")
Back to Journals » Cancer Management and Research » Volume 11
Genetic And Epigenetic Regulation Of E-Cadherin Signaling In Human Hepatocellular Carcinoma
Authors Fan X, Jin S , Li Y, Khadaroo PA, Dai Y, He L, Zhou D, Lin H
Received 1 August 2019
Accepted for publication 27 September 2019
Published 16 October 2019 Volume 2019:11 Pages 8947—8963
DOI https://doi.org/10.2147/CMAR.S225606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Xiaoxiao Fan,1,* Shengxi Jin,1,2,* Yirun Li,1 Parikshit Asutosh Khadaroo,2,3 Yili Dai,1,2 Lifeng He,1 Daizhan Zhou,1 Hui Lin1
1Department of General Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 3School of Public Health and Preventive Medicine, Monash University, Melbourne, VIC, Australia
*These authors contributed equally to this work
Correspondence: Hui Lin; Daizhan Zhou
Department of General Surgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, 3 East Qingchun Road, Hangzhou 310016, People’s Republic of China
Tel +86-13738055489; +86-21-61569704
Fax +86-571-86044817; +86-21-61569704
Email [email protected]; [email protected]
Abstract: E-cadherin is well known as a growth and invasion suppressor and belongs to the large cadherin family. Loss of E-cadherin is widely known as the hallmark of epithelial-to-mesenchymal transition (EMT) with the involvement of transcription factors such as Snail, Slug, Twist and Zeb1/2. Tumor cells undergoing EMT could migrate to distant sites and become metastases. Recently, numerous studies have revealed how the expression of E-cadherin is regulated by different kinds of genetic and epigenetic alteration, which are implicated in several crucial transcription factors and pathways. E-cadherin signaling plays an important role in hepatocellular carcinoma (HCC) initiation and progression considering the highly mutated frequency of CTNNB1 (27%). Combining the data from The Cancer Genome Atlas (TCGA) database and previous studies, we have summarized the roles of gene mutations, chromosome instability, DNA methylation, histone modifications and non-coding RNA in E-cadherin in HCC. In this review, we discuss the current understanding of the relationship between these modifications and HCC. Perspectives on E-cadherin-related research in HCC are provided.
Keywords: E-cadherin, HCC, genetic alterations, epigenetic alterations
Introduction
Liver cancer is highly prevalent in Asia, and especially in China. It is estimated to be the sixth most commonly diagnosed form of cancer and the fourth cause of cancer-related death in 2018.1 Hepatocellular carcinoma (HCC) is a typical liver cancer, accounting for approximately 70–85% of all cases.2 Recent studies have revealed that HCC development is the result of an accumulation of genetic and epigenetic alterations, which are implicated in several crucial pathways and processes, such as the cadherin pathway.
There are at least nine subfamilies of cadherin involved in cancer, as follows: classical type I (represented by E-cadherin CDH1); classical type II (represented by CDH11); desmosomal cadherin (represented by DSG2); seven-domain (7D) cadherin (CDH16 and CDH17); truncated cadherin (also known as T-cadherin); clustered protocadherins (PCDHs), represented by PCDHɑ6; non-clustered protocadherin is divided into two subfamilies which are called the δ1 subfamily and δ2 subfamily; and the cadherin-related protein (represented by FAT1, FAT4).3
E-cadherin (CDH1) is reported to be a key growth and invasion suppressor in cancer. The mature E-cadherin protein in humans consists of a single transmembrane domain, and a cytoplasmic domain, which connects with the catenin complex, and five cadherin repeats in the extracellular domain, named EC1–EC5.4,5 The EC1 subdomain differs from the others in that it contains a histidine-alanine-valine sequence, which is triggered by the binding of calcium at the EC1–EC2 interface to create the trans dimerization which is essential for adhesion between adjacent cells.6 As for the cytoplasmic domain, early studies have proposed a model for an E-cadherin/β-catenin complex (CCC).7,8 The cytoplasmic domain of E-cadherin helps to establish cell–cell adhesion through binding to the cytoskeleton via the catenin protein family containing ɑ-catenin, β-catenin and γ-catenin.9 Besides E-cadherin and catenin, there are also other proteins, for example the P120 arm-family protein, that act as key regulators of the E-cadherin/β-catenin complex.10,11 In the current review, we focus on the genetic and epigenetic regulatory mechanisms involved in E-cadherin signaling in cancers, mainly in HCC.
The Function And Detection Of E-Cadherin In Cancer
E-cadherin has received increasing attention in the past few years because of its extensive impact in human cancers, such as gastric cancer,12,13 breast cancer,14 HCC15 and renal cell carcinomas.16 In the progression of human cancer, loss of E-cadherin is widely known as the hallmark of epithelial-to-mesenchymal transition (EMT), which is considered to be a strong signal of tumor progression and metastasis.17 Tumor cells undergoing EMT act as cancer stem-like cells, migrate to distant sites and become metastases.18 Contact inhibition of locomotion (CIL) is a phenomenon whereby cells cease moving after cell–cell contact. The adhesive properties of E-cadherin hold healthy cells together and maintain CIL. However, because of the dysregulation of E-cadherin in malignant cells, CIL is disrupted which facilitates cancer cell migration to distant sites.19,20 Contact inhibition of proliferation (CIP) refers to a phenomenon in which the ability of cell proliferation is affected by cell density.19 CIP is important for maintaining tissue morphogenesis. However, when CIP is disrupted because of the loss of E-cadherin, it can lead to uncontrolled growth and tumor initiation. Although the underlying regulatory mechanism for CIP remains unclear, some mechanisms have been proposed to explain it. For example, the E-cadherin extracellular domain is thought to bind to receptor tyrosine kinases, including the EGF receptor (EGFR) and c-Met, to decrease rates of cell growth.21,22 Besides migration and proliferation, E-cadherin is also reported to affect cell contraction through the cadherin–actomyosin system.23
The comprehensive biological functions of E-cadherin have been partly attributed to the interaction between E-cadherin and multiple signaling pathways, such as the Wnt/β-catenin pathway, the PI3K/AKT pathway, the hippo pathway and the NFκB signaling pathway.24,25 E-cadherin in densely packed epithelial monolayers can inhibit the access of EGF to the EGFR as well as down-stream signaling from the EGFR via Merlin.22 However, E-cadherin is also regulated by numerous pathways. Studies in a variety of cancers have documented the complex relationship between β-catenin signaling and E-cadherin-mediated cell–cell adhesion. Nuclear β-catenin signaling plays a central role in human cancer,26 including prostate cancer,27 breast cancer,28 colon cancer29 and HCC.30 There is a well-established model which demonstrates how β-catenin is involved in cancer initiation, progression, dormancy, immunity and cancer stem cell maintenance.31 First, a reduction in cadherin levels can release β-catenin bound at the cell surface. As a result, nuclear β-catenin signaling is enhanced.32 However, reduced E-cadherin levels do not always lead to constitutive β-catenin signaling activation.33 The β-catenin-TCF has been reported to regulate the Axin2-GSK3β-Snail1 axis to influence the expression of E-cadherin.34 In vitro, the E-cadherin promoter region was found to be bound by LEF-1/β-catenin complexes, which indicates a negative feedback loop in Wnt/β-catenin signaling and E-cadherin expression.35 The complex link between E-cadherin and β-catenin signaling requires further exploration in future research.
The expression of E-cadherin can be detected at the transcriptional level by monitoring CDH1 mRNA levels, or directly at the protein level. RT-qPCR is widely used for mRNA detection.36 In addition, RNA-seq37 and probes38 are also frequently used to detect mRNA. Several methods have been employed for the detection of proteins, including Western blotting, immunohistochemistry and immunofluorescence methods. E-cadherin as a transmembrane linker protein sometimes is hard to be detected because of the hydrophobicity of membrane proteins. To solve this problem, nanodiscs for solubilizing membrane proteins have been developed.39 With its help, the traditional detection techniques, such as electron microscopy, small‐angle X‐ray scattering and mass spectroscopy method, could detect E-cadherin more easily. Interestingly, researchers have developed a novel electrochemical label-free immunosensor to identify the expression of E-cadherin by modifying indium-tin oxide electrodes with anti-E-cadherin monoclonal antibodies.40
The Impact Of Genetic Alterations On E-Cadherin Signaling Regulation
Cancer progression has long been regarded as an outcome of genetic alterations. Point mutations, deletions, insertions, amplifications and translocations in oncogenes and suppressors are believed to be responsible for HCC development.41 As an important factor in HCC progression, genetic alterations also play a large role in E-cadherin signaling regulation.
Gene Mutations
The cluster of the CDH1 is located at chromosome 16q22.1. Researchers have identified its structure using cDNA probes covering the whole protein-coding sequence. The whole length of the CDH1 gene contains 16 well-conserved exons ranging in size between 115 and 2245 bp and 15 introns ranging from 120 bp to 65 kb.42 Recently, germ-line mutation of CDH1, which means any detectable variation within germ cells, has been found in hereditary diffuse gastric cancers13 and lobular breast cancer.43 A total of 69 different kinds of CDH1 somatic mutation have been reported. The mutation types mainly consist of splice site mutations and truncation mutations which are caused by insertions, deletions, and nonsense mutations.44 These kinds of mutation have been found to commonly occur in lobular breast cancer45 and diffuse gastric cancer,46,47 but rarely in HCC.44,48 In most cases, these mutations, which occur in combination with a loss of heterozygosity (LOH) of the wild‐type allele, have been reported to downregulate the expression of E-cadherin in HCC. Betty L. et al, observed 64% of allele loss for CDH1 in hepatitis B virus-positive Chinese HCC patients.49 Yu et al, analyzed the retained E-cadherin allele in nine tumor samples harboring LOH in both 16q and 16p markers and detected a silent polymorphism at codon 692 (GCC→GCT) in two samples, without any other nucleotide changes.50 These findings indicate that the mechanism of E-cadherin inactivation in hepatoma is more likely attributable to the loss of one copy of the E‐cadherin gene rather than the mutation of the CDH1 gene itself.
To clarify the genetic network of E-cadherin expression, we systematically reviewed previous studies and found 71 genes which were reported to be significantly related to E-cadherin regulation. We consulted the Cancer Genome Atlas (TCGA) database and found that 26 genes were frequently altered (>2%) in HCC. These 26 genes are presented in Figure 1.51
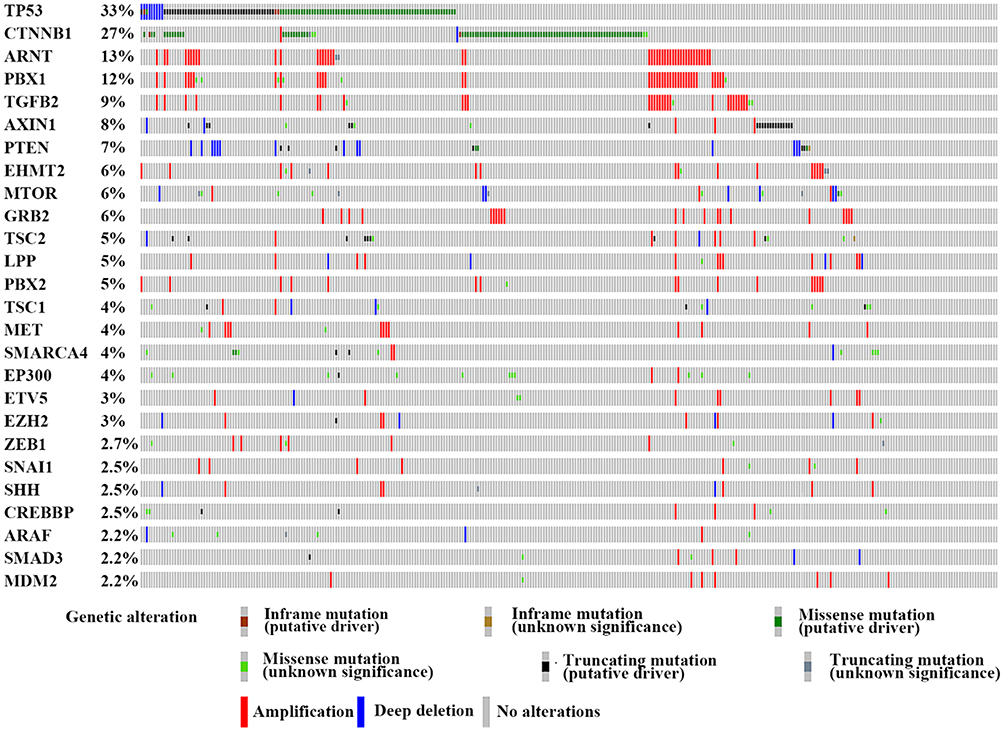 |
Figure 1 The genetic alteration of the genes involved in the E-cadherin regulation process. We proceeded to the TCGA database and finally identified 26 genes which were frequently altered (>2%) in HCC. Notes: This figure is from the cBioPortal website (http://www.cbioportal.org/); TCGA data set. Cerami E, Gao J, Dogrusoz U, et al The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404.51 Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.211 |
The most frequently altered gene was P53. The genetic alteration of this gene was observed in HCC in approximately 33% of clinical cases. One SNP array analysis of 125 HCC patients revealed that 20.8% of TP53 mutations were inactivating mutations.52 Recently, it was found that TP53 mutant cell lines showed a smaller cell size in a hepatocellular cancer spheroid formation test, which resulted from the lower CDH1 expression levels.53 Moreover, evidence demonstrated the mutant-p53 could affect the expression of Twist1 and Smad interacting protein 1 (SIP1) and further regulate E-cadherin in adrenocortical carcinoma.54 In ovarian tumor cells, p53 has been reported to repress E-cadherin expression by recruiting DNA (cytosine-5)-methyltransferase 1(DNMT1) to the CDH1 promotor through activation of the PI3K/Akt pathway.55
CTNNB1 encodes for β-catenin and is another high-frequency mutation gene in HCC and primarily demonstrates missense mutations. The Wnt-β-catenin pathway is often activated in HCC through CTNNB1 mutations (27% of HCC cases in TCGA data set, Figure 1) which is believed to have a strong impact on E-cadherin expression (Figure 2).56,57 In a study of renal interstitial fibrosis (RIF), the expression of E-cadherin was repressed through downregulation of miRNA-200a which directly targets the CTNNB1 3ʹUTR.58 In a study of PS341 (Bortezomib), which can inhibit the transcriptional activation of CTNNB1, Yang et al, reported a positive relationship between E-cadherin and PS341 levels.59 As the encoding gene of β-catenin, CTNNB1 can change the expression of E-cadherin via altered β-catenin protein levels. There is, however, a lack of direct evidence to clarify the impact of different CTNNB1 mutation types on E-cadherin expression in HCC.
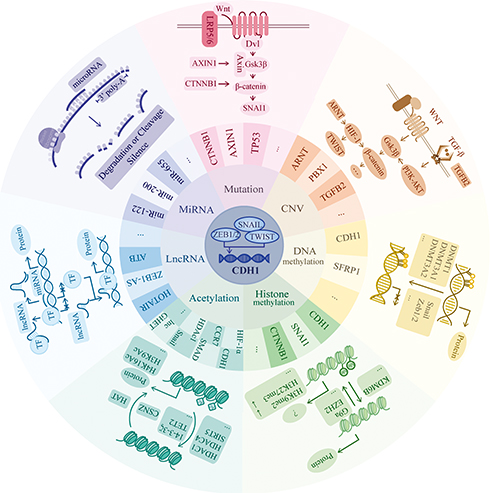 |
Figure 2 Genetic and epigenetic regulation mechanisms. CDH1 expression is mainly driven by Snail, zinc-finger E-box-binding (ZEB) and TWIST, which is one type of basic helix-loop-helix (bHLH) transcription factors. They bind to the E-box of CDH1 promotor to repress the expression. In this review, we focus on seven parts of regulatory pathways including mutation, CNV, DNA methylation, histone methylation, acetylation, lncRNA and miRNA. In mutation aspect, we mainly list the involvement of gene CTNNB1 and AXIN1, which both participate in the WNT/β-catenin pathway. In the WNT/β-catenin pathway, when the signaling is “on.” Axin removes from the destruction complex and β-catenin moves into the nucleus to bind to the target DNA such as SNAI1 to activate the transcription. In CNV part, we mainly list the involvement of gene ARNT, PBX1 and TGFB2. They mainly related to the WNT-β catenin and TGF-β pathway. In TGF-β pathway, TGF-β induces the SMAD pathway to activate the expression of β-catenin. In non-SMADs pathway, TGF-β regulates the Gsk-3β to change the expression of β-catenin via PI3K-Akt pathway. The HIF1ɑ factor is reported to directly regulate TWIST to have an impact on E-cadherin expression as well as Snail factor. In DNA methylation aspect, it always serves as the down-regulator of target gene expression when there exists hypermethylation area at the promotor region. Here, we list 2 genes which are reported to be regulated by DNA methylation and the recruiting factors such as Snail and Zeb1/2 as well as the recruited DNMTs. In histone methylation part, its methylation, typically at the tail of H3 and H4 histones, will epigenetically influence the gene expression due to the variant space structure. Both the H3K9me2 and H3K27me3 downregulates the expression of CDH1. In acetylation part, the balance between acetylation and deacetylation change the positive charge mainly from the lysine, thus, highly acetylated histones form more accessible chromatin and tend to be associated with active transcription. In lncRNA part, we list two main progression of its function: Decoy and Guide. LncRNAs decoy the target by binding and titrating away a protein or RNA target to negatively regulate the expression. And guide by directing the localization of specific proteins to their target to form a complex. In miRNA part, miRNAs function via base-pairing with complementary sequences (mostly in the 3ʹ-UTR) within mRNA molecules to degrade or cleave the target mRNA to silence the transcription. “ … ” represents other factors which are not listed in the figure. |
AXIN1 has been identified to be a concentration-limiting factor responsible for the destruction of β-catenin. In the Wnt/β-catenin pathway, when Wnt does not bind to the receptor, AXIN, GSK and APC form a destruction complex to accelerate the destruction of β-catenin. Conversely, when the Wnt pathway is active, AXIN is removed from the destruction complex which results in β-catenin moving into the nucleus and binding to target DNA to activate transcription. However, we had to emphasize that previous studies have also indicated that CTNNB1 and AXIN gene alterations are mutually exclusive.52 Zucman-Rossi et al, suggested that the loss of function of AXIN1 is not equivalent to the gain of function of β-catenin in HCC.60 In view of the complex network of AXIN relationships, more research on this topic is required.
Chromosome Instability
Chromosome instability (CIN) is an interesting aspect of genetic variation in CDH1. CIN is identified as the rate (cell to cell variability) of changed karyotypes of a given cell population, including gaining or losing whole chromosomes or fractions of chromosomes or altering the structure of chromosomes.61 Substantial evidence has demonstrated the participation of aneuploidy in the development of HCC.62–64 Gao et al, demonstrated the role of 16q loss in the downregulation of E-cadherin in human ovarian cancer cells (OV-5P) by isolating mesenchymal variants from clonal epithelial populations.65 In addition, it has been reported that trisomic epithelial cancer cells always demonstrate TWIST1-positive status in human colorectal cancer.66 We also identified a few genes which are highly amplified in HCC tissue and have a great impact on the expression of E-cadherin. ARNT (13% amplification frequency in TCGA dataset) encodes the aryl hydrocarbon receptor nuclear translocator protein, which has been identified as the beta subunit of a heterodimeric transcription factor, hypoxia-inducible factor 1 (HIF1). The HIF1ɑ factor is reported to directly regulate TWIST and further alter E-cadherin expression67 as well as that of Snail factor.68 PBX1 encodes the Pre-B-cell leukemia transcription factor 1 (PBX1) and is found to occur at a high frequency of amplification in HCC (12% in TCGA dataset). Risolino et al, reported that PBX1 modulated TGF-β-related E-cadherin expression by inducing SMAD3.69 Besides PBX1, another gene demonstrating a high frequency of amplification status is TGFβ2 (9% in TCGA dataset), a cytokine that interacts with TGF-β receptors and functions as the activator of the TGF-β pathway. Various studies have identified the roles of TGF-β-activated small mothers against decapentaplegic (Smad) and non-Smad signaling in E-cadherin expression.70 However, some of these highly amplified genes in HCC are still poorly explored (Figure 1). Fully understanding the genetic mechanisms of E-cadherin signaling regulation will allow us to develop a better understanding of the biological processes involved in HCC initiation and metastasis.
The Important Transcription Factors (SNAIL, TWIST And ZEB1/2) Involved In E-Cadherin Signaling Regulation
The CDH1 regulatory sequence is 1.2 kb in length, containing four E-box regions that can be recognized by transcription factors.71 E-box is a DNA response element with a palindromic canonical sequence of CACGTG.72 In recent studies, the E boxes in the human E-cadherin promoter were demonstrated to play a key role in the epithelial-specific expression of E-cadherin.73
Snail is a family of transcription factors encoded by the SNAI1 gene.74 As the most studied E-cadherin suppressors, snail reads the CDH1 promoter with its DNA-binding zinc fingers, and uses the SNAG domain to recruit transcription factors, chromatin enzymes and cofactors such as the LSD1/HDAC complex, G9a, Suv39H1 and SIN3A to epigenetically regulate the expression of CDH1. It is hypothesized that snail binds LSD1/HDAC to create the initial repressive chromatin environment by modifying histones such as H3K4. Then, snail binds to G9a and Suv39H1 to promote the recruitment of DNA methyltransferases (DNMTs) to perform DNA methylation.75 Snail has also been reported to be a co-regulator with enhancer of zeste homolog 2 (EZH2) in a repression model of the EZH2/HDAC1/2/Snail complex.76 Slug (also called snail2) is encoded by the SNAI2 gene. Slug has the same SNAG domain as Snail. However, slug is markedly different from snail in terms of its proline-serine rich central region, especially in the specific SLUG domain. The unique Snail2 function remains unexplored to date.77
Twist1 is a basic helix-loop-helix transcription factor encoded by the TWIST1 gene. Together with twist2, the twist family proteins are overexpressed in a variety of human tumors and play a role in cancer.78 Twist can directly or indirectly cause the transcriptional repression of E-cadherin through the E-box elements on the E-cadherin promoter.79 SETD8, which belongs to the domain-containing methyltransferase family and specifically targets H4K20 for monomethylation, functionally interplays with Twist and regulates E-cadherin expression.80 Interestingly, previous studies on whether Twist interacts with Snail/slug protein and further affects E-cadherin signaling are contradictory. In chromatin immunoprecipitation (ChIP) assays, Twist1 was revealed to recruit Snail/slug proteins through the interaction between the twist1 protein and the C-terminal zinc fingers of snail/slug.81 Twist1 is reported to downregulate snail mRNA expression in KYSE-30 cells.82 However, Yang et al, failed to observe an induction of Snail in human mammary epithelial cells when Twist expression was altered, and they suggested that Twist and Snail may function independently in affecting E-cadherin expression.79
ZEB1 and its mammalian paralog ZEB2 both belong to the ZEB family within the zinc-finger class of homeodomain transcription factors. Among these, ZEB1 has been demonstrated to repress E-cadherin transcription by binding to two E-box sequences in its promoter region and interacting with the SWI/SNF family protein BRG1. It seems that the involvement of BRG1 is required for the regulation of ZEB1.83 ZEB2 also represses the activity of the E-cadherin promoter by binding to a conserved E-box sequence.84 However, ZEB2 plays a role in the TGFβ signaling pathway and is activated by full-length SMADs.85 In addition, a previous study also reported that a complex of Cdc42 GTPase-activating protein (CdGAP) and ZEB2 mediated the repression of E-cadherin expression.86
Epigenetic Mechanisms Play An Important Role In E-Cadherin Expression
Epigenetics is the study of heritable phenotype changes that do not involve alterations in the DNA sequence,87 including four DNA modifications, 16 classes of histone modifications88 and non-coding RNA (Figure 2). Recently, growing evidence suggests that epigenetic alterations play important roles in CDH1 expression and indeed lead to the development of HCC.89
DNA Methylation In The Regulation Of CDH1 Expression
Typically, DNA methylation occurs at the 5ʹ-position of the cytosine (5mC) of CpG dinucleotides catalyzed by DNMTs.90 It generally serves as a down-regulator of target gene expression when a hypermethylation area exists at the promotor region.91 Recent studies have revealed the aberrant 49% methylation of CDH1 in HCC using bisulphite genomic sequencing and ChIP assays.92,93 Similarly, a study focused on methylation status during multistep hepatocarcinogenesis also reported that the frequency of E-cadherin hypermethylation in premalignant lesions is less than 7%, whereas the frequency remarkably increased to 30% in HCC.94 These findings suggest a significant driving role of CDH1 hypermethylation in the development of HCC. DNMT1, DNMT3A1 and DNMT3A2 are reported to selectively induce hypermethylation of the CDH1 promoter,95 and the hypermethylation of CpG islands is highly associated with E-cadherin suppression at both the mRNA and protein levels.96 Moreover, several studies have provided consistent evidence for the role of DNMT1 depletion in restoring E-cadherin expression via demethylation,97 and increasing the methylation level of CpG islands around the promoter region can result in hepatocarcinogenesis through a reduction in E-cadherin expression.15,98
Hepatitis B virus (HBV) and hepatitis C virus (HCV) infection are regarded as one of the most important risk factors in the progression of HCC. In China, most HCC cases are associated with HBV infection.99 In HBxAg-positive cells, E-cadherin was found to be suppressed, and this phenomenon was associated with hypermethylation of the E-cadherin promoter.96 This kind of E-cadherin suppression is dependent on DNMT1 because both the RNA and protein levels of DNMT1 were significantly increased in HBx-expressing cells, and the expression of E-cadherin was completely restored after treatment with a DNMT inhibitor (5′-Aza-2′dC).100 In the study of HCV, 5′-Aza-2′dC can also reverse the E-cadherin suppressive function caused by the HCV core protein. The expression levels of both DNMT1 and DNMT 3B were dramatically increased in HCV core-expressing cells which lead to the suppression of E-cadherin.101 However, the detailed mechanism through which the hepatitis virus interacts with methyltransferase remains obscure.
The transcription factor Snail is one of the most studied regulators of CDH1 DNA methylation in HCC.75 Lim et al, clearly demonstrated that Snail plays a central role in CpG methylation of the E-cadherin promoter by recruiting DNMT1 to a specific site, which maintains DNA methylation and is implicated in tumorigenesis.102 Furthermore, Snail is crucial in maintaining the aberrant methylation status of the CDH1 promoter in HCC, as DNMT1 cannot regulate the progression of methylation at the CDH1 promoter alone without snail participation.103
Histone Methylation In The Regulation Of CDH1 Expression
Histone methylation typically takes place at arginine or lysine amino acid residues at the protein sequence. The former is controlled by protein arginine methyltransferases and the latter by protein lysine methyltransferases (PKMTs).104 Histones package and histone methylation, typically at the tail of the H3 and H4 histones, will epigenetically influence gene expression because of the variant space structure.105,106 Trimethylation of H3 lysine 4 (H3K4me3) and trimethylation of H3 lysine 36 (H3K36me3) are associated with active transcription, whereas trimethylation of H3 lysine 27 (H3K27me3), di- and trimethylation of H3 lysine 9 (H3K9me2/3) and trimethylation of H4 lysine 20 (H4K20me3) are associated with repressed gene expression.107,108
Interestingly, a web of interactions appears to tightly coordinate DNA and histone methylation in E-cadherin regulation networks. For example, a previous study reported that the expression levels of G9a, which is recruited by Snail, are positively correlated with the H3K9me2 levels of CDH1.109 However, the relationship between H3K9me2 and E-cadherin expression is more complicated than once thought. In breast cancer, knockdown of G9a can restore E-cadherin expression by suppressing H3K9me2 and blocking DNA methylation. However, in HCC, E-cadherin expression is unchanged following knockdown of G9a. This contradiction may be explained by the involvement of other pathways, including TGF-β signaling in HCC.110 TGF-β is reported to be a strong regulator of epigenetics. It can promote SNAI1 expression by removing histone H3 lysine trimethylation marks via the induction of lysine demethylase 6B (KDM6B) levels in breast cancer.111 Feng et al, explored the epigenetic function of basil polysaccharide and demonstrated that it could suppress the expression of G9a, H3K9me2 and HIF-1ɑ, which regulated β-catenin112 and twist,113 and further increased the expression of E-cadherin in HCC. However, the causal relationship was not fully explored in their study.114
In addition, another histone methylation modification type, H3K27me3, is also reported to be correlated with EZH2 and E-cadherin expression. EZH2 is a PKMT and is capable of mono-, di-, and especially trimethylation of Lys-27 on histone 3 (H3K27).115 A close relationship between H3K27me3 and EZH2 has been reported in HCC.116 In a study of non-tumorigenic hepatocytes, EZH2 was demonstrated to directly interact with Snail with the help of lncRNA HOTAIR.117 Interestingly, Li et al, have discovered that CLDN14, as a novel direct target of EZH2-mediated H3K27me3, is involved in the Wnt/β-catenin pathway to regulate E-cadherin expression.118
Histone Acetylation And Deacetylation Of CDH1 Itself And Other Factors
Typically, acetylation targets Lys residues in the amino-terminal tails of core histone proteins, which is catalyzed by histone acetyl transferases and histone deacetylases (HDACs). The acetylation of histones changes histone positive charge, mainly at the lysine position. Therefore, highly acetylated histones can lead to more accessible chromatin and this is associated with active transcription.119 In HCC, many studies have demonstrated that HDAC1, which is highly expressed in HCC, is essential for different kinds of classical pathway to change the transcription of CDH1. Lei et al,120 reported that HDAC1 might downregulate the promoter activity of E-cadherin in regulating the TGF-β pathway. However, because of the complexity of the TGF-β pathway, the specific target of HDAC1 and the underlying mechanism remain unknown. It is currently thought that HDAC1 interacts with snail and further regulates E-cadherin expression. In a pancreatic cancer study, silencing of E-cadherin was mediated by a transcriptional repressor complex containing Snail and recruited HDAC1/2.99 Early studies demonstrated that DNMT and HDAC appear to act as synergistic regulators in silencing target genes.121,122 Snail recruits related enzymes which may cooperate in the regulation progression. Besides snail protein, it has also been found that ZEB1 can bind to its target gene (including CDH1) and induce a decrease in histone H3K27 acetylation.123 In addition, HDAC1 is reported to directly regulate the H3K9Ac and H4K16Ac levels of the CDH1 promoter to downregulate the expression of E-cadherin through the ten-eleven-translocation (TET) family protein TET2.124
Xu et al, observed that histone deacetylase inhibitors could influence the histone stability of the CDH1 gene and upregulate SNAI1 expression. The above-mentioned inhibitors can upregulate the expression of COP9 signalosome 2 (CSN2) to combine with Snail and exposes its acetylation site. As a result, the hyperacetylation of Snail represses its degradation and downregulates the expression of E-cadherin.125,126 The modification of snail has received increasing attention in recent research. P300, which belongs to a coactivator family together with CREB-binding protein, contains a histone acetyltransferase (PAT/HAT) domain, and is newly identified that it could acetylate Snail and further lead to Snail transcriptional repression.127 In HCC, knockdown of p300 expression can also recover E-cadherin expression.128 Furthermore, the expression of Twist was also decreased by the suppressive binding of H3K27ac to the Twist promoter, which is induced by inhibition of the PI3K/Akt and Wnt/β-catenin signaling pathways.129
lncRNAs Function In E-Cadherin Signaling Through Several Crucial Pathways And Factors
Non-coding RNAs (ncRNA) are human genes that do not code for proteins. ncRNAs are divided into two groups: small ncRNAs (<200 nt, including miRNAs, siRNAs, piRNAs) and long ncRNAs (longer than 200 nt). The latter function as various types of gene expression regulator, including functioning as molecular signals, molecular decoys (by binding and titrating away a protein or RNA target to negatively regulate expression), protein guides (which direct the localization of specific proteins to their target to form a complex) and scaffolds (to bind relevant molecular components).130 Recently, it has been revealed that the dysregulation of several lncRNAs can affect the expression of E-cadherin in the pathogenesis of HCC (Table 1), with the involvement of some important signal transduction pathways. Here, we have summarized the lncRNAs involved in E-cadherin signaling.
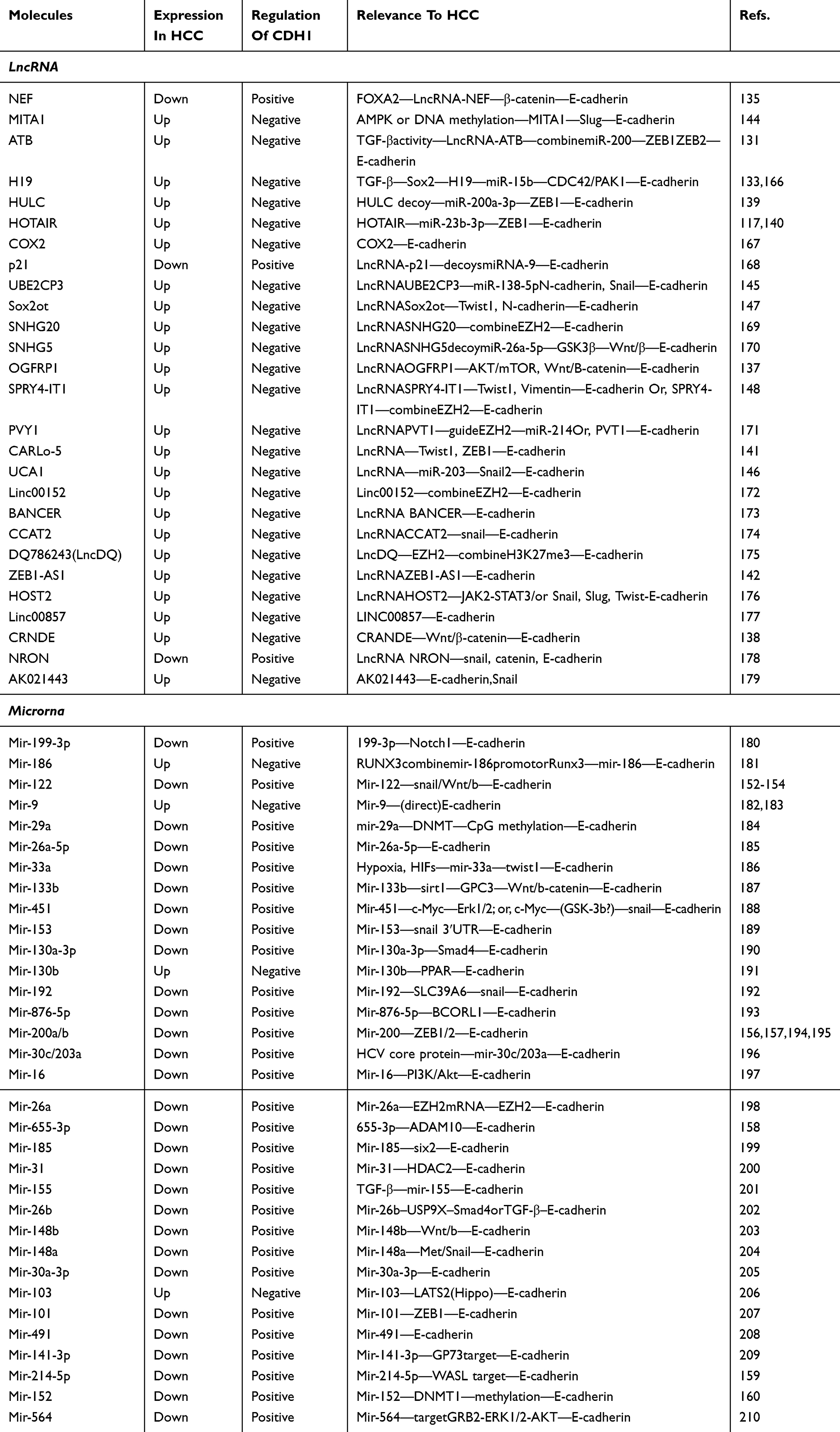 |
Table 1 The lncRNA And microRNA Involved In E-Cadherin Expression In HCC |
LncRNAs can regulate E-cadherin expression by affecting the Wnt/β-catenin pathway. lncRNA ATB131 and lncRNA H19132 were found to be regulated by TGF-β signaling and further function as oncogenic regulators to decrease the expression of E-cadherin in HCC. LncRNA ATB downregulates E-cadherin expression by sharing the response elements of miR-200 with ZEB1 and ZEB2,131 whereas lncRNA H19 activates the CDC43/PAK1 signaling pathway by targeting miR-15b.133 Activation of the Wnt/β-catenin pathway has been identified to affect E-cadherin expression. Ge et al, demonstrated that lncRNA HOTAIR, a HOX transcript antisense intergenic RNA, could activate the Wnt pathway in esophageal squamous cell carcinoma cells by inhibiting WIF-1 expression.134 In HCC, lncRNA HOTAIR was found to be recruited by the Snail protein, and mediated the Snail/EZH2 interaction, as mentioned above.117 Recently, lncRNA-NEF was reported to upregulate E-cadherin expression through the suppression of Wnt/β-catenin.135 Similarly, lncRNA SNHG5 decoys miR-26-5p to activate the Wnt/β-catenin pathway and downregulate E-cadherin expression.136 Researchers have also reported that lncRNA OGFRP21137 and lncRNA CRANDE138 can influence E-cadherin expression through the Wnt/β-catenin pathway in HCC, but the specific mechanisms underlying these phenomena remain to be explored.
Certain lncRNAs can significantly affect E-cadherin expression by directly targeting important transcription factors in E-cadherin signaling. ZEB1 and ZEB2, zinc-finger and homeodomain proteins are important components of a network of transcription factors that control E-cadherin expression. Several lncRNAs, including ATB,131 HULC,139 HOTAIR,140 CARLo-5141 and ZEB-AS1,142 are reported to target ZEB1 and ZEB2 and interact with paired E-box-like promoter elements to control transcriptional activity.143 In addition, researchers have found that certain lncRNAs function via E-box repressors such as snail, slug and twist1, to downregulate the expression of E-cadherin. The AMPK-associated lncRNA MITA1 was reported to upregulate slug expression.144 lncRNA UBE2CP3 and lncRNA UCA1 can suppress the expression of E-cadherin by upregulating Snail expression,145,146 while lncRNA Sox2ot,147 lncRNA CARLo-5141 and SPRY4-IT1148 can suppress the expression of E-cadherin by activating Twist.
MicroRNAs Can Demonstrate A Suppressive Or Oncogenetic Role In HCC By Affecting E-Cadherin Expression
As mentioned above, microRNAs (abbreviated to miRNAs) are a large ncRNA family. In a difference from lncRNAs, miRNAs function via base-pairing with complementary sequences (mostly in the 3ʹ-UTR) within mRNA molecules to silence mRNA transcription.149 Altered expression levels of various types of miRNA have been observed in human cancers, especially in HCC.150
miR-122 is reported to play a critical role in liver diseases such as steatohepatitis, fibrosis and HCC.151 Hsu et al, reported that miR-122 regulates polyploidization in murine liver, which is thought to be a key step in the progression of HCC.152 Jin et al, reported that mir-122 may affect the expression of E-cadherin by regulating the snail and Wnt/β-catenin pathway.153,154 Conversely, Liang et al, demonstrated that miR-122 can be sequestered by HBx-LINE1, a hybrid RNA transcript of human LINE1 and the HBV-encoded X gene generated in tumor cells in HBV-positive HCC, to down-regulate the expression of E-cadherin.155
Various miRNAs demonstrate an oncogenic role by altering different pathways. However, there is increasing evidence to suggest that miRNAs play critical roles as tumor suppressors. miR-200 has been identified as a marker for E-cadherin-positive cancer cells. Sun-Mi Park et al, have reported that miR-200 directly targeted ZEB1 and ZEB2 to upregulate E-cadherin in 60 cell lines in a drug screening panel of human cancer cell lines at the National Cancer Institute (NCI60).156 In HCC, Ding et al, proposed the miR-200b could regulate E-cadherin signaling by ZEB1.157 Wu et al, observed that miR-655-3p was significantly down-regulated in HCC tissues and HCC cell lines. They reported that miR-655-3p directly targets a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), which indirectly regulates the β-catenin signaling pathway and upregulates E-cadherin protein levels during HCC progression.158 Li et al, discovered that miR-214-5p targets Wiskott-Aldrich Syndrome Like (WASL) protein and indirectly upregulates the expression of E-cadherin. Restoring WASL expression could reverse the inhibition effect of miR-214-5p on E-cadherin.159 Additionally, Huang et al,160 have reported an epigenetic function for miRNA-152 as a tumor suppressor. They found that miR-152 levels are frequently correlated with DNMT1 mRNA expression in HBV-related HCCs, and DNMT1 can inhibit the hypermethylation of E-cadherin to increase E-cadherin expression in HCC.
E-Cadherin Is A Promising Indicator And Target For The Treatment Of HCC
In view of the aberrant E-cadherin expression that is associated with the initiation and progression of HCC, E-cadherin may serve as a promising prognostic indicator. A study of the expression levels of E-cadherin in 41 HCC samples suggested that the survival rate in patients with negative expression of E-cadherin was significantly lower.161 A meta-analysis of 2439 patients in 30 studies also demonstrated the predictive potential of E-cadherin for the prognosis of HCC patients.162 Epigenetic drugs represent an attractive therapeutic strategy. Panobinostat, a novel pan-HDAC-inhibitor has demonstrated substantial efficacy in several preclinical models of cancer, and is reported to upregulate CDH1 and demonstrated the highest preclinical efficacy when combined with sorafenib in HCC models.163 Apart from epigenetic drugs, gene therapy approaches have gained more attention. The miRNAs mentioned above could potentially serve as a component of E-cadherin gene therapy because of their specificity and ease of handling. Therefore, the gene of interest in the form of a tumor suppressor short-interfering RNA can be efficiently delivered to cells using viruses such as lentiviruses, adenoviruses or adeno-associated viruses.164 However, viral carriers can cause severe side effects, which might limit the application of viral vectors in future clinical practice. Therefore, other non-viral-based transgene vectors165 are worthy of exploration.
Conclusion
Previous research in different cancer types has revealed the basic regulatory framework for E-cadherin signaling. Patients with HCC tend to have a high-risk of multi-centric tumor occurrence and E-cadherin is a crucial factor in this process. Here, we present an overview of E-cadherin regulatory mechanisms, focusing on genetic and epigenetic alterations. Based in part on the data from the TCGA dataset, we can find some highly altered genes associated with E-cadherin which are poorly explored in HCC. An exhaustive exploration of the epigenetic regulatory mechanisms of E-cadherin, including DNA methylation, histone modifications and ncRNA, will help us to fully understand the special biological processes involved in HCC initiation and metastasis.
Acknowledgments
We are very grateful to the institutions that have supported our research. This work was supported by the National Natural Science Foundation of China [81872297 and 81874059] and the Zhejiang Province Analysis and Test Technology Project [2018C37062].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45(4):529–538. doi:10.1016/j.jhep.2006.05.013
3. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. 2014;14(2):121–134. doi:10.1038/nrc3647
4. Harrison OJ, Bahna F, Katsamba PS, et al. Two-step adhesive binding by classical cadherins. Nat Struct Mol Biol. 2010;17(3):348–357. doi:10.1038/nsmb.1784
5. Nardone V, Lucarelli AP, Dalle Vedove A, et al. Crystal structure of human E-Cadherin-EC1EC2 in complex with a peptidomimetic competitive inhibitor of cadherin homophilic interaction. J Med Chem. 2016;59(10):5089–5094. doi:10.1021/acs.jmedchem.5b01487
6. Beavon IR. The E-cadherin-catenin complex in tumour metastasis: structure, function and regulation. Eur J Cancer. 2000;36(13Spec No):1607–1620. doi:10.1016/s0959-8049(00)00158-1
7. van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65(23):3756–3788. doi:10.1007/s00018-008-8281-1
8. Wijnhoven BP, Dinjens WN, Pignatelli M. E-cadherin-catenin cell-cell adhesion complex and human cancer. Br J Surg. 2000;87(8):992–1005. doi:10.1046/j.1365-2168.2000.01513.x
9. Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–261. doi:10.1146/annurev.cellbio.22.010305.104241
10. Epifano C, Megias D, Perez-Moreno M. p120-catenin differentially regulates cell migration by Rho-dependent intracellular and secreted signals. EMBO Rep. 2014;15(5):592–600. doi:10.1002/embr.201337868
11. Anastasiadis PZ, Reynolds AB. Regulation of Rho GTPases by p120-catenin. Curr Opin Cell Biol. 2001;13(5):604–610. doi:10.1016/s0955-0674(00)00258-1
12. Ascano JJ, Frierson H
13. Guilford P, Hopkins J, Harraway J, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392(6674):402–405. doi:10.1038/32918
14. Horne HN, Oh H, Sherman ME, et al. E-cadherin breast tumor expression, risk factors and survival: pooled analysis of 5,933 cases from 12 studies in the Breast Cancer Association Consortium. Sci Rep. 2018;8(1):6574. doi:10.1038/s41598-018-23733-4
15. Kanai Y, Ushijima S, Hui AM, et al. The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer. 1997;71(3):355–359. doi:10.1002/(sici)1097-0215(19970502)71:3<355::aid-ijc8>3.0.co;2-x
16. Zhang X, Yang M, Shi H, et al. Reduced E-cadherin facilitates renal cell carcinoma progression by WNT/beta-catenin signaling activation. Oncotarget. 2017;8(12):19566–19576. doi:10.18632/oncotarget.15361
17. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166(1):21–45. doi:10.1016/j.cell.2016.06.028
18. Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15(5):311–325. doi:10.1038/nrd.2015.13
19. Mendonsa AM, Na TY, Gumbiner BM. E-cadherin in contact inhibition and cancer. Oncogene. 2018;37(35):4769–4780. doi:10.1038/s41388-018-0304-2
20. Perrais M, Chen X, Perez-Moreno M, Gumbiner BM. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell. 2007;18(6):2013–2025. doi:10.1091/mbc.e06-04-0348
21. Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. Embo J. 2004;23(8):1739–1748. doi:10.1038/sj.emboj.7600136
22. Kourtidis A, Lu R, Pence LJ, Anastasiadis PZ. A central role for cadherin signaling in cancer. Exp Cell Res. 2017;358(1):78–85. doi:10.1016/j.yexcr.2017.04.006
23. Lecuit T, Yap AS. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol. 2015;17(5):533–539. doi:10.1038/ncb3136
24. McCrea PD, Maher MT, Gottardi CJ. Nuclear signaling from cadherin adhesion complexes. Curr Top Dev Biol. 2015;112:129–196.
25. Hu QP, Kuang JY, Yang QK, Bian XW, Yu SC. Beyond a tumor suppressor: soluble E-cadherin promotes the progression of cancer. Int J Cancer. 2016;138(12):2804–2812. doi:10.1002/ijc.29982
26. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4(5):a008052. doi:10.1101/cshperspect.a008052
27. Schneider JA, Logan SK. Revisiting the role of Wnt/beta-catenin signaling in prostate cancer. Mol Cell Endocrinol. 2018;462(Pt A):3–8. doi:10.1016/j.mce.2017.02.008
28. Wang Z, Zhang H, Hou J, et al. Clinical implications of beta-catenin protein expression in breast cancer. Int J Clin Exp Pathol. 2015;8(11):14989–14994.
29. Rahmani F, Avan A, Hashemy SI, Hassanian SM. Role of Wnt/beta-catenin signaling regulatory microRNAs in the pathogenesis of colorectal cancer. J Cell Physiol. 2018;233(2):811–817. doi:10.1002/jcp.25897
30. Vilchez V, Turcios L, Marti F, Gedaly R. Targeting Wnt/beta-catenin pathway in hepatocellular carcinoma treatment. World J Gastroenterol. 2016;22(2):823–832. doi:10.3748/wjg.v22.i2.823
31. Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8(20):33972–33989. doi:10.18632/oncotarget.15687
32. Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27(55):6920–6929. doi:10.1038/onc.2008.343
33. van de Wetering M, Barker N, Harkes IC, et al. Mutant E-cadherin breast cancer cells do not display constitutive Wnt signaling. Cancer Res. 2001;61(1):278–284.
34. Yook JI, Li XY, Ota I, et al. A Wnt-Axin2-GSK3beta cascade regulates snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398–1406. doi:10.1038/ncb1508
35. Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59(1):3–10. doi:10.1016/0925-4773(96)00597-7
36. Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nat Protoc. 2006;1(3):1559–1582. doi:10.1038/nprot.2006.236
37. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi:10.1038/nrg2484
38. Wetmur JG. DNA probes: applications of the principles of nucleic acid hybridization. Crit Rev Biochem Mol Biol. 1991;26(3–4):227–259. doi:10.3109/10409239109114069
39. Rouck JE, Krapf JE, Roy J, Huff HC, Das A. Recent advances in nanodisc technology for membrane protein studies (2012–2017). FEBS Lett. 2017;591(14):2057–2088. doi:10.1002/1873-3468.12706
40. Ben Ismail M, Carreiras F, Agniel R, et al. Application of APTES-Anti-E-cadherin film for early cancer monitoring. Colloids Surf B Biointerfaces. 2016;146:550–557. doi:10.1016/j.colsurfb.2016.06.048
41. Li S, Mao M. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett. 2013;340(2):247–253. doi:10.1016/j.canlet.2012.09.027
42. Berx G, Staes K, van Hengel J, et al. Cloning and characterization of the human invasion suppressor gene E-cadherin (CDH1). Genomics. 1995;26(2):281–289. doi:10.1016/0888-7543(95)80212-5
43. Corso G, Intra M, Trentin C, Veronesi P, Galimberti V. CDH1 germline mutations and hereditary lobular breast cancer. Fam Cancer. 2016;15(2):215–219. doi:10.1007/s10689-016-9869-5
44. Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12(4):226–237. doi:10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D
45. Da Silva L, Parry S, Reid L, et al. Aberrant expression of E-cadherin in lobular carcinomas of the breast. Am J Surg Pathol. 2008;32(5):773–783. doi:10.1097/PAS.0b013e318158d6c5
46. Melo S, Figueiredo J, Fernandes MS, et al. Predicting the functional impact of CDH1 missense mutations in hereditary diffuse gastric cancer. Int J Mol Sci. 2017;18(12):2687. doi:10.3390/ijms18122687
47. Tamura G, Sakata K, Nishizuka S, et al. Inactivation of the E-cadherin gene in primary gastric carcinomas and gastric carcinoma cell lines. Jpn J Cancer Res. 1996;87(11):1153–1159. doi:10.1111/j.1349-7006.1996.tb03125.x
48. Wang G, Huang CH, Zhao Y, et al. Genetic aberration in primary hepatocellular carcinoma: correlation between p53 gene mutation and loss-of-heterozygosity on chromosome 16q21-q23 and 9p21-p23. Cell Res. 2000;10(4):311–323. doi:10.1038/sj.cr.7290058
49. Slagle BL, Zhou YZ, Birchmeier W, Scorsone KA. Deletion of the E-cadherin gene in hepatitis B virus-positive Chinese hepatocellular carcinomas. Hepatology. 1993;18(4):757–762. doi:10.1002/hep.1840180402
50. Wei Y, Van Nhieu JT, Prigent S, Srivatanakul P, Tiollais P, Buendia MA. Altered expression of E-cadherin in hepatocellular carcinoma: correlations with genetic alterations, beta-catenin expression, and clinical features. Hepatology. 2002;36(3):692–701. doi:10.1053/jhep.2002.35342
51. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
52. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–698. doi:10.1038/ng.2256
53. Pomo JM, Taylor RM, Gullapalli RR. Influence of TP53 and CDH1 genes in hepatocellular cancer spheroid formation and culture: a model system to understand cancer cell growth mechanics. Cancer Cell Int. 2016;16:44. doi:10.1186/s12935-016-0318-1
54. Bulzico D, Torres DC, Ferreira GM, et al. A novel TP53 Mutation Associated with TWIST1 and SIP1 expression in an aggressive adrenocortical carcinoma. Endocr Pathol. 2017;28(4):326–331. doi:10.1007/s12022-017-9482-7
55. Cheng JC, Auersperg N, Leung PC. Inhibition of p53 represses E-cadherin expression by increasing DNA methyltransferase-1 and promoter methylation in serous borderline ovarian tumor cells. Oncogene. 2011;30(37):3930–3942. doi:10.1038/onc.2011.117
56. Liu J, Ruan B, You N, et al. Downregulation of miR-200a induces EMT phenotypes and CSC-like signatures through targeting the beta-catenin pathway in hepatic oval cells. PLoS One. 2013;8(11):e79409. doi:10.1371/journal.pone.0079409
57. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
58. Gong Y, Qin Z, Zhou B, Chen H, Shi Z, Zhang J. MicroRNA-200a inhibits transforming growth factor beta1-induced proximal tubular epithelial-mesenchymal transition by targeting beta-catenin. Nephron. 2017;137(3):237–249. doi:10.1159/000479168
59. Yang Z, Liu S, Zhu M, et al. PS341 inhibits hepatocellular and colorectal cancer cells through the FOXO3/CTNNB1 signaling pathway. Sci Rep. 2016;6:22090. doi:10.1038/srep22090
60. Zucman-Rossi J, Benhamouche S, Godard C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26(5):774–780. doi:10.1038/sj.onc.1209824
61. Heng HH, Bremer SW, Stevens JB, et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013;32(3–4):325–340. doi:10.1007/s10555-013-9427-7
62. Wilkinson PD, Delgado ER, Alencastro F, et al. The polyploid state restricts hepatocyte proliferation and liver regeneration in mice. Hepatology. 2019;69(3):1242–1258. doi:10.1002/hep.30286
63. Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432(7015):338–341. doi:10.1038/nature03099
64. Sheltzer JM, Amon A. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet. 2011;27(11):446–453. doi:10.1016/j.tig.2011.07.003
65. Gao C, Su Y, Koeman J, et al. Chromosome instability drives phenotypic switching to metastasis. Proc Natl Acad Sci U S A. 2016;113(51):14793–14798. doi:10.1073/pnas.1618215113
66. Celesti G, Di Caro G, Bianchi P, et al. Presence of Twist1-positive neoplastic cells in the stroma of chromosome-unstable colorectal tumors. Gastroenterology. 2013;145(3):647–657 e615. doi:10.1053/j.gastro.2013.05.011
67. Yang MH, Wu MZ, Chiou SH, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. doi:10.1038/ncb1691
68. Yang SW, Zhang ZG, Hao YX, et al. HIF-1alpha induces the epithelial-mesenchymal transition in gastric cancer stem cells through the snail pathway. Oncotarget. 2017;8(6):9535–9545. doi:10.18632/oncotarget.14484
69. Risolino M, Mandia N, Iavarone F, et al. Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111(36):E3775–E3784. doi:10.1073/pnas.1407074111
70. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–172. doi:10.1038/cr.2009.5
71. Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24(56):8277–8290. doi:10.1038/sj.onc.1208991
72. Chaudhary J, Skinner MK. Basic helix-loop-helix proteins can act at the E-box within the serum response element of the c-fos promoter to influence hormone-induced promoter activation in sertoli cells. Mol Endocrinol. 1999;13(5):774–786. doi:10.1210/mend.13.5.0271
73. Behrens J, Lowrick O, Klein-Hitpass L, Birchmeier W. The E-cadherin promoter: functional analysis of a G.C-rich region and an epithelial cell-specific palindromic regulatory element. Proc Natl Acad Sci U S A. 1991;88(24):11495–11499. doi:10.1073/pnas.88.24.11495
74. Paznekas WA, Okajima K, Schertzer M, Wood S, Jabs EW. Genomic organization, expression, and chromosome location of the human SNAIL gene (SNAI1) and a related processed pseudogene (SNAI1P). Genomics. 1999;62(1):42–49. doi:10.1006/geno.1999.6010
75. Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the snail story. Curr Pharm Des. 2014;20(11):1698–1705. doi:10.2174/13816128113199990512
76. Tong ZT, Cai MY, Wang XG, et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and snail to inhibit E-cadherin. Oncogene. 2012;31(5):583–594. doi:10.1038/onc.2011.254
77. Molina-Ortiz P, Villarejo A, MacPherson M, et al. Characterization of the SNAG and SLUG domains of Snail2 in the repression of E-cadherin and EMT induction: modulation by serine 4 phosphorylation. PLoS One. 2012;7(5):e36132. doi:10.1371/journal.pone.0036132
78. Zhao Z, Rahman MA, Chen ZG, Shin DM. Multiple biological functions of twist1 in various cancers. Oncotarget. 2017;8(12):20380–20393. doi:10.18632/oncotarget.14608
79. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi:10.1016/j.cell.2004.06.006
80. Yang F, Sun L, Li Q, et al. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. Embo J. 2012;31(1):110–123. doi:10.1038/emboj.2011.364
81. Lander R, Nasr T, Ochoa SD, Nordin K, Prasad MS, Labonne C. Interactions between twist and other core epithelial-mesenchymal transition factors are controlled by GSK3-mediated phosphorylation. Nat Commun. 2013;4:1542. doi:10.1038/ncomms2543
82. Forghanifard MM, Ardalan Khales S, Farshchian M, Rad A, Homayouni-Tabrizi M, Abbaszadegan MR. Negative regulatory role of TWIST1 on SNAIL gene expression. Pathol Oncol Res. 2017;23(1):85–90. doi:10.1007/s12253-016-0093-2
83. Sanchez-Tillo E, Lazaro A, Torrent R, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29(24):3490–3500. doi:10.1038/onc.2010.102
84. Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–1278. doi:10.1016/s1097-2765(01)00260-x
85. Bassez G, Camand OJ, Cacheux V, et al. Pleiotropic and diverse expression of ZFHX1B gene transcripts during mouse and human development supports the various clinical manifestations of the “Mowat-Wilson” syndrome. Neurobiol Dis. 2004;15(2):240–250. doi:10.1016/j.nbd.2003.10.004
86. He Y, Northey JJ, Pelletier A, et al. The Cdc42/Rac1 regulator CdGAP is a novel E-cadherin transcriptional co-repressor with Zeb2 in breast cancer. Oncogene. 2017;36(24):3490–3503. doi:10.1038/onc.2016.492
87. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. doi:10.1101/gad.1787609
88. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi:10.1016/j.cell.2012.06.013
89. Matsumura T, Makino R, Mitamura K. Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin Cancer Res. 2001;7(3):594–599.
90. Zlotorynski E. Epigenetics: DNA methylation prevents intragenic transcription. Nat Rev Mol Cell Biol. 2017;18(4):212–213. doi:10.1038/nrm.2017.18
91. Traube FR, Carell T. The chemistries and consequences of DNA and RNA methylation and demethylation. RNA Biol. 2017;14(9):1099–1107. doi:10.1080/15476286.2017.1318241
92. Chiba T, Yokosuka O, Fukai K, et al. Identification and investigation of methylated genes in hepatoma. Eur J Cancer. 2005;41(8):1185–1194. doi:10.1016/j.ejca.2005.02.014
93. Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163(3):1101–1107. doi:10.1016/S0002-9440(10)63469-4
94. Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163(4):1371–1378. doi:10.1016/S0002-9440(10)63495-5
95. Arzumanyan A, Friedman T, Kotei E, Ng IO, Lian Z, Feitelson MA. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. 2012;31(5):563–572. doi:10.1038/onc.2011.255
96. Liu J, Lian Z, Han S, et al. Downregulation of E-cadherin by hepatitis B virus X antigen in hepatocellullar carcinoma. Oncogene. 2006;25(7):1008–1017. doi:10.1038/sj.onc.1209138
97. Qiu X, Qiao F, Su X, Zhao Z, Fan H. Epigenetic activation of E-cadherin is a candidate therapeutic target in human hepatocellular carcinoma. Exp Ther Med. 2010;1(3):519–523. doi:10.3892/etm_00000082
98. Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A. 1995;92(16):7416–7419. doi:10.1073/pnas.92.16.7416
99. Tanaka M, Katayama F, Kato H, et al. Hepatitis B and C virus infection and hepatocellular carcinoma in China: a review of epidemiology and control measures. J Epidemiol. 2011;21(6):401–416. doi:10.2188/jea.JE20100190
100. Lee JO, Kwun HJ, Jung JK, Choi KH, Min DS, Jang KL. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24(44):6617–6625. doi:10.1038/sj.onc.1208827
101. Arora P, Kim EO, Jung JK, Jang KL. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 2008;261(2):244–252. doi:10.1016/j.canlet.2007.11.033
102. Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279(46):48350–48359. doi:10.1074/jbc.M403427200
103. Lim SO, Gu JM, Kim MS, et al. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135(6):
104. Cheng X. Structural and functional coordination of DNA and histone methylation. Cold Spring Harb Perspect Biol. 2014;6(8):a018747. doi:10.1101/cshperspect.a018747
105. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. doi:10.1038/nbt.1685
106. Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell. 2002;109(7):801–806. doi:10.1016/s0092-8674(02)00798-5
107. Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J Hematol Oncol. 2016;9(1):49. doi:10.1186/s13045-016-0279-9
108. Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–181. doi:10.1016/j.cell.2005.01.001
109. Dong C, Wu Y, Yao J, et al. G9a interacts with snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122(4):1469–1486. doi:10.1172/JCI57349
110. Yokoyama M, Chiba T, Zen Y, et al. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget. 2017;8(13):21315–21326. doi:10.18632/oncotarget.15528
111. Ramadoss S, Chen X, Wang CY. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem. 2012;287(53):44508–44517. doi:10.1074/jbc.M112.424903
112. Monga SP. beta-catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology. 2015;148(7):1294–1310. doi:10.1053/j.gastro.2015.02.056
113. Zhang J, Tian XJ, Xing J. Signal transduction pathways of EMT induced by TGF-beta, SHH, and WNT and their crosstalks. J Clin Med. 2016;5(4):41. doi:10.3390/jcm5040041
114. Feng B, Zhu Y, Su Z, et al. Basil polysaccharide attenuates hepatocellular carcinoma metastasis in rat by suppressing H3K9me2 histone methylation under hepatic artery ligation-induced hypoxia. Int J Biol Macromol. 2018;107(Pt B):2171–2179. doi:10.1016/j.ijbiomac.2017.10.088
115. Tan JZ, Yan Y, Wang XX, Jiang Y, Xu HE. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin. 2014;35(2):161–174. doi:10.1038/aps.2013.161
116. Cai MY, Hou JH, Rao HL, et al. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol Med. 2011;17(1–2):12–20. doi:10.2119/molmed.2010.00103
117. Battistelli C, Cicchini C, Santangelo L, et al. The snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene. 2017;36(7):942–955. doi:10.1038/onc.2016.260
118. Li CP, Cai MY, Jiang LJ, et al. CLDN14 is epigenetically silenced by EZH2-mediated H3K27ME3 and is a novel prognostic biomarker in hepatocellular carcinoma. Carcinogenesis. 2016;37(6):557–566. doi:10.1093/carcin/bgw036
119. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16(4):258–264. doi:10.1038/nrm3931
120. Lei W, Zhang K, Pan X, et al. Histone deacetylase 1 is required for transforming growth factor-beta1-induced epithelial-mesenchymal transition. Int J Biochem Cell Biol. 2010;42(9):1489–1497. doi:10.1016/j.biocel.2010.05.006
121. Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21(1):103–107. doi:10.1038/5047
122. Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25(3):338–342. doi:10.1038/77124
123. Roche J, Nasarre P, Gemmill R, et al. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancers (Basel). 2013;5(2):334–356. doi:10.3390/cancers5020334
124. Yang G, Zeng X, Wang M, Wu A. The TET2/E-cadherin/beta-catenin regulatory loop confers growth and invasion in hepatocellular carcinoma cells. Exp Cell Res. 2018;363(2):218–226. doi:10.1016/j.yexcr.2018.01.011
125. Xu W, Liu H, Liu ZG, et al. Histone deacetylase inhibitors upregulate snail via smad2/3 phosphorylation and stabilization of snail to promote metastasis of hepatoma cells. Cancer Lett. 2018;420:1–13. doi:10.1016/j.canlet.2018.01.068
126. Derynck R, Zhang YE. Smad-dependent and smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi:10.1038/nature02006
127. Chang R, Zhang Y, Zhang P, Zhou Q. Snail acetylation by histone acetyltransferase p300 in lung cancer. Thorac Cancer. 2017;8(3):131–137. doi:10.1111/1759-7714.12408
128. Yokomizo C, Yamaguchi K, Itoh Y, et al. High expression of p300 in HCC predicts shortened overall survival in association with enhanced epithelial mesenchymal transition of HCC cells. Cancer Lett. 2011;310(2):140–147. doi:10.1016/j.canlet.2011.06.030
129. Song Y, Li ZX, Liu X, Wang R, Li LW, Zhang Q. The Wnt/beta-catenin and PI3K/Akt signaling pathways promote EMT in gastric cancer by epigenetic regulation via H3 lysine 27 acetylation. Tumour Biol. 2017;39(7):1010428317712617. doi:10.1177/1010428317712617
130. He Y, Meng XM, Huang C, et al. Long noncoding RNAs: novel insights into hepatocelluar carcinoma. Cancer Lett. 2014;344(1):20–27. doi:10.1016/j.canlet.2013.10.021
131. Yuan JH, Yang F, Wang F, et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25(5):666–681. doi:10.1016/j.ccr.2014.03.010
132. Zhang J, Han C, Ungerleider N, et al. A transforming growth factor-beta and H19 signaling axis in tumor-initiating hepatocytes that regulates hepatic carcinogenesis. Hepatology. 2018.
133. Zhou Y, Fan RG, Qin CL, Jia J, Wu XD, Zha WZ. LncRNA-H19 activates CDC42/PAK1 pathway to promote cell proliferation, migration and invasion by targeting miR-15b in hepatocellular carcinoma. Genomics. 2018. doi:10.1016/j.ygeno.2018.12.009
134. Ge XS, Ma HJ, Zheng XH, et al. HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci. 2013;104(12):1675–1682.
135. Liang WC, Ren JL, Wong CW, et al. LncRNA-NEF antagonized epithelial to mesenchymal transition and cancer metastasis via cis-regulating FOXA2 and inactivating Wnt/beta-catenin signaling. Oncogene. 2018;37(11):1445–1456. doi:10.1038/s41388-017-0041-y
136. Li Y, Guo D, Zhao Y, et al. Long non-coding RNA SNHG5 promotes human hepatocellular carcinoma progression by regulating miR-26a-5p/GSK3beta signal pathway. Cell Death Dis. 2018;9(9):888. doi:10.1038/s41419-018-1111-y
137. Chen W, You J, Zheng Q, Zhu YY. Downregulation of lncRNA OGFRP1 inhibits hepatocellular carcinoma progression by AKT/mTOR and Wnt/beta-catenin signaling pathways. Cancer Manag Res. 2018;10:1817–1826. doi:10.2147/CMAR.S164911
138. Zhu L, Yang N, Du G, et al. LncRNA CRNDE promotes the epithelial-mesenchymal transition of hepatocellular carcinoma cells via enhancing the Wnt/beta-catenin signaling pathway. J Cell Biochem. 2018.
139. Li SP, Xu HX, Yu Y, et al. LncRNA HULC enhances epithelial-mesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget. 2016;7(27):42431–42446. doi:10.18632/oncotarget.9883
140. Yang T, He X, Chen A, Tan K, Du X. LncRNA HOTAIR contributes to the malignancy of hepatocellular carcinoma by enhancing epithelial-mesenchymal transition via sponging miR-23b-3p from ZEB1. Gene. 2018;670:114–122. doi:10.1016/j.gene.2018.05.061
141. Dou C, Sun L, Jin X, et al. Long non-coding RNA CARLo-5 promotes tumor progression in hepatocellular carcinoma via suppressing miR-200b expression. Oncotarget. 2017;8(41):70172–70182. doi:10.18632/oncotarget.19597
142. Lan T, Chang L, Wu L, Yuan Y. Downregulation of ZEB2-AS1 decreased tumor growth and metastasis in hepatocellular carcinoma. Mol Med Rep. 2016;14(5):4606–4612. doi:10.3892/mmr.2016.5836
143. Caramel J, Ligier M, Puisieux A. Pleiotropic roles for ZEB1 in cancer. Cancer Res. 2018;78(1):30–35. doi:10.1158/0008-5472.CAN-17-2476
144. Ma M, Xu H, Liu G, et al. MITA1, a novel energy stress-inducible lncRNA, promotes hepatocellular carcinoma metastasis. Hepatology. 2019. doi:10.1002/hep.30602
145. Cao SW, Huang JL, Chen J, et al. Long non-coding RNA UBE2CP3 promotes tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget. 2017;8(39):65370–65385. doi:10.18632/oncotarget.18524
146. Xiao JN, Yan TH, Yu RM, et al. Long non-coding RNA UCA1 regulates the expression of snail2 by miR-203 to promote hepatocellular carcinoma progression. J Cancer Res Clin Oncol. 2017;143(6):981–990. doi:10.1007/s00432-017-2370-1
147. Sun J, Wei X, Xu L. Upregulation of lncRNA Sox2ot indicates a poor prognosis for patients with hepatocellular carcinoma and promotes cell invasion. Oncol Lett. 2018;16(1):1189–1195. doi:10.3892/ol.2018.8725
148. Zhou M, Zhang XY, Yu X. Overexpression of the long non-coding RNA SPRY4-IT1 promotes tumor cell proliferation and invasion by activating EZH2 in hepatocellular carcinoma. Biomed Pharmacother. 2017;85:348–354. doi:10.1016/j.biopha.2016.11.035
149. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi:10.1038/nrg2843
150. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–222. doi:10.1038/nrd.2016.246
151. Roderburg C, Trautwein C. Cell-specific functions of miRNA in the liver. J Hepatol. 2017;66(3):655–656. doi:10.1016/j.jhep.2016.09.015
152. Hsu SH, Delgado ER, Otero PA, et al. MicroRNA-122 regulates polyploidization in the murine liver. Hepatology. 2016;64(2):599–615. doi:10.1002/hep.28573
153. Jin Y, Wang J, Han J, Luo D, Sun Z. MiR-122 inhibits epithelial-mesenchymal transition in hepatocellular carcinoma by targeting snail1 and Snail2 and suppressing WNT/beta-cadherin signaling pathway. Exp Cell Res. 2017;360(2):210–217. doi:10.1016/j.yexcr.2017.09.010
154. Wang N, Wang Q, Shen D, Sun X, Cao X, Wu D. Downregulation of microRNA-122 promotes proliferation, migration, and invasion of human hepatocellular carcinoma cells by activating epithelial-mesenchymal transition. Onco Targets Ther. 2016;9:2035–2047. doi:10.2147/OTT.S92378
155. Liang HW, Wang N, Wang Y, et al. Hepatitis B virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J Hepatol. 2016;64(2):278–291. doi:10.1016/j.jhep.2015.09.013
156. Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. doi:10.1101/gad.1640608
157. Ding W, Dang H, You H, et al. miR-200b restoration and DNA methyltransferase inhibitor block lung metastasis of mesenchymal-phenotype hepatocellular carcinoma. Oncogenesis. 2012;1:e15. doi:10.1038/oncsis.2012.15
158. Wu G, Zheng K, Xia S, et al. MicroRNA-655-3p functions as a tumor suppressor by regulating ADAM10 and beta-catenin pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2016;35(1):89. doi:10.1186/s13046-016-0444-6
159. Li H, Wang H, Ren Z. MicroRNA-214-5p inhibits the invasion and migration of hepatocellular carcinoma cells by targeting wiskott-aldrich syndrome like. Cell Physiol Biochem. 2018;46(2):757–764. doi:10.1159/000488734
160. Huang J, Wang Y, Guo Y, Sun S. Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology. 2010;52(1):60–70. doi:10.1002/hep.23660
161. Wu LQ, Lu Y, Lu HJ, Lv ZH. Can E-cadherin and CD34 be used as indicators of prognosis for hepatocellular carcinoma patients? Clin Chem Lab Med. 2008;46(8):1122–1126. doi:10.1515/CCLM.2008.231
162. Chen J, Zhao J, Ma R, Lin H, Liang X, Cai X. Prognostic significance of E-cadherin expression in hepatocellular carcinoma: a meta-analysis. PLoS One. 2014;9(8):e103952. doi:10.1371/journal.pone.0103952
163. Lachenmayer A, Toffanin S, Cabellos L, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol. 2012;56(6):1343–1350. doi:10.1016/j.jhep.2012.01.009
164. Catela Ivkovic T, Voss G, Cornella H, Ceder Y. microRNAs as cancer therapeutics: a step closer to clinical application. Cancer Lett. 2017;407:113–122. doi:10.1016/j.canlet.2017.04.007
165. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. doi:10.1038/nrg3763
166. Zhang J, Han C, Ungerleider N, et al. A transforming growth factor-beta and H19 signaling axis in tumor-initiating hepatocytes that regulates hepatic carcinogenesis. Hepatology. 2019;69(4):1549–1563. doi:10.1002/hep.30153
167. Ye Y, Xu Y, Lai Y, et al. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J Cell Biochem. 2018;119(3):2951–2963. doi:10.1002/jcb.26509
168. Ding G, Peng Z, Shang J, Kang Y, Ning H, Mao C. LincRNA-p21 inhibits invasion and metastasis of hepatocellular carcinoma through miR-9/E-cadherin cascade signaling pathway molecular mechanism. Onco Targets Ther. 2017;10:3241–3247. doi:10.2147/OTT.S134910
169. Liu J, Lu C, Xiao M, Jiang F, Qu L, Ni R. Long non-coding RNA SNHG20 predicts a poor prognosis for HCC and promotes cell invasion by regulating the epithelial-to-mesenchymal transition. Biomed Pharmacother. 2017;89:857–863. doi:10.1016/j.biopha.2017.01.011
170. Heller G, Altenberger C, Steiner I, et al. DNA methylation of microRNA-coding genes in non-small-cell lung cancer patients. J Pathol. 2018;245(4):387–398. doi:10.1002/path.5079
171. Gou X, Zhao X, Wang Z. Long noncoding RNA PVT1 promotes hepatocellular carcinoma progression through regulating miR-214. Cancer Biomark. 2017;20(4):511–519. doi:10.3233/CBM-170331
172. Deng X, Zhao XF, Liang XQ, Chen R, Pan YF, Liang J. Linc00152 promotes cancer progression in hepatitis B virus-associated hepatocellular carcinoma. Biomed Pharmacother. 2017;90:100–108. doi:10.1016/j.biopha.2017.03.031
173. Zhou T, Gao Y. Increased expression of LncRNA BANCR and its prognostic significance in human hepatocellular carcinoma. World J Surg Oncol. 2016;14(1):8. doi:10.1186/s12957-015-0757-5
174. Xu Y, Wang B, Zhang F, et al. Long non-coding RNA CCAT2 is associated with poor prognosis in hepatocellular carcinoma and promotes tumor metastasis by regulating snail2-mediated epithelial-mesenchymal transition. Onco Targets Ther. 2017;10:1191–1198. doi:10.2147/OTT.S127100
175. Zeng B, Lin Z, Ye H, et al. Upregulation of LncDQ is Associated with poor prognosis and promotes tumor progression via epigenetic regulation of the EMT Pathway in HCC. Cell Physiol Biochem. 2018;46(3):1122–1133. doi:10.1159/000488841
176. Wu Y, Yuan T, Wang WW, et al. Long noncoding RNA HOST2 promotes epithelial-mesenchymal transition, proliferation, invasion and migration of hepatocellular carcinoma cells by activating the JAK2-STAT3 signaling pathway. Cell Physiol Biochem. 2018;51(1):301–314. doi:10.1159/000495231
177. Xia C, Zhang XY, Liu W, et al. LINC00857 contributes to hepatocellular carcinoma malignancy via enhancing epithelial-mesenchymal transition. J Cell Biochem. 2018.
178. Yao Z, Xiong Z, Li R, Liang H, Jia C, Deng M. Long non-coding RNA NRON is downregulated in HCC and suppresses tumour cell proliferation and metastasis. Biomed Pharmacother. 2018;104:102–109. doi:10.1016/j.biopha.2018.05.006
179. Yang J, Li J, Liu B, et al. Long noncoding RNA AK021443 promotes cell proliferation and migration by regulating epithelial-mesenchymal transition in hepatocellular carcinoma cells. DNA Cell Biol. 2018;37(5):481–490. doi:10.1089/dna.2017.4030
180. Giovannini C, Fornari F, Dallo R, et al. MiR-199-3p replacement affects E-cadherin expression through notch1 targeting in hepatocellular carcinoma. Acta Histochem. 2018;120(2):95–102. doi:10.1016/j.acthis.2017.12.004
181. Gou Y, Zhai F, Zhang L, Cui L. RUNX3 regulates hepatocellular carcinoma cell metastasis via targeting miR-186/E-cadherin/EMT pathway. Oncotarget. 2017;8(37):61475–61486. doi:10.18632/oncotarget.18424
182. Tan HX, Wang Q, Chen LZ, et al. MicroRNA-9 reduces cell invasion and E-cadherin secretion in SK-Hep-1 cell. Med Oncol. 2010;27(3):654–660. doi:10.1007/s12032-009-9264-2
183. Drakaki A, Hatziapostolou M, Polytarchou C, et al. Functional microRNA high throughput screening reveals miR-9 as a central regulator of liver oncogenesis by affecting the PPARA-CDH1 pathway. BMC Cancer. 2015;15:542. doi:10.1186/s12885-015-1584-3
184. Kogure T, Kondo Y, Kakazu E, Ninomiya M, Kimura O, Shimosegawa T. Involvement of miRNA-29a in epigenetic regulation of transforming growth factor-beta-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Hepatol Res. 2014;44(8):907–919. doi:10.1111/hepr.12188
185. Chang L, Li K, Guo T. miR-26a-5p suppresses tumor metastasis by regulating EMT and is associated with prognosis in HCC. Clin Transl Oncol. 2017;19(6):695–703. doi:10.1007/s12094-016-1582-1
186. Guo XF, Wang AY, Liu J. HIFs-MiR-33a-twsit1 axis can regulate invasiveness of hepatocellular cancer cells. Eur Rev Med Pharmacol Sci. 2016;20(14):3011–3016.
187. Tian Z, Jiang H, Liu Y, et al. MicroRNA-133b inhibits hepatocellular carcinoma cell progression by targeting Sirt1. Exp Cell Res. 2016;343(2):135–147. doi:10.1016/j.yexcr.2016.03.027
188. Huang JY, Zhang K, Chen DQ, et al. MicroRNA-451: epithelial-mesenchymal transition inhibitor and prognostic biomarker of hepatocelluar carcinoma. Oncotarget. 2015;6(21):18613–18630. doi:10.18632/oncotarget.4317
189. Xia W, Ma X, Li X, et al. miR-153 inhibits epithelial-to-mesenchymal transition in hepatocellular carcinoma by targeting snail. Oncol Rep. 2015;34(2):655–662. doi:10.3892/or.2015.4008
190. Liu Y, Li Y, Wang R, et al. MiR-130a-3p regulates cell migration and invasion via inhibition of Smad4 in gemcitabine resistant hepatoma cells. J Exp Clin Cancer Res. 2016;35:19. doi:10.1186/s13046-016-0444-6
191. Tu K, Zheng X, Dou C, et al. MicroRNA-130b promotes cell aggressiveness by inhibiting peroxisome proliferator-activated receptor gamma in human hepatocellular carcinoma. Int J Mol Sci. 2014;15(11):20486–20499. doi:10.3390/ijms151120486
192. Lian J, Jing Y, Dong Q, et al. miR-192, a prognostic indicator, targets the SLC39A6/SNAIL pathway to reduce tumor metastasis in human hepatocellular carcinoma. Oncotarget. 2016;7(3):2672–2683. doi:10.18632/oncotarget.6603
193. Xu Q, Zhu Q, Zhou Z, et al. MicroRNA-876-5p inhibits epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma by targeting BCL6 corepressor like 1. Biomed Pharmacother. 2018;103:645–652. doi:10.1016/j.biopha.2018.04.037
194. Hung CS, Liu HH, Liu JJ, et al. MicroRNA-200a and −200b mediated hepatocellular carcinoma cell migration through the epithelial to mesenchymal transition markers. Ann Surg Oncol. 2013;20(Suppl 3):S360–368. doi:10.1245/s10434-012-2482-4
195. Wang J, Yang X, Ruan B, et al. Overexpression of miR-200a suppresses epithelial-mesenchymal transition of liver cancer stem cells. Tumour Biol. 2015;36(4):2447–2456. doi:10.1007/s13277-014-2856-2
196. Liu D, Wu J, Liu M, Yin H, He J, Zhang B. Downregulation of miRNA-30c and miR-203a is associated with hepatitis C virus core protein-induced epithelial-mesenchymal transition in normal hepatocytes and hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2015;464(4):1215–1221. doi:10.1016/j.bbrc.2015.07.107
197. Wu WL, Wang WY, Yao WQ, Li GD. Suppressive effects of microRNA-16 on the proliferation, invasion and metastasis of hepatocellular carcinoma cells. Int J Mol Med. 2015;36(6):1713–1719. doi:10.3892/ijmm.2015.2379
198. Ma DN, Chai ZT, Zhu XD, et al. MicroRNA-26a suppresses epithelial-mesenchymal transition in human hepatocellular carcinoma by repressing enhancer of zeste homolog 2. J Hematol Oncol. 2016;9:1. doi:10.1186/s13045-015-0229-y
199. Zhu SM, Chen CM, Jiang ZY, et al. MicroRNA-185 inhibits cell proliferation and epithelial-mesenchymal transition in hepatocellular carcinoma by targeting six2. Eur Rev Med Pharmacol Sci. 2016;20(9):1712–1719.
200. Kim HS, Lee KS, Bae HJ, et al. MicroRNA-31 functions as a tumor suppressor by regulating cell cycle and epithelial-mesenchymal transition regulatory proteins in liver cancer. Oncotarget. 2015;6(10):8089–8102.
201. Li DP, Fan J, Wu YJ, Xie YF, Zha JM, Zhou XM. MiR-155 up-regulated by TGF-beta promotes epithelial-mesenchymal transition, invasion and metastasis of human hepatocellular carcinoma cells in vitro. Am J Transl Res. 2017;9(6):2956–2965.
202. Shen G, Lin Y, Yang X, Zhang J, Xu Z, Jia H. MicroRNA-26b inhibits epithelial-mesenchymal transition in hepatocellular carcinoma by targeting USP9X. BMC Cancer. 2014;14:393. doi:10.1186/1471-2407-14-393
203. Zhang JG, Shi Y, Hong DF, et al. MiR-148b suppresses cell proliferation and invasion in hepatocellular carcinoma by targeting WNT1/beta-catenin pathway. Sci Rep. 2015;5:8087. doi:10.1038/srep08087
204. Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM. MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting met/snail signaling. Oncogene. 2014;33(31):4069–4076. doi:10.1038/onc.2013.369
205. Wang W, Lin H, Zhou L, et al. MicroRNA-30a-3p inhibits tumor proliferation, invasiveness and metastasis and is downregulated in hepatocellular carcinoma. Eur J Surg Oncol. 2014;40(11):1586–1594. doi:10.1016/j.ejso.2013.11.008
206. Han LL, Yin XR, Zhang SQ. miR-103 promotes the metastasis and EMT of hepatocellular carcinoma by directly inhibiting LATS2. Int J Oncol. 2018;53(6):2433–2444.
207. Zhao S, Zhang Y, Zheng X, et al. Loss of MicroRNA-101 promotes epithelial to mesenchymal transition in hepatocytes. J Cell Physiol. 2015;230(11):2706–2717. doi:10.1002/jcp.24995
208. Zhou Y, Li Y, Ye J, et al. MicroRNA-491 is involved in metastasis of hepatocellular carcinoma by inhibitions of matrix metalloproteinase and epithelial to mesenchymal transition. Liver Int. 2013;33(8):1271–1280. doi:10.1111/liv.12190
209. Hou X, Yang L, Jiang X, et al. Role of microRNA-141-3p in the progression and metastasis of hepatocellular carcinoma cell. Int J Biol Macromol. 2019;128:331–339. doi:10.1016/j.ijbiomac.2019.01.144
210. Liang C, Xu Y, Ge H, et al. miR-564 inhibits hepatocellular carcinoma cell proliferation and invasion by targeting the GRB2-ERK1/2-AKT axis. Oncotarget. 2017;8(64):107543–107557. doi:10.18632/oncotarget.22504
211. Gao J, Aksoy BA, Dogrusoz U. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.