")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Future management of human obesity: understanding the meaning of genetic susceptibility
Authors Jenkins A, Campbell L
Received 27 June 2014
Accepted for publication 18 August 2014
Published 5 December 2014 Volume 2014:4 Pages 219—232
DOI https://doi.org/10.2147/AGG.S53594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr John Martignetti
Arthur B Jenkins,1,2 Lesley V Campbell2,3
1School of Medicine, University of Wollongong, Wollongong, NSW, Australia; 2Diabetes and Obesity Research Program, Garvan Institute of Medical Research, Sydney, NSW, Australia; 3Diabetes Centre and Department of Endocrinology, St Vincent's Hospital, Sydney, NSW, Australia
Abstract: Gene–environment interactions are central to the expression of obesity. The condition is strongly heritable (ie, genetic), and most of the variation in obesity levels between countries and between individuals can be explained by the effects of obesogenic environments on individual genetic susceptibilities. The nature of the obesogenic environmental influences is not clear in detail, but they correlate closely with measures of affluence. The causes of variation in genetic susceptibility are also not clearly defined, but their general nature has become clearer. The failure of genome-wide association studies or large linkage studies to identify or replicate causative genetic variants, together with the segregation of obesity-related traits in families, implicates a heterogenetic mechanism in which rare, dominantly or additively expressed genetic variants are responsible for most of common obesity. The search for rare causative variants continues with some successes, but those identified contribute very little to the overall burden and, assuming heterogenetics, there are many more to find. The time when genomic risk factors provide more information than do currently available markers, such as family history, is a long way off. Genomic studies to date have contributed little, if anything, to the prevention and treatment of common obesity and its associated disorders. This contrasts with the obvious and immediate potential implications of the well-established overall genetic basis of obesity, which have not yet been exploited in the clinical or public health arenas. Genomic studies, which have helped to define the genetic basis of common obesity mainly by exclusion, will in the future play an increasingly important role in understanding and managing obesity, but only with parallel studies of the physiological, behavioral, and economic influences.
Keywords: obesity phenotypes, obesogenic environment, genomics, pathophysiology, treatment, prevention
Background and aims
According to the World Health Organization projections, by 2015 about 2.3 billion adults in the world will be overweight and over 700 million will be obese, and economic costing predicts that obesity-related expenditure in USA will approach US$100 billion per annum.1 It is now widely, if not universally, accepted that the rising national and global prevalence of overweight and obesity since the 1980s can be understood as the effects of increasingly obesogenic environments (OEs), which are correlated with measures of affluence and food availability,2 in genetically predisposed individuals.3,4
Obesity is assessed by the body mass index (BMI, kg/m2) in most epidemiological and genetic studies, often together with measures of central fat distribution such as waist circumference because of adverse metabolic and health associations. However, neither BMI nor other anthropometric measurements are by themselves sufficiently accurate or precise as measures of body fatness in cross-sectional studies.5–8 Further progress in our understanding of the genetic underpinnings of obesity will depend on, among other things, the use of more informative phenotypes.
The large number of potential proximal environmental factors responsible for promoting overconsumption and/or underexpenditure of energy and their relative importance is under lively debate (eg, Luke and Cooper, associated commentaries, and response9), but the evidence of a primary role of food availability and marketing is persuasive for most.2 Preventative public health efforts targeting food consumption and/or physical activity have not yet been successful10,11 and can appear weak against the pressure for commercial profit: eg, the long campaign directed at reducing consumption of sugar-sweetened beverages while consumption has increased fivefold since 1950.12 Similarly, individual weight loss programs have had very limited, if any, success as defined by long-term (>2 year) reduced weight maintenance.10,11,13 Past pharmaceutical approaches have been equally disappointing, both in terms of modest treatment effects and unacceptable side effects.14,15 At present, the most successful treatments for obesity are surgical procedures, still with limited application for most obese and overweight people.14,16
The classic twin adoption studies of Stunkard showed that both fatness and thinness were highly heritable and not influenced by the adopting family members’ adiposity.17 Numerous genetic studies since, in a variety of populations and using various measures of adiposity, have been consistent with those fundamental findings.4 The nature of the genetic variation responsible for this heritability is becoming clear even though the particular genetic variants are not. Evidence of segregation of obesity-related phenotypes in families18–20 and the failure to account for the heritability in genome-wide association studies (GWAS)3,21 and linkage studies22 strongly favor a predominant role for rare variants with large effects expressed under the influence of OE. The physiological mechanism(s) responsible has/have not been established, but the genetic and physiological information currently available remain consistent with the neurobehavioral hypothesis (NBH) proposed by O’Rahilly and Farooqi.23 In NBH, common obesity is the result of widespread genetic susceptibility to environmental cues related to food intake,18,24 mediated by appetite-regulating pathways within the hypothalamus. The mechanisms linking OE to the expression of obesogenic gene variant effects are not yet clear.
We aim here to review the recent evidence that leads us to accept the working model of the obesity epidemic summarized above, to discuss the physiological, clinical, and public health implications of the model, and to discuss ways in which future genomic research could enhance our understanding of causes and potential treatments of obesity.
Obesity phenotypes have not been adequate in most genetic and genomic studies
Common obesity
We have clinical and phenotypic modeling backgrounds but claim no technical or theoretical expertise in genomics. However, we argue that the outstanding problems in the genetics of obesity are intimately connected with our areas of interest and that further progress in genomic studies is limited by the inadequacies of the phenotypes used in most studies. The choice of phenotypic markers for expensive, large-scale genomic studies is usually restricted to anthropometric measures, BMI, and/or circumferences (waist, hip). Neither these measures nor the simplistic clinical management phenotypes derived from them adequately represent the level of body fatness,5–8 and their continuing use contributes to the current uncertainties.6,25
More direct measures of body fatness based on skinfold thickness, bioelectric impedance analysis,26 dual-energy X-ray absorptiometry (DEXA), or hydrodensitometry27 have been used in a limited number of genomic studies without obvious benefits regarding gene discovery. With the more direct measures (skinfolds, DEXA, hydrodensitometry), the high cost, or other logistic requirements of phenotyping have restricted the applications to relatively small samples. While bioelectric impedance analysis is cheaper, its confounding by variability in hydration levels degrades its accuracy regarding body fat content.28,29
The concept of increased adiposity or body fatness appears intuitively simple but is not easy to define or measure accurately in humans. The direct measures mentioned earlier might provide effective approaches to measurement apart from their logistical problems, but even if an accurate measure of, eg, total fat mass is obtained, it is not clear a priori how to express it as a biologically meaningful index of increased adiposity. The common clinical usage of percent body fat cutoffs is arbitrary.30 Analyzing continuous measures such as the percent body fat or fat mass indices implies assumptions about how human adiposity affects, or is affected by, disease processes. Using either BMI or percent body fat as covariates can lead to erroneous conclusions in a genetic context.31 One possible approach is the use of multivariate phenotypic constructs obtained from techniques such as principal components or factor analysis, which partition correlations between multiple input variables into latent factors. These procedures eliminate uncorrelated components of the input variables, which can reduce error variance and hence increase statistical power. Similar improvements in power are expected in other types of multivariate analysis such as structural equation modeling.32,33 Recently, we reported such an analysis of body composition data using factor analysis in a small-scale study (n~200). We extracted a factor (Adiposity), which captured more precisely the segregation in families of obesity-related traits than did either BMI or measured percent body fat.18 However, the detailed phenotypic characterization required for that particular analysis may be impractical for large-scale genomic studies. The expected power benefits from multivariate phenotypes are most apparent when there are strong correlations between the individual phenotypes, as is the case with many phenotypes obtained in genomic studies of obesity, but this is not the only requirement for derivation of useful phenotypes. The aim must be to construct biologically informative phenotypes which, in principle, could be achieved either by applying prior knowledge to the development of explicit physiological models composed of measurable variables31 or by careful consideration of the biological and statistical properties of the available raw phenotypes.18 It is also important to consider the origins of the correlations between phenotypic markers, so as to specify the correct functional forms of the relationships, and especially to avoid correlated errors when choosing phenotypes for inclusion. For example, in our own study,18 which used a combination of anthropometric and DEXA measurements, we used log-transformations to accommodate the dimensional relationships between input variables and avoided inclusion of multiple phenotypes based on the same primary measurement (eg, fat mass and percent fat) and therefore sharing correlated errors. These precautions, which amount to standard requirements for the extraction of reliable multivariate constructs, have generally not been applied in multivariate genomic or genetic studies.20,34
The choice of phenotype(s) for genomic studies of common obesity is unresolved. If, as we discuss in the following sections, the potential utility of large-scale association studies has been exhausted, more expensive phenotypic measures may be justified in smaller scale case–control or linkage studies. It seems likely to us that any single phenotypic measure would contain less genetic information than carefully designed multivariate constructs. For example, multivariate constructs derived from commonly obtained anthropometric measurements (weight, height, and, waist–hip circumferences) may be worth considering. However, we expect that the additional costs of direct measures of body fat would be more than repaid by the increased power and interpretability of the resulting analyses.
Sub-obesity phenotypes
Clinically, obesity and/or overweight are not homogeneous phenotypes. There are prospects for identifying useful genetic markers for those at greater or lesser risk of adverse health consequences of obesity, which would assist in developing or targeting treatments. There is growing evidence that a substantial subset of obese individuals (~25%), defined by a lack of risk factors for comorbidities, may be protected from some or all of the adverse consequences of obesity.35 There is an established association between abdominal, especially visceral, fat deposition and adverse health consequences at least partly independent of total adiposity, which may be under independent genetic influences.36,37 Identification of genetic determinants of these clinical phenotypes, we submit, depends on the development of genetically informative quantitative phenotypes using the principles summarized earlier.
There are no effective nonsurgical treatments or preventative strategies for obesity
Despite much nutritional and physical activity research, no lifestyle regimen has been reported to maintain substantial weight loss in the long term.38 This result is consistent with the strong feedback of metabolic and appetite-regulating hormones to the hypothalamus when weight loss does occur in the obese,39 a finding consistent with the predictions of the NBH. Similarly, public health initiatives promoting lifestyle changes at the population level have been minimally, if at all, effective in reducing obesity levels.11 There is almost no other area in drug development that is replete with as many failures and withdrawals as seen in obesity.40 Current novel obesity agents include a 5HT2c receptor agonist (Lorcaserin), a combination of phentermine and extended-release topiramate (Qysmia), both of which have modest treatment effects and side effects of concern to regulatory agencies41 and a peripherally acting drug (Beloranib, a methionine aminopeptidase inhibitor) currently in Phase III trials, so far without major side effects.42 Of those three, only Beloranib offers a prospect of broad usage, but to date, results of treatment have only been reported out to 12 weeks. Currently, only bariatric surgery achieves long-term weight loss: by physical restriction (banding), by increasing gastrointestinal satiety hormone release (gastric bypass), and/or by diverting nutrients.43,44
Despite this lack of effective treatments and strategies, some governments are being advised to consider financial penalties for obese patients in regard to health insurance, justified by the belief that obesity is a personal choice.45 In the USA, the National Institutes of Health has funded the Childhood Obesity Prevention and Treatment Research (COPTR) consortium formed to prevent obesity in preschoolers and treat obesity in 1,700 13–17 year olds.46 Reports of recent childhood lifestyle interventions in randomized trials showed no significant weight effect despite employing the currently regarded optimum methodology.47 For example, 12-month follow-up of shared care management in 3–10 year olds in a randomized trial showed no better BMI outcomes for intervention than untreated controls (intervention effect −0.1 [−0.7, 0.5] kg/m2).48 Against this background of failure, the possible emotional impact of frequent measurements of body size and weight of a child (and mother)49,50 should not be ignored when considering large-scale projects such as COPTR.46 It is also not possible at present to determine which young people are “healthy” obese, and arguably, who do not require medical intervention for improved metabolic health.35 In summary, the treatment issue in obesity is the lack of proven long-term effective treatments at both the individual and population levels.
Genetic variants contributing to common obesity are expressed under obesogenic environmental influences
Gene–environment interactions are central to the expression of obesity. The rapid rise in prevalence in developed countries over the last three decades, coupled with the strong heritability of obesity, allows no other conclusion.51 Obesity rates continue to rise in developing countries, but are at or near saturation in developed countries.52,53 While the identity and strengths of the proximal factors comprising OE are not agreed upon, it is clear that indices of affluence either within (eg, socioeconomic status [SES]) or between (eg, gross domestic product [GDP]) populations can be powerful proxies for total OE.54–58 The effect of OE by those measures on obesity levels is not linear, with evidence of saturation in developed in developed countries56 consistent with reports of diminishing national trends in some developed countries. The apparent plateau level of national BMI at high GDP (~26 kg/m2)56 is well below a notional full expression of clinically defined obesity (BMI≥30 kg/m2), indicating that substantially <100% of individuals are susceptible to OE. Within countries, the situation is more complex.54,55 In multiple samples from 36 developing countries, a positive effect of individual SES explained 74% of the within-country variance in BMI,58 but in developed countries the relationship is often reversed, with low SES groups showing the highest obesity prevalence.54,55 The reasons for this apparent reversal of effect are not clear. Likely contributors include the influence educational level on obesogenic behaviors and negative effects of obesity on SES.59 There are also methodological issues regarding confounding variables, particularly associations between measures of SES and ethnicity. However, in the most extensively studied, if not necessarily representative, country60 (USA), the association between one measure of SES (education) and BMI has weakened over the period 1970–2000.61 It may be that SES now plays a more minor role compared to the national level of affluence in the most developed countries.
Measures of affluence explain approximately 50% of the between-country variance in BMI and approximately 70% of the within-country BMI variance in developing countries.58 No gene-discovery studies (GWAS or linkage or candidate) that we are aware of have utilized any markers of OE as covariates. Omitting such a powerful conditional determinant from genetic studies, perhaps unavoidable with the uncertainties of identity and measurement, must lead to a reduced power to detect genetic associations and to artifactual associations if population stratification, either within or between countries, leads to confounding between OE and genetic markers.
The mechanistic link(s) at the molecular level between OE and obesity development are not clear. The physiological evidence implicates hypothalamic appetite-regulating pathways with their various sensor and effector systems,23,24 and the genomic evidence (see “Obesogenic genetic variants”) still supports the basic structure of the NBH. In this scheme, reduced (or increased) function in various components leads to either an increased drive to eat in the presence of OE and/or, equivalently, a decreased restraint of an appropriate drive in response to feedback signals. In either case, without counteracting changes in energy expenditure, the expected result is maintenance of larger fat stores at a level that either balances the increased drive with higher feedback signals or normalizes the feedback signal itself in the dysfunctional pathway or parallel (redundant) pathways.
There is currently great interest in the possibility that epigenetic changes to gene function could provide a molecular link between OE and obesogenic gene variant effects. This proposed mechanism is a key element in the hypothesis of the developmental origins of health and disease (DOHaD), which proposes that a substantial fraction of the rising prevalence of obesity (and other complex diseases) is due to exposures to OE in utero.62,63 Environmental conditions, including nutrient availability, can epigenetically influence expression of genes involved in energy metabolism64,65 and at least one component of NBH (proopiomelanocortin [POMC]66) in humans. Epigenetic DNA methylation may be enriched and more variable in obese compared to nonobese humans.67 These and many other studies, predominantly in animal models, have clearly established the potential of DOHaD, but much less evidence exists of any substantial contribution to endemic human obesity. Direct evidence in humans comes from studies of the effects of maternal obesity on offspring phenotypes, studies of which are very difficult to design with appropriate controls; most do not account for the genetic influences on obesity which will confound any effects of in utero environment. The strongest human evidence is in a series of studies of the effects of maternal bariatric surgery on obesity in children, using a within-subject design (pre- and post-surgery) to control for genetic influences. Children conceived after surgery had substantially lower levels of various obesity-related phenotypes compared to those conceived before surgery68 and showed an altered DNA methylation pattern in white blood cells.69 However, there are reasons to doubt that these findings are applicable to endemic obesity. Both studies used morbidly obese subjects (BMI ≈46 kg/m2) undergoing the most drastic form of bariatric surgery (biliopancreatic diversion) which resulted in massive weight loss (BMI ≈30 kg/m2), which itself results in nutritional deficiencies in utero.70 The incidence of folate deficiency is a particular concern for the interpretation of the DNA methylation pattern.62 In contrast, a large hospital-records-based within-subject study of the effects of much less severe, purely restrictive bariatric surgery (BMI change from 36 kg/m2 to 32 kg/m2) found no evidence for reduced adiposity in children conceived after surgery, with trends in the opposite direction.71 Other studies were designed to isolate any nongenetic effect of maternal adiposity by use of maternal FTO genotype as an instrumental variable72 or by comparing influences of maternal and paternal adiposity on offspring adiposity.73 Both studies concluded that any maternal effect on offspring adiposity is weak and unlikely to explain any substantial fraction of the obesity epidemic. So while DOHaD and its proposed epigenetic mechanisms are based on undoubtedly exciting emerging biological insights, we found no persuasive evidence for an important role of DOHaD in endemic human obesity.
Heritability and mode of inheritance of obesity susceptibility
Obesity is highly heritable, but estimates of the degree of heritability have varied substantially between studies (h2~40%–80%). The source(s) of this variability have not been established, but a leading candidate is the effects of variations and trends in OE. For example, twin studies generally give higher heritability estimates than do sibling studies or multigenerational family studies, perhaps because OE is likely to vary less between twins than between generations or siblings of different ages.19 Similarly, as obesity levels have risen over time in response to rising OE, so have the measured genetic contributions to the overall variance, and hence h2.74 Many studies have detected evidence of segregation and/or multimodality of adiposity-related traits but the implied modes of inheritance have varied. Early studies (reviewed in Price et al19) found evidence of recessive expression of higher adiposity, while more recent studies have tended to favor dominant or additive models of expression.18–20 As argued by Price et al,19 part of this variation may be due to secular trends in gene–OE interactions,74 which have the effect of simulating recessive inheritance in multigenerational studies. The use of extreme obesity phenotypes would also tend to favor a recessive pattern if the true mode is additive and the heterozygous phenotype is either obscured by definition75 or is indistinguishable in the data.76,77 The use of inappropriate phenotypes can also obscure signals of dominance or additivity. Our own demonstration of segregation of Adiposity in families with type 2 diabetes mellitus (T2DM)18 was obtained only with a multivariate phenotype based on direct measures of body compartments (Figure 1A), and was obscured in body fat percent and BMI data (Figure 1B), despite the close correlations between Adiposity and body fat percent or BMI (R2=0.87, Figure 1C). The genetic information is contained within the apparent noise in Figure 1C. In summary, published studies are either not inconsistent with, or give explicit support to, dominant or additive expression of genetic susceptibility to increased adiposity. Because of its use of an informative and biologically plausible phenotype and a highly enriched sample (family history positive compared to a carefully matched family history negative control group) from a developed (OE-saturated) country, and despite its small sample size (n=202), we submit our study18 as the best current indicator of the mode of inheritance of susceptibility to common obesity.
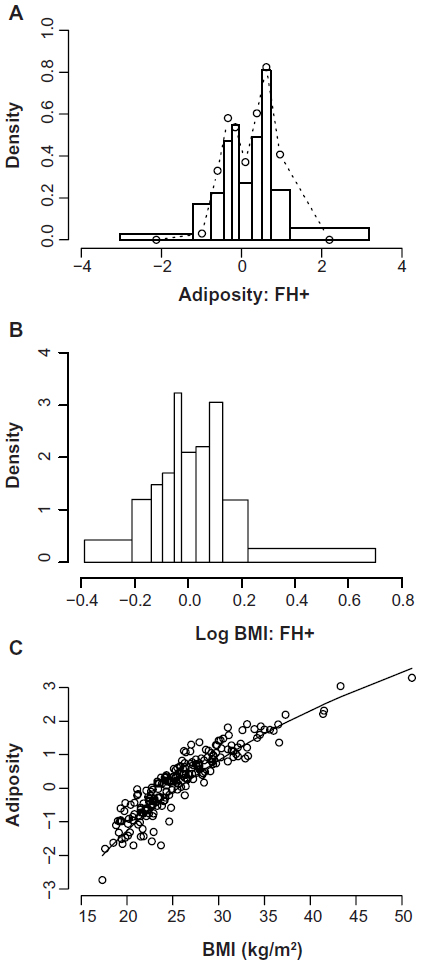 | Figure 1 Distributions of age- and sex-adjusted obesity-related phenotypes in healthy adults with a family history of T2DM (FH+, n=80). |
The bimodal distribution of Adiposity in individuals with a positive family history of T2DM (FH+, Figure 1A) is consistent with segregation in families of rare obesity susceptibility variants with major effects, contrary to the predictions of a polygenic model in which variants at many loci have cumulative, small effects on the phenotype. Previous studies of the transmission of obesity-related phenotypes have also generally not favored the polygenic model. However, a recent simulation study based on extensive identity by descent and BMI data in families reaches the conclusion that polygenic inheritance of BMI is capable of explaining the failure of replication of hits in linkage studies, possibly by random sampling of chromosomal blocks containing multiple obesogenic variants of small effects, and therefore questions the conclusion that a heterogenetic model is favored by the exclusion of competing hypotheses.78 Their conclusion is based on the equivalence of various polygenetic and heterogenetic models, rather than on statistical evidence favoring any particular model. They prefer the polygenic model for reasons of parsimony, although it seems to us the least parsimonious of their mechanisms. The heterogenetic models tested were very stringently defined in terms of private variants in each family, ie, a minor allele fraction (MAF) of ~0.01% vide the usual definition of rare (MAF <1%) based on the current power of GWAS.79 Whether the result could apply to more realistic situations in which, for example, interactions between genes and environment were considered and the heterogenetic model was less stringently defined is not yet clear, and we take the current evidence for segregation of obesity-related phenotypes to strongly favor the heterogenetic model.
Our analysis18 was insensitive to the nature of the inherited obesogenic variants, but we can conclude that they are rare to have escaped detection in large-scale studies even of BMI, even allowing for confounding by OE, and must have large effects to account for the separation between modes of Adiposity (0.93 standard deviation units [SD]). The two factors extracted by Tayo et al,20 which are likely to partition the variance in Adiposity, show similar effect sizes (0.84–0.89 SD). The BMI equivalent of the effect size in Adiposity is approximately 4 kg/m2, and other segregation studies have reported similarly large effects although direct comparisons are hindered by the variety of phenotypes analyzed (BMI,19 fat mass,80 percent body fat,81 skinfolds82), the different populations sampled, and the genetic models tested (additive, dominant, mixed). Our major effects accounted for 91% for the age- and sex-adjusted Adiposity variance, but this high value is a function of the enrichment of susceptibility in our sample (positive family history) as well as the high national GDP and hence OE. The residual variance (9%; 95% CI, 4%–69%) contains the effects of all other determinants of Adiposity including polygenes, the environment, and interactions between them. While the point estimate indicates minimal contribution from all these sources in our sample, the wide confidence intervals (probably related to small sample size) do not rule out a substantial contribution in this or other samples. However, taken with the other estimates of heritability of obesity-related phenotypes and their strengths and limitations referred to above, our result supports a predominant role of segregating rare variants in the transmission of obesity susceptibility.
Obesogenic genetic variants
The genetic determinants responsible for endemic obesity remain largely undiscovered. Over 50 loci have been associated with BMI and/or categorical obesity in GWAS and meta-analyses,3,21,83 but the combined (additive) effects of all variants account for only a small fraction of the phenotypic variation (1%–2%), leaving most of the heritability unexplained. Possible sources of this missing heritability have been well discussed by Hebebrand et al25: nonspecific phenotypes, measurement error, small effects sizes, rare variants, inflated heritability estimates, developmental aspects, gene–gene and gene–environment interactions, to which we add common copy number variations (CNVs). While GWAS have not systematically targeted CNVs, a recent study of linkage disequilibrium between common single nucleotide variants (SNVs) and common CNVs concluded that “[…] for complex traits, the heritability void left by genome-wide association studies will not be accounted for by common CNVs.”84 While all of those sources of error no doubt contribute to the current heritability void, the evidence of segregation of obesity-related traits points to a predominant role of rare variants or a heterogenetic model of transmission. Under a heterogenetic model, estimates of heritability are not a useful way of combining evidence from gene-discovery studies, and it is more instructive to examine the effect sizes attributable to identified gene variants. Figure 2 illustrates the distribution of effect sizes (when reported in or readily transformable to BMI units, n=47); GWAS-identified results are extracted from recent reviews3,21,83 or the studies referred to therein. The sources of the effect sizes of other variants identified in various candidate designs are discussed in points “1. FTO” to “7. 16p11.2”. Compared to our estimates of the segregating effect size (4.1 kg/m2),18 most effects, including all identified in GWAS, are vanishingly small and can be ruled out as contributors to segregation, although they, and other similar but unknown loci, presumably contribute to the background variability. The exclusions should include FTO variants if based on effect size alone, but as we discuss in “1. FTO”, their emerging mechanism of action may provide a model for other larger effects in the same region. Also, most GWAS effects on BMI are so small that there can be no confidence that they represent effects on body fat as opposed nonfat compartments. That leaves only the outliers in Figure 2 as candidates for the types of variant that could contribute to a segregation signal.
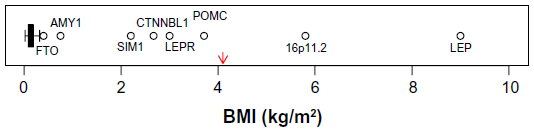 | Figure 2 Boxplot of published heterozygote effect sizes (when reported in or transformable to BMI units) of obesity-associated genetic variants (n=47). |
- FTO: common single-nucleotide polymorphisms (SNPs) in the FTO region have the largest effect size of any variants detected in GWAS, but that effect (0.4 kg/m2) is too small to make a significant contribution to the segregation signal that we and others detect. However, it may be that other rarer variants in the same region could contribute, and recent advances regarding the mechanisms of action of the FTO variants provide a model that exemplifies the potential of the NBH. Obesity-associated sequences within FTO are functionally connected through noncoding RNA with an increased expression of the homeobox gene IRX3, deletion of which results in reduced fat mass in mice.85 Studies in humans have found associations between obesity-associated FTO variants and levels of the satiety hormone leptin (negative),86 the hunger hormone ghrelin (positive),86,87 and brain responsivity to food cues (positive).87 So while common FTO SNPs do not contribute substantially to common obesity susceptibility, they do illustrate the potential of other rarer variants affecting the same processes.
- AMY1: CNVs in AMY1 encoding salivary amylase have been associated with variations in BMI (low copy number associated with higher BMI) in two independent samples.88 The effect size plotted in Figure 2 (0.75 kg/m2) is based on the maximum possible negative difference in copy number (−5) from the mean of the normal-weight distributions in Falchi et al.88 This effect size is small compared to our estimate,18 but could contribute to a segregation signal in larger scale studies. The CNV in AMY1 was investigated in relation to BMI after an initial screen of effects on gene expression in adipose tissue,88 but the salivary amylase product is also expressed in saliva under the influence of copy number89 and in various other tissues. The mechanisms linking AMY1 expression and BMI have not been established, but AMY1 is known to have a role in sensory perception of starch in foods as well as being regulated by autonomic nervous system (ANS) activity,90 providing potential links to and from hypothalamic appetite-regulating centers and therefore possible consistency with the NBH.
- SIM1: SIM1 is a homologue of a transcription factor in mice, which is known to be involved in the development of hypothalamic appetite regulation centers, and haplo-insufficiency results in a hyperphagic obesity in mice and humans. Common SNPs in and near SIM1 regions were associated with BMI in a candidate gene study in Pima Indians.91 The risk alleles were common in Pima Indians (~0.6), but less so in Europeans (~0.3) with no association with BMI in the European sample. Perhaps linked functional variants have originated in or become enriched (through genetic drift or selection) in Pima Indians. The potential mechanism is clear, and consistent with the NBH, and the effect size (2.2 kg/m2) would be detectable in a reasonably powered linkage study.
- CTNNBL1: multiple SNPs mainly in intronic regions of CTNNBL1 were associated with BMI and fat mass in a sample of unrelated US Caucasians and were supported in a French Caucasian case–control study of categorical obesity,92 but not in a central European sample93 or a Danish sample.94 Failure of replication is an expected feature of a heterogenetic model in which different populations or smaller groups (down to families) could be expected to have different genetic origins of obesity susceptibility. The effect size of 2.7 kg/m2 would be detectable in a reasonably powered linkage study, but there are reasons to doubt the specificity of the continuous phenotypes for body fatness. Liu et al92 used directly measured fat mass as a control on the BMI analyses, but did not control for effects of body size on fat mass, and so both phenotypes contain information about nonfat body compartments. Consistent with this potential confounding, the replication study by Andreasen et al94 found no association with BMI, but significant associations with weight and height. CTNNBL1 variants may be associated with increased risk of obesity, but the measured effect size on BMI includes an unknown but probably substantial contribution from genetic effects on body size. Confirmation using an unbiased phenotype is needed. The functions of CTNNBL1 are not established, and there is no clear potential link to the NBH, but it is expressed in at least one region of the human central nervous system (CNS) (cortex) and variants have been associated with measures of CNS function (memory).95
- POMC: POMC is a complex pro-peptide, the products of which are secreted by neurons that are critically involved in appetite regulation. Congenital deficiency of POMC is the cause of a rare form of monogenic obesity, and haplo-insufficiency of POMC is linked to increased BMI in affected families.96 The effects of POMC deficiency were important elements in the development of the NBH.23 The heterozygote effect size in Figure 2 (3.7 kg/m2) is an approximation based the original report in SD units (+1.3) multiplied by the average SD in males and females (2.85 kg/m2) in the reference data97 used by Farooqi et al.96
- LEP and LEPR: leptin acting through the leptin receptor functions as a feedback signal from adipose tissue fat stores with strong suppressive effects on appetite through the hypothalamic leptin–melanocortin signaling pathway. Both homozygous leptin and leptin receptor deficiencies cause rare monogenic severe obesity in humans driven by hyperphagia, and both are crucial elements in the development of the NBH. Heterozygous loss-of-function variants in both genes are associated with substantial effects on body fatness.98,99 In both cases, the phenotype affected was directly measured by body fat percent adjusted for age, sex, height, and weight, and the effects were substantial: +20% body fat for LEP98 and +6% for LEPR,99 which roughly translate to effects in BMI units in our sample18 of +9 and +3 kg/m2, respectively.
- 16p11.2: independent associations with obesity of large deletions at two locations in 16p11.2 have been detected following a strategy of resequencing at loci known to be associated with rare forms of extreme obesity,100,101 and one of the locations was replicated using continuous BMI in a population sample101 represented in Figure 2. The BMI effect size in Figure 2 is based on a very small sample (4) of heterozygous carriers in that one population, but the combined effect on BMI in all populations sampled was +1.1 SD units (n=8), similar to our effect size on Adiposity (+0.93 SD).18 The responsible causal loci are not clear, but the deleted region contains SH2B1, which has established links to hyperphagia and obesity in humans and animals acting through leptin signaling pathways102 consistent with the NBH.
As judged by effect size, only the POMC, LEP, and LEPR heterozygous loss-of-function mutations and the deletions at 16p11.2 stand out as possible examples of the types of variants which could contribute to a heterogenetic mechanism of susceptibility to common obesity, although contributions from the others cannot be excluded (except perhaps CTNNBL1). While we do not suppose that the outlier loci identified in Figure 2 are necessarily representative of all causative variants, it is noteworthy that their mean effect size (+3.9 kg/m2) is very close to our estimate of average effect size (+4.1 kg/m2).18 The gene products of the first three have key roles in appetite regulation, as does one candidate for the 16p11.2 effect, and as do other gene products identified as causative agents in severe monogenic obesity (PSK1, MC4R, SIM1).21 Other causes of severe (syndromic) obesity associated with generalized developmental CNS disturbances are also linked to disruption of appetite regulation (Bardet–Biedl syndrome, Prader–Willi syndrome, pseudohypoparathyroidism, WAGR syndrome).21 When taken together with our evidence of increased sensitivity to food-related cues in healthy family-history-positive individuals,24 the genomic evidence continues to point to hypothalamic appetite regulation pathways as the most likely location of rare variants driving the susceptibility to obesity in response to increased OE as proposed in the NBH. The effects of common variants in FTO illustrate how such mechanisms can operate, and have increased interest in the potential roles of noncoding RNAs.
The search for rare variants with large effects on obesity susceptibility is in progress and has resulted in some success,101 as have related approaches in other complex diseases.103,104 However, the contributions of the newly identified variants to population susceptibilities are very small, and if, as we conclude, most of the obesity susceptibility is due to rare variants, there are many more to find. There are formidable problems in design and statistical analysis to be solved before identification of rare variants becomes a routine procedure, and we may currently be able to find only the “low-hanging fruit” that are amenable to current techniques.101 The variants’ effects must be large, and it might therefore be hoped that simple phenotypes such as BMI would be adequate for reliable discovery, more so than in GWAS where the small effect sizes must be interpreted with caution due to biases in BMI. However, even large effects can be generated or obscured by BMI bias; measured biases in different ethnic groups range from −6 kg/m2 to +4.5 kg/m2 at equivalent body fat percentage105,106 and in our ethnically unselected sample from −3.6 kg/m2 to +6.7 kg/m2 (95% CI −3.1, 4.3) at equivalent Adiposity (Figure 1C).18 There is a clear need for more informative phenotypes in these studies.
Implications of a heterogenetic mechanism for gene discovery, treatment, and prevention
GWAS studies have reached their potential
The results obtained from GWAS studies have excluded a major causative role for common genetic variants in obesity, directly for common SNPs and indirectly for common CNVs.84 The GWAS design does not have the power to detect rare causative variants in any feasible sample size. Power and accuracy would undoubtedly be improved by the use of better phenotypes than BMI, but it is unlikely that the improvements would be enough to capture many rare variants. The increased costs associated with better phenotyping would be better used in smaller scale studies following the types of approaches recommended by Walters et al.101
Many variants may produce similar effects on obesity-related traits
Under a heterogenetic model, the separation between modes of Adiposity in Figure 1A represents the average effect size of the causative variants. Our analysis treats it as a fixed (ie, constant) effect, and while we were unable to model the range of dispersion of effects, nor any heterogeneity of background variability, the data are consistent with a very narrow range of effect sizes superimposed on a constant background variability. It might be thought implausible that many heterogenetic variants could all result in similar effects sizes, but it is possible for that pattern to arise from the properties of the affected physiological system(s). For example, the NBH proposes that appetite regulation systems in the hypothalamus are the target of most obesogenic variants; those systems are very complex involving a large number of sensory, effector, and integrative processes, all interacting with and subject to feedback from other central regulatory systems such as the ANS, and affected by systems modulating mood and other behavioral traits. The overall properties of this system, or network, of sensors, integrators, and effectors are not clearly understood, but it is likely that it exhibits features of redundancy in that a complete or partial failure of any component is partly compensated for by the remaining intact links (an analogy can be found in the properties of the glucoregulatory system107). Such a system could produce the segregation behavior that we and others observe.
Can phenotypic convergence help to locate new therapeutic targets?
Under some possible structures of the physiological system(s) involved, there could be clustering of the effects of multiple variants on potentially targetable subsystems. However, appetite regulation is such a central process with links to and from many other systems (ANS, mood, etc) that it makes specific targeting without undesirable side effects potentially very difficult, as can been seen in the history of drug development in this area.40 Recent interest in the melanocortin-4 receptor as a potentially targetable component of the NBH appears to be an illustration of continuing problems of this nature. Antagonist compounds to melanocortin-4 receptor are being developed for targeting depression and anxiety,108 while agonists are being developed to target obesity.109 This is reminiscent of the history of Rimonabant, a drug developed to target obesity though antagonism of cannabinoid receptors in satiety pathways but which was withdrawn due to side effects on depression.40 An improved understanding of the properties of the appetite-regulating system as a control system could affect strategies for targeting. If, for example, the system is strongly hierarchical with lateral connections to other systems such as the ANS mainly at high levels in the hierarchy, that could be quite different in its implications from a more distributed network-like structure with many lateral connections. If a hierarchical structure could be defined, there may be obvious potential targets at the mid-level in the system, below the lateral links. A network structure presents a much more challenging problem.
Similarities and differences in obesity susceptibility in populations
Obesity and overweight vary between populations due to effects of and interactions between genetic and environmental factors. Some small genetically isolated populations have developed very high prevalences of obesity in response to recent environmental changes (eg, Pima Indians,110 Nauruans,111 Australian Aboriginals112). There are also marked, though usually less extreme, differences in obesity prevalences between large national populations,113 which, as discussed earlier, can be assigned mainly to the effects of OE on genetically susceptible individuals. The genetic determinants of the variation in susceptibility between individuals or groups are not clear, except for a very small number of individuals with severe monogenic obesity. There are currently no known genetic variants or combinations of variants that are helpful as indicators of risk, in either individuals or populations. Genetic risk scores calculated by adding known obesogenic variant loads have minimal predictive power.114 A very rough calculation based on current estimates of effect sizes (Figure 2) indicates that approximately 40 different variants per individual would be needed to account for obesity risk using a multiplicative (epistatic) model.
The origin of the undefined heterogenetic variation responsible for the varying phenotypic responses to OE is also unclear. Plausible theories include the effects of positive or negative selection, or of genetic drift, the pros and cons of which have been extensively discussed115–117 without resolution. A strong case can be made for selection, which brings together many features of obesity.117 The need for Homo sapiens to supply adequate glucose for a large brain to metabolize in recurrent famine situations and for the female to store sufficient fat to carry a pregnancy successfully and feed the baby could have ensured selection of genes that prioritized high-energy nutrients in the sparse environments and encouraged rapid ingestion and fat storage. It would be interesting to know whether the CNV in the salivary amylase gene (AMY1), which affects detection of starch and associates with higher BMI, has been subject to pre-agricultural selection. Parallel selection for rapid immune and stress responsiveness is also possible.117 Those “survivor” genes selected as a result of responsiveness to environmental nutrient cues could, with current OE, be predicted to increase the prevalence of obesity. However, there is at present very little direct evidence from genomic studies which bears on this question. A few studies report signals of selection at obesity susceptibility loci identified in GWAS118,119 but no clear pattern has yet emerged. If however, as we conclude, causative obesogenic variants are rare, unknown, and not captured in GWAS, the detected selection signals have marginal relevance to endemic obesity and we do not yet have the genomic data to test selection hypotheses.
Opportunity for more targeted prevention and customized treatment of obesity
The results of genomic studies to date have contributed very little, if anything of immediate relevance to the prevention and treatment of common obesity and its associated disorders. This contrasts, in our view, with the obvious and immediate potential implications of the well-established genetic basis for the disorder, which have not yet been exploited in the clinical or public health arenas. It seems to us that the failure, to date, to explain obesity susceptibility by genomic studies is, in some quarters, being interpreted incorrectly as questioning the overall genetic basis of the disorder. It may be that accumulation of causal variants would therefore amplify the genetic message, but the time when risk information from genomic data would be more informative than, say, family history appears to be a long way off. Similarly, we can find no evidence that known genomic markers are able to predict responses to currently available treatments. Again, this contrasts with the potential for known nongenomic risk factors, like family history, to predict the need for and responses to interventions.120
In the future, it is probable that information from genomic and physiological studies will identify new therapeutic targets that ideally would avoid the side effects responsible for the recurrent failure of drug development.40 In the short term, discovery of genomic markers that would help to identify subsets of patients who would either respond to currently available treatments or who are at greater or lesser risk of the common side effects or comorbidities of obesity would be an advance. A recent analysis found that the benefit of reduced availability of junk food in schools was restricted to children with a family history of overweight.120 This highlights one of the potential benefits of genetic risk classification of individuals, but there is much to do before genomic information provides better risk prediction than do currently available markers such as family history. A family history of T2DM may be a particularly powerful discriminator as it identifies a group with history of obesity with pathogenic consequences.18,24
There is strong desire in many quarters to intervene early, in children or in utero, in part because childhood obesity is a strong predictor of adult obesity and its consequences. As we now understand, this relationship reflects the shared genetic makeup of child and adult and is unlikely to be causative. The drive for aggressive early interventions ought to be diminished by the knowledge of the genetic basis of the disorder and by the lack of effectiveness of past interventions and the potential adverse consequences of interventions.49,50
Implications for enhanced patient care such as quality of life
An acceptance by science, medicine, and the public that common obesity is strongly heritable would in itself be a therapeutic advance, which should lead to significant changes to current approaches:
- Development of a more sympathetic approach to current patient management by clinicians who would then accept that genetic predisposition to susceptibility to the current OE underlies the repeated failures of “will-power”.121
- Lessening of repetitive weight loss attempts undertaken despite the inevitable regain and the personal sense of failure that this entails.
- Early life intervention should be undertaken only where evidence of both benefit and lack of harm has been demonstrated in studies. There is a real possibility of permanent mental and physical harm if useless interventions are repeated in ever younger populations.49,50
- Similarly, public health interventions to prevent obesity that are unsuccessful must be altered or abandoned. The public influences on obesity are mainly in the domains of economics and marketing, not in modifiable personal behaviors. Investment in ineffective behavioral messages targeting obesity is futile and serves only to maintain a false picture of the problem ignoring the genetic element, and thereby maintaining the dominant public51 and clinical121 view of obesity as a personal failing.
Studies to define the condition of healthy obesity and its maintenance will allow less intervention, and hence increased quality of life, if no adverse associations or health conditions are found. There are no established protective markers yet, but a recent report of a low-frequency (1.5%) variant in the Cyclin D2 gene, which increases body mass but decreases diabetes risk, may be an encouraging sign122; 1.5% could represent a useful proportion of the healthy obese (~25%).
Conclusion
As genomic naïves, it appears to us that in the immediate future we must aim or hope for two things:
- Identification of points of convergence in phenotypic pathways by better measurement and assessment of collateral links to other physiological systems. We should not aim to personalize treatments to an individual gene, which seems very unrealistic in this domain, and should be targeting the effects of groups of genes by satiety induction.
- Identification, discrimination, and quantitation of obesogenic environmental factors and understanding how they interact with obesogenic genetic variants to increase their effect. There may be opportunities to counteract personal exposures to elements of OE or to identify more realistic targets for public health interventions.
We believe that genomic studies, which have helped to define the genetic basis of common obesity mainly by exclusion, will in the future play an increasingly important role in the understanding and management of obesity, but not without parallel studies of the physiological, behavioral, and economic influences, and especially not without the use of more informative phenotypes than BMI.
Disclosure
The authors report no conflicts of interest in this work.
References
Wolf AM, Colditz GA. Current estimates of the economic cost of obesity in the United States. Obes Res. 1998;6(2):97–106. | |
Swinburn BA, Sacks G, Hall KD, et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378(9793):804–814. | |
El-Sayed Moustafa JS, Froguel P. From obesity genetics to the future of personalized obesity therapy. Nat Rev Endocrinol. 2013;9(7):402–413. | |
O’Rahilly S, Farooqi IS. Human obesity as a heritable disorder of the central control of energy balance. Int J Obes. 2008;32(Suppl 7):55–61. | |
Baumgartner RN, Heymsfield SB, Roche AF. Human body composition and the epidemiology of chronic disease. Obes Res. 1995;3(1):73–95. | |
Muller MJ, Bosy-Westphal A, Krawczak M. Genetic studies of common types of obesity: a critique of the current use of phenotypes. Obes Rev. 2010;11(8):612–618. | |
Okorodudu DO, Jumean MF, Montori VM, et al. Diagnostic performance of body mass index to identify obesity as defined by body adiposity: a systematic review and meta-analysis. Int J Obes. 2010;34(5):791–799. | |
Molarius A, Seidell JC. Selection of anthropometric indicators for classification of abdominal fatness – a critical review. Int J Obes. 1998;22(8):719–727. | |
Luke A, Cooper RS. Physical activity does not influence obesity risk: time to clarify the public health message. Int J Epidemiol. 2013;42(6):1831–1836. | |
Jain A. Treating obesity in individuals and populations. BMJ. 2005;331(7529):1387–1390. | |
Hill JO. Can a small-changes approach help address the obesity epidemic? Am J Clin Nutr. 2009;89(2):477–484. | |
Bray GA, Popkin BM. Dietary sugar and body weight: have we reached a crisis in the epidemic of obesity and diabetes? Health be damned! Pour on the sugar. Diabetes Care. 2014;37(4):950–956. | |
Dombrowski SU, Knittle K, Avenell A, Arajo-Soares V, Sniehotta F. Long term maintenance of weight loss with non-surgical interventions in obese adults: systematic review and meta-analyses of randomised controlled trials. BMJ. 2014;348:g2646. | |
Gray LJ, Cooper N, Dunkley A, et al. A systematic review and mixed treatment comparison of pharmacological interventions for the treatment of obesity. Obes Rev. 2012;13(6):483–498. | |
Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA. 2014;311(1):74–86. | |
Picot J, Jones J, Colquitt JL, Loveman E, Clegg AJ. Weight loss surgery for mild to moderate obesity: a systematic review and economic evaluation. Obes Surg. 2012;22(9):1496–1506. | |
Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322(21):1483–1487. | |
Jenkins AB, Batterham M, Samocha-Bonet D, Tonks K, Greenfield JR, Campbell LV. Segregation of a latent high adiposity phenotype in families with a history of type 2 diabetes mellitus implicates rare obesity-susceptibility genetic variants with large effects in diabetes-related obesity. PLoS One. 2013;8:e70435. | |
Price RA, Charles MA, Pettitt DJ, Knowler WC. Obesity in Pima Indians: genetic segregation analyses of body mass index complicated by temporal increases in obesity. Hum Biol. 1994;66(2):251–274. | |
Tayo BO, Harders R, Luke A, Zhu X, Cooper RS. Latent common genetic components of obesity traits. Int J Obes. 2008;32(12):1799–1806. | |
O’Rahilly S. Human genetics illuminates the paths to metabolic disease. Nature. 2009;462(7271):307–314. | |
Saunders CL, Chiodini BD, Sham P, et al. Meta-analysis of genome-wide linkage studies in BMI and obesity. Obesity. 2007;15(9):2263–2275. | |
O’Rahilly S, Farooqi IS. Human obesity: a heritable neurobehavioral disorder that is highly sensitive to environmental conditions. Diabetes. 2008;57(11):2905–2910. | |
Samocha-Bonet D, Campbell LV, Viardot A, et al. A family history of type 2 diabetes increases risk factors associated with overfeeding. Diabetologia. 2010;53:1700–1708. | |
Hebebrand J, Volckmar AL, Knoll N, Hinney A. Chipping away the ‘missing heritability’: GIANT steps forward in the molecular elucidation of obesity – but still lots to go. Obes Facts. 2010;3(5):294–303. | |
Chen G, Adeyemo AA, Johnson T, et al. A genome-wide scan for quantitative trait loci linked to obesity phenotypes among West Africans. Int J Obes. 2005;29(3):255–259. | |
Pérusse L, Rice T, Chagnon YC, et al. A genome-wide scan for abdominal fat assessed by computed tomography in the Quebec Family Study. Diabetes. 2001;50(3):614–621. | |
Dehghan M, Merchant AT. Is bioelectrical impedance accurate for use in large epidemiological studies? Nutr J. 2008;7:26. | |
Bohm A, Heitmann BL. The use of bioelectrical impedance analysis for body composition in epidemiological studies. Eur J Clin Nutr. 2013;67(Suppl 1):79–85. | |
Ho-Pham LT, Campbell LV, Nguyen TV. More on body fat cutoff points. Mayo Clin Proc. 2011;86(6):584. | |
Jenkins AB, Samaras K, Gordon M, Snieder H, Spector T, Campbell LV. Lack of heritability of circulating leptin concentration in humans after adjustment for body size and adiposity using a physiological approach. Int J Obes Relat Metab Disord. 2001;25:1625–1632. | |
Schmitz S, Cherny SS, Fulker DW. Increase in power through multivariate analyses. Behav Genet. 1998;28(5):357–363. | |
Allison DB, Thiel B, St Jean P, Elston RC, Infante MC, Schork NJ. Multiple phenotype modeling in gene-mapping studies of quantitative traits: power advantages. Am J Hum Genet. 1998;63(4):1190–1201. | |
He LN, Liu YJ, Xiao P, et al. Genomewide linkage scan for combined obesity phenotypes using principal component analysis. Ann Hum Genet. 2008;72(pt 3):319–326. | |
Samocha-Bonet D, Chisholm DJ, Tonks K, Campbell LV, Greenfield JR. Insulin-sensitive obesity in humans – a ‘favorable fat’ phenotype? Trends Endocrinol Metab. 2012;23(3):116–124. | |
Samaras K, Nguyen TV, Jenkins AB, Kelly PJ, Campbell LV. Clustering of insulin resistance, total and central abdominal fat: same genes or same environment? Twin Res. 1999;2:218–225. | |
Malis C, Rasmussen EL, Poulsen P, et al. Total and regional fat distribution is strongly influenced by genetic factors in young and elderly twins. Obes Res. 2005;13(12):2139–2145. | |
Look AHEAD Research Group; Wadden TA, Bantle JP, Blackburn G, et al. Eight-year weight losses with an intensive lifestyle intervention: the look AHEAD study. Obesity. 2014;22(1):5–13. | |
Sumithran P, Prendergast LA, Delbridge E, et al. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011;365(17):1597–1604. | |
George M, Rajaram M, Shanmugam E. New and emerging drug molecules against obesity. J Cardiovasc Pharmacol Ther. 2014;19(1):65–76. | |
Woloshin S, Schwartz LM. The new weight-loss drugs, lorcaserin and phentermine-topiramate: slim pickings? JAMA Intern Med. 2014; 174(4):615–619. | |
Joharapurkar AA, Dhanesha NA, Jain MR. Inhibition of the methionine aminopeptidase 2 enzyme for the treatment of obesity. Diabetes Metab Syndr Obes. 2014;7:73. | |
Ionut V, Burch M, Youdim A, Bergman RN. Gastrointestinal hormones and bariatric surgery-induced weight loss. Obesity. 2013;21(6):1093–1103. | |
O’Brien PE, MacDonald L, Anderson M, Brennan L, Brown WA. Long-term outcomes after bariatric surgery: fifteen-year follow-up of adjustable gastric banding and a systematic review of the bariatric surgical literature. Ann Surg. 2013;257(1):87–94. | |
Harrison D. Overweight people could pay more for health insurance: NIB Chief Mark Fitzgibbon. Sydney Morning Herald. May 5, 2014. | |
Pratt CA, Boyington J, Esposito L, et al. Childhood obesity prevention and treatment research (COPTR): interventions addressing multiple influences in childhood and adolescent obesity. Contemp Clin Trials. 2013;36(2):406–413. | |
Small L, Bonds-McClain D, Melnyk B, Vaughan L, Gannon AM. The preliminary effects of a primary care-based randomized treatment trial with overweight and obese young children and their parents. J Pediatr Health Care. 2014;28(3):198–207. | |
Wake M, Lycett K, Clifford SA, et al. Shared care obesity management in 3–10 year old children: 12 month outcomes of HopSCOTCH randomised trial. BMJ. 2013;346:3092. | |
Richardson SS, Daniels CR, Gillman MW, et al. Don’t blame the mothers. Nature. 2014;512:131–132. | |
Schwartz MB, Puhl R. Childhood obesity: a societal problem to solve. Obes Rev. 2003;4(1):57–71. | |
Jou C. The biology and genetics of obesity – a century of inquiries. N Engl J Med. 2014;370(20):1874–1877. | |
Rokholm B, Baker JL, Sorensen TI. The levelling off of the obesity epidemic since the year 1999 – a review of evidence and perspectives. Obes Rev. 2010;11(12):835–846. | |
Olds T, Maher C, Zumin S, et al. Evidence that the prevalence of childhood overweight is plateauing: data from nine countries. Int J Pediatr Obes. 2011;6(5–6):342–360. | |
McLaren L. Socioeconomic status and obesity. Epidemiol Rev. 2007;29:29–48. | |
Sobal J, Stunkard AJ. Socioeconomic status and obesity: a review of the literature. Psychol Bull. 1989;105(2):260–275. | |
Egger G, Swinburn B, Amirul Islam FM. Economic growth and obesity: an interesting relationship with world-wide implications. Econ Hum Biol. 2012;10(2):147–153. | |
Ezzati M, Vander Hoorn S, Lawes CM, et al. Rethinking the “diseases of affluence” paradigm: global patterns of nutritional risks in relation to economic development. PLoS Med. 2005;2(5):e133. | |
Hruschka DJ, Brewis AA. Absolute wealth and world region strongly predict overweight among women (ages 18–49) in 360 populations across 36 developing countries. Econ Hum Biol. 2013;11(3):337–344. | |
Hruschka DJ. Do economic constraints on food choice make people fat? A critical review of two hypotheses for the poverty-obesity paradox. Am J Hum Biol. 2012;24(3):277–285. | |
Su D, Esqueda OA, Li L, Pagan JA. Income inequality and obesity prevalence among OECD countries. J Biosoc Sci. 2012;44(4):417–432. | |
Zhang Q, Wang Y. Trends in the association between obesity and socioeconomic status in US adults: 1971 to 2000. Obes Res. 2004;12(10):1622–1632. | |
Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. | |
Gluckman PD, Hanson MA, Buklijas T. A conceptual framework for the developmental origins of health and disease. J Dev Orig Health Dis. 2010;1(01):6–18. | |
Lavebratt C, Almgren M, Ekstrom TJ. Epigenetic regulation in obesity. Int J Obes. 2012;36(6):757–765. | |
Slomko H, Heo HJ, Einstein FH. Epigenetics of obesity and diabetes in humans. Endocrinology. 2012;153(3):1025–1030. | |
Stevens A, Begum G, White A. Epigenetic changes in the hypothalamic pro-opiomelanocortin gene: a mechanism linking maternal undernutrition to obesity in the offspring? Eur J Pharmacol. 2011;660(1):194–201. | |
Xu X, Su S, Barnes VA, et al. A genome-wide methylation study on obesity: differential variability and differential methylation. Epigenetics. 2013;8(5):522–533. | |
Smith J, Cianflone K, Biron S, et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metabol. 2009;94(11):4275–4283. | |
Guénard F, Deshaies Y, Cianflone K, Kral JG, Marceau P, Vohl M-C. Differential methylation in glucoregulatory genes of offspring born before vs after maternal gastrointestinal bypass surgery. Proc Natl Acad Sci U S A. 2013;110(28):11439–11444. | |
Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition. 2010;26(11–12):1031–1037. | |
Willmer M, Berglind D, Sorensen TIA, Naeslund E, Tynelius P, Rasmussen F. Surgically induced interpregnancy weight loss and prevalence of overweight and obesity in offspring. PLoS One. 2013;8(12):e82247. | |
Lawlor DA, Timpson NJ, Harbord RM, et al. Exploring the developmental overnutrition hypothesis using parental-offspring associations and FTO as an instrumental variable. PLoS Med. 2008;5(3):33. | |
Veena SR, Krishnaveni GV, Karat SC, Osmond C, Fall CH. Testing the fetal overnutrition hypothesis; the relationship of maternal and paternal adiposity to adiposity, insulin resistance and cardiovascular risk factors in Indian children. Public Health Nutr. 2013;16(9):1656–1666. | |
Rokholm B, Silventoinen K, Angquist L, Skytthe A, Kyvik KO, Sorensen TI. Increased genetic variance of BMI with a higher prevalence of obesity. PLoS One. 2011;6(6):20816. | |
Jiao H, Arner P, Dickson SL, et al. Genetic association and gene expression analysis identify FGFR1 as a new susceptibility gene for human obesity. J Clin Endocrinol Metabol. 2011;96(6):962–966. | |
Comuzzie AG, Blangero J, Mahaney MC, et al. Major gene with sex-specific effects influences fat mass in Mexican Americans. Genet Epidemiol. 1995;12(5):475–488. | |
Hasstedt SJ, Hoffman M, Leppert MF, Elbein SC. Recessive inheritance of obesity in familial non-insulin-dependent diabetes mellitus, and lack of linkage to nine candidate genes. Am J Hum Genet. 1997;61(3):668–677. | |
Hemani G, Yang J, Vinkhuyzen A, et al. Inference of the genetic architecture underlying BMI and height with the use of 20,240 sibling pairs. Am J Hum Genet. 2013;93(5):865–875. | |
Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425. | |
Lecomte E, Herbeth B, Nicaud V, Rakotovao R, Artur Y, Tiret L. Segregation analysis of fat mass and fat-free mass with age- and sex-dependent effects: the Stanislas Family Study. Genet Epidemiol. 1997;14(1):51–62. | |
Rice T, Borecki IB, Bouchard C, Rao DC. Segregation analysis of fat mass and other body composition measures derived from underwater weighing. Am J Hum Genet. 1993;52(5):967–973. | |
Beunen G, Maes HH, Vlietinck R, et al. Univariate and multivariate genetic analysis of subcutaneous fatness and fat distribution in early adolescence. Behav Genet. 1998;28(4):279–288. | |
Choquet H, Meyre D. Molecular basis of obesity: current status and future prospects. Curr Genomics. 2011;12(3):154–168. | |
Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712. | |
Smemo S, Tena JJ, Kim KH, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–375. | |
Benedict C, Axelsson T, Söderberg S, et al. The fat mass and obesity-associated gene (FTO) is linked to higher plasma levels of the hunger hormone ghrelin and lower serum levels of the satiety hormone leptin in older adults. Diabetes. 2014;63(11):3955–3959. | |
Karra E, O’Daly OG, Choudhury AI, et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest. 2013;123(8):3539–3551. | |
Falchi M, El-Sayed Moustafa JS, Takousis P, et al. Low copy number of the salivary amylase gene predisposes to obesity. Nat Genet. 2014;46(5):492–497. | |
Perry GH, Dominy NJ, Claw KG, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39(10):1256–1260. | |
Santos JL, Saus E, Smalley SV, et al. Copy number polymorphism of the salivary amylase gene: implications in human nutrition research. J Nutrigenet Nutrigenomics. 2012;5(3):117–131. | |
Traurig M, Mack J, Hanson RL, et al. Common variation in SIM1 is reproducibly associated with BMI in Pima Indians. Diabetes. 2009; 58(7):1682–1689. | |
Liu YJ, Liu XG, Wang L, et al. Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum Mol Genet. 2008;17(12):1803–1813. | |
Vogel CI, Greene B, Scherag A, et al. Non-replication of an association of CTNNBL1 polymorphisms and obesity in a population of Central European ancestry. BMC Med Genet. 2009;10:14. | |
Andreasen CH, Mogensen MS, Borch-Johnsen K, et al. Studies of CTNNBL1 and FDFT1 variants and measures of obesity: analyses of quantitative traits and case-control studies in 18,014 Danes. BMC Med Genet. 2009;10:17. | |
Papassotiropoulos A, Stefanova E, Vogler C, et al. A genome-wide survey and functional brain imaging study identify CTNNBL1 as a memory-related gene. Mol Psychiatry. 2013;18(2):255–263. | |
Farooqi IS, Drop S, Clements A, et al. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. 2006;55(9):2549–2553. | |
Cole TJ, Freeman JV, Preece MA. Body mass index reference curves for the UK, 1990. Arch Dis Child. 1995;73(1):25–29. | |
Farooqi IS, Keogh JM, Kamath S, et al. Partial leptin deficiency and human adiposity. Nature. 2001;414(6859):34–35. | |
Farooqi IS, Wangensteen T, Collins S, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356(3):237–247. | |
Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463(7281):671–675. | |
Walters RG, Coin LJ, Ruokonen A, et al. Rare genomic structural variants in complex disease: lessons from the replication of associations with obesity. PLoS One. 2013;8(3):58048. | |
Doche ME, Bochukova EG, Su HW, et al. Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J Clin Invest. 2012;122(12):4732–4736. | |
Johansen CT, Wang J, Lanktree MB, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42(8):684–687. | |
Momozawa Y, Mni M, Nakamura K, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43(1):43–47. | |
Deurenberg P, Yap M, Van Staveren WA. Body mass index and percent body fat: a meta analysis among different ethnic groups. Int J Obes. 1998;22:1164–1171. | |
Rush EC, Freitas I, Plank LD. Body size, body composition and fat distribution: comparative analysis of European, Maori, Pacific Island and Asian Indian adults. Br J Nutr. 2009;102(4):632–641. | |
Boyle PJ, Zrebiec J. Physiological and behavioral aspects of glycemic control and hypoglycemia in diabetes. South Med J. 2007;100(2):175–182. | |
Chaki S, Okubo T. Melanocortin-4 receptor antagonists for the treatment of depression and anxiety disorders. Curr Top Med Chem. 2007;7(11):1145–1151. | |
Jeon MK, Cheon HG. Promising strategies for obesity pharmacotherapy: melanocortin-4 (MC-4) receptor agonists and melanin concentrating hormone (MCH) receptor-1 antagonists. Curr Top Med Chem. 2009;9(6):504–538. | |
Valencia ME, Bennett PH, Ravussin E, Esparza J, Fox C, Schulz LO. The Pima Indians in Sonora, Mexico. Nutr Rev. 1999;57(5 pt 2):55–57. | |
Zimmet P, Arblaster M, Thoma K. The effect of westernization on native populations. Studies on a Micronesian community with a high diabetes prevalence. Aust N Z J Med. 1978;8(2):141–146. | |
O’Dea K. Westernisation, insulin resistance and diabetes in Australian aborigines. Med J Aust. 1991;155(4):258–264. | |
Finucane MM, Stevens GA, Cowan MJ, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet. 2011;377(9765):557–567. | |
Li S, Zhao JH, Luan J, et al. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am J Clin Nutr. 2010;91(1):184–190. | |
Prentice AM, Hennig BJ, Fulford AJ. Evolutionary origins of the obesity epidemic: natural selection of thrifty genes or genetic drift following predation release? Int J Obes. 2008;32(11):1607–1610. | |
Speakman JR. Evolutionary perspectives on the obesity epidemic: adaptive, maladaptive, and neutral viewpoints. Annu Rev Nutr. 2013;33:289–317. | |
Campbell LV. The thrifty gene hypothesis: maybe everyone is right? Int J Obes. 2008;32(4):723–724. | |
Klimentidis YC, Abrams M, Wang J, Fernandez JR, Allison DB. Natural selection at genomic regions associated with obesity and type-2 diabetes: East Asians and sub-Saharan Africans exhibit high levels of differentiation at type-2 diabetes regions. Hum Genet. 2011;129(4):407–418. | |
Southam L, Soranzo N, Montgomery SB, et al. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes- and obesity-susceptibility variants? Diabetologia. 2009;52(9):1846–1851. | |
Anderson PM, Butcher KF. Reading, writing and refreshments: are school finances contributing to children’s obesity? J Hum Resour. 2006;41(3):467–494. | |
Majdan JF. Memoirs of an obese physician. Ann Intern Med. 2010; 153(10):686–687. | |
Steinthorsdottir V, Thorleifsson G, Sulem P, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46(3):294–298. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.