. To avoid false positives, all tested gene sets underwent Bonferroni correction (P < 0.05/10,678 = 4.68 × 10−6). Pathway enrichment analysis was conducted using the Metascape web tool (metascape.org), based on MSigDB to determine the functions of mapped genes.19 Additionally, using 54 GTEx tissues, we performed tissue-specific enrichment analysis for the genome-wide pleiotropy results generated by PLACO. We also calculated the average expression levels (log2-transformed) of all identified pleiotropic genes across all 54 GTEx tissues and conducted tissue-specific testing using differentially expressed genes (DEGs) for each tissue (upregulated and downregulated DEGs were pre-calculated based on the sign of the t-statistics).
SNP-Level Pleiotropy and Locus Identification
In this study, we used GWAS summary data and performed pleiotropic analysis using PLACO under the composite null hypothesis to identify genetic shared associations between COPD and multiple cardiovascular diseases at the SNP level. PLACO is a statistical method specifically designed for detecting gene pleiotropy. Variants were scored based on the squared Z-value, and those with Z2 > 80 were excluded. To explore trait correlations, we calculated and combined the Z-value correlation matrix. The intersection-union test (IUT) framework was then applied to test the null hypothesis of no pleiotropy, yielding the final pleiotropic P-value. Significant pleiotropic variants were defined as SNPs with P < 5 × 10−8. To avoid redundancy within the same LD block, we performed LD pruning using the European reference panel from the 1000 Genomes Project, defining independent variants with r2 < 0.1.20
To assess the biological significance of pleiotropic SNPs, we used the FUMA tool for functional mapping and annotation. SNPs with strong LD (r2 ≥ 0.6) within a 250 kb window were clustered to define genomic risk loci, while independent lead SNPs were defined with r2 < 0.1. Functional annotations of independent significant SNPs and their LD proxies were performed using ANNOVAR, CADD, RegulomeDB, and ChromHMM.21 Finally, we conducted Bayesian colocalization analysis to identify shared risk loci associated with both COPD and cardiovascular traits.22
Prioritization of Druggable Targets
To explore potential therapeutic implications, we integrated GWAS summary statistics with expression quantitative trait loci (eQTL) data using summary-data-based Mendelian randomization (SMR).23 eQTLs are genetic variants associated with gene expression levels, helping to explain individual differences in gene expression. By analyzing the associations between specific SNPs and gene expression, eQTL studies identify genetic variants that may influence gene expression. In the SMR method, GWAS and eQTL data are combined to investigate how SNPs affect complex traits, including disease susceptibility.24 The SMR and HEIDI methods were used to assess pleiotropic associations between gene expression and complex traits. SMR evaluates whether the effect of a SNP on a phenotype is mediated through gene expression. If a SNP is associated with both gene expression and the trait, it suggests pleiotropy. The HEIDI test checks whether this association is due to colocalization, meaning the SNP affects both gene expression and the trait via the same causal variant. If colocalization is confirmed, it provides a clearer understanding of the genetic mechanisms behind the complex trait.25 Genes showing significant results in SMR (P < 0.01) and no heterogeneity in HEIDI (P > 0.05) were considered likely causal and potential targets for COPD–CVD comorbidity.
Statistical Power and Multiple-Testing Correction
We evaluated the statistical power of our genome-wide genetic correlation analyses using established LDSC/HDL-based power approximations. Given the effective sample sizes from the COPD and cardiovascular GWAS and the density of common SNPs after quality control (HapMap3 variants), our study was sufficiently powered to detect modest genetic correlations; under these conditions, the design is expected to achieve an estimated power of approximately 0.80 for detecting genetic correlations as small as |rg| ≈ 0.10–0.15 at a two-sided α level of 0.05. The large sample sizes and broad SNP coverage also ensured adequate power to identify gene- and pathway-level associations of small-to-moderate effect sizes after multiple-testing correction.
Unless otherwise stated, multiple-testing correction was performed within each analysis type rather than across all tests globally. Bonferroni thresholds for MAGMA gene-level and gene-set enrichment analyses were therefore calculated separately based on the number of genes and gene sets tested, respectively. PLACO results were evaluated at the conventional genome-wide significance level (P < 5 × 10−8), and SMR results were filtered using the predefined combination of SMR (P < 0.01) and HEIDI (P > 0.05) criteria. This strategy balances control of type I error with adequate power to detect true pleiotropic and putatively causal signals.
Results
Shared Genetic Architecture Between COPD and CVDs
We first examined the genome-wide genetic correlations between COPD and cardiovascular traits. Results from LDSC and high-definition likelihood HDL were highly concordant (Table 1). Both methods consistently demonstrated significant positive genetic correlations between COPD and HTN, CHD, HF (P < 0.05).
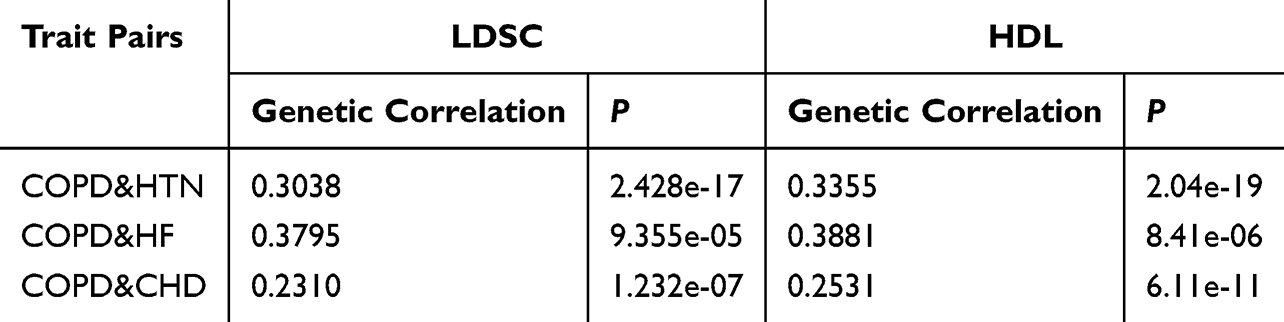 |
Table 1 Genetic Correlation Results for Trait Pairs Based on LDSC and HDL |
Gene-Level Enrichment Analysis
Using the MAGMA framework implemented via the FUMA platform, we identified 4,729 genes significantly enriched in COPD–CVD associations (P < 0.05), of which 277 remained significant after FDR correction (Figure 2, Tables S1 and S2 in Supplementary Material). Pathway analysis revealed that these genes were involved in several key biological processes, including calcium-permeable nicotinic acetylcholine receptor signaling, RUNX3-regulated Notch signaling, and acetylcholine binding and downstream events (Figure 3 and Table S3 in Supplementary Material). Moreover, tissue-specific enrichment analyses demonstrated significant aggregation of these genes in the cerebellum, pituitary, uterus, and lung, suggesting multi-organ involvement in COPD–CVD comorbidity.
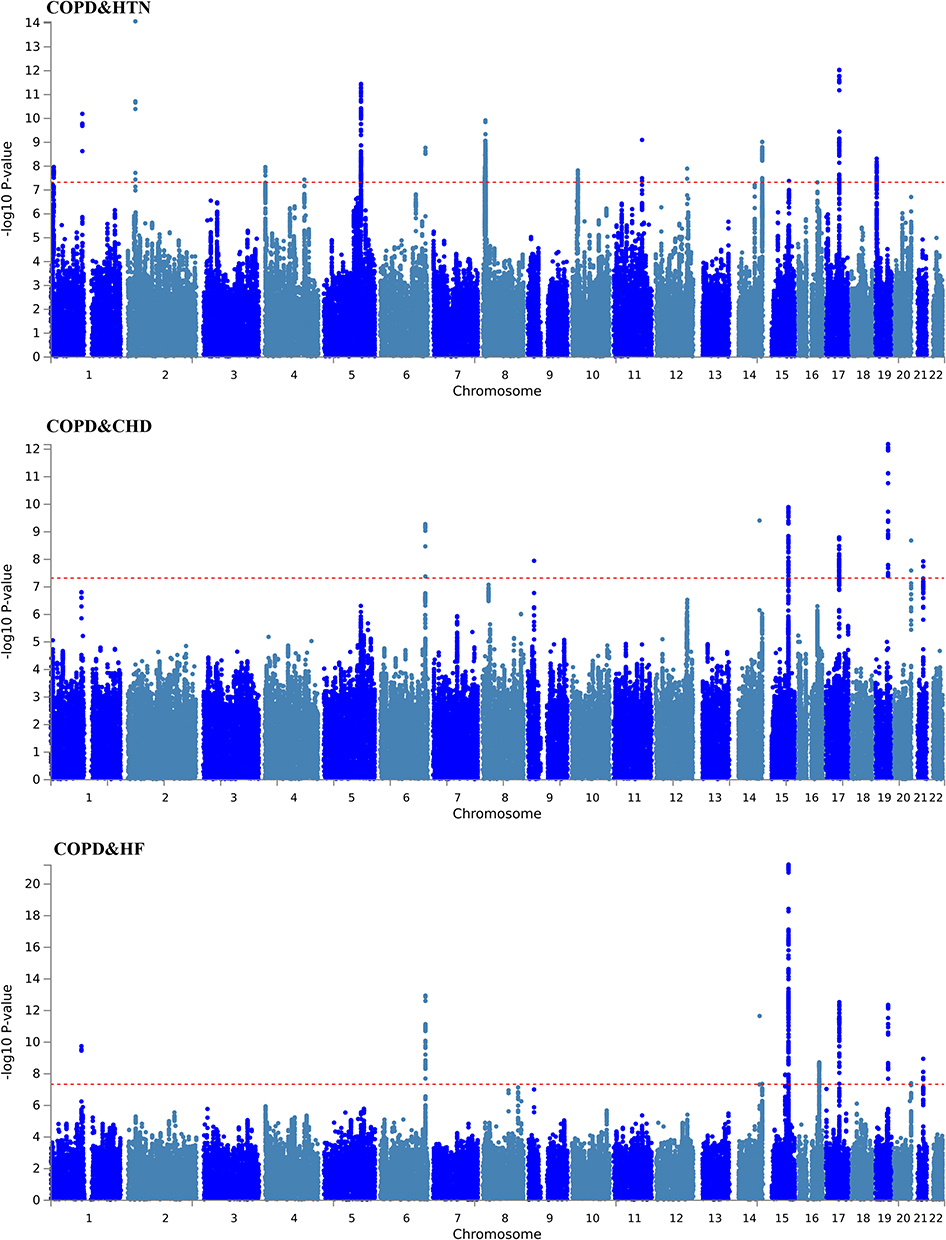 |
Figure 2 Results of Gene Enrichment. Abbreviations: COPD, Chronic obstructive pulmonary disease; HTN, including hypertension; CHD, coronary heart disease; HF, heart failure. |
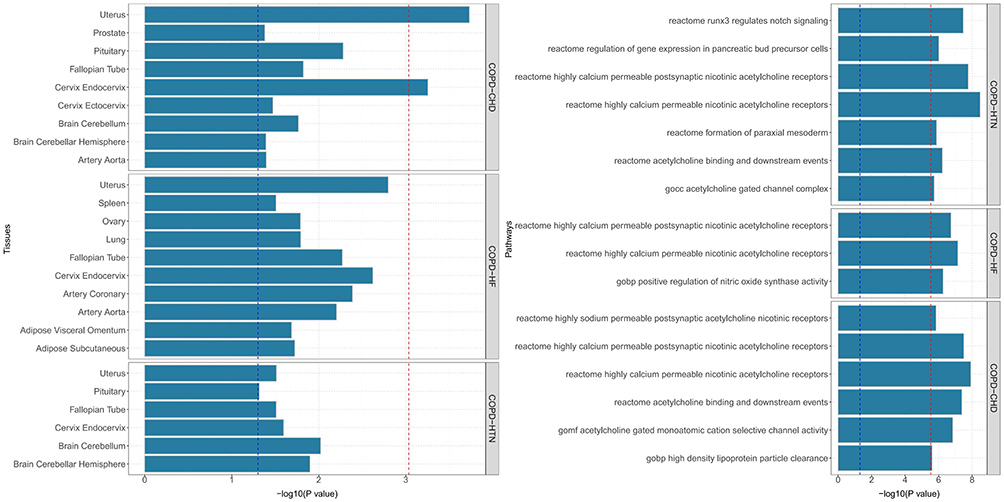 |
Figure 3 MAGMA gene set and tissue specificity of genome-wide pleiotropy results. Abbreviations: COPD, Chronic obstructive pulmonary disease; HTN, including hypertension; CHD, coronary heart disease; HF, heart failure. |
SNP-Level Pleiotropy and Risk Loci
To further pinpoint pleiotropic variants underlying the observed genetic correlations, we applied the PLACO method. A total of 94 genome-wide significant pleiotropic SNPs (P < 5×10−8) were identified across COPD and cardiovascular traits (Table S4 in Supplementary Material). Functional mapping with FUMA revealed 40 pleiotropic genomic risk loci (Table S5 in Supplementary Material). Colocalization analysis confirmed six loci with strong evidence of shared association (PP.H4 > 0.7), including rs16948048, rs2671654, rs12601665, and rs7225606 (Table S6 in Supplementary Material). Notably, the 15q25.1 region was repeatedly observed across multiple trait pairs (COPD–HF, COPD–HTN, COPD–CHD), underscoring its pivotal role in COPD–CVD comorbidity (Table S7 in Supplementary Material) (Figure 4).
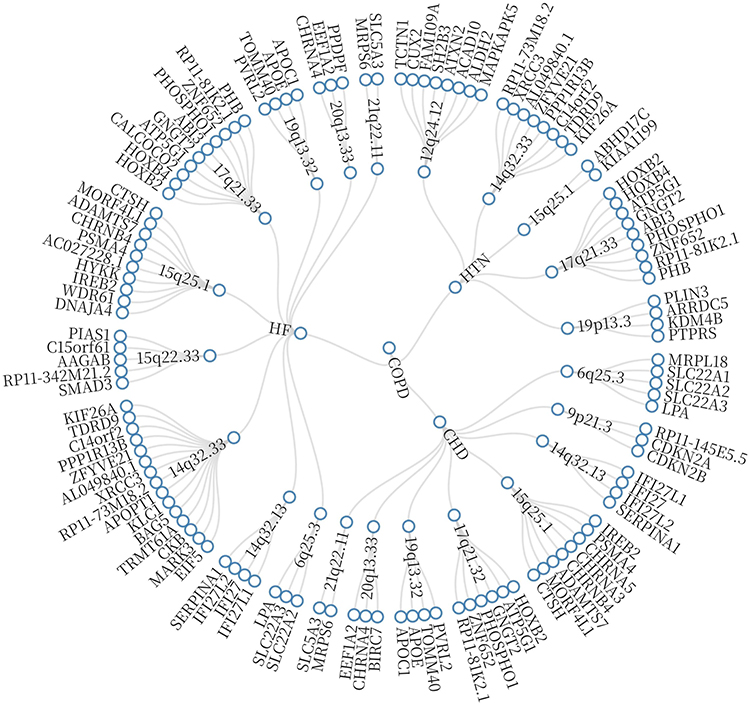 |
Figure 4 Shared pleiotropic regions across COPD and cardiovascular disease combinations. Abbreviations: COPD, Chronic obstructive pulmonary disease; HTN, including hypertension; CHD, coronary heart disease; HF, heart failure. |
Prioritization of Therapeutic Targets in European Populations
Using the SMR approach, we first identified 626 genes as potential intervention targets within complex trait signals (P_SMR < 0.01, P_HEIDI > 0.05) (Table S8 in Supplementary Material). Combining PLACO, MAGMA, and FUMA results further prioritized a panel of pleiotropic genes with strong cross-tissue signals, including ZNF652, XRCC3, XKR6, SOX7, SLC22A5, RP1L1, and C5orf56 (Table S9 in Supplementary Material). These loci represent potential molecular entry points for therapeutic interventions (Figure 5).
 |
Figure 5 Prioritized candidate genes with pleiotropic effects across COPD and cardiovascular diseases. |
Discussion
In this study, we systematically applied multiple genetic approaches to comprehensively examine the shared genetic basis between COPD and three major cardiovascular diseases (hypertension, coronary heart disease, and heart failure). Our results demonstrated robust positive genetic correlations and convergent pleiotropic signals across gene-, pathway-, and tissue-level analyses.
Traditionally, COPD has been considered a respiratory disorder, whereas CVDs are primarily attributed to metabolic abnormalities and vascular dysfunction. However, both LDSC and HDL analyses confirmed significant genetic correlations between COPD and HTN, CHD, and HF (P < 0.05), suggesting that their coexistence is not merely phenotypic coincidence but reflects an intrinsic genetic overlap. This observation is consistent with epidemiological data showing markedly elevated cardiovascular risk among COPD patients,26 thereby reinforcing the molecular rationale for comorbidity.
At the genome-wide level, multiple shared susceptibility regions were identified. Notably, the 15q25.1 locus, repeatedly highlighted across trait combinations, harbors CHRNA3/5 and IREB2, both of which have been consistently implicated in smoking behavior, reduced lung function, and increased CVD risk.27,28 Variants in CHRNA3/5, encoding nicotinic acetylcholine receptor subunits, not only predispose individuals to nicotine dependence and lung cancer but also influence autonomic regulation of vascular tone and inflammatory responses. Functional studies indicate that CHRNA3/5 variants enhance airway inflammation and alveolar damage, while in vascular tissues they regulate calcium influx and nitric oxide (NO) synthesis, thereby modulating atherosclerosis development.7,29–31 Collectively, CHRNA3/5 may contribute to COPD–CVD comorbidity through a “smoking susceptibility–airway inflammation–vascular dysfunction” pathway.
IREB2, a key regulator of cellular iron homeostasis, further underscores the oxidative stress dimension of comorbidity. Functional impairment of this gene disrupts iron balance, leading to excessive reactive oxygen species (ROS) production and downstream tissue injury. In pulmonary tissues, aberrant iron accumulation exacerbates alveolar damage and accelerates lung function decline, while in cardiovascular systems it promotes cardiomyocyte apoptosis, mitochondrial dysfunction, and atherosclerotic lesion progression.32,33 Thus, IREB2 emerges as a pivotal mediator linking iron metabolism dysregulation to COPD–CVD pathology.
Gene-set enrichment further highlighted RUNX3-regulated Notch signaling and nicotinic acetylcholine receptor pathways as common molecular networks. Notch signaling plays an essential role in both immune regulation and vascular homeostasis. Abnormal activation of Notch promotes Th1/Th17 differentiation, airway epithelial injury, and mucus hypersecretion in COPD.34,35 In parallel, Notch signaling drives endothelial proliferation, macrophage polarization, and plaque formation in atherosclerosis,36 supporting its role as a molecular hub connecting pulmonary and vascular inflammation. Similarly, the nicotinic acetylcholine receptor pathway links COPD and CVD via the shared exposure of smoking: nicotine binding to CHRNA3/5 not only perpetuates nicotine dependence and airway inflammation but also perturbs calcium signaling and endothelial function, contributing to vascular remodeling and coronary pathology.37,38
Tissue-specific analyses revealed enrichment not only in lung and heart, but also in whole blood, cerebellum, and pituitary, pointing toward a potential “lung–heart–brain axis”. This multi-organ axis has important implications: cerebellar signals may indicate dysregulation of autonomic nervous control, which in turn influences bronchial reactivity, heart rate, and vascular tone;39 pituitary enrichment implicates the hypothalamic–pituitary–adrenal (HPA) axis, a key regulator of systemic inflammation and stress response. Dysregulation of the HPA axis, including glucocorticoid resistance observed in COPD40 and altered cortisol rhythms in chronic inflammation,41,42 could increase cardiovascular vulnerability. Moreover, immune–neuroendocrine crosstalk may propagate systemic inflammation into the CNS, exacerbating neuroendocrine stress and mood disturbances, which further impair cardiovascular outcomes.43
Finally, integration of PLACO and SMR analyses prioritized several pleiotropic genes and druggable targets, including ZNF652, XRCC3, SLC22A5, and SOX7. ZNF652, a zinc-finger transcription factor, has been implicated in blood pressure regulation and vascular smooth muscle function,44 suggesting cross-organ transcriptional regulation in comorbidity. XRCC3, a DNA repair gene, carries variants such as Thr241Met that increase susceptibility to both coronary heart disease45 and lung cancer,46 highlighting impaired DNA repair as a shared mechanism. SLC22A5, encoding the carnitine transporter OCTN2, is crucial for fatty acid oxidation; its dysfunction disrupts myocardial energetics and has been linked to both Crohn’s disease47 and heart failure.48 SOX7, a member of the SOX transcription factor family, cooperates with SOX17 and SOX18 to regulate angiogenesis and endothelial specification,49 while its deficiency leads to vascular malformations and dysfunction.50 Together, these pleiotropic genes represent promising entry points for therapeutic intervention in COPD–CVD comorbidity.
Strengths and Limitations
This study has two major strengths. First, by integrating large-scale GWAS summary statistics of COPD and major cardiovascular diseases, we achieved sufficient statistical power to detect shared genetic signals. Second, through complementary analytic methods, including LDSC, HDL, MAGMA, PLACO, and SMR, we systematically assessed genetic correlation, pleiotropy, and potential druggable targets, which enhanced the robustness of our findings.
Despite these strengths, several limitations warrant consideration. First, the study was based primarily on summary-level GWAS data from European populations; therefore, the generalizability of our findings to other ethnic groups remains uncertain. Future studies involving multi-ethnic cohorts are needed to validate and extend these results. Second, although genetic colocalization and enrichment analyses suggest potential shared mechanisms, these approaches cannot establish causality. To further validate our findings, we plan in future studies to replicate these key signals in independent cohorts and to functionally characterize the most promising candidate genes using experimental models: at the cellular level, gene knockout or overexpression combined with transcriptomic and proteomic profiling will help elucidate downstream regulatory networks; at the organismal level, gene-edited animal models will provide direct evidence for the roles of these genes in disease progression. Third, possible sample overlap between GWAS datasets cannot be entirely ruled out, which may bias estimates of genetic correlation. Finally, this study did not consider environmental exposures, lifestyle factors, or epigenetic regulation, all of which likely contribute to the complex interplay between COPD and cardiovascular diseases and should be integrated into future research.
Conclusion
This study demonstrates significant genetic overlap between COPD and major CVDs, including hypertension, coronary heart disease, and heart failure. By integrating large-scale GWAS data with LDSC, HDL, MAGMA, PLACO, and SMR, we identified shared risk regions such as 15q25.1 (CHRNA3/5, IREB2) and 4q22 (SOX7), along with pleiotropic genes like ZNF652, XRCC3, and SLC22A5. Tissue enrichment analyses suggested potential common mechanisms underlying comorbidity.
These findings highlight shared pathways involving neurotransmitter signaling, vascular development, and energy metabolism, offering new insights into the genetic mechanisms linking COPD and CVDs. Our results emphasize opportunities for precision prevention and therapeutic intervention, while future studies in diverse populations and functional validation are needed to confirm these findings for clinical application.
Data Sharing Statement
The dataset generated during and analyzed during the current study are available from the corresponding author on reasonable request.
Ethical Approval
In accordance with Article 32 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Beings issued by the National Science and Technology Ethics Committee of the People’s Republic of China, this study was exempted from ethical review because the data analyzed pose no harm to human participants, do not involve sensitive personal information or commercial interests, and were obtained from open and legally accessible databases.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to beaccountable for all aspects of the work.
Funding
This research was supported by Jiangxi Provincial Natural Science Foundation Youth Project (20252BAC200561) and Science and Technology Research Project of Jiangxi Provincial Department of Education (GJJ2400826).
Disclosure
The authors affirm that they have no commercial or financial affiliations that could be perceived as potential conflicts of interest. This includes any relationships or financial interests that could influence or bias the study’s results and conclusions.
The authors declare no conflicts of interest.
References
1. Raherison C, Girodet PO. Epidemiology of COPD. Eur Respir Rev. 2009;18(114):213–13. doi:10.1183/09059180.00003609
2. Müllerova H, Agusti A, Erqou S, Mapel DW. Cardiovascular comorbidity in COPD: systematic literature review. Chest. 2013;144(4):1163–1178. doi:10.1378/chest.12-2847
3. Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Respir Med. 2015;3(8):631–639. doi:10.1016/S2213-2600(15)00241-6
4. Sin DD, Anthonisen NR, Soriano JB, Agusti AG. Mortality in COPD: role of comorbidities. Eur Respir J. 2006;28(6):1245–1257. doi:10.1183/09031936.00133805
5. Feary JR, Rodrigues LC, Smith CJ, Hubbard RB, Gibson JE. Prevalence of major comorbidities in subjects with COPD and incidence of myocardial infarction and stroke: a comprehensive analysis using data from primary care. Thorax. 2010;65(11):956–962. doi:10.1136/thx.2009.128082
6. Maclay JD, MacNee W. Cardiovascular disease in COPD: mechanisms. Chest. 2013;143(3):798–807. doi:10.1378/chest.12-0938
7. Hobbs BD, de Jong K, Lamontagne M, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017;49(3):426–432. doi:10.1038/ng.3752
8. Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–1130. doi:10.1038/ng.3396
9. Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. doi:10.1038/ng.3406
10. Ning Z, Pawitan Y, Shen X. High-definition likelihood inference of genetic correlations across human complex traits. Nat Genet. 2020;52(8):859–864. doi:10.1038/s41588-020-0653-y
11. de Leeuw CA, Mooij JM, Christiaan A, et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Computational Biology. 2015;11(4):e1004219. doi:10.1371/journal.pcbi.1004219
12. Lu H, Qiao J, Shao Z, Wang T, Huang S, Zeng P. A comprehensive gene-centric pleiotropic association analysis for 14 psychiatric disorders with GWAS summary statistics. BMC Med. 2021;19(1):314. doi:10.1186/s12916-021-02186-z
13. van Oort S, Beulens JWJ, van Ballegooijen AJ, Grobbee DE, Larsson SC. Association of Cardiovascular Risk Factors and Lifestyle Behaviors With Hypertension: a Mendelian Randomization Study. Hypertension. 2020;76(6):1971–1979. doi:10.1161/HYPERTENSIONAHA.120.15761
14. He D, Lu H, Ou X, et al. Exposure to major coronary heart disease events reduces lung cancer risk: a mendelian randomization study based on a European population. BMC Cancer. 2025;25(1):152. doi:10.1186/s12885-025-13485-6
15. Zhang P, Xin Y, Yuan H, Liu Z. Identification of the crucial roles of BAXhigh NK cells in human derived mesenchymal stem cell therapy for chronic heart failure patients. Pathol Res Pract. 2025;269:155924. doi:10.1016/j.prp.2025.155924
16. Cui G, Li S, Ye H, et al. Gut microbiome and frailty: insight from genetic correlation and mendelian randomization. Gut Microbes. 2023;15(2):2282795. doi:10.1080/19490976.2023.2282795
17. Hart T, Chandrashekhar M, Aregger M, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163(6):1515–1526. doi:10.1016/j.cell.2015.11.015
18. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
19. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
20. Hu YQ, Jin XJ, Lei SF, et al. Inflammatory bowel disease and osteoporosis: common genetic effects, pleiotropy, and causality. Hum Immunol. 2024;85(5):110856. doi:10.1016/j.humimm.2024.110856
21. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi:10.1038/s41467-017-01261-5
22. Lin J, Zhou J, Xu Y. Potential drug targets for multiple sclerosis identified through Mendelian randomization analysis. Brain. 2023;146(8):3364–3372. doi:10.1093/brain/awad070
23. Wu J, Mei Y, Li X, et al. PRCP is a promising drug target for intracranial aneurysm rupture supported via multi-omics analysis. Stroke Vasc Neurol. 2024;10(2):1.
24. Sun X, Chen B, Qi Y, et al. Multi-omics Mendelian randomization integrating GWAS, eQTL and pQTL data revealed GSTM4 as a potential drug target for migraine. J Headache Pain. 2024;25(1):117. doi:10.1186/s10194-024-01828-w
25. Dong XX, Chen DL, Chen HM, et al. DNA methylation biomarkers and myopia: a multi-omics study integrating GWAS, mQTL and eQTL data. Clin Clin Epigenet. 2024;16(1):157. doi:10.1186/s13148-024-01772-1
26. Sá-Sousa A, Rodrigues C, Jácome C, et al. Cardiovascular Risk in Patients with Chronic Obstructive Pulmonary Disease: a Systematic Review. J Clin Med. 2024;13(17):5173. doi:10.3390/jcm13175173
27. Zinellu A, Zinellu E, Pau MC, et al. A systematic review and meta-analysis of homocysteine concentrations in chronic obstructive pulmonary disease. Clin Exp Med. 2023;23(3):751–758. doi:10.1007/s10238-022-00833-0
28. Lee YJ, Choi S, Kwon SY, et al. A Genome-Wide Association Study in Early COPD: identification of One Major Susceptibility Loci. Int J Chron Obstruct Pulmon Dis. 2020;15:2967–2975. doi:10.2147/COPD.S269263
29. Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–642. doi:10.1038/nature06846
30. Cooke JP, Ghebremariam YT. Endothelial nicotinic acetylcholine receptors and angiogenesis. Trends Cardiovasc Med. 2008;18(7):247–253. doi:10.1016/j.tcm.2008.11.007
31. Barrie ES, Hartmann K, Lee SH, et al. The CHRNA5/CHRNA3/CHRNB4 Nicotinic Receptor Regulome: genomic Architecture, Regulatory Variants, and Clinical Associations. Hum Mutat. 2017;38(1):112–119. doi:10.1002/humu.23135
32. DeMeo DL, Mariani T, Bhattacharya S, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85(4):493–502. doi:10.1016/j.ajhg.2009.09.004
33. Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823(9):1468–1483. doi:10.1016/j.bbamcr.2012.05.010
34. Guo R, Han D, Song X, et al. Context-dependent regulation of Notch signaling in glial development and tumorigenesis. Sci Adv. 2023;9(45):eadi2167. doi:10.1126/sciadv.adi2167
35. Tsokos GC. Notch notches lupus. Kidney Int. 2020;97(2):251–253. doi:10.1016/j.kint.2019.10.018
36. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28(1):219–242. doi:10.1183/09031936.06.00053805
37. Cooke JP. Angiogenesis and the role of the endothelial nicotinic acetylcholine receptor. Life Sci. 2007;80(24–25):2347–2351. doi:10.1016/j.lfs.2007.01.061
38. Hardin M, Zielinski J, Wan ES, et al. CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am J Respir Cell Mol Biol. 2012;47(2):203–208. doi:10.1165/rcmb.2012-0011OC
39. van Reedt Dortland AK, Vreeburg SA, Giltay EJ, et al. The impact of stress systems and lifestyle on dyslipidemia and obesity in anxiety and depression. Psychoneuroendocrinology. 2013;38(2):209–218. doi:10.1016/j.psyneuen.2012.05.017
40. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645. doi:10.1016/j.jaci.2012.12.1564
41. Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267(9):1244–1252. doi:10.1001/jama.1992.03480090092034
42. Vgontzas AN, Chrousos GP. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: multiple interactions and disturbances in sleep disorders. Endocrinol Metab Clin North Am. 2002;31(1):15–36. doi:10.1016/s0889-8529(01)00005-6
43. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi:10.1038/nrn2297
44. International Consortium for Blood Pressure Genome-Wide Association Studies, Ehret GB, Munroe PB, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–109. doi:10.1038/nature10405.
45. Zhu ML, Zhao XY, Guo YL, et al. Association of DNA repair gene XRCC3 Thr241Met polymorphism with risk of coronary artery disease: a meta-analysis. Mol Biol Rep. 2012;39(2):1903–1910. doi:10.1007/s11033-011-0916-4
46. Zienolddiny S, Campa D, Lind H, et al. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis. 2006;27(3):560–567. doi:10.1093/carcin/bgi236
47. Peltekova VD, Wintle RF, Rubin LA, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36(5):471–475. doi:10.1038/ng1339
48. Grube M, Meyer Zu Schwabedissen HE, Präger D, et al. Uptake of cardiovascular drugs into cardiomyocytes by organic cation transporter novel type 2 (OCTN2). Circulation. 2006;113(9):1114–1122. doi:10.1161/CIRCULATIONAHA.105.580688
49. Zhou Y, Williams J, Smallwood PM, Nathans J. Sox7, Sox17, and Sox18 cooperatively regulate vascular development in mice. PLoS One. 2015;10(12):e0143650. doi:10.1371/journal.pone.0143650
50. Costa G, Mazan A, Gandillet A, et al. SOX7 regulates arterial specification in hemogenic endothelium. Blood. 2012;119(22):5461–5472. doi:10.1182/blood-2011-10-387928
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Efficacy, Safety, Tolerability, and Real-World Data of Patiromer for the Treatment of Hyperkalemia
Colbert G, Sannapaneni S, Lerma EV
Drug, Healthcare and Patient Safety 2022, 14:87-96
Published Date: 14 July 2022
Baseline Characteristics, Risk Factors and Etiology of Heart Failure Among Patients Hospitalized at a Teaching Hospital in Somalia: Cross-Sectional Study
Farah Yusuf Mohamud M, Jeele MOO, Cetinkaya O, Goitom Sereke S, Bongomin F, AM Ahmed M
Research Reports in Clinical Cardiology 2022, 13:63-71
Published Date: 13 September 2022
Optimal Management of Heart Failure and Chronic Obstructive Pulmonary Disease: Clinical Challenges
Cuthbert JJ, Pellicori P, Clark AL
International Journal of General Medicine 2022, 15:7961-7975
Published Date: 25 October 2022
Development and Validation of a Coronary Heart Disease Risk Prediction Model in Snorers with Hypertension: A Retrospective Observed Study
Wang M, Wang M, Zhu Q, Yao X, Heizhati M, Cai X, Ma Y, Wang R, Hong J, Yao L, Sun L, Yue N, Ren Y, Li N
Risk Management and Healthcare Policy 2022, 15:1999-2009
Published Date: 28 October 2022
Efficacy and Safety of QiShen YiQi Dripping Pills in the Treatment of Coronary Heart Disease Complicating Chronic Heart Failure (Syndrome of Qi Deficiency with Blood Stasis): Study Protocol for a Randomized, Placebo-Controlled, Double-Blind and Multi-Centre Phase II Clinical Trial
He X, Jiang Y, Li S, Liu D, Li Z, Han X, Zhang X, Dong X, Liu H, Huang J, Wang X, Long W, Ni S, Yang Z, Ye T
International Journal of General Medicine 2023, 16:6177-6188
Published Date: 28 December 2023